")
Back to Journals » International Journal of Nanomedicine » Volume 12
Co-delivery of paclitaxel and TOS-cisplatin via TAT-targeted solid lipid nanoparticles with synergistic antitumor activity against cervical cancer
Authors Liu B, Han L, Liu J, Han S, Chen Z, Jiang L
Received 16 June 2016
Accepted for publication 1 September 2016
Published 31 January 2017 Volume 2017:12 Pages 955—968
DOI https://doi.org/10.2147/IJN.S115136
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Linlin Sun
Bo Liu, Li Han, Junyan Liu, Shumei Han, Zhen Chen, Lixi Jiang
Department of Internal Medicine Oncology, Shandong Cancer Hospital Affiliated to Shandong University, Shandong Academy of Medical Science, Ji’nan, People’s Republic of China
Background: Cervical cancer is a major world health problem for women. Currently, cancer research focuses on improving therapy for cervical cancer using various treatment options such as co-delivery of chemotherapeutic agents by nanocarriers.
Purpose: The aim of this study was to develop trans-activating transcriptional activator (TAT)-modified solid lipid nanoparticles (SLNs) for co-delivery of paclitaxel (PTX) and α-tocopherol succinate-cisplatin prodrug (TOS-CDDP) (TAT PTX/TOS-CDDP SLNs) in order to achieve synergistic antitumor activity against cervical cancer.
Methods: Lipid prodrug of CDDP (TOS-CDDP) and TAT-containing polyethylene glycol-distearoyl-phosphatidylethanolamine (TAT-PEG-DSPE) were synthesized. TAT PTX/TOS-CDDP SLNs were prepared by emulsification and solvent evaporation method. Physicochemical characteristics of SLNs such as size, morphology, and release profiles were explored. In vitro and in vivo studies were carried out to assess the efficacy of their antitumor activity in target cells.
Results: TAT PTX/TOS-CDDP SLNs could be successfully internalized by HeLa cells and showed a synergistic effect in the suppression of cervical tumor cell growth. They exhibited high tumor tissue accumulation, superior antitumor efficiency, and much lower toxicity in vivo.
Conclusion: The present study indicates that the co-delivery system provides a promising platform as a combination therapy for the treatment of cervical cancer, and possibly other types of cancer as well.
Keywords: cervical cancer, lipid-based prodrug, combination therapy, solid lipid nanoparticles, cell-penetrating peptide
Introduction
Cervical cancer is a major world health problem for women and is the fourth most common cancer in women worldwide, with 85% of cases occurring in developing countries.1,2 The global yearly incidence of cervical cancer in 2012 was 528,000, and the annual death rate was 266,000.3 The treatment of cervical cancer is represented by radiotherapy or surgery for early-stage disease and concurrent chemoradiation for advanced-stage patients. However, these conventional treatment strategies are highly aggressive or nonspecific and often accompanied by serious side effects.4 Currently, cancer research focuses on improving therapy for cervical cancer using various treatment options such as co-delivery of chemotherapeutic agents by nanocarriers, gene therapy, recombinant protein therapy, etc.5–7
Platinum-based combination chemotherapy regimen such as cisplatin/paclitaxel (CDDP/PTX) or carboplatin/PTX has been considered as the first-line therapy for cervical cancer.8–10 A randomized phase III study compared CDDP/PTX regimen with cisplatin alone in 264 women with metastatic, recurrent, or persistent cervical cancer. Results showed that CDDP/PTX regimen had a higher response rate (36% vs 19%) and improved progression-free survival (PFS).11 Another phase III study compared four cisplatin-doublet regimens (CDDP/PTX, CDDP/topotecan, CDDP/gemcitable, and CDDP/vinorelbine); results suggest that CDDP/PTX is superior to the other regimens.8 However, it is difficult to combine free drugs to obtain optimal antitumor effect due to their different biochemical properties and pharmacokinetics.12,13 Thus, there is a call for developing a drug delivery system that could optimize the synergistic anticancer effects while reducing toxic side effects. Nanotechnology-based combination drug delivery to tumor tissues has emerged as an effective strategy. Most nanotechnology-based combination drug delivery systems comprise drug compounds with similar water solubility.13 It is necessary to engineer a nano-sized platform to co-delivery of a hydrophobic drug (PTX) and a water- and oil-insoluble drug (CDDP). Also, a prodrug is formed to avoid a rapid escape of the water-soluble drug from the carrier.14,15
Lipid prodrug strategy has received considerable attention in the field of nanoparticulate systems.16,17 Lipid-based prodrug nanocarriers have several advantages, such as balancing the pharmacokinetics of drugs, higher drug loading and stability in vivo, good biocompatibility, controllable drug release, etc.18,19 A lipid prodrug of PTX has been designed by conjugation to squalene.15 Results showed that PTX lipid-based prodrug nanoparticles (NPs) exhibited the following features: high drug loading (23.5 wt.% PTX loading vs 10% into liposomes and 0.98% into polymer NPs), no “burst release” of the drug, more effective than the free drug, and higher maximal tolerated doses in animal models. In this study, we designed a novel CDDP lipid prodrug, α-tocopherol succinate-cisplatin prodrug (TOS-CDDP).
TOS, a succinic acid ester of α-tocopherol, is the most effective form of vitamin E derivatives exhibiting nontoxic and biocompatible characteristics. This compound has been found to inhibit the proliferation of various cancer cells in vitro and in vivo,20–26 including cervical cancer cells, breast cancer cells, prostate cancer cells, gastric cancer cells, and so on. TOS, a mitochondria-targeting drug, has the synergic effect on nuclear DNA-damaging drugs such as CDDP.25 The dual drug conjugates (TOS-CDDP) were anticipated to improve the lipophilic property, enable easy encapsulation into lipid NPs, and improve their in vivo stability.
Lipid-based nanocarriers have been shown to have great potential for co-delivery of multidrugs.27–29 They display distinctive features such as high drug loading, capability for modifications and conjugations, and so forth.27–29 Solid lipid nanoparticles (SLNs) were established as alternative lipid-based nanocarriers such as emulsions, micelles, liposomes, and polymeric NPs. SLNs are composed of 0.1%–30% (w/w) lipids dispersed in an aqueous solution of 0.5%–5.0% (w/w) surfactant as stabilizing agent.30 SLNs provide the following advantages: physical stability, low cytotoxicity, protection of the incorporated drug from degradation, and controlled release.31
Targeted drug delivery, which can carry drugs to specific organs or tissues, is another highly desirable treatment technique in cancer nanotechnology.32–34 The cell-penetrating peptide (CPP) trans-activating transcriptional activator (TAT) is commonly used in payload delivery into cells both in the natural L-configuration and as a D-amino acid peptide.35 The improved activity and penetration of drugs delivered with TAT can be made use of to improve the efficacy of the standard drug dose, attenuate side effect, and overcome drug resistance.36,37
The aim of this study was to develop TAT-modified SLNs for co-delivery of PTX and TOS-CDDP (TAT PTX/TOS-CDDP SLNs) in order to achieve synergistic antitumor activity against cervical cancer. Lipid prodrug of CDDP (TOS-CDDP) and TAT-containing polyethylene glycol-distearoyl-phosphatidylethanolamine (TAT-PEG-DSPE) was synthesized. Physicochemical characteristics of SLNs such as size, morphology, and release profiles were explored. In vitro and in vivo studies were carried out to assess the efficacy of their antitumor activity in target cells.
Materials
Chemicals and regents
CDDP, PTX, glyceryl monostearate (GMS), and α-tocopherol succinate were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). DSPE-PEG-Maleimide (average molecular weight 3.4 kDa) was purchased from Taiyuan Pegchem Technology Co., Ltd. (Shanxi, People’s Republic of China). GMS and soybean lecithin (SBL) were purchased from TCI (Shanghai) Development Co., Ltd. (Shanghai, People’s Republic of China). TAT peptide with terminal cysteine (Cys-AYGRKKRRQRRR) was obtained from Chinese Peptide Co., Ltd. (Zhejiang, People’s Republic of China). N,N′-dicyclohexyl-carbodimide (DCC) and 4-dimethylaminopyridine (DMAP) were obtained from GL Biochem., Ltd. (Shanghai, People’s Republic of China). All other chemicals were of analytical grade or higher.
Cells and animals
Human cervix adenocarcinoma cell line (HeLa cells) was obtained from the American type culture collection (Manassas, VA, USA) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Sigma-Aldrich Co.) supplemented with 10% fetal bovine serum (FBS) (Fisher Chemicals, Fairlawn, NJ, USA) and 100 U/mL penicillin and 100 mg/mL streptomycin (Sigma-Aldrich Co.) in a 5% CO2 fully humidified atmosphere.
BALB/c nude mice (4–6 weeks old, 18–22 g weight) were purchased from the Medical Animal Test Center of Shandong University (Ji’nan, People’s Republic of China) and were maintained under specific pathogen-free conditions. Animals were treated according to the ethical guidelines of Shandong University after obtaining Shandong Univeristy Institutional Ethical Committee approval. The animal experiments were carried out following the internationally accepted animal care guidelines for laboratory use of the European Community (EEC Directive of 1986; 86/609/EEC).
Methods
Synthesis and characterization of TOS-CDDP
For the synthesis of TOS-CDDP, cis,cis,trans-diamminedichlorodihydroxyplatimum (c,c,t-[Pt(NH3)2Cl2(OH)2]) was synthesized (Figure 1).38 First, cis-CDDP (0.5 mmol) was dissolved in 10 mL of water and heated on a water bath and maintained at 50°C. Hydrogen peroxide (H2O2, 0.05 mol) was added into CDDP solution by stirring for 1 h at 50°C, and the reaction was continued with stirring for 12 h at room temperature. Then, the solvent was removed by evaporation, and the product was washed with water, ethyl alcohol, and ether, and then vacuum dried.
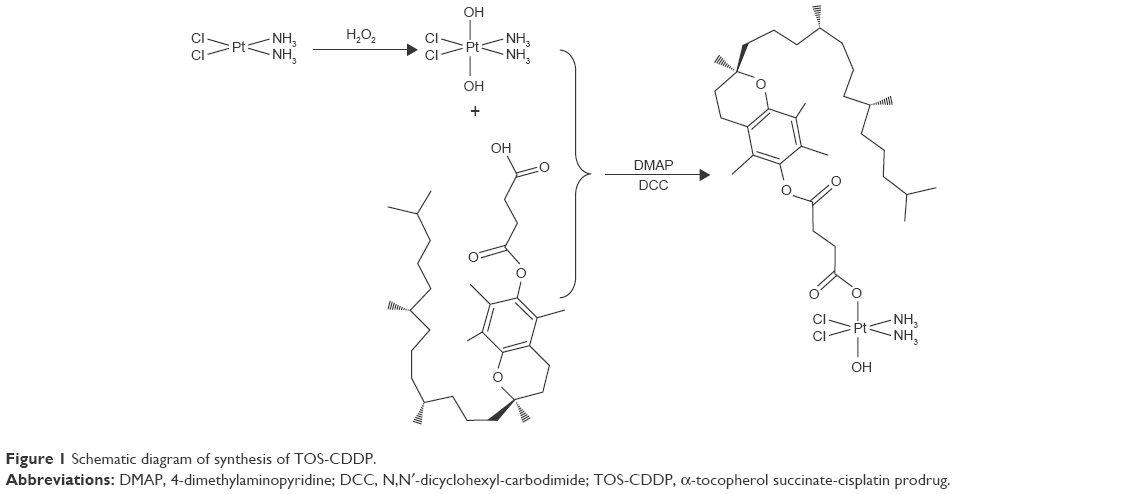 | Figure 1 Schematic diagram of synthesis of TOS-CDDP. |
TOS, c,c,t-[Pt(NH3)2Cl2(OH)2], DCC, and DMAP (1:10:10:0.1) were dissolved in dimethyl sulfoxide (DMSO) and stirred under nitrogen atmosphere at room temperature for 24 h.39 The mixture was filtered to remove the by-product. TOS-CDDP was obtained by freeze-drying.
Synthesis and characterization of TAT-PEG-DSPE
The TAT-PEG-DSPE conjugate was synthesized as follows.40,41 DSPE-PEG-Mal and Cys-TAT (molar ratio 1:2) were dissolved in chloroform and reacted by stirring overnight. The solvent was removed by evaporation, and the product was freeze-dried. TAT-PEG-DSPE was obtained by separating the product by gel filtration chromatography.
Preparation of TAT PTX/TOS-CDDP SLNs
Table 1 summarizes all kinds of SLNs prepared in this study and their abbreviations. TAT PTX/TOS-CDDP SLNs were prepared using emulsification and solvent evaporation method (Figure 2).27 Briefly, PTX (20 mg), TOS-CDDP (10 mg), SLB (50 mg), and GMS (100 mg) were dissolved in chloroform (5 mL) to produce an oil phase. TAT-PEG-DSPE and Tween 80 (1%) were dissolved in double distilled water (45 mL) to form aqueous phase. Oil phase was added to the aqueous phase containing 1% Tween 80 and homogenized (20,000 rpm; 3 min). The resulting TAT PTX/TOS-CDDP SLNs were purified by dialysis against phosphate-buffered saline (PBS) for 4 h.
 | Table 1 Abbreviations of all kinds of SLNs prepared |
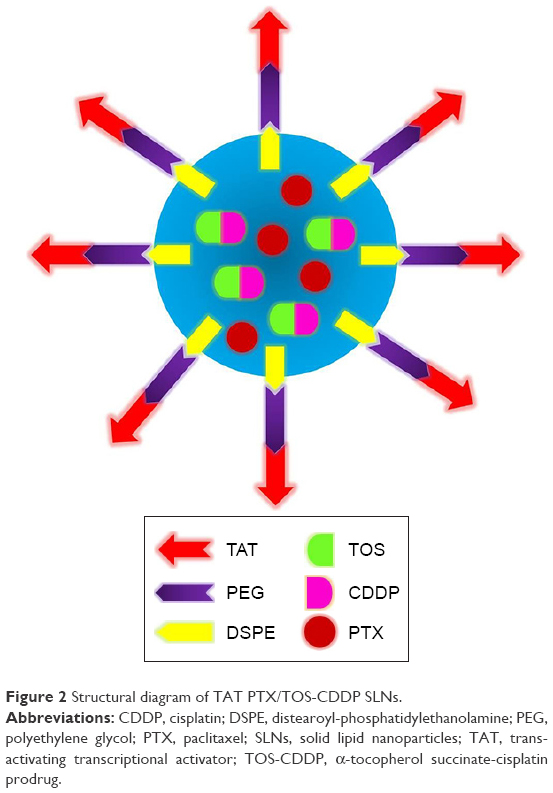 | Figure 2 Structural diagram of TAT PTX/TOS-CDDP SLNs. |
PTX and TOS-CDDP co-loaded SLNs do not contain TAT-PEG-DSPE ligands (PTX/TOS-CDDP SLNs) and were prepared by the same method as TAT PTX/TOS-CDDP SLNs, except for the aqueous phase: Tween 80 (1%) was dissolved in double distilled water (45 mL) to form aqueous phase.
PTX-loaded SLNs (PTX SLNs) were prepared by the same method as PTX/TOS-CDDP SLNs, except for the oil phase: PTX (40 mg), SLB (50 mg), and GMS (100 mg) were dissolved in chloroform (5 mL) to produce an oil phase.
TOS-CDDP-loaded SLNs (TOS-CDDP SLNs) were prepared by the same method as PTX/TOS-CDDP SLNs, except for the oil phase: TOS-CDDP (20 mg), SLB (50 mg), and GMS (100 mg) were dissolved in chloroform (5 mL) to produce an oil phase.
CDDP-loaded SLNs (CDDP SLNs) were prepared by the same method as TOS-CDDP SLNs, except for the oil phase: CDDP (20 mg), SLB (50 mg), and GMS (100 mg) were dissolved in chloroform (5 mL) to produce an oil phase.
Blank SLNs were prepared by the same method as PTX/TOS-CDDP SLNs, except for the oil phase: SLB (50 mg) and GMS (100 mg) were dissolved in chloroform (5 mL) to produce an oil phase.
Fluorescent couramin 6-loaded SLNs of all kinds were prepared by the same method as mentioned previously, with the addition of 20 mg couramin 6 into each formula. Couramin 6-loaded SLNs were named with “couramin 6” after the systems, for example, TAT PTX/TOS-CDDP SLNs (couramin 6).
Characterization of surface morphology
Surface morphology of TAT PTX/TOS-CDDP SLNs was investigated by transmission electron microscopy (TEM), using JEM-1200EX transmission electron microscope (JEOL Ltd., Tokyo, Japan).42,43 Samples were prepared by placing a drop of sample onto a copper grid and air-drying, following negative staining with one drop of 2% aqueous solution of sodium phosphotungstate for contrast enhancement. The air-dried samples were then directly examined under the microscope.
Measurement of particle size and zeta potential
Particle size (mean volume diameter) and polydispersity index (PDI) of all formulations were measured by photon correlation spectroscopy using Zetasizer Nano ZS (Malvern Instruments, Malvern, UK).44 To obtain information concerning the stabilities of the prepared samples, their zeta potentials were determined with Zetasizer Nano ZS, and the medium used was double-distilled water.
Determination of drug loading and entrapment efficiency
Content of PTX and CDDP in various formulations was analyzed by high-performance liquid chromatography (LC-20A; Shimadzu, Kyoto, Japan) at 227 nm and by UV spectrophotometry (UV-1800; Shimadzu) at 273 nm, respectively.45,46 The drug-loading content (DL) was defined as the weight (W) ratio of the loaded drug to the drug-loaded SLNs. The drug encapsulation efficiency (EE) was calculated from the weight (W) ratio between the drug incorporated in SLNs and that fed in the fabrication:
DL (%) = (Winitial drug − Wfree drug)/Winitial drug and lipid ×100 |
EE (%) = (Winitial drug − Wfree drug)/Winitial drug ×100 |
Evaluation of plasma stability
To test the plasma stability of the samples, 10 μL of TAT PTX/TOS-CDDP SLNs was diluted in 1 mL of plasma.47 Samples were then separately incubated at 37°C for 24 h, separately. At scheduled times (0, 2, 4, 8, 12, 24 h), 1 mL of each sample was diluted with 2 mL of tetrahydrofuran (THF) and the mixture was bath sonicated for 5 min, followed by centrifugation at 10,000 rpm for 5 min. The variation trends of the EE for both PTX and CDDP were calculated by the method as described in the “Determination of drug loading and entrapment efficiency” section.
In vitro release profile of PTX and CDDP
The drug released from the various formulations was captured using the dialysis bag method.48 The drug-loaded SLNs (0.5 mL) were put into the dialysis bag, which was immersed in 20 mL 0.2M PBS (pH 7.4 or 5.0, 37°C, 100 rpm shaking) containing 0.1% (v/v) Tween 80. At each time point, the dialysis bag was relocated in a new dissolution medium to maintain a sink condition. The concentration of PTX or CDDP released from the dialysis bag was analyzed by the method as described in the “Determination of drug loading and entrapment efficiency” section.
Cellular uptake of SLNs
To evaluate cellular uptake efficiency of SLNs, coumarin 6 was used as a model fluorescent molecule, which can be encapsulated into various SLNs for quantitative investigation on cellular uptake by HeLa cells.7 The cells were equilibrated with Hank’s buffered salt solution at 37°C for 1 h before coumarin 6-loaded SLNs were added at a concentration of 200 mg/mL. After incubation for 2, 4, and 8 h, the medium was removed and the wells were washed three times with cold PBS solution. Then, cells were washed once with 1 mL of PBS and were detached with trypsin/ethylenediaminetetraacetic acid (EDTA). Then the cells were centrifuged at 1,500 rpm, 4°C for 5 min, the supernatant was discarded, and the cells were re-suspended in 300 μL of PBS and directly introduced to a BD FACSCalibur flow cytometer (Becton, Dickinson and Company, Franklin lakes, NJ, USA).
In vitro tumor cell inhibition and drugs’ synergistic effect
HeLa cells (5,000 cells/well) were seeded in 96-well microtiter plate and incubated overnight in a 5% CO2 incubator at 37°C for attachment.18 Cells were then treated with blank SLNs, various drugs loaded SLNs formulations, and free drug solutions in different concentrations (0.1, 0.4, 1.6, 6.4, 25, 100 μM) for 48 h. Free drugs were dissolved in DMSO to make a stock solution of 5 mM concentration, named PTX/TOS-CDDP solution, PTX solution, and TOS-CDDP solution. Serial dilutions of free drugs in DMSO were made from this stock solution and 2 μL of each free drug solution was added to cells to obtain desired final concentrations. About 20 μL of MTT reagent (5 mg/mL) was added to each well and incubated for 4 h at 37°C. Formazan crystals were then solubilized in 100 μL of the solubilization buffer (10% SDS in 0.01 M HCl) and incubated overnight. Absorbance was measured with spectrophotometer at 550 nm. The percent cell viability was calculated considering the untreated cells as 100% viability. Half maximal inhibitory concentration (IC50) values of samples were calculated.
Tumor cell proliferation inhibition behaviors of free drugs and drug-loaded SLNs were evaluated against HeLa cells following the similar procedure, and 48 h incubation time was applied. The inhibitory concentration (ICx) values were determined using Origin 8.0 (OriginLab, Northampton, MA, USA) according to the fitted data. The Combination Index (CI) was measured according to the Chou and Talalay’s method.49 To distinguish synergistic, additive, or antagonistic cytotoxic effects, the following equation was used:
CIx = (D)1/(Dx)1 + (D)2/(Dx)2 |
(Dx)1 and (Dx)2 represent the ICx value of PTX and CDDP alone, respectively. (D)1 and (D)2 represent the concentration of PTX and CDDP in the combination system at the ICx value. CI >1 represents antagonism, CI =1 represents additive, and CI <1 represents synergism. In this study, IC50 (inhibitory concentration to produce 50% cell death) was applied.
In vivo antitumor efficacy
The antitumor efficacy of drug-loaded SLNs was evaluated in comparison to free drugs on cervical cancer-bearing mice. Practically, 200 μL of the HeLa cell suspension, equivalent to 1×106 cells, were injected subcutaneously into mice toward the upper portion of the right flank, to develop a solid tumor model.15 Tumors were allowed to grow until they reached a volume around 100 mm3 before initiating the treatment. Tumor length and width were measured with calipers, and the tumor volume was calculated using the following equation:
Tumor volume (V) = length × width2/2 |
Tumor-bearing nude mice were randomly divided into 10 groups of 8 each, and all groups received intravenous injections on days 0, 3, 6, 9, and 12 in the lateral tail vein of either 1) TAT PTX/TOS-CDDP SLNs, 2) PTX/TOS-CDDP SLNs, 3) PTX SLNs, 4) TOS-CDDP SLNs, 5) CDDP SLNs, 6) PTX/TOS-CDDP solution, 7) PTX solution, 8) TOS-CDDP solution, 9) blank SLNs, or 10) 0.9% saline. All treatments were performed at the same dose, at 20 μg PTX and/or 10 μg TOS-CDDP per kg, except the blank SLNs and 0.9% saline. The mice were monitored regularly for changes in tumor volume (V) and tumor weight (W). The antitumor efficacy of each formulation was evaluated by tumor inhibition rate (TIR), which was calculated by the following equation:
TIR (%) = (Wcontrol group − Wtreated group)/Wcontrol group ×100 |
Body weight changes of different groups were also recorded.
In vivo tissue distribution study
Tumor-bearing nude mice were randomly divided into two groups of eight each. TAT PTX/TOS-CDDP SLNs and PTX/TOS-CDDP solutions were administered to the mice by tail vein injection separately. For the in vivo organ distribution study, each sample was investigated at 0.5, 1, 4, 24, and 48 h after intravenous injection. At predetermined time intervals, mice were sacrificed and the tumor, heart, liver, spleen, lung, and kidney of mice were collected. Tissues were initially weighed and homogenized with physiological saline to determine the amount of PTX or CDDP in each tissue. The concentration of PTX or CDDP released from the dialysis bag was analyzed by the method as described in the “Determination of drug loading and entrapment efficiency” section.
Statistical analysis
All experiments were performed at least three times and expressed as mean ± standard deviation. Statistical significances were analyzed using the Student’s t-test. P<0.05 was considered statistically significant.
Results
Synthesis of TOS-CDDP and TAT-PEG-DSPE
TOS-CDDP prodrug was first synthesized. The chromatography spectra graph is shown in Figure 3. TOS-CDDP was prepared through the reaction between the carboxylic group of TOS and the hydroxyl group of c,c,t-[Pt(NH3)2Cl2(OH)2]. TAT-PEG-DSPE was synthesized by forming an amido linkage between DSPE-PEG and TAT. The chemical structures of TOS-CDDP and TAT-PEG-DSPE were confirmed using 1H-NMR spectroscopy. TOS-CDDP, δ (ppm): 2.06 (–NH2), 2.56 (–CH2–C=O–O–), 2.41 (–OH). TAT-PEG-DSPE, δ (ppm): 2.39 (–CH2C=O, –NHC=O), 3.16 (–CH2N–), 4.11 (–NHC=O).
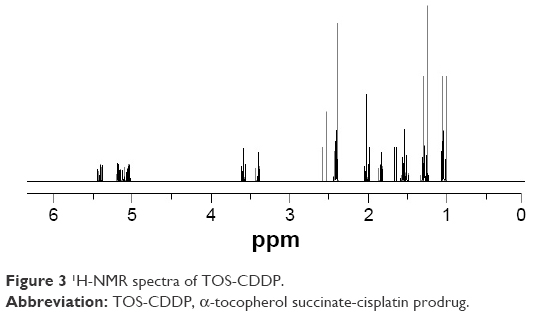 | Figure 3 1H-NMR spectra of TOS-CDDP. |
Characterization of TAT PTX/TOS-CDDP SLNs
Surface morphology
TEM image of TAT PTX/TOS-CDDP SLNs exhibited a core-shell type of spherical surface morphology (Figure 4). The black core and gray shell structure are clearly visible in the picture. The size scales in the image show the diameter of TAT PTX/TOS-CDDP SLNs to be about 100 nm.
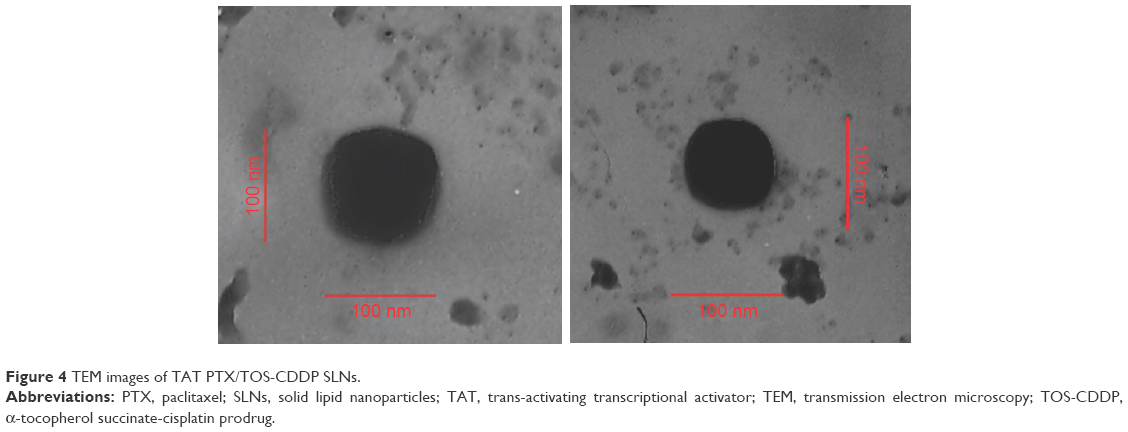 | Figure 4 TEM images of TAT PTX/TOS-CDDP SLNs. |
Particle size and zeta potential
Particle size, PDI, and zeta potential of all formulations were investigated and summarized (Table 2). It can be seen that the blank SLNs had a size of 83 nm and a slightly negative surface charge of −12 mV. Size of TAT PTX/TOS-CDDP SLNs increased to 109 nm, and zeta potential decreased to −31 mV. Drug-loaded SLNs do not contain TAT-PEG-DSPE ligands. They exhibited no obvious change in size but a slight decrease in surface charge was noted.
 | Table 2 Characterization of all formulations |
Drug loading and entrapment efficiency
The EE of PTX and CDDP of all formulations were about 90%. The PTX DL of samples was between 3.5% and 5.9%, The CDDP DL of samples was between 2.3% and 4.4%. The results indicated that PTX and CDDP were successfully loaded into the SLNs. The encapsulation capacity of SLNs was good, and this could be useful for drug delivery and therapeutic effect.
Plasma stability
The stability of TAT PTX/TOS-CDDP SLNs in plasma was determined by monitoring the PTX and CDDP encapsulation ability in the presence of plasma during 24 h (Figure 5). The results illustrated that PTX and CDDP were well protected in the SLNs. At 12 h, over 80% of intact drugs were recovered from SLNs samples. After 24 h, more than 70% of the drugs were still encapsulated in SLNs.
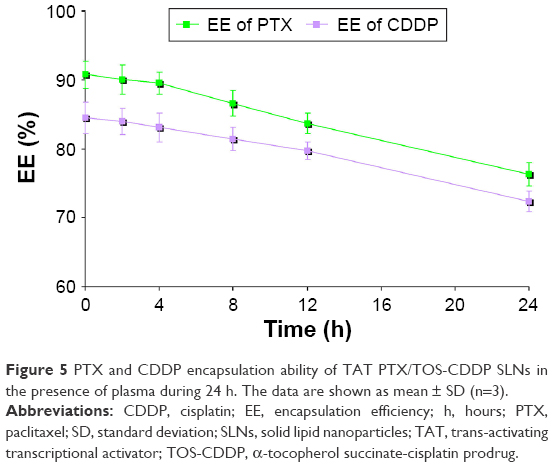 | Figure 5 PTX and CDDP encapsulation ability of TAT PTX/TOS-CDDP SLNs in the presence of plasma during 24 h. The data are shown as mean ± SD (n=3). |
In vitro release
In vitro release profile of PTX and CDDP from SLNs is shown in Figures 6 and 7. Drug release from TAT PTX/TOS-CDDP SLNs was slower than from PTX/TOS-CDDP SLNs (P<0.05). Drug release at pH 5.0 was faster than at pH 7.4 (P<0.05). The PTX release from PTX/TOS-CDDP SLNs at pH 5.0 was the most rapid, where over 80% of the drug was released within 24 h. CDDP released from TAT PTX/TOS-CDDP SLNs at pH 7.4 exhibited the most sustained release behavior, with complete release occurring at 96 h.
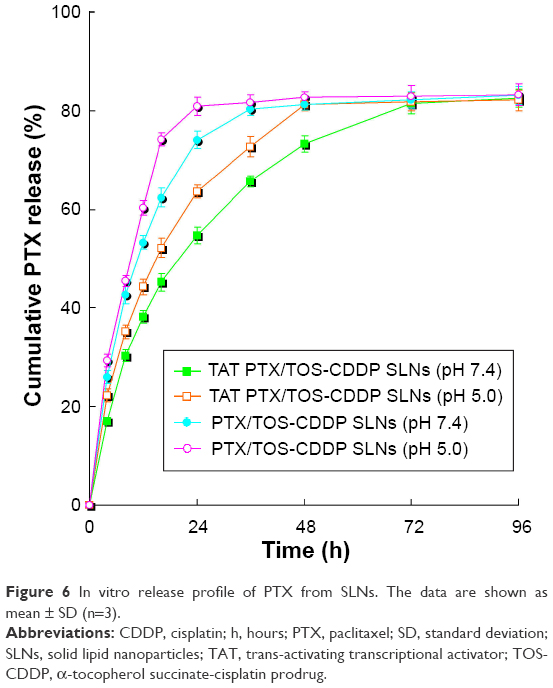 | Figure 6 In vitro release profile of PTX from SLNs. The data are shown as mean ± SD (n=3). |
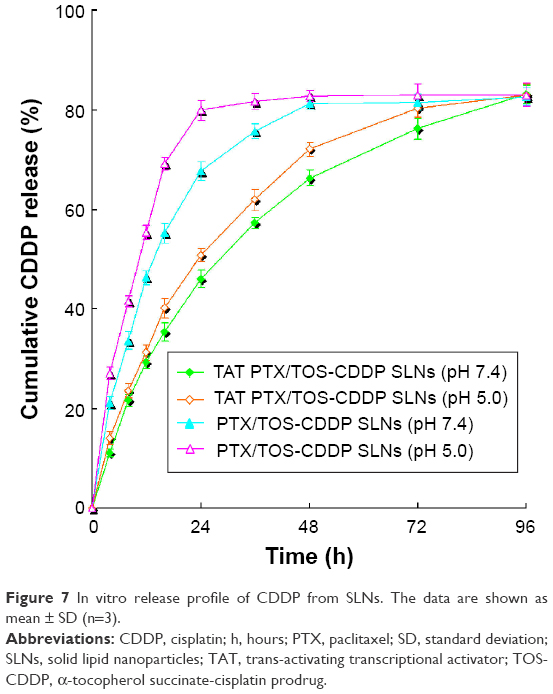 | Figure 7 In vitro release profile of CDDP from SLNs. The data are shown as mean ± SD (n=3). |
Cellular uptake
Cellular uptake efficiency of the coumarin 6-loaded SLNs was tested, and the data are presented in Figure 8. As showed in the figure, cellular uptake efficiency of TAT PTX/TOS-CDDP SLNs was significantly higher than other SLNs (P<0.05). Cellular uptake efficiency of TAT PTX/TOS-CDDP SLNs reached over 80% at 4 h posttreatment. Cellular uptake of PTX/TOS-CDDP SLNs, blank SLNs, and other samples showed no obvious difference when compared with each other (P>0.05).
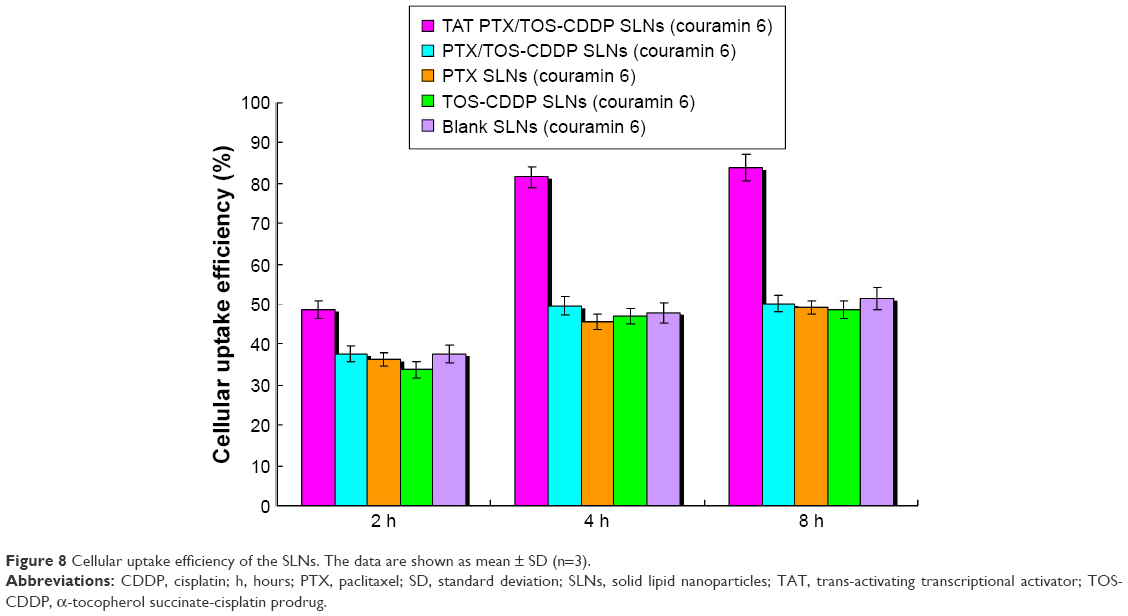 | Figure 8 Cellular uptake efficiency of the SLNs. The data are shown as mean ± SD (n=3). |
In vitro tumor cell inhibition
Cytotoxicity of the blank SLNs was first evaluated in HeLa cells. As showed in Figure 9, cell viabilities of HeLa cells treated with blank SLNs at all the tested concentrations, up to 1 mg/mL, after 48 h of incubation were above 80%, indicating that SLNs system had good biocompatibility. IC50 values and CI50 values of free drug solutions and drug-loaded SLNs are summarized in Table 3. The IC50 values of drug-loaded SLNs were larger than their free drug solution counterparts. CI50 value of PTX/TOS-CDDP solution was 1.284, which indicates that mixing of PTX and TOS-CDDP into one solution could not lead to synergism efficiency. CI50 values of TAT PTX/TOS-CDDP SLNs and PTX/TOS-CDDP SLNs were 0.646 and 0.861, respectively, which shows that they exhibit a significant synergism effect.
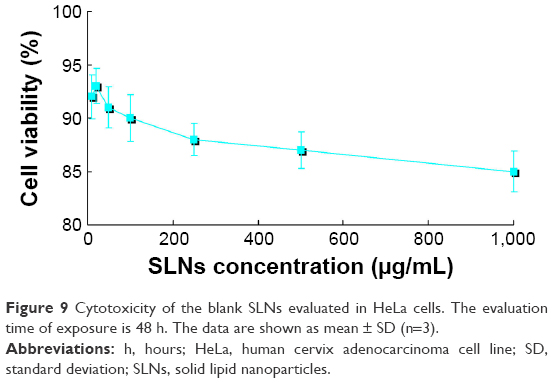 | Figure 9 Cytotoxicity of the blank SLNs evaluated in HeLa cells. The evaluation time of exposure is 48 h. The data are shown as mean ± SD (n=3). |
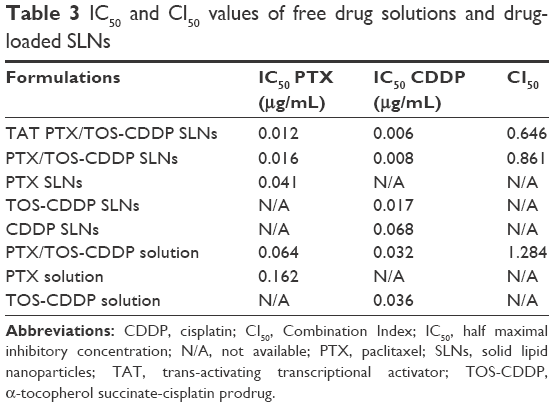 | Table 3 IC50 and CI50 values of free drug solutions and drug-loaded SLNs |
In vivo antitumor activity
Figure 10 indicates that the growth of cervical tumors is not affected by treatment with blank SLNs compared with saline-treated tumors. Although the drug solutions and drug-loaded SLNs treatments have been shown to significantly reduce the tumor volume, the TAT PTX/TOS-CDDP SLNs system was by far the most efficient (P<0.05). Three weeks after treatment, the tumors from each treatment group were weighted and tumor inhibition efficiency was calculated. Figure 11 shows the TIR results. TAT PTX/TOS-CDDP SLNs showed the highest TIR (72.2%), while TIR of PTX/TOS-CDDP SLNs was 58.9%. Other formulations showed lower TIR. Relative body weight loss was also monitored throughout the different treatments. Absolute weight loss was observed in the control and drug solution-treated mice but not after giving drug-loaded SLNs treatment.
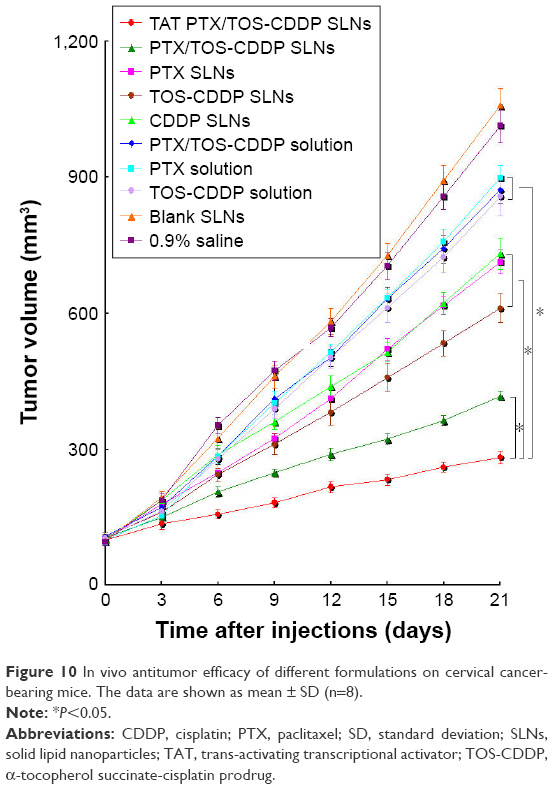 | Figure 10 In vivo antitumor efficacy of different formulations on cervical cancer-bearing mice. The data are shown as mean ± SD (n=8). |
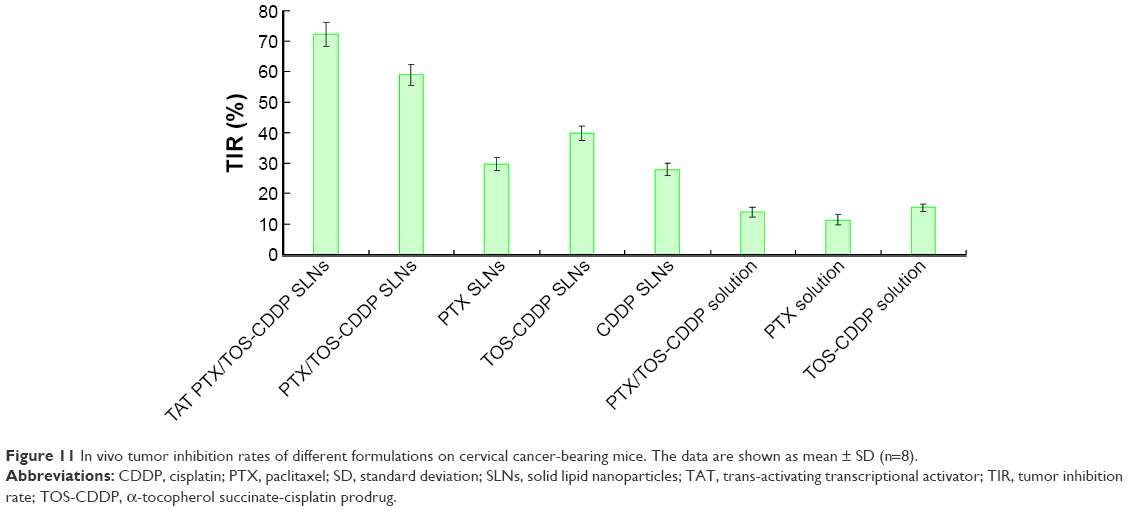 | Figure 11 In vivo tumor inhibition rates of different formulations on cervical cancer-bearing mice. The data are shown as mean ± SD (n=8). |
In vivo tissue distribution
In vivo PTX or CDDP tissue distribution results of TAT PTX/TOS-CDDP SLNs and PTX/TOS-CDDP solution are shown in Figures 12 and 13, respectively. The concentration of PTX and CDDP in tumor, lungs, and liver was higher following the injection of TAT PTX/TOS-CDDP SLNs than after the injection of PTX/TOS-CDDP solution. However, the drug concentration in heart and kidney was lower in TAT PTX/TOS-CDDP SLNs group than in PTX/TOS-CDDP solution group. Drug concentrations in the tumor tissue remained relatively stable at all time points until 24 h and 48 h after injection in TAT PTX/TOS-CDDP SLNs group, whereas the drug concentrations in PTX/TOS-CDDP solution group reduced to a great extent.
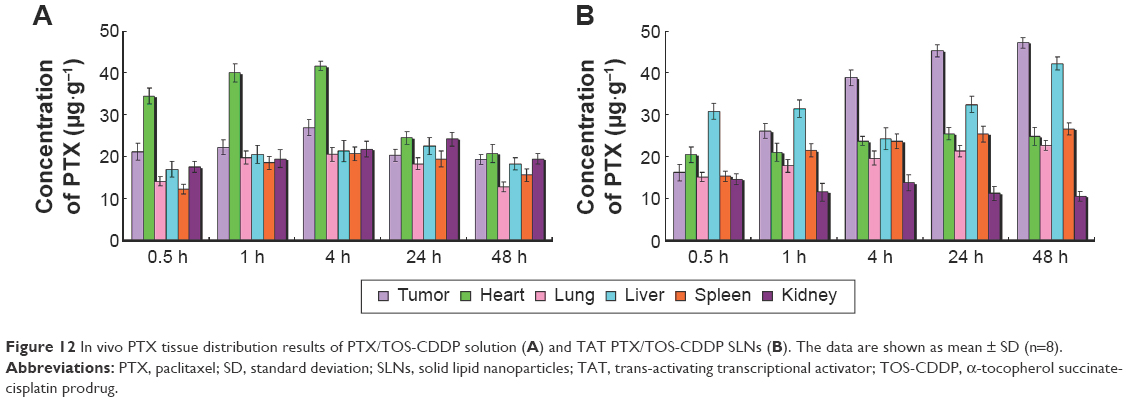 | Figure 12 In vivo PTX tissue distribution results of PTX/TOS-CDDP solution (A) and TAT PTX/TOS-CDDP SLNs (B). The data are shown as mean ± SD (n=8). |
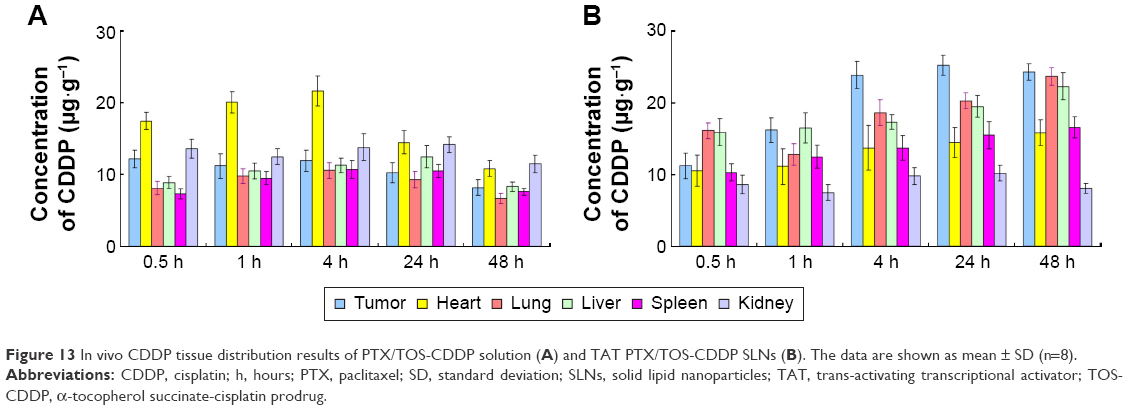 | Figure 13 In vivo CDDP tissue distribution results of PTX/TOS-CDDP solution (A) and TAT PTX/TOS-CDDP SLNs (B). The data are shown as mean ± SD (n=8). |
Discussion
The aim of this study was to develop nanomedicine in order to achieve synergistic antitumor activity against cervical cancer. Our previous study had taken advantage of folate-contained, gene-loaded NPs for the treatment of cervical cancer and achieved remarkable antitumor effect.50 In the present study, TAT containing PTX and TOS-CDDP prodrug co-loaded SLNs were constructed and evaluated.
At the start of the research, TOS-CDDP prodrug and TAT-PEG-DSPE ligand were synthesized. TAT peptide, a cationic classical CPP, is known to efficiently facilitate and enhance the cellular uptake of a large variety of cargoes such as drugs, genes, peptides, proteins, liposomes, and particles in a nontoxic fashion.51 To combine the anticancer effects of CDDP and TOS, the dual drug conjugates (TOS-CDDP) were developed.52
SLNs were prepared using emulsification and a solvent evaporation method. Two of the most important parameters of carriers are particle size and zeta potential.43 They have been demonstrated to play important roles in determining cellular and tissue uptake efficiency and toxic effect on cells. The zeta potential and size of particles not only determine their colloidal stability but also influence the effectiveness of their interaction with negatively charged cell membranes, which are the pivotal steps for successful cellular uptake. Size of TAT PTX/TOS-CDDP SLNs was above 100 nm. The size of TAT PTX/TOS-CDDP SLNs was larger than blank SLNs (P<0.05); however, the size of blank SLNs, CDDP SLNs, TOS-CDDP SLNs, PTX SLNs, and PTX/TOS-CDDP SLNs exhibited no obvious differences (P>0.05). This means the loading of PTX and/or CDDP did not increase the size of the particles, but the addition of TAT ligands increased the diameter of the particles. The surface charge of TAT PTX/TOS-CDDP SLNs was −31 mV, which is lower than the blank SLNs (P<0.05), suggesting that the loading of drugs and inclusion of TAT ligands decreased the charge of the SLNs. TEM image showed the core–shell structure of TAT PTX/TOS-CDDP SLNs, which could be explained by a TAT-PEG outer layer on the surface of the drug-loaded SLNs core. The size scale bars in Figure 4 show the size of TAT PTX/TOS-CDDP SLNs to be about 100 nm, which was in accordance with the size measurement study.
For successful drug delivery, it is important that drug-loaded carriers retain the drug for a certain time period after administration.48 Slower drug release from TAT PTX/TOS-CDDP SLNs than from PTX/TOS-CDDP SLNs may be attributed to the presence of TAT-PEG-PE, in which PEG contributes for sustained release. Also, the release of PTX and CDDP from SLNs was greatly affected by the environmental acidity. Drug release at acidic pH was faster than at neutral conditions. The rapid drug release at pH 5.0 might be attributed to the significantly increased protonation degree of the carboxyl groups in TAT PTX/TOS-CDDP SLNs polymer backbone at a lower pH value, resulting in the weakening of electrostatic interactions between the SLNs and drug molecules. The acidic condition may also accelerate the degradation of the ester bond, allowing CDDP to be released more freely. In summary, the sustained-release pattern of drug-loaded SLNs could be helpful with the in vivo drug delivery, and the faster release in the acidic condition may allow for enhanced release of the drugs in the tumor tissue.
The therapeutic effects of the drug-loaded SLNs would depend on internalization and sustained retention of the NPs by the cancer cells.7 The research on in vitro cellular uptake could provide some circumstantial evidence on the advantages of the NP formulation in comparison with the free drug. Coumarin 6, a fluorescent probe, was used to represent the drug in the NP formulation to analyze the cellular uptake of SLNs. Cellular uptake efficiency of TAT PTX/TOS-CDDP SLNs was significantly higher than other SLNs. This could be attributed to enhanced cancer cell-specific adherence of the TAT ligands.53 The improved activity and penetration of drugs delivered with TAT can be utilized to improve the efficacy of the standard drug dose, attenuate side effect, and overcome drug resistance.
The biocompatibility of the blank SLNs was evaluated using MTT assay. Figure 9 shows that the cell viabilities of HeLa cells treated with the SLNs after 48 h incubation were over 80%. This result illustrated that the constructed SLNs delivery system may have little influence on the cancer cells. To determine the potential of the dual drug-loaded SLNs for use as cancer therapeutics, the inhibition efficacy of drug-loaded SLNs in tumor cells in vitro was evaluated in HeLa cervical cancer cell line. To verify the synergistic effect of the co-delivery system, the in vitro antitumor effects of free drug solutions and drug-loaded NLCs against HeLa cells were tested. To understand the effect of dual drugs on cytotoxicity in HeLa cells, the Chou and Talalay’s method was used to determine whether the drug combination effect was synergistic, additive, or antagonistic. CI50 value of PTX/TOS-CDDP solution did not show synergism efficiency, while CI50 of TAT PTX/TOS-CDDP SLNs and PTX/TOS-CDDP SLNs exhibited significant synergism effect. The reason might be attributed to differences in the cell uptake pathways of free drugs and drug-loaded SLNs, and the controlled release manner of drug-loaded SLNs. In cell culture medium, most free drugs could quickly display their effects after being transported into cells via passive diffusion. However, the drug-loaded NPs were mainly taken up by cells via the endocytosis pathway and then exerted the antitumor activity after the drug molecules were released from the NPs.54 CI50 value of the TAT PTX/TOS-CDDP SLNs was the lowest (0.646), indicating co-delivery of PTX and TOS-CDDP by TAT-modified SLNs had evident superiority as compared with free drug combination and unmodified PTX/TOS-CDDP SLNs.
The antitumor efficacy of the drug-loaded SLNs was then investigated in vivo on a human cervical carcinoma xenograft model in mice and compared to the other treatment combinations. The TAT PTX/TOS-CDDP SLNs showed considerable improvement in antitumor activity, as compared to PTX/TOS-CDDP SLNs and other formulations. Treatments with saline or blank SLNs alone displayed no antitumor activity on human cervical cancer. Clearly, the dual drug-loaded SLNs exhibited a better antitumor effect than single drug-loaded SLNs, suggesting a synergistic effect of PTX and CDDP. Noteworthy, TAT PTX/TOS-CDDP SLNs displayed improved anticancer activity and lower toxicity compared to the parent drugs injected alone or in combination, which could be observed by the body weight lost. Significant weight loss associated with free drug solutions indicated the toxicity of these treatments. On the contrary, the mice treated with the drug-loaded SLNs showed a slight increase in their body weight, which supports both the efficient anticancer activity of the systems and the absence of adverse effects. In summary, TAT PTX/TOS-CDDP SLNs displayed a more efficient anticancer pharmacological activity with almost complete suppression of tumor growth and absence of toxicity related to weight loss. Such increased anticancer activity and decreased toxicity of TAT PTX/TOS-CDDP SLNs may be explained by the biodistribution data. The administration of TAT PTX/TOS-CDDP SLNs led to a dramatic increase of drug accumulation in the tumor tissue, as compared with the free drug solutions. This phenomenon may be explained by the fact that solid tumors have leaky micro-vasculatures and the nano-sized particles could passively target the tumor due to the enhanced permeability and retention (EPR) effects.55 The EPR effects prevented the entry of the system into the normal cell but favored selective entry into the tumor, which resulted in the efficient drug accumulation in tumor tissue. Specific distribution of the drug in the tumor tissue compared with the other tissues could decrease the side effects and lead to better antitumor therapeutic efficiency. At the same time, in vivo drug distribution in heart and kidney may cause systemic toxicity. The drug distribution of PTX/TOS-CDDP solution was higher than TAT PTX/TOS-CDDP SLNs, and this may cause systemic toxicity.56
Conclusion
This novel PTX and TOS-CDDP loaded, TAT-modified nanoparticulate drug delivery system achieved the functional goals of the system, that is, improved efficacy with combination therapy and ligand–receptor mediated targeting. TAT PTX/TOS-CDDP SLNs could be successfully internalized by HeLa cells. Co-delivery of two drugs showed a synergistic effect in suppressing the cervical tumor cell growth. TAT PTX/TOS-CDDP SLNs exhibited high tumor tissue accumulation, superior antitumor efficiency, and much lower toxicity in vivo. The present study indicates that the co-delivery system provides a promising platform as a combination therapy for the treatment of cervical cancer, and possibly other types of cancer as well. This strategy of combination therapy deserves further investigation for the treatment of different tumors.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. | ||
Kamangar F, Dores GM, Anderson WF. Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. J Clin Oncol. 2006;24(14):2137–2150. | ||
Cervical Cancer: Estimated Incidence, Mortality and Prevalence Worldwide in 2012. International Agency for Research on Cancer and World Health Organization; 2012. Available from: http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx. Accessed March 16, 2016. | ||
Hani U, Osmani RA, Bhosale RR, Shivakumar HG, Kulkarni PK. Current perspectives on novel drug delivery systems and approaches for management of cervical cancer: a comprehensive review. Curr Drug Targets. 2016;17(3):337–352. | ||
Vivero-Escoto JL, Slowing II, Lin VS. Tuning the cellular uptake and cytotoxicity properties of oligonucleotide intercalator-functionalized mesoporous silica nanoparticles with human cervical cancer cells HeLa. Biomaterials. 2010;31(6):1325–1333. | ||
Zheng Y, Chen H, Zeng X, et al. Surface modification of TPGS-b-(PCL-ran-PGA) nanoparticles with polyethyleneimine as a co-delivery system of TRAIL and endostatin for cervical cancer gene therapy. Nanoscale Res Lett. 2013;8(1):161. | ||
Zeng X, Tao W, Mei L, Huang L, Tan C, Feng SS. Cholic acid-functionalized nanoparticles of star-shaped PLGA-vitamin E TPGS copolymer for docetaxel delivery to cervical cancer. Biomaterials. 2013;34(25):6058–6067. | ||
Monk BJ, Sill MW, McMeekin DS, et al. Phase III trial of four cisplatin-containing doublet combinations in stage IVB, recurrent, or persistent cervical carcinoma: a Gynecologic Oncology Group study. J Clin Oncol. 2009;27(28):4649–4655. | ||
Lorusso D, Petrelli F, Coinu A, Raspagliesi F, Barni S. A systematic review comparing cisplatin and carboplatin plus paclitaxel-based chemotherapy for recurrent or metastatic cervical cancer. Gynecol Oncol. 2014;133(1):117–123. | ||
Mabuchi S, Isohashi F, Yokoi T, et al. A phase II study of postoperative concurrent carboplatin and paclitaxel combined with intensity-modulated pelvic radiotherapy followed by consolidation chemotherapy in surgically treated cervical cancer patients with positive pelvic lymph nodes. Gynecol Oncol. 2016;141(2):240–246. | ||
Moore DH, Blessing JA, McQuellon RP, et al. Phase III study of cisplatin with or without paclitaxel in stage IVB, recurrent, or persistent squamous cell carcinoma of the cervix: a gynecologic oncology group study. J Clin Oncol. 2004;22(15):3113–3119. | ||
Hu CM, Aryal S, Zhang L. Nanoparticle-assisted combination therapies for effective cancer treatment. Ther Deliv. 2010;1(2):323–334. | ||
Parhi P, Mohanty C, Sahoo SK. Nanotechnology-based combinational drug delivery: an emerging approach for cancer therapy. Drug Discov Today. 2012;17(17–18):1044–1052. | ||
Zhang W, Li C, Shen C, et al. Prodrug-based nano-drug delivery system for co-encapsulate paclitaxel and carboplatin for lung cancer treatment. Drug Deliv. 2015:1–6. | ||
Maksimenko A, Alami M, Zouhiri F, et al. Therapeutic modalities of squalenoyl nanocomposites in colon cancer: an ongoing search for improved efficacy. ACS Nano. 2014;8(3):2018–2032. | ||
Lockhart JN, Stevens DM, Beezer DB, Kravitz A, Harth E. Dual drug delivery of tamoxifen and quercetin: regulated metabolism for anticancer treatment with nanosponges. J Control Release. 2015;220(Pt B):751–757. | ||
Sagnella SM, Gong X, Moghaddam MJ, et al. Nanostructured nanoparticles of self-assembled lipid pro-drugs as a route to improved chemotherapeutic agents. Nanoscale. 2011;3(3):919–924. | ||
Qu CY, Zhou M, Chen YW, Chen MM, Shen F, Xu LM. Engineering of lipid prodrug-based, hyaluronic acid-decorated nanostructured lipid carriers platform for 5-fluorouracil and cisplatin combination gastric cancer therapy. Int J Nanomedicine. 2015;10:3911–3920. | ||
Mura S, Bui DT, Couvreur P, Nicolas J. Lipid prodrug nanocarriers in cancer therapy. J Control Release. 2015;208:25–41. | ||
Duhem N, Danhier F, Pourcelle V, et al. Self-assembling doxorubicin-tocopherol succinate prodrug as a new drug delivery system: synthesis, characterization, and in vitro and in vivo anticancer activity. Bioconjug Chem. 2014;25(1):72–81. | ||
Prasad KN, Kumar B, Yan XD, Hanson AJ, Cole WC. Alpha-tocopheryl succinate, the most effective form of vitamin E for adjuvant cancer treatment: a review. J Am Coll Nutr. 2003;22(2):108–117. | ||
Pussinen PJ, Lindner H, Glatter O, et al. Lipoprotein-associated alpha-tocopheryl-succinate inhibits cell growth and induces apoptosis in human MCF-7 and HBL-100 breast cancer cells. Biochim Biophys Acta. 2000;1485(2–3):129–144. | ||
Kline K, Yu W, Sanders BG. Vitamin E and breast cancer. J Nutr. 2004;134(12 Suppl):3458S–3462S. | ||
Zhang X, Peng X, Yu W, et al. Alpha-tocopheryl succinate enhances doxorubicin-induced apoptosis in human gastric cancer cells via promotion of doxorubicin influx and suppression of doxorubicin efflux. Cancer Lett. 2011;307(2):174–181. | ||
Mallick A, More P, Ghosh S, et al. Dual drug conjugated nanoparticle for simultaneous targeting of mitochondria and nucleus in cancer cells. ACS Appl Mater Interfaces. 2015;7(14):7584–7598. | ||
Iyer R, Croucher JL, Chorny M, et al. Nanoparticle delivery of an SN38 conjugate is more effective than irinotecan in a mouse model of neuroblastoma. Cancer Lett. 2015;360(2):205–212. | ||
Venishetty VK, Komuravelli R, Kuncha M, Sistla R, Diwan PV. Increased brain uptake of docetaxel and ketoconazole loaded folate-grafted solid lipid nanoparticles. Nanomedicine. 2013;9(1):111–121. | ||
Wu M, Fan Y, Lv S, Xiao B, Ye M, Zhu X. Vincristine and temozolomide combined chemotherapy for the treatment of glioma: a comparison of solid lipid nanoparticles and nanostructured lipid carriers for dual drugs delivery. Drug Deliv. 2015:1–6. | ||
Shi S, Han L, Deng L, et al. Dual drugs (microRNA-34a and paclitaxel)-loaded functional solid lipid nanoparticles for synergistic cancer cell suppression. J Control Release. 2014;194:228–237. | ||
Wissing SA, Kayser O, Müller RH. Solid lipid nanoparticles for parenteral drug delivery. Adv Drug Deliv Rev. 2004;56(9):1257–1272. | ||
Weber S, Zimmer A, Pardeike J. Solid Lipid Nanoparticles (SLN) and Nanostructured Lipid Carriers (NLC) for pulmonary application: a review of the state of the art. Eur J Pharm Biopharm. 2014;86(1):7–22. | ||
Zhu D, Tao W, Zhang H, et al. Docetaxel (DTX)-loaded polydopamine-modified TPGS-PLA nanoparticles as a targeted drug delivery system for the treatment of liver cancer. Acta Biomater. 2016;30:144–154. | ||
He L, Lai H, Chen T. Dual-function nanosystem for synergetic cancer chemo-/radiotherapy through ROS-mediated signaling pathways. Biomaterials. 2015;51:30–42. | ||
Han L, Tang C, Yin C. Dual-targeting and pH/redox-responsive multi-layered nanocomplexes for smart co-delivery of doxorubicin and siRNA. Biomaterials. 2015;60:42–52. | ||
Ruoslahti E. Tumor penetrating peptides for improved drug delivery. Adv Drug Deliv Rev. Epub 2016 Apr 1. | ||
Pan L, Liu J, He Q, Wang L, Shi J. Overcoming multidrug resistance of cancer cells by direct intranuclear drug delivery using TAT-conjugated mesoporous silica anoparticles. Biomaterials. 2013;34(11):2719–2730. | ||
Liang DS, Su HT, Liu YJ, Wang AT, Qi XR. Tumor-specific penetrating peptides-functionalized hyaluronic acid-d-α-tocopheryl succinate based nanoparticles for multi-task delivery to invasive cancers. Biomaterials. 2015;71:11–23. | ||
Kelland LR, Barnard CF, Evans IG, et al. Synthesis and in vitro and in vivo antitumor activity of a series of trans platinum antitumor complexes. J Med Chem. 1995;38(16):3016–3024. | ||
Mi Y, Zhao J, Feng SS. Vitamin E TPGS prodrug micelles for hydrophilic drug delivery with neuroprotective effects. Int J Pharm. 2012;438(1–2):98–106. | ||
Qin Y, Chen H, Zhang Q, et al. Liposome formulated with TAT-modified cholesterol for improving brain delivery and therapeutic efficacy on brain glioma in animals. Int J Pharm. 2011;420(2):304–312. | ||
Koren E, Apte A, Sawant RR, Grunwald J, Torchilin VP. Cell-penetrating TAT peptide in drug delivery systems: proteolytic stability requirements. Drug Deliv. 2011;18(5):377–384. | ||
Ye J, Wang A, Liu C, Chen Z, Zhang N. Anionic solid lipid nanoparticles supported on protamine/DNA complexes. Nanotechnology. 2008;19(28):285708. | ||
Zhu Y, Liang G, Sun B, Tian T, Hu F, Xiao Z. A novel type of self-assembled nanoparticles as targeted gene carriers: an application for plasmid DNA and antimicroRNA oligonucleotide delivery. Int J Nanomedicine. 2016;11:399–410. | ||
Sütő B, Berkó S, Kozma G, et al. Development of ibuprofen-loaded nanostructured lipid carrier-based gels: characterization and investigation of in vitro and in vivo penetration through the skin. Int J Nanomedicine. 2016;11:1201–1212. | ||
Gao M, Xu Y, Qiu L. Enhanced combination therapy effect on paclitaxel-resistant carcinoma by chloroquine co-delivery via liposomes. Int J Nanomedicine. 2015;10:6615–6632. | ||
Singh M, Bhatnagar P, Mishra S, Kumar P, Shukla Y, Gupta KC. PLGA-encapsulated tea polyphenols enhance the chemotherapeutic efficacy of cisplatin against human cancer cells and mice bearing Ehrlich ascites carcinoma. Int J Nanomedicine. 2015;10:6789–6809. | ||
Immordino ML, Brusa P, Rocco F, Arpicco S, Ceruti M, Cattel L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J Control Release. 2004;100(3):331–346. | ||
Chen LC, Chen YC, Su CY, Hong CS, Ho HO, Sheu MT. Development and characterization of self-assembling lecithin-based mixed polymeric micelles containing quercetin in cancer treatment and an in vivo pharmacokinetic study. Int J Nanomedicine. 2016;11:1557–1566. | ||
Lv S, Tang Z, Li M, et al. Co-delivery of doxorubicin and paclitaxel by PEG-polypeptide nanovehicle for the treatment of non-small cell lung cancer. Biomaterials. 2014;35(23):6118–6129. | ||
Liu B, Han SM, Tang XY, Han L, Li CZ. Cervical cancer gene therapy by gene loaded PEG-PLA nanomedicine. Asian Pac J Cancer Prev. 2014;15(12):4915–4918. | ||
Liu C, Liu F, Feng L, Li M, Zhang J, Zhang N. The targeted co-delivery of DNA and doxorubicin to tumor cells via multifunctional PEI-PEG based nanoparticles. Biomaterials. 2013;34(10):2547–2564. | ||
Arias JL, Reddy LH, Othman M, et al. Squalene based nanocomposites: a new platform for the design of multifunctional pharmaceutical theragnostics. ACS Nano. 2011;5(2):1513–1521. | ||
Dong X, Liu L, Zhu D, Zhang H, Leng X. Transactivator of transcription (TAT) peptide-chitosan functionalized multiwalled carbon nanotubes as a potential drug delivery vehicle for cancer therapy. Int J Nanomedicine. 2015;10:3829–3840. | ||
Prabaharan M, Grailer JJ, Pilla S, Steeber DA, Gong SQ. Amphiphilic multi-arm-block copolymer conjugated with doxorubicin via pH-sensitive hydrazone bond for tumor-targeted drug delivery. Biomaterials. 2009;30(29):5757–5766. | ||
Choi J, Ko E, Chung HK, et al. Nanoparticulated docetaxel exerts enhanced anticancer efficacy and overcomes existing limitations of traditional drugs. Int J Nanomedicine. 2015;10:6121–6132. | ||
He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31(13):3657–3666. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.