")
Back to Journals » Journal of Pain Research » Volume 12
Co-crystals as a new approach to multimodal analgesia and the treatment of pain
Authors Almansa C, Frampton CS, Vela JM, Whitelock S, Plata-Salamán CR
Received 9 March 2019
Accepted for publication 22 July 2019
Published 4 September 2019 Volume 2019:12 Pages 2679—2689
DOI https://doi.org/10.2147/JPR.S208082
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Erica Wegrzyn
Carmen Almansa,1 Christopher S Frampton,2 José Miguel Vela,1 Steve Whitelock,3 Carlos R Plata-Salamán4
1Esteve Pharmaceuticals, S.A., Parc Cientific Barcelona, Barcelona 08028, Spain; 2Rbar3 Ltd, Cambridge, UK; 3Mundipharma Research Ltd, Cambridge, UK; 4Esteve Pharmaceuticals, S.A., Torre Esteve, Barcelona 08038, Spain
Correspondence: Carmen Almansa
Esteve Pharmaceuticals, S.A., Parc Cientific Barcelona, C/Baldiri Reixac 4–8, Barcelona 08028, Spain
Tel +34 93 446 6148
Fax +34 93 446 6432
Email [email protected]
Abstract: Pain is highly prevalent, but frequently untreated or under-treated, and health care professionals are faced with a range of treatment challenges. Multimodal therapy is recommended and can be achieved using open combinations (ie, concomitant administration) of individual agents, fixed-dose combinations (FDCs), or multimodal agents (ie, single agents with multiple mechanisms of action). Co-crystallization of active pharmaceutical ingredients (APIs) offers another approach, with the potential to provide drugs with unique properties and advantages for therapeutic applications compared to combinations. API–API co-crystals are single-entity forms that offer a unique possibility of improving the physicochemical properties of both constituent APIs, as well as permitting their synchronous release. Consequently, this may positively impact on their pharmacokinetic (PK) properties and profiles, providing a potential advantage over FDCs and translating into improved clinical efficacy and safety profiles. We report here a revision of the literature concerning API–API co-crystals for the treatment of pain. It becomes apparent that identifying APIs with complementary mechanisms of action that can be adequately co-crystallized in an appropriate molecular ratio applicable for therapeutic use is challenging. In addition, API–API co-crystals normally result in a mere increased exposure of an API without defined clinical benefits (since, to maintain the benefit-risk, the dose needs to be proportionally reduced to adjust for the increased exposure). An exception to this is the co-crystal of tramadol-celecoxib (CTC), that represents a unique concept in co-crystal technology. In CTC neither of its three active components that have complementary mechanisms of action (ie, the two enantiomers of tramadol and celecoxib) show increased exposure levels versus commercially available single-entity reference products, but rather show a change in their PK profile with potential clinical advantages. CTC is in Phase III clinical development for the treatment of pain.
Keywords: co-crystal, efficacy, pain, multimodal, pharmacokinetics, physicochemical properties, safety
Introduction
Pain is highly prevalent across clinical settings, including in primary care, emergency departments, and medical and surgical wards.1–5 Health care professionals involved in the management of patients with pain navigate a plethora of treatment challenges, including lack of analgesic efficacy, tolerability issues, and abuse liability. As a result, pain management remains suboptimal, with many patients remaining un- or under-treated.1,6–9 Inadequate treatment can lead to numerous deleterious effects on patients’ health and well-being, as well as progression from acute to chronic pain.8,10–12
One possible solution to these issues which has gained substantial traction over recent decades is multimodal therapy,10 defined by the International Association for the Study of Pain as “the concurrent use of separate therapeutic interventions with different mechanisms of action within one discipline aimed at different pain mechanisms”.13 As pain involves multiple pathways and mediators, such an approach is likely to be more efficacious than using an analgesic with a single mode of action and also has the potential to reduce side effects and be opioid-sparing; accordingly, its use is recommended for pain management.7,14–16 Multimodal analgesia can incorporate a diverse range of drug treatments although careful consideration must be given to the mechanism(s) of action and side effect profile of each component.7,17
Multimodal engagement can be achieved via a range of different methods (Figure 1). To date, these have included treatment with open combinations (ie, concomitant administration) of two or more drugs, fixed-dose combinations (FDCs), or multimodal agents (ie, single agents with multiple mechanisms of action18). Each of these approaches has inherent benefits and limitations. For example, the use of open combinations allows individualized treatment, but this somewhat ad hoc approach can lead to issues, including suboptimal dosing and decreased compliance.14 Compared with open combination, FDCs provide a more standardized approach and can reduce pill burden, improve compliance, and decrease costs;14,19,20 however, they do not provide additional clinical advantages beyond those of the constituent active pharmaceutical ingredients (APIs). Finally, designing single multimodal agents is highly challenging, largely due to the difficulty in achieving selectivity for desired targets while avoiding off-target activity. Consequently, such agents have often been discovered by serendipity rather than by design.21,22
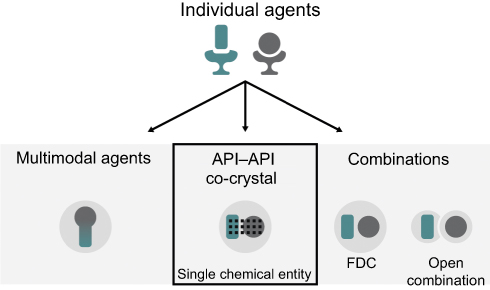 |
Figure 1 Potential methods for delivering multimodal drug therapy. Abbreviations: API, active pharmaceutical ingredient; FDC, fixed-dose combination. |
A new method for achieving multimodal pharmacotherapy involves the development of a certain type of pharmaceutical co-crystal. In simple terms, a pharmaceutical co-crystal is formed by an API and a second component contained within a single, unique crystal structure. The majority of currently available pharmaceutical co-crystals contain a single API and a non-active co-crystal former (coformer) in the same crystal structure, ie, they are API–coformer(s) co-crystals.23 However, it is the development of co-crystals containing more than one API (“API–API co-crystals”) that offers hope in terms of the novel delivery of multimodal therapy. Such co-crystals also offer the potential to provide benefits beyond the simple combination of the constituent APIs therein (as would be achieved, for example, by the use of a FDC), as well as a reduced pill burden and improved compliance. In this article, we review the potential role of co-crystals in the treatment of pain, with a special focus on API–API co-crystals. We begin by providing a background on co-crystal terminology and regulatory status, as well as a summary of advances made in fields outside of the pain arena.
Co-crystal terminology
Historically, co-crystal nomenclature has suffered from a lack of consensus,23,24 but in a 2012 perspectives article, Aitipamula et al sought to bring some clarity to the debate and defined co-crystals as
solids that are crystalline single phase materials composed of two or more different molecular and/or ionic compounds generally in a stoichiometric ratio which are neither solvates nor simple salts.25
The US Food and Drug Administration (FDA) defines co-crystals as
crystalline materials composed of two or more different molecules, one of which is the API, in a defined stoichiometric ratio within the same crystal lattice that are associated by nonionic and noncovalent bonds.26
With these definitions in mind, it is important to note that co-crystals differ from other solid-state drug forms, including salts. Table 1 shows a glossary of relevant terms used in this paper. The design of co-crystals is a complex process, and requires consideration of supramolecular interactions,23 which are weak donor–acceptor interactions that link individual molecules within a crystal structure of a material. Unlike salts, the components of the crystal lattice in co-crystals do not depend on ionic bonds.24 Instead, co-crystal formation is based on weaker interactions such as π–π-stacking, hydrogen bonds, and van der Waals interactions.27,28
API–coformer co-crystals
Historically, drug development utilizing co-crystal technology has been under-researched, and the few pharmaceutical co-crystals that were in development contained a single API with coformer(s).23,24,26 Co-crystals were therefore seen by regulatory bodies as a type of drug formulation involving an API and a coformer, with the latter considered to be an excipient. Hence, co-crystals were considered to be pharmaceutical intermediates20,23,29 and therefore were required to be manufactured in a pharmaceutical establishment under Current Good Manufacturing Practices (CGMP) applicable to dosage forms. In recent years, however, regulatory guidance has evolved alongside scientific advances in solid-state forms. In the USA, the FDA has recently re-classified co-crystals from a regulatory perspective so that they are considered similar to a polymorph of an API.26 The European Medicines Agency (EMA) published a reflection paper in 2015 in which co-crystals are considered in the same manner as hydrates, solvates, salts, and polymorphs, with co-crystals of a previously licensed drug(s) not classified as new active substances unless they have different efficacy and/or safety than the constituent APIs. The EMA considers that the formation of co-crystals is normally subject to compliance with CGMP for APIs, as is the case for salts.24
Co-crystallization of an API within an API–coformer co-crystal has several potential physicochemical and pharmacokinetic (PK) benefits, compared with the API alone.23,24,26,30–34 For example, over 100 co-crystal studies have shown improved solubility and/or dissolution rates,23 properties that can lead to optimized oral absorption and enhanced bioavailability.31 Other promising effects include changes in drug biodistribution, permeability, and metabolism.31,34 The physicochemical properties of an API – for example, chemical and physical stability, photostability, melting points, particle morphology and size, tableting and compaction, and tendency to form agglomerates23,34 – can also be optimized via co-crystallization, leading to easier manufacturing processes. In recent years, there has been renewed interest in the field of pharmaceutical co-crystals, but to date there are relatively few examples that have been successfully commercialized. The antidepressant escitalopram, originally thought to be a salt, is now known to be a salt/co-crystal complex (escitalopram oxalate–oxalic acid; Cipralex® [Lundbeck Ltd, Valby, Denmark], Lexapro® [Allergan, Irvine, CA, USA]), and the sodium-glucose cotransporter 2 inhibitors ipragliflozin–L-proline (Suglat® [Astellas Pharma, Inc., Tokyo, Japan]) and ertugliflozin–L-pyroglutamic acid (Steglatro™ [Merck and Co., Inc., Whitehouse Station, NJ, USA]) – approved treatments for type 2 diabetes – are co-crystals.23,33–39 In the case of ipragliflozin–L-proline, the co-crystal was developed in order to improve drug quality, since ipragliflozin changed to a non-stoichiometric hydrate under certain hygrothermal conditions. The L-proline co-crystal provided a form with superior stability and constant quality with good reproducibility.40 Specific benefits of co-crystallization with a coformer have also been reported for other APIs. For example, the anticonvulsant valproic acid, which is liquid at room temperature, was co-crystallized with sodium valproate to form sodium valproate–valproic acid (Depakote® [Abbvie Inc., North Chicago, IL, USA]), a product that is solid at room temperature and provides enhanced stability compared with salts of valproic acid, including sodium valproate itself.23,41 A co-crystal of the anti-emetic aprepitant (aprepitant–L-proline monohydrate; Figure 2A), which has a significantly improved dissolution rate compared with aprepitant, has also been patented.42 In addition, co-crystals of the muscle relaxant metaxalone have been reported to improve preclinical in vivo bioavailability.23 While such improvements, conferred by co-crystallization, provide benefits with respect to the ease of drug development and – due to improved bioavailability – may permit administration of lower doses of the API therein, API–coformer co-crystals do not offer additional clinical benefits (ie, the clinical efficacy and safety profiles of the constituent APIs are unlikely to be altered). In contrast, and as we discuss in the following sections, API–API co-crystals have the potential to provide an improved clinical profile compared with commercially available single-entity reference products administered alone or in free combination.
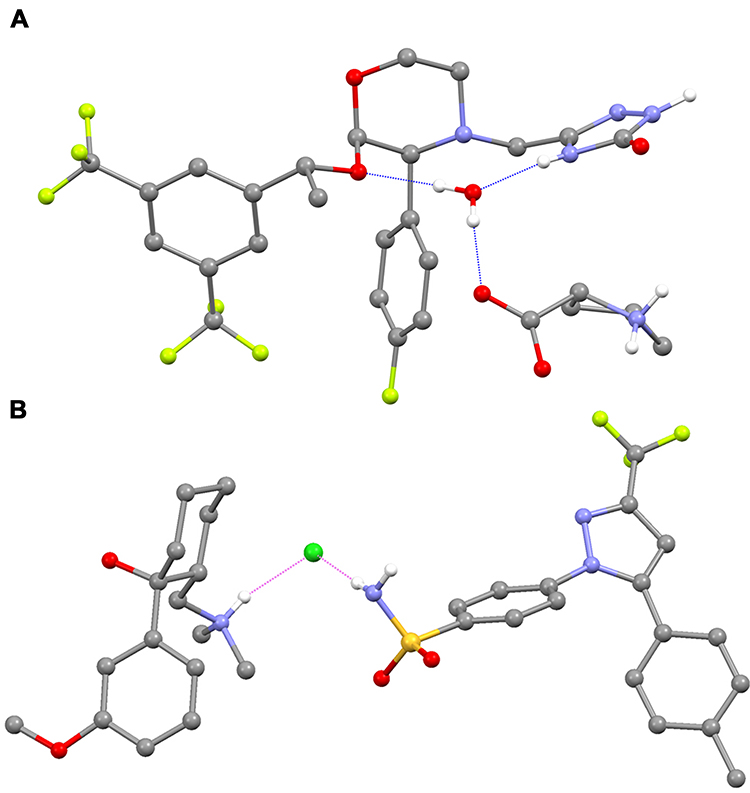 |
Figure 2 Structure of (A) an example API–coformer co-crystal (aprepitant–L-proline monohydrate)42 and (B) an example API–API co-crystal (CTC). Notes: Figure B is reproduced with permission from Almansa C, Mercè R, Tesson N, et al. Co-crystal of tramadol hydrochloride–celecoxib (ctc): a novel API–API co-crystal for the treatment of pain. Cryst Growth Des. 2017;17:1884–1892.59 Available from: https://pubs.acs.org/action/doSearch?AllField=10.1021%2Facs.cgd.6b01848. © 2017 American Chemical Society; further permissions related to the material excerpted should be directed to the ACS.Abbreviations: API, active pharmaceutical ingredient; CTC, co-crystal of tramadol-celecoxib in a 1:1 molecular ratio. |
API–API co-crystals
API–API co-crystals are unique, single-entity, solid forms28 and as such they differ from open combinations or FDCs. Co-crystals containing more than one active ingredient were first commercialized in the agrochemical arena.43 BASF SE (Ludwigshafen, Germany) developed Faban®, which contains the two fungicides pyrimethanil and dithianon. Compared with individual reference products, Faban® decreases pyrimethanil volatility and achieves greater disease control.43 In the pharmaceutical field, API–API co-crystals have the potential to deliver multimodal therapy and to provide benefits over FDCs (Figure 3). In terms of physicochemical and formulation-related benefits, API–API co-crystals may offer unique dissolution profiles, possibly leading to increased bioavailability and other clinically relevant changes to the PK of one or more of the component APIs.31,44,45 API–API co-crystals differentially offer synchronized release of both active components, potentially favoring unique pharmacodynamic interactions and synergistic effects20 in terms of clinical efficacy. As is the case for FDCs and single multimodal agents,34,46 API–API co-crystals may reduce the number of prescriptions and associated administrative costs, and increase patient compliance. Finally, API–API co-crystals may also provide a way to counter problems often associated with the development of FDCs in terms of stability, solubility differences and chemical interactions between individual APIs; for example, by improving stability and tabletability.20,23,47,48
Although their potential benefits are numerous, challenges also exist in the development of API–API co-crystals. It is difficult, for example, to identify APIs with appropriate relative potencies, such that optimal dosing can be achieved within the necessary stoichiometric constraints of a co-crystal; furthermore, not all drugs can be co-crystallized.20,23 Such challenges mean that API–API co-crystals are even scarcer than API–coformer co-crystals, with few appearing to have progressed beyond patent application or early development stages.20 Recently, the successful chemical synthesis of a 5-fluorocytosine–5-fluorouracil co-crystal was reported in the field of oncology, which the research team suggest might provide a starting point for further exploring the potential utility of API–API co-crystals in the therapy area.49 Only one marketed compound, sacubitril/valsartan (Entresto® [Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA]), which has been defined as a supramolecular entity (ie, a chemical entity composed of more than one type of molecule), can be considered an API–API co-crystal.23,50 Entresto®, which is used in the treatment of heart failure, comprises a complex containing anionic forms of sacubitril and valsartan, sodium cations and water molecules in the molar ratio of 1:1:3:2.5, respectively.50–53 In Entresto®, valsartan’s bioavailability is increased compared with commercially available single-entity reference products (ie, other tablet formulations). Approved 26, 51,and 103 mg valsartan doses in Entresto® are equivalent to 40, 80, and 160 mg valsartan doses, respectively, of commercially available reference product.51,54 Hence, in the case of Entresto®, dosing is adjusted such that the administered dose of valsartan is reduced accordingly.
API–API co-crystals in the treatment of pain
In the context of pain and multimodal analgesia, it is particularly important that the individual APIs within an API–API co-crystal have complementary mechanisms of action. For example, ensuring inclusion of an API with anti-inflammatory properties is likely to provide therapeutic benefits, as many pain states have an inflammatory component; indeed, anti-inflammatory agents are a recommended part of multimodal treatment regimens.7,16,55 API selection also presents the opportunity to include both centrally and peripherally mediated forms of analgesia.56,57
Few API–API co-crystals for the treatment of pain appear to have progressed beyond the patent application or early development stage.20 Therefore, at present, multimodal analgesia is primarily achieved via the use of open combinations or FDCs. Currently available FDCs include those containing NSAIDs, opioids and/or paracetamol, for example tramadol/dexketoprofen, tramadol/paracetamol, hydrocodone/paracetamol, oxycodone/paracetamol, codeine/paracetamol, etodolac/paracetamol, ibuprofen/paracetamol, hydrocodone/ibuprofen, and aspirin/paracetamol.15,58 API–API co-crystals patented or reportedly in early development in the pain arena include ethenzamide–gentisic acid, aceclofenac–paracetamol, paracetamol–indomethacin and mefenamic acid, tramadol–paracetamol, tramadol–naproxen, and meloxicam–aspirin.20 Although it did not progress to clinical development, meloxicam–aspirin represents an informative case study, illustrating the ability of API–API co-crystals to improve API bioavailability. In a rat PK study, the oral bioavailability of meloxicam from a meloxicam–aspirin co-crystal was higher than from meloxicam alone (69% versus 16%, respectively). Maximum plasma concentrations of meloxicam were also increased after administration of the co-crystal, although time to achieve maximum plasma concentration was similar versus the reference meloxicam. The authors of this study speculated that these effects could lead to a more rapid onset of analgesia, proposing that plasma levels of meloxicam after co-crystal administration might reach those required for therapeutic effect sooner than from reference meloxicam.45
Co-crystal of tramadol-celecoxib (CTC) is the only API–API co-crystal in late-stage clinical development for the treatment of acute pain. CTC is a unique, single-entity59 that contains two APIs belonging to well-known and complementary analgesic classes: rac-tramadol.HCl and celecoxib (in a 1:1 molecular ratio [1:1.27 weight ratio]; Figure 2B). Together, these two APIs recruit four of the most relevant mechanisms of action for targeting pain (Figure 4). Tramadol is a centrally acting mu-opioid receptor agonist, as is its active O-desmethyl metabolite (M1). Additionally, the dextrorotatory enantiomer of tramadol is an inhibitor of serotonin reuptake, whereas the levorotatory enantiomer of tramadol inhibits noradrenaline reuptake. Tramadol therefore recruits three analgesic mechanisms of action, and the single-entity product is indicated for the treatment of moderate to severe pain.60 Celecoxib is a NSAID that preferentially inhibits cyclooxygenase-2 (COX-2) over COX-161 and is indicated for the relief of chronic pain in osteoarthritis, rheumatoid arthritis and ankylosing spondylitis, as well as for acute pain and primary dysmenorrhea.62,63 Of note, celecoxib may have an improved cardiac safety profile compared with some other COX-2 inhibitors, and moderate doses are non-inferior to ibuprofen or naproxen with regards to cardiovascular safety.64,65 Combining tramadol and celecoxib provides both peripherally and centrally mediated analgesia, and has the potential to improve efficacy and safety profiles compared with existing pain therapies.59,66
Analytical studies demonstrated that the intrinsic dissolution rates (IDRs) of rac-tramadol.HCl and celecoxib are modified by co-crystallization within CTC ( in a 1:1 molecular ratio). Specifically, the IDR of celecoxib from CTC is faster than from celecoxib alone, while that of tramadol from CTC is slower than from rac-tramadol.HCl alone.59 These modified physicochemical properties appear to confer modified preclinical and clinical profiles to the constituent APIs (as proposed in Figure 3). In a rat study, for example, a suspension of CTC exerted supra-additive anti-nociceptive effects (Figure 5), without potentiating adverse effects in separate single-dose safety studies, suggesting potential for an improved clinical benefit–risk ratio, compared with the commercially available, single-entity reference products.67 In single- and multiple-dose Phase I clinical studies, the PK profiles of both tramadol and celecoxib were modified after oral administration of CTC, compared with the administration of the reference products alone or in open combination (Figure 6).66,68 These findings were consistent with the modified IDRs seen in analytical studies.59 It is possible that these modified PK profiles may provide clinical benefits, such as an improved safety profile for tramadol, due to the reduction in its peak levels, and an earlier onset of celecoxib-mediated analgesia. This is a unique finding, which provides a differential profile in the case of CTC; one that is not only based on increasing the bioavailability of the constituent APIs. Evidence from a Phase II study of acute moderate to severe pain following extraction of two or more impacted third molars requiring bone removal appears to confirm that this may translate into an improved clinical profile. In this study, CTC tablets (tramadol/celecoxib) 44 mg/56 mg, 66 mg/84 mg, and 88 mg/112 mg had significantly improved efficacy compared with 100 mg immediate-release tramadol.69 The highest dose of CTC tested contains 88 mg rac-tramadol.HCl; thus, the co-crystal may potentially provide improved efficacy at lower tramadol doses. Overall, CTC was well tolerated in completed Phase I and Phase II clinical studies.66,68,69 Taken together, these preclinical and clinical data on CTC support the theory that API–API co-crystals may provide improved clinical properties (Figure 3).
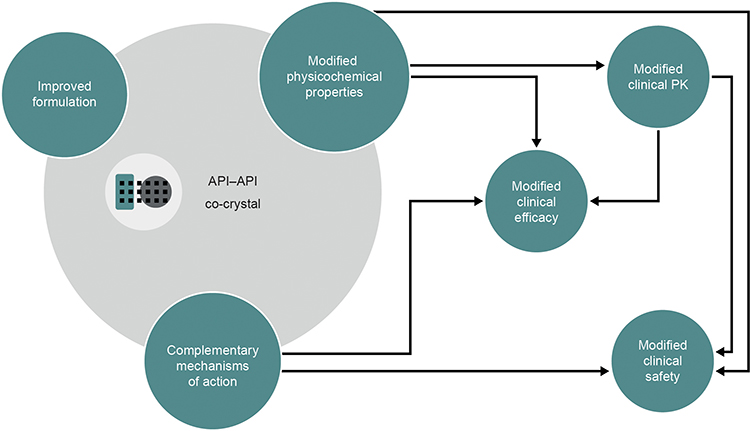 |
Figure 3 The potential impact of an API–API co-crystal on the physicochemical and clinical properties of the constituent APIs. Abbreviations: API, active pharmaceutical ingredient; PK, pharmacokinetics. |
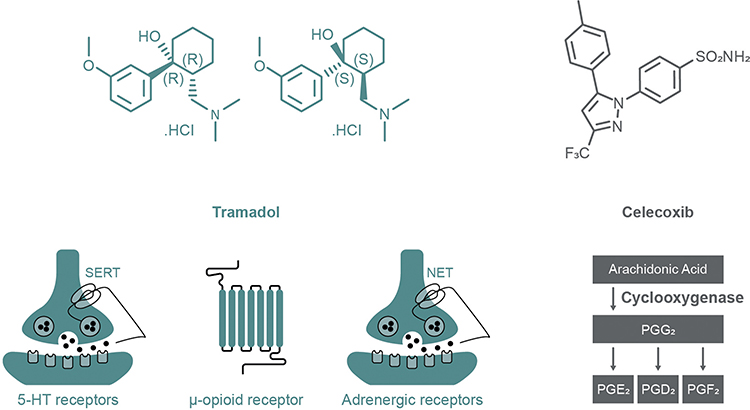 |
Figure 4 Mechanisms of action of the constituent APIs within the API–API co-crystal CTC. Abbreviations: 5-HT, 5-hydroxytryptamine; API, active pharmaceutical ingredient; CTC, co-crystal of tramadol-celecoxib; NET, norepinephrine transporter; PG, prostaglandin; SERT, serotonin transporter. |
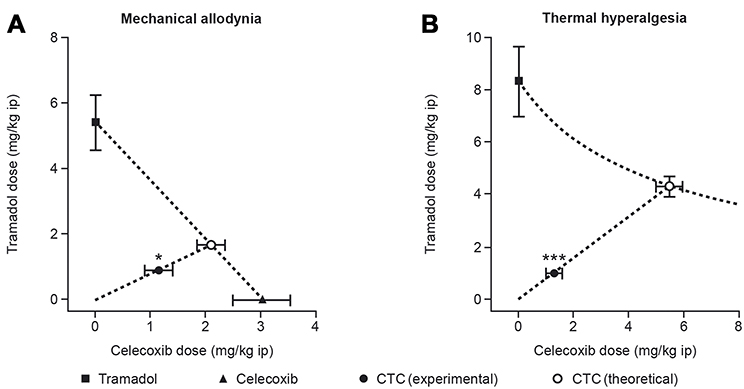 |
Figure 5 The anti-nociceptive effects of an API–API co-crystal (a suspension of CTC) in a rat model of acute postsurgical pain (n=8–10 per group). *p<0.05, ***p<0.001 significant difference between the theoretical additive dose and the experimental additive dose (Student’s t-test). Notes: Isobolographic analysis of the effects of a suspension of CTC in a rat pain model showed supra-additive effects on both mechanical allodynia (A) and thermal hyperalgesia (B) endpoints. Figures A and B are reproduced from Merlos M, Portillo-Salido E, Brenchat A, et al. Administration of a co-crystal of tramadol and celecoxib in a 1:1 molecular ratio produces synergistic antinociceptive effects in a postoperative pain model in rats. Eur J Pharmacol. 2018;833:370–378.67 © 2018 The Authors. Published by Elsevier B.V. License available at: https://creativecommons.org/licenses/by-nc-nd/4.0/legalcode. Abbreviations: API, active pharmaceutical ingredient; CTC, co-crystal of tramadol-celecoxib in a 1:1 molecular ratio; ip, intraperitoneal. |
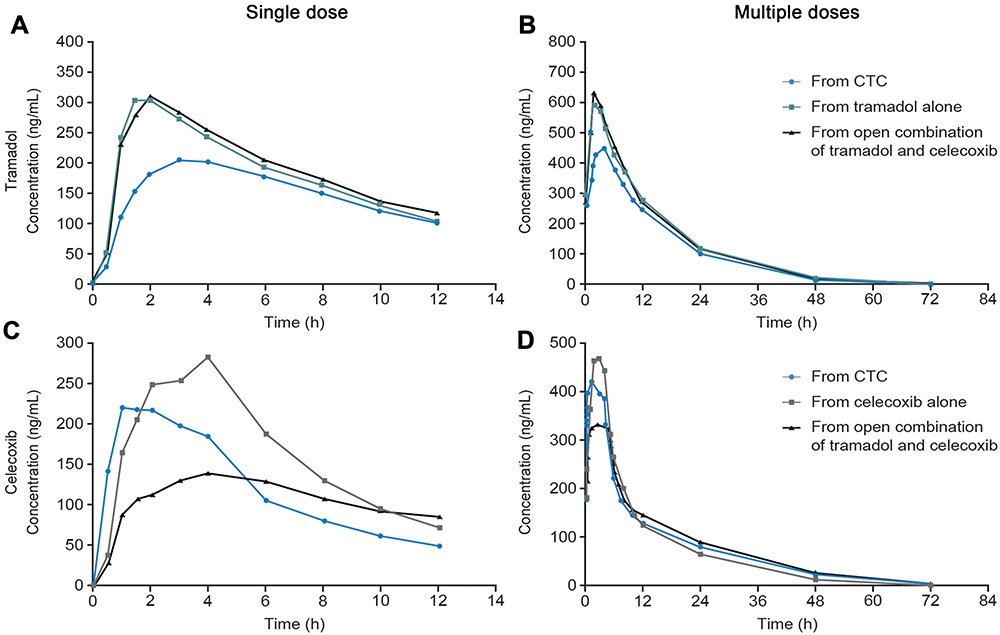 |
Figure 6 The clinical pharmacokinetics of tramadol and celecoxib after the administration of the API–API co-crystal CTC, compared with the individual, commercially available, single-entity reference products administered alone or in open combination (n=28 celecoxib alone; n=28 open combination of immediate-release tramadol and celecoxib; n=29 CTC; n=30 immediate-release tramadol alone).68. Notes: The pharmacokinetic profiles of tramadol (A) and (B) and celecoxib (C) and (D) were modified after administration of CTC, compared with administration of the commercially available, single-entity reference products alone or in open combination. Figures A–D are reproduced with minor modifications (minor changes to the size of individual points, axis title fonts, format of units on y-axes, and minor changes to the key) from Videla S, Lahjou M, Vaque A, et al. Pharmacokinetics of multiple doses of co-crystal of tramadol-celecoxib: findings from a 4-way randomized open-label Phase I clinical trial. Br J Clin Pharmacol. 2018;84(1):64–78. © 2017 The Authors. British Journal of Clinical Pharmacology published by John Wiley & Sons Ltd on behalf of British Pharmacological Society. License available at: https://creativecommons.org/licenses/by-nc-nd/4.0/legalcode. Abbreviations: API, active pharmaceutical ingredient; CTC, co-crystal of tramadol-celecoxib. |
Future perspectives
API–API co-crystals have the potential to impact clinical practice and the future clinical management of pain. For example, modified physicochemical properties and complementary mechanisms of action may lead to synchronized release, synergistic analgesic effects without potentiating adverse events, and modified clinical profiles. These may in turn permit lower doses of individual APIs, thus reducing tolerability issues and associated costs. However, after reviewing the current literature and development status of API–API co-crystals in general and in the pain field in particular, it becomes apparent that identifying APIs with complementary mechanisms of action that can be used at adequate doses for obtaining suitable co-crystals is challenging. Consequently, the number of analgesic API–API co-crystals developed in the future is likely to continue to be limited.
Conclusions
Pharmaceutical co-crystals that contain a single API, co-crystallized with non-active coformer(s) provide a means to improve the API’s physicochemical and PK properties and can permit the development of drugs that would otherwise be difficult to formulate. However, although a few API-coformer co-crystals have reached the market, none of them is in the field of pain.
API–API co-crystals may provide additional advantages, such as the possibility of improved clinical efficacy and/or safety. With respect to pain management, the use of an API–API co-crystal could also deliver improved, multimodal analgesia. This approach also offers benefits over the use of FDCs, which can be subject to difficulties in development. Despite these benefits, and a number of patent applications, few API–API co-crystals have reached late-stage clinical development, and the development of such agents remains challenging. CTC is the only API–API co-crystal in late-stage development for the treatment of pain and is currently in Phase III development.70–72 In contrast to other API–API co-crystals developed to date, findings with CTC suggest a correlation between differentiated physicochemical profiles (of individual APIs) and beneficial changes in clinical PK that may translate into improved clinical efficacy and safety profiles.
 |
Table 1 Glossary of terms |
Abbreviations
5-HT, 5-hydroxytryptamine; API, active pharmaceutical ingredient; CGMP, Current Good Manufacturing Practices; COX, cyclooxygenase; CTC, co-crystal of tramadol-celecoxib (in a 1:1 molecular ratio); EMA, European Medicines Agency; FDA, US Food and Drug Administration; FDC, fixed-dose combination; IDR, intrinsic dissolution rate; ip, intraperitoneal; M1, O-desmethyl tramadol; NET, norepinephrine transporter; PG, prostaglandin; PK, pharmacokinetics; SERT, serotonin transporter.
Acknowledgments
Medical writing support for this review article was provided by Hannah Mace, MPharmacol, CMPP, at Aspire Scientific (Bollington, UK), and was funded by Mundipharma Research Ltd (Cambridge, UK). Mundipharma Research and Esteve Pharmaceuticals, S.A. have been collaborating on the development of co-crystal of tramadol-celecoxib.
Disclosure
Carmen Almansa, José Miguel Vela, and Carlos R Plata-Salamán are employees of Esteve Pharmaceuticals S.A. Steve Whitelock is an employee of Mundipharma Research Ltd. Carlos R Plata-Salamán has been named as an inventor in a patent application filed by Laboratorios del Dr Esteve, S.A., entitled “Co-crystals of Tramadol and Coxibs" (WO 2011044962). The authors report no other conflicts of interest in this work.
References
1. Faculty of Pain Medicine. The Royal College of Anaesthetists. Core standards for pain management services in the UK; 2015. Available from: https://www.rcoa.ac.uk/system/files/FPM-CSPMS-UK2015.pdf.
2. Mura P, Serra E, Marinangeli F, et al. Prospective study on prevalence, intensity, type, and therapy of acute pain in a second-level urban emergency department. J Pain Res. 2017;10:2781–2788. doi:10.2147/JPR.S134133
3. Gregory J, McGowan L. An examination of the prevalence of acute pain for hospitalised adult patients: a systematic review. J Clin Nurs. 2016;25(5–6):583–598. doi:10.1111/jocn.2016.25.issue-5pt6
4. Fayaz A, Croft P, Langford RM, Donaldson LJ, Jones GT. Prevalence of chronic pain in the UK: a systematic review and meta-analysis of population studies. BMJ Open. 2016;6(6):e010364. doi:10.1136/bmjopen-2015-010364
5. Leadley RM, Armstrong N, Lee YC, Allen A, Kleijnen J. Chronic diseases in the European Union: the prevalence and health cost implications of chronic pain. J Pain Palliat Care Pharmacother. 2012;26(4):310–325. doi:10.3109/15360288.2012.736933
6. Moore RA, Derry S, Aldington D, Wiffen PJ. Single dose oral analgesics for acute postoperative pain in adults - an overview of Cochrane reviews. Cochrane Database Syst Rev. 2015;(9):CD008659.
7. Chou R, Gordon DB, de Leon-Casasola OA, et al. Management of postoperative pain: a clinical practice guideline from the American Pain Society, the American Society of Regional Anesthesia and Pain Medicine, and the American Society of Anesthesiologists’ Committee on Regional Anesthesia, Executive Committee, and Administrative Council. J Pain. 2016;17(2):131–157. doi:10.1016/j.jpain.2015.12.006
8. Sinatra R. Causes and consequences of inadequate management of acute pain. Pain Med. 2010;11(12):1859–1871. doi:10.1111/j.1526-4637.2010.00983.x
9. Borsook D, Hargreaves R, Bountra C, Porreca F. Lost but making progress–where will new analgesic drugs come from? Sci Transl Med. 2014;6(249):249sr243. doi:10.1126/scitranslmed.3008320
10. Gan TJ. Poorly controlled postoperative pain: prevalence, consequences, and prevention. J Pain Res. 2017;10:2287–2298. doi:10.2147/JPR.S134133
11. Kehlet H, Jensen TS, Woolf CJ. Persistent postsurgical pain: risk factors and prevention. Lancet. 2006;367(9522):1618–1625. doi:10.1016/S0140-6736(06)68700-X
12. Meissner W, Coluzzi F, Fletcher D, et al. Improving the management of post-operative acute pain: priorities for change. Curr Med Res Opin. 2015;31(11):2131–2143. doi:10.1185/03007995.2015.1092122
13. International Association for the Study of Pain. Task force on multimodal pain treatment defines terms for chronic pain care; 2017. Available from: https://www.iasp-pain.org/PublicationsNews/NewsDetail.aspx?ItemNumber=6981.
14. Raffa RB, Pergolizzi JV
15. O’Brien J, Pergolizzi JV, van der Laar M, et al. Fixed-dose combinations at the front line of multimodal pain management: perspective of the nurse-prescriber. Nurs Res Rev. 2013;3:9–22. doi:10.2147/NRR.S36876
16. Elvir-Lazo OL, White PF. The role of multimodal analgesia in pain management after ambulatory surgery. Curr Opin Anesthesiol. 2010;23(6):697–703. doi:10.1097/ACO.0b013e32833fad0a
17. Dale R, Stacey B. Multimodal treatment of chronic pain. Med Clin North Am. 2016;100(1):55–64. doi:10.1016/j.mcna.2015.08.012
18. Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem. 2005;48(21):6523–6543. doi:10.1021/jm049494r
19. Dhillon S. Tramadol/paracetamol fixed-dose combination: a review of its use in the management of moderate to severe pain. Clin Drug Investig. 2010;30(10):711–738. doi:10.2165/11205830-000000000-00000
20. Thipparaboina R, Kumar D, Chavan RB, Shastri NR. Multidrug co-crystals: towards the development of effective therapeutic hybrids. Drug Discov Today. 2016;21(3):481–490. doi:10.1016/j.drudis.2016.02.001
21. Gattrell W, Johnstone C, Patel S, Smith CS, Scheel A, Schindler M. Designed multiple ligands in metabolic disease research: from concept to platform. Drug Discov Today. 2013;18(15–16):692–696. doi:10.1016/j.drudis.2013.02.006
22. Geldenhuys WJ, Youdim MB, Carroll RT, Van der Schyf CJ. The emergence of designed multiple ligands for neurodegenerative disorders. Prog Neurobiol. 2011;94(4):347–359. doi:10.1016/j.pneurobio.2011.04.010
23. Duggirala NK, Perry ML, Almarsson O, Zaworotko MJ. Pharmaceutical cocrystals: along the path to improved medicines. Chem Commun (Camb). 2016;52(4):640–655. doi:10.1039/c5cc09289b
24. European Medicines Agency. Reflection paper on the use of cocrystals of active substances in medicinal products; 2015. Available from: https://www.ema.europa.eu/documents/scientific-guideline/reflection-paper-use-cocrystals-active-substances-medicinal-products_en.pdf.
25. Aitipamula S, Banerjee R, Bansal AK, et al. Polymorphs, salts, and cocrystals: what’s in a name? Cryst Growth Des. 2012;12(5):2147–2152. doi:10.1021/cg3002948
26. US Food and Drug Administration. Guidance for industry. Regulatory classification of pharmaceutical co-crystals; 2018. Available from: https://www.fda.gov/downloads/Drugs/Guidances/UCM281764.pdf.
27. Lara-Ochoa F, Espinosa-Pérez G. Cocrystals definitions. Supramol Chem. 2007;19(8):553–557. doi:10.1080/10610270701501652
28. Sekhon BS. Drug-drug co-crystals. Daru. 2012;20(1):45. doi:10.1186/2008-2231-20-45
29. US Food and Drug Administration. Guidance for industry. Regulatory classification of pharmaceutical co-crystals; 2013. Available from: https://www.fda.gov/downloads/Drugs/Guidances/UCM281764.pdf.
30. Thakuria R, Delori A, Jones W, Lipert MP, Roy L, Rodriguez-Hornedo N. Pharmaceutical cocrystals and poorly soluble drugs. Int J Pharm. 2013;453(1):101–125. doi:10.1016/j.ijpharm.2012.10.043
31. Shan N, Perry ML, Weyna DR, Zaworotko MJ. Impact of pharmaceutical cocrystals: the effects on drug pharmacokinetics. Expert Opin Drug Metab Toxicol. 2014;10(9):1255–1271. doi:10.1517/17425255.2014.865723
32. Steed JW. The role of co-crystals in pharmaceutical design. Trends Pharmacol Sci. 2013;34(3):185–193. doi:10.1016/j.tips.2012.12.003
33. Brittain HG. Pharmaceutical cocrystals: the coming wave of new drug substances. J Pharm Sci. 2013;102(2):311–317. doi:10.1002/jps.23402
34. Bolla G, Nangia A. Pharmaceutical cocrystals: walking the talk. Chem Commun (Camb). 2016;52(54):8342–8360. doi:10.1039/c5cc09289b
35. Astellas Pharma Inc. Approval of Suglat® tablets, a selective SGLT2 inhibitor for treatment of type 2 diabetes, in Japan; 2014. Available from: https://www.astellas.com/en/corporate/news/detail/approval-of-suglat-tablets-a-s.html.
36. Ohkura T. Ipragliflozin: a novel sodium-glucose cotransporter 2 inhibitor developed in Japan. World J Diabetes. 2015;6(1):136–144. doi:10.4239/wjd.v6.i1.136
37. Markham A. Ertugliflozin: first global approval. Drugs. 2018;78(4):513–519. doi:10.1007/s40265-018-0878-6
38. Mascitti V, Maurer TS, Robinson RP, et al. Discovery of a clinical candidate from the structurally unique dioxa-bicyclo[3.2.1]octane class of sodium-dependent glucose cotransporter 2 inhibitors. J Med Chem. 2011;54(8):2952–2960. doi:10.1021/jm200049r
39. Frampton C Cocrystal clear solutions; 2010. Available from: https://www.soci.org/Chemistry-and-Industry/CnI-Data/2010/5/Cocrystal-clear-solutions.
40. Imamura M, Nakanishi K, Shiraki R, Onda K, Sasuga D, Yuda M, inventors. Cocrystal of C-glycoside derivative and L-proline. Patent US8097592B2. 2007.
41. Sherman B, inventor. Solid substances comprising valproic acid and sodium valproate. U.S. Patent 6077542. 2000.
42. Holland J, Frampton C, Chorlton A, Gooding D, inventors. Aprepitant l-proline solvates - compositions and cocrystals. WO2013076659A1. 2012.
43. Viertelhaus M, Hafner A. Co-crystals and their advantages for APIs with challenging properties. Chim Oggi. 2015;33(5):23–26.
44. Kuminek G, Cao F, de Oliveira Da Rocha AB, Cardoso SG, Rodríguez-Hornedo N. Cocrystals to facilitate delivery of poorly soluble compounds beyond-rule-of-5. Adv Drug Deliv Rev. 2016;101:143–166. doi:10.1016/j.addr.2016.04.022
45. Cheney ML, Weyna DR, Shan N, Hanna M, Wojtas L, Zaworotko MJ. Coformer selection in pharmaceutical cocrystal development: a case study of a meloxicam aspirin cocrystal that exhibits enhanced solubility and pharmacokinetics. J Pharm Sci. 2011;100(6):2172–2181. doi:10.1002/jps.22434
46. Collier R. Reducing the “pill burden”. CMAJ. 2012;184(2):E117–118. doi:10.1503/cmaj.109-4076
47. Maeno Y, Fukami T, Kawahata M, et al. Novel pharmaceutical cocrystal consisting of paracetamol and trimethylglycine, a new promising cocrystal former. Int J Pharm. 2014;473(1–2):179–186. doi:10.1016/j.ijpharm.2014.07.008
48. Desai D, Wang J, Wen H, Li X, Timmins P. Formulation design, challenges, and development considerations for fixed dose combination (FDC) of oral solid dosage forms. Pharm Dev Technol. 2013;18(6):1265–1276. doi:10.3109/10837450.2012.660699
49. Da Silva CCP, de Melo CC, Souza MS, Diniz LF, Carneiro RL, Ellena J. 5-fluorocytosine/5-fluorouracil drug-drug cocrystal: a new development route based on mechanochemical synthesis. J Pharm Innov. 2018; 14:50–56.
50. Feng L, Karpinski P, Sutton P, et al. LCZ696: a dual-acting sodium supramolecular complex. Tetrahedron Lett. 2012;53(3):275–276. doi:10.1016/j.tetlet.2011.11.029
51. Novartis Pharmaceuticals Corporation. Entresto prescribing information; 2017. Available from: https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/entresto.pdf.
52. European Medicines Agency. Entresto assessment report; 2015. Available from: https://www.ema.europa.eu/documents/assessment-report/entresto-epar-public-assessment-report_en.pdf.
53. Novartis. Novartis’ new heart failure medicine LCZ696 approved by FDA to reduce risk of cardiovascular death and heart failure hospitalization; now called Entresto™ (sacubitril/valsartan); 2015. Available from: http://www.multivu.com/players/English/7488651-novartis-fda-approval-entresto/.
54. Novartis Europharm Limited. Entresto summary of product characteristics; 2017. Available from: https://www.ema.europa.eu/documents/product-information/entresto-epar-product-information_en.pdf.
55. Polomano RC, Fillman M, Giordano NA, Vallerand AH, Nicely KL, Jungquist CR. Multimodal analgesia for acute postoperative and trauma-related pain. Am J Nurs. 2017;117(3 Suppl 1):S12–26. doi:10.1097/01.NAJ.0000526748.61302.b6
56. Zukowski M, Kotfis K. The use of opioid adjuvants in perioperative multimodal analgesia. Anaesthesiol Intensive Ther. 2012;44(1):42–46.
57. Maxwell C, Nicoara A. New developments in the treatment of acute pain after thoracic surgery. Curr Opin Anaesthesiol. 2014;27(1):6–11. doi:10.1097/ACO.0000000000000029
58. Varrassi G, Hanna M, Macheras G, et al. Multimodal analgesia in moderate-to-severe pain: a role for a new fixed combination of dexketoprofen and tramadol. Curr Med Res Opin. 2017;33(6):1165–1173. doi:10.1080/03007995.2017.1301903
59. Almansa C, Mercè R, Tesson N, Farran J, Tomàs J, Plata-Salamán CR. Co-crystal of tramadol hydrochloride–celecoxib (ctc): a novel API–API co-crystal for the treatment of pain. Cryst Growth Des. 2017;17(4):1884–1892. doi:10.1021/acs.cgd.6b01848
60. Grond S, Sablotzki A. Clinical pharmacology of tramadol. Clin Pharmacokinet. 2004;43(13):879–923. doi:10.2165/00003088-200443130-00004
61. Penning TD, Talley JJ, Bertenshaw SR, et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-methylphenyl)-3- (trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, celecoxib). J Med Chem. 1997;40(9):1347–1365. doi:10.1021/jm960803q
62. McCormack PL. Celecoxib: a review of its use for symptomatic relief in the treatment of osteoarthritis, rheumatoid arthritis and ankylosing spondylitis. Drugs. 2011;71(18):2457–2489. doi:10.2165/11208240-000000000-00000
63. Frampton JE, Keating GM. Celecoxib: a review of its use in the management of arthritis and acute pain. Drugs. 2007;67(16):2433–2474. doi:10.2165/00003495-200767160-00008
64. Wadman M. The pain game. Nature. 2007;448(7152):400–401. doi:10.1038/nature05984
65. Nissen SE, Yeomans ND, Solomon DH, et al. Cardiovascular safety of celecoxib, naproxen, or ibuprofen for arthritis. N Engl J Med. 2016;375(26):2519–2529. doi:10.1056/NEJMoa1611593
66. Videla S, Lahjou M, Vaque A, et al. Single-dose pharmacokinetics of co-crystal of tramadol-celecoxib: results of a four-way randomized open-label phase I clinical trial in healthy subjects. Br J Clin Pharmacol. 2017;83(12):2718–2728. doi:10.1111/bcp.13395
67. Merlos M, Portillo-Salido E, Brenchat A, et al. Administration of a co-crystal of tramadol and celecoxib in a 1:1 molecular ratio produces synergistic antinociceptive effects in a postoperative pain model in rats. Eur J Pharmacol. 2018;833:370–378. doi:10.1016/j.ejphar.2018.06.022
68. Videla S, Lahjou M, Vaque A, et al. Pharmacokinetics of multiple doses of co-crystal of tramadol-celecoxib: findings from a 4-way randomized open-label Phase I clinical trial. Br J Clin Pharmacol. 2018;84(1):64–78. doi:10.1111/bcp.13428
69. López-Cedrún J, Videla S, Burgueño M, et al. Co-crystal of tramadol-celecoxib in patients with moderate to severe acute post-surgical oral pain: a dose-finding, randomised, double-blind, placebo- and active-controlled, multicentre, phase II trial. Drug R D. 2018;18(2):137–148. doi:10.1007/s40268-018-0235-y
70. ClinicalTrials.gov. Efficacy and safety in a randomised acute pain study of MR308 (tramadol/celecoxib). (STARDOM1); 2018. Available from: https://clinicaltrials.gov/ct2/show/NCT02982161?term=MR308&rank=1.
71. ClinicalTrials.gov. Efficacy and safety in a randomised acute pain study of MR308: STARDOM2. (STARDOM2); 2018. Available from: https://clinicaltrials.gov/ct2/show/NCT03062644?term=MR308&rank=2.
72. ClinicalTrials.gov. Co-crystal E-58425 vs tramadol and celecoxib for moderate to severe acute pain after bunionectomy. Phase III clinical trial; 2018. Available from: https://clinicaltrials.gov/ct2/show/NCT03108482.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.