")
Back to Journals » Journal of Inflammation Research » Volume 15
Co-Application of C16 and Ang-1 Improves the Effects of Levodopa in Parkinson Disease Treatment
Authors Fu XX, Wang J , Cai HY, Jiang H, Jiang JZ, Chen HH, Han S
Received 1 April 2022
Accepted for publication 29 June 2022
Published 7 July 2022 Volume 2022:15 Pages 3797—3814
DOI https://doi.org/10.2147/JIR.S368291
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Xiao-Xiao Fu,1,* Jin Wang,1,* Hua-Ying Cai,1 Hong Jiang,1 Jin-Zhan Jiang,2 Hao-Hao Chen,2 Shu Han1
1Institute of Anatomy and Sir Run Run Shaw Hospital, Medical College, Zhejiang University, Hangzhou, People’s Republic of China; 2Medical Molecular Biology Laboratory, School of Medicine, Jinhua Polytechnic, Jinhua, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shu Han, Institute of Anatomy, Sir Run Run Shaw Hospital, Medical College, Zhejiang University, 866 Yuhangtang Road, Hangzhou, People’s Republic of China, Tel +86-571-88208160, Fax +86-571-88208094, Email [email protected] Hao-Hao Chen, Medical Molecular Biology Laboratory, School of Medicine, Jinhua Polytechnic, 1188 Wuzhou Steet, Jinhua, People’s Republic of China, Tel +86-579-82265128, Fax +86-579-82265110, Email [email protected]
Background: Levodopa is regarded as a standard medication in Parkinson disease (PD) treatment. However, long-term administration of levodopa leads to levodopa-induced dyskinesia (LID), which can markedly affect patient quality of life. Previous studies have shown that neuroinflammation in the brain plays a role in LID and increases potential neuroinflammatory mediators associated with the side effects of levodopa.
Objective: The treatment effect of C16 (a peptide that competitively binds integrin αvβ 3 and inhibits inflammatory cell infiltration) and angiopoietin-1 (Ang-1; a vascular endothelial growth factor vital for blood vessel protection), along with levodopa, was evaluated in a rodent model of PD.
Methods: We administered a combination of C16 and Ang-1 in a rodent model of PD induced by MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine). Seventy-five mice were randomly divided into five treatment groups: control, vehicle, levodopa, C16+Ang-1, and levodopa+C16+Ang-1. Behavioral, histological, and electrophysiological experiments were used to determine neuron function and recovery.
Results: The results showed that C16+Ang-1 treatment alleviated neuroinflammation in the CNS and promoted the recovery effects of levodopa on neural function.
Conclusion: Our study suggests that C16+Ang-1 can compensate for the shortcomings of levodopa, improve the CNS microenvironment, and ameliorate the effects of levodopa. This treatment strategy could be developed as a combinatorial therapeutic in the future.
Keywords: levodopa-induced dyskinesia, Parkinson’s disease, C16+Ang-1 treatment, neuroinflammation, microenvironment
Introduction
As a chronic and progressive neurodegenerative disease, Parkinson disease (PD) is characterized by the death of dopaminergic neurons in the substantia nigra pars compacta (SNpc). Lack of dopamine in the basal ganglion cells leads to typical Parkinsonian symptoms, including tremor, rigidity, bradykinesia, and postural instability.1 Administration of levodopa, which is a dopamine substitute, was regarded as a big breakthrough in PD treatment. Since its discovery, levodopa has been considered a standard medication in the treatment of PD; almost all PD patients receive levodopa during their illness course.1 However, long-term administration of levodopa can lead to many side effects, such as motor fluctuations and dyskinesias. Until now, the underlying mechanisms for these complications have remained unclear. Dyskinesia (abnormal involuntary movements) has also been shown to affect a majority of PD patients receiving levodopa, which limits its clinical application.2
Aging is considered a primary risk factor for the development of PD. Aging-related neuroinflammation has been shown to lead to neuronal loss. Although the underlying mechanisms are not fully elucidated, microglia and further release of pro-inflammatory factors may be involved.3 It has been demonstrated that decreasing inflammation could potentially reduce dopaminergic neuronal death. Inflammation in PD may also correlate with nigral cell death. Interactions between neurons, microglia, and astrocytes have an important role in maintaining the response of neurons to levodopa post-synaptically. In support of this, levodopa-induced dyskinesia (LID) in rodents has been shown to be correlated with glial cells and neuroinflammation.4 In a rat model of LID, there was an increase in inflammatory markers in the striatum, such as neuronal cyclooxygenase-2 (COX2) and glial inducible nitric oxide synthase (iNOS). Gliosis, which is commonly seen in astrocytes and microglia in PD brains, occurred after long-term treatment of levodopa. Thus, inflammation is an important factor in PD and is considered to be an underlying cause of neuronal degeneration and requires further investigation.4,5
Previous studies have shown that malfunction of the blood-brain barrier (BBB) can lead to vascular extravasation and infiltration of peripheral immune cells, which are the major steps of the neuroinflammatory response.6 Angiopoietin-1 (Ang-1), which is a member of the vascular endothelial growth factor family, functions to establish and maintain vascular integrity, maturation, and stabilization.7 Moreover, Ang-1 has been shown to inhibit blood vessel leakage and reduce inflammatory cell infiltration.8,9 Interestingly, Ang-1 has been used to alleviate the impacts of lipopolysaccharide (LPS), by reducing pro-inflammatory cytokines in the striatum of LID mice.1,8
C16 (KAFDITYVRLKF) is a short, active peptide that can selectively and competitively bind to integrin αvβ3 and influence the integrin-dependent binding of leukocytes and endothelial cells, which are needed for inflammatory cell transmigration.9 Our previous work has shown that C16 and Ang-1 can synergistically improve the anti-inflammatory response in rats of experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis (MS).10 In this study, we investigated if the co-application of C16 and Ang-1 could also improve the inflammatory microenvironment in the central nervous system (CNS) in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD model. Thus, we sought to understand if the combinatorial treatment can improve the effects of levodopa in the treatment of PD.
Materials and Methods
Animals
Male C57/BL6 (12 weeks, weighing 25–30 g) were provided by Vital River Lab Animal Technology (Beijing, China) and maintained in a standard laboratory environment with a 12 h light-dark cycle and 40–60% humidity at 22 ± 1°C. All animals were given water and food ad libitum. The experiments were performed abiding by the NIH guidelines and approved by the Ethics of Committee of Zhejiang University Medical College (SRRSH202102016, date of approval: 10-Feb-2021).
A total of 75 mice were randomly divided into 5 groups (n = 15 per group): control, vehicle, levodopa, C16+Ang-1, and levodopa+C16+Ang-1. Except for the control group, mice were intraperitoneally (i.p.) administrated MPTP (in saline; Sigma-Aldrich, St Louis, MO, USA) at a concentration of 30 mg/kg for 5 consecutive days. Mice in the control group were injected i.p. with saline.11 All drug treatments began on the next day following MPTP injections. C16 (20 mg/kg) and/or Ang-1 (4 mg/Kg) in 1 mL phosphate buffered saline (PBS) were injected intravenously once daily for three weeks. Mice in the vehicle and control groups were intravenously administered 1 mL PBS.10 Levodopa was administered at 6 mg/kg along with 6 mg/kg benserazide i.p. once a day for 21 consecutive days.1
Open Field Test
The open field test was used to evaluate locomotor activity. Mice were placed in the open field test 21 days after MPTP injection. The test lasted for 7 days, 6 days for training and the last day for measurement. One mouse was placed in the open arena (32 × 44×44 cm) with a video camera fixed over the arena for monitoring. The 5-min activity of the animal was recorded under weak illumination (~ 40 W). Data of mean velocity, total distance traveled, and activity were analyzed using the EthoVision video tracking system (Noldus, Wageningen, Netherlands).
Pole Climbing Test
A ball (1 cm diameter) was fixed on top of a pole (height 50 cm, diameter 1 cm) and wrapped in gauze to prevent the mouse from slipping while climbing up the pole. The mice were placed on the top of the pole. The total time a mouse took to transit from the top to bottom was recorded and analyzed.12
Forepaw Gripping Ability
Each mouse was trained on a grip strength meter (GSM; TSE-Systems) for 2 days, 5 min each day. The training involved suspending each mouse by the tail just above the bar of the GSM and pulling it away from the bar by its tail in one smooth motion until its forepaw grip on the GSM bar was released. The force (in grams) at the instant before the mouse’s forepaw grip on the bar was released was measured using the GSM.12 The average of four measurements was used for analysis.
Novel Object Recognition (NOR) Test
The NOR test was used to evaluate the ability of a mouse to recognize a relatively novel object. The test was done in a sound isolated room in a chamber (60 × 40 × 50 cm). There were three phases in the test procedure: habituation (1 d), familiarization (1 d), and test phase (1 d). Novelty preference was assessed by the difference in the time each animal spent near the object in the novel and familiar locations (NL and F, respectively), and the discrimination score was defined as (NL−F)/(NL+F).13
Muscular Coordination Test
The rotarod test was used to evaluate muscular coordination in the mice. The test was performed daily for 8 days beginning 21 days after levodopa treatment. Animals were trained three times before MPTP administration. Each animal was put on a rotarod machine (KN-75, Natsume Seisakusho) at the speed of 15 rpm. The time that an animal took to fall off the device was recorded.14
Electrophysiological Analysis
A two-lead telemetry-based EMG transmitter (F20-EET, Data Sciences International, St. Paul, MN, USA) was used in the electrophysiological experiments. Three weeks after PD induction, animals were anesthetized, and the electrodes were subcutaneously embedded into the hindlimbs using blunt dissection. The electrodes were inserted through the biceps and quadriceps with a small incision in the thigh, followed by sutures. DataQuest Acquisition hardware (Data Sciences International) was used to record the EMG data, and LabChart (ADInstuments, Colorado Springs, CO, USA) was used for data processing. The transmitter frequency was set at 455 kHz to decrease interfering signals.15
Tissue Preparation and Processing
All animals were sacrificed six weeks after MPTP injection. After anesthetization with sodium pentobarbital, the animals were perfused with cold saline intracardially, and perfused with 4% paraformaldehyde (pH = 7.4) in sequence. Afterward, the brain tissues were put in 4% paraformaldehyde for 4 h, followed by being immersed in 30% sucrose for 48 h. Tissue dissection (10 µm, coronal sections) was performed with a cryostat and freezing microtome (Leica; Buffalo Grove, IL, USA). The sections were used in the following histopathological and immunofluorescent analyses.
Histology
To observe the neuronal structure, Nissl staining was performed. A total of 100 μL of 1% cresyl violet solution was dropped onto the tissue on the slide. The slides were kept for 2 h at 37°C and washed in phosphate-buffered saline (PBS) for 3×10 min. After dehydration with alcohol and xylene, the slides were mounted with resin and observed under light microscope. The samples were analyzed using an optical microscope with a 400× magnification. Only neurons with large, multipolar and pyramid-shapes, a well-defined nucleus, and adequate endoplasmic reticulum were considered as viable and healthy cells. Sections were observed by an experienced pathologist who was blinded to the experiment. At least five view fields per section were randomly selected and examined.
Transmission Electron Microscopy (TEM)
A proportion of striatum and nigra substance was fixed in 2.5% glutaraldehyde solution, immersed in 1% osmium tetroxide at 4°C, and then washed 3 times with 0.1 M PB. After fixation, the following steps were performed using an EM processor with agitation at room temperature. The tissues were dehydrated in graded ethanol (30%, 50%, 70%, 80%, 90%, 95%) for 5 min, followed by changes of absolute ethanol (3×10 min). After 2×15 min in 1, 2-propylene oxide (PO), tissues were immersed in a 1:1 PO:Epon mixture for 1 h. These tissues were then incubated overnight in pure Epon and embedded in pure Epon at 60°C for 3 days. The Epon-embedded tissues were cut into 90 nm sections with a diamond knife on an ultracut microtome and collected on a 200-mesh copper grid. Lead citrate (approximately 3%) and 8% uranyl acetate were filtered before use. The grids were stained with lead citrate droplets for 20 min in a Petri dish, washed three times with distilled water, and were then ready for electron microscopy analysis.9
Evans Blue (EB) Staining
EB staining was used to indicate edema, BBB vascular permeability, and blood vessel integrity (n = 3). Briefly, mice were anesthetized and infused with 4 mL/kg 2% EB solution through the right femoral vein at 37°C for 5 min. Two hours later, 300 mL saline was perfused to wash out the residual dye from the blood vessels. Under an ultraviolet light filter, the sections were visualized. Red staining indicated high vascular permeability in the BBB and vessels. Staining intensity was evaluated using Image J (NIH, Bethesda, MD, USA).
Tissues were then homogenized in N,N-dimethylformamide (Sigma-Aldrich) and placed in the dark for 72 h at room temperature before centrifugation at 10,000× g for 25 min. The suspension was measured at OD610 with a spectrophotometer (Molecular Devices OptiMax, USA). The content of EB dye (μg/g) was calculated according to the standard curve.9
Immunofluorescence
Tissue sections were encircled using a water-repellent pen (Invitrogen, Carlsbad, CA, USA) and then immersed in 0.01 mol/L Tris-saline buffer for 10 min. Afterward, the sections were permeabilized and blocked with 0.3% Triton X-100 containing 10% goat serum for 0.5 h. The sections were incubated with respective antibodies at 4°C overnight: mouse/rabbit anti-synaptophysin (Syn, 1:500; Abcam, Cambridge, MA, USA), anti-acetylcholinesterase (ACHE, 1:500; Thermo Fisher Scientific, Waltham, MA, USA), anti-tyrosine hydroxylase (TH) (1:500; R&D Systems, Minneapolis, MN, USA), anti-glial fibrillary acidic protein (GFAP), anti-CD-3, anti-γ-aminobutyric acid (GABA)-transporter (GAT-1), anti-ZO-1 (zonal occludens-1) (1:500; Cayman Chemical, Ann Arbor, MI, USA), anti-leucine-rich-repeat kinase 2 (LRRK2), anti-Iba-1 (1:500; Santa Cruz, CA, USA), or anti-caspase 3 (1:500; Cayman Chemical). After three washes, the sections were stained with a FITC/TRIFC-conjugated goat anti-rabbit/mouse IgG secondary antibody (1:200; Thermo Fisher Scientific, Waltham, MA, USA) for 60 min at 37°C and mounted with mounting medium (Gel Mount antifade aqueous, Southern Biotech). An inactive antibody was used as a negative control. At least five sections of the striatum and nigra substance with three visual fields on each section were randomly selected and counted. Images were acquired with a fluorescence microscope (Buffalo Grove) and analyzed using Image J software.
Inflammatory Cell Infiltration
An experienced pathologist imaged and analyzed tissue sections stained with the pan-specific leukocyte marker CD3. For each section, five randomly selected fields were analyzed. Inflammatory cell infiltration was graded as previously described:13 0, without inflammation; 1, limited cellular infiltration around blood vessels and meninges; 2, mild infiltration (1–10 inflammatory cells/slide in parenchymal tissues); 3, moderate infiltration (11–100 inflammatory cells/slide in parenchymal tissues); 4, severe infiltration (> 100 inflammatory cells/slide in parenchymal tissues).
ELISA
Animals were sacrificed and the blood samples were put at 4°C by adding heparin and immediate centrifugation at 1000×g for 20 min and then 10,000×g for 10 min. Samples were stored at −80°C until further investigation. To determine the serum cytokine levels, samples were put into 96-well plates that were pre-coated with anti-interleukin (IL)-10, anti-IL-6, anti-reactive oxygen species (ROS), anti-tumor necrosis factor-α (TNF-α), and anti-GABA (R&D Systems) antibodies. The plate was kept for 1 h at 37°C and then treated with a secondary antibody conjugated to horse radish peroxidase (HRP) (1:2000; Bio-Rad, CA, USA) at 37°C for 60 min. Absorption at 450 nm was measured (Model 680; Bio-Rad). Data were analyzed using GraphPad Prism 4.0 (GraphPad Prism Software, CA, USA).
Western Blot Analysis
Mice were sacrificed and approximately 10 mm thick tissue samples of stratum and substance nigra were taken for protein lysate. Protein samples from the lysate were loaded on 12% SDS-PAGE for electrophoresis and then transferred onto PVDF membranes. After blocking nonspecific binding sites with 5% bovine serum albumin, each membrane was incubated for 12 h at room temperature with primary rabbit polyclonal mouse/rabbit anti-ACHE (1:1500; Thermo Fisher Scientific,), anti-TH (1:1500; R&D Systems), anti-GFAP, anti-CD-3, anti-GAT-1 and anti-ZO-1 (1:1500; Cayman Chemical), anti-LRRK2, anti-Iba-1 (1:1500; Santa Cruz), or anti-caspase 3 (1:1000; Cayman Chemical), followed by incubation with a peroxidase-conjugated goat anti-rabbit/mouse antibody (1:5000; Santa Cruz, CA) and ECL detection. To normalize protein bands to the loading control, membranes were washed with TBST and re-probed with rabbit anti-β-actin antibody (1:5000; AbCam, MA). For the negative control, the primary antibody was omitted.
Statistical Analysis
Data are presented as mean ± standard deviation (SD) unless otherwise indicated. All data were analyzed using SPSS 13.0. Differences between groups were analyzed using two-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test. A P value < 0.05 was considered significant. All statistical graphs were plotted using GraphPad Prism 4.0 (GraphPad Prism Software, Inc., San Diego, CA). Statistical analysis was also performed by a statistician who was blinded to the study design.
Results
C16+Ang-1 Improves the Effects of Levodopa on Alleviating Functional Impairment
We first assessed functional impairment in mice following MPTP treatment. As shown in the open field test, there was a significant decrease in the traveled distance (Figure 1A) and mean velocity (Figure 1B) in the vehicle group compared to the control group. Furthermore, the average pole climbing time was prolonged (Figure 1C). In the rotarod test, mice in the vehicle group showed less time on the device compared to those in control group (Figure 1D). Moreover, the forepaw grip strength of mice in the vehicle group significantly decreased compared to the control group (Figure 1E). C16+Ang-1 or levodopa alone ameliorated the functional impairment in the vehicle group, while C16+Ang-1 exhibited better effects, indicated by the increased distance traveled and increased mean velocity in the open field test, increased average grip strength of forepaws, less climbing time, and increased time in the rotarod test (Figure 1A–E). In the NOR test, MPTP-injected mice had fewer discrimination scores, which meant similar time spent exploring the novel and familiar objects, suggesting spatial cognition impairment in PD mice. However, C16+Ang-1, levodopa, and C+A+L treatment rescued the memory impairment (Figure 1F).
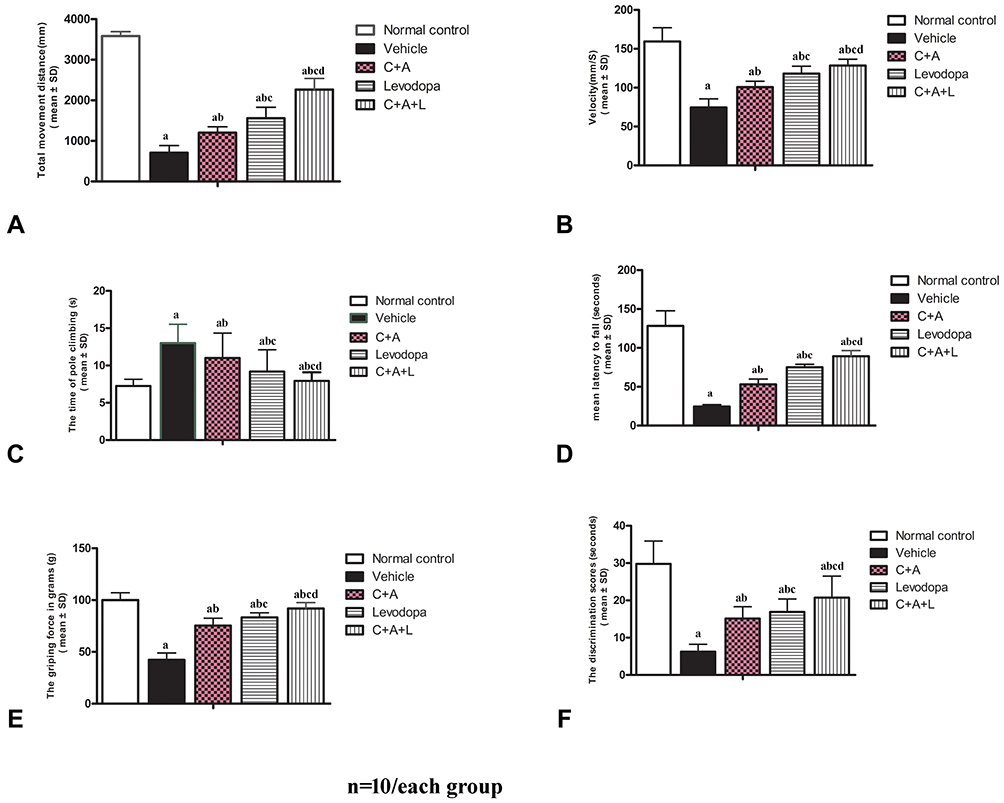 |
Figure 1 C16+Ang-1 alleviated functional impairment. Mice were treated with MPTP to establish the PD model. Mice were treated with either C16+Ang-1, levodopa, or C+A+L. Motor and cognitive function were assessed. (A and B) The total distance travelled (A) and the mean velocity (B) of mice in each group in the open field test. (C) The average pole climbing time in the pole climbing test. (D) Time staying on the device in the rotarod test. (E) Average grip strength of forepaws. (F) Discrimination scores (%) in the NOR test. a, P < 0.05 versus control; b, P < 0.05 versus vehicle; c, P < 0.05 versus C16+Ang-1 group; d, P < 0.05 versus levodopa group. |
C16+Ang-1 and C+A+L Treatment Alleviates Muscle Synchronous Contraction Compared to Levodopa
Dystonia and PD share many pathophysiological features. Dystonia is characterized by involuntary contractions in the opposing muscles, leading to the continual abnormality in postures and/or twisting movements. Dystonia can be induced in rodents by MPTP (Figure 2). In the control group, the quadriceps femoris (agonist) contracted, but the bicep femoris (antagonist) relaxed (Figure 2A). In the vehicle group, there was high wave amplitude (uV) in both muscles, an indicator of synchronous contraction, compared to the control group, indicating dystonia (Figure 2B). In the C16+Ang-1 (Figure 2C) and C+A+L (Figure 2E) groups, the antagonist did not contract when the agonist was exogenously stimulated (Figure 2F); levodopa treatment (Figure 2D) only partly reversed this phenomenon.
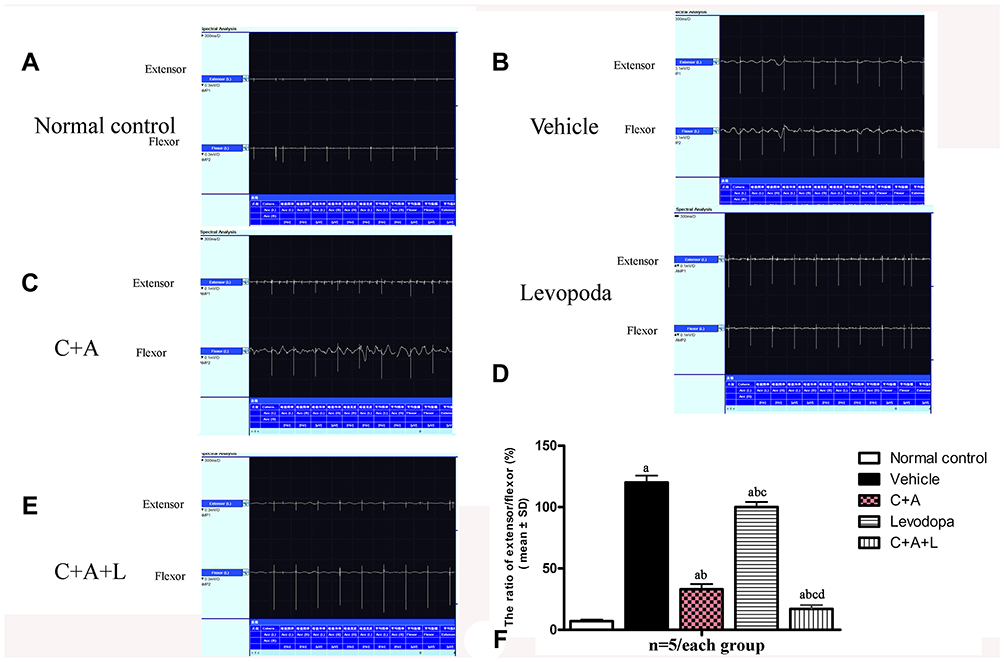 |
Figure 2 C16+Ang-1 and C+A+L alleviated muscle synchronous contraction. Wave amplitude (uV) was measured. (A) control; (B) MPTP; (C) MPTP and C16+Ang-1; (D) MPTP injection and levodopa; (E) MPTP and C+A+L; (F) synchronous contraction amplitude of extensor/flexor in each group. a, P < 0.05 versus control; b, P < 0.05 versus vehicle group; c, P < 0.05 versus C16+Ang-1 treated group; d, P < 0.05 versus levodopa-treated group. |
C16+Ang-1 Treatment Alleviates Inflammation and Suppresses Activated Astrocytes
The PD mice showed increased levels of serum pro-inflammatory cytokines including IL-6, TNF-α, and ROS (Figure 3A–C). The IL-6, TNF-α, and ROS levels were significantly decreased in the C16+Ang-1 and C+A+L groups, but not in the levodopa group (Figure 3A–C). Furthermore, the downregulation of anti-inflammatory cytokine IL-10 following MPTP treatment was also restored in the C16+Ang-1 and C+A+L groups, but not in the levodopa group (Figure 3D).
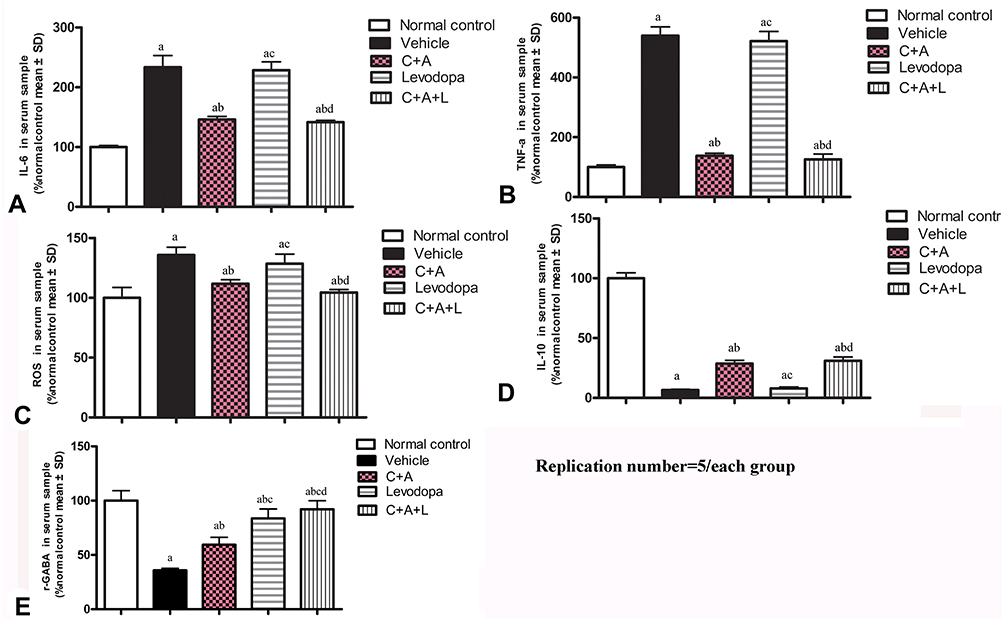 |
Figure 3 C16+Ang-1 treatment alleviated inflammation. (A–D): IL-6 (A), TNF-α (B), ROS (C), IL-10 (D), and γ-GABA (E) were measured a, P < 0.05 versus control; b, P < 0.05 versus vehicle group; c, P < 0.05 versus C16+Ang-1 treated group; d, P < 0.05 versus levodopa-treated group. |
CD3 immunostaining (green, Figure 4A–E) and Nissel staining (Figure 4F–J) indicated inflammatory cell infiltration in the MPTP-treated mice (Figure 4B and G) compared to the control group (Figure 4A and F). Inflammation in the PD mice was attenuated in the C16+Ang-1 (Figure 4C and H) and C+A+L (Figure 4E and J) groups, but not in the levodopa group (Figure 4D and I).
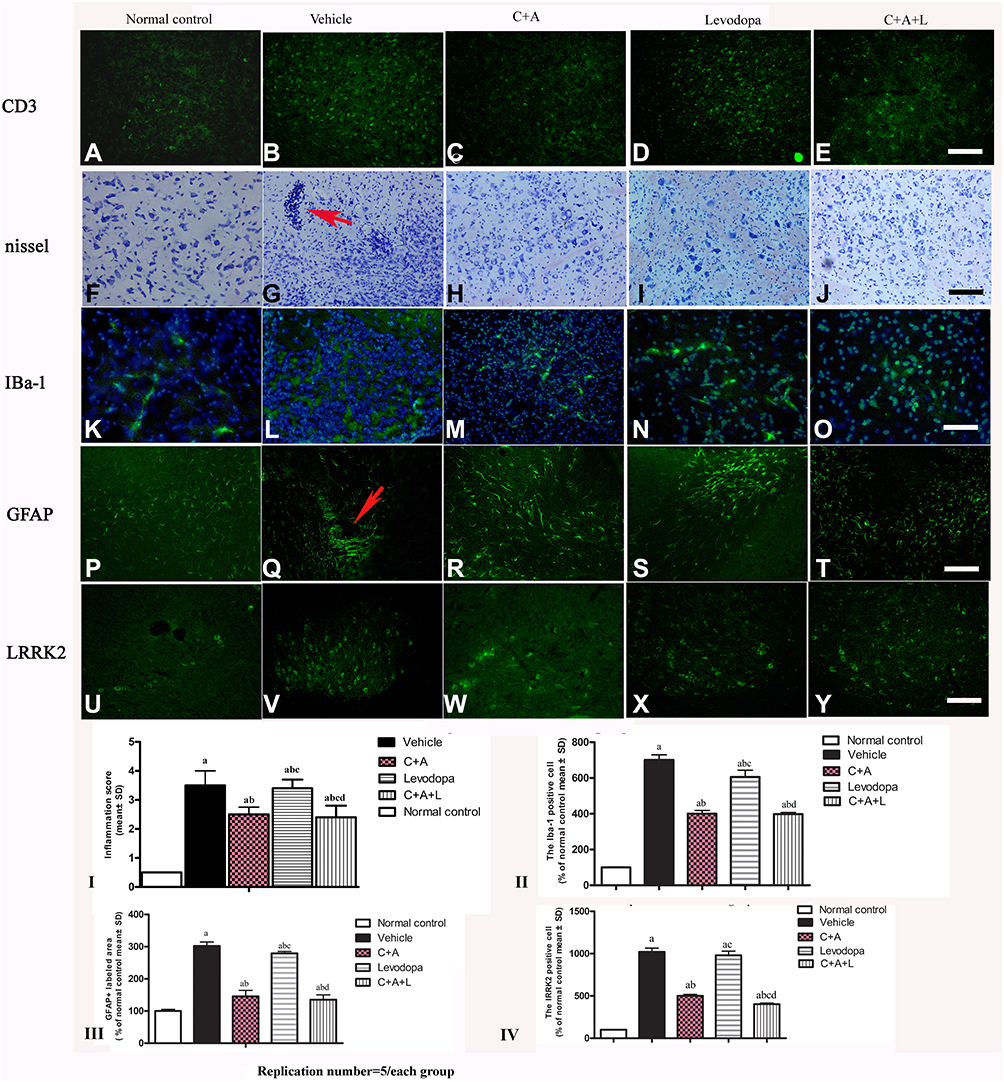 |
Figure 4 (A–J) CD3 and Nissel immunostaining. (A–E) CD3 immunostaining (green); (F–J) Nissel staining. (A and F) control; (B and G) MPTP, arrow in G denoted “perivascular cuffing” of inflammatory cells infiltration. (C and H) MPTP and C16+Ang-1; (D and I) MPTP and levodopa; (E and J) MPTP and C+A+L; (K–O) C16+Ang-1 treatment suppressed activated astrocytes as indicated by Iba-1 immunostaining. Iba-1 immunostaining (green) in the corpus striatum is shown. (K) control; (L) MPTP; (M) MPTP and C16+Ang-1; (N) MPTP and levodopa; (O) MPTP and C+A+L; (P–T): C16+Ang-1 suppressed activated astrocytes as indicated by GFAP immunostaining. (P) control; (Q) MPTP, arrow showed the reactive astrogliosis forms glia scar; (R) MPTP and C16+Ang-1; (S) MPTP and levodopa; (T) MPTP and C+A+L; (U–Y) Detection of LRRK2 using immunofluorescence. IRRK2 (green) in the corpus striatum is shown. (U) control; (V) MPTP; (W) MPTP and C16+Ang-1; (X) MPTP and levodopa; (Y) MPTP and C+A+L; (I–IV): Quantification of inflammatory scores (I), Iba-1+ cells (II), GFAP+ cells (III) and IRRK2+ cells (IV). a, P < 0.05 versus control; b, P < 0.05 versus vehicle group; c, P < 0.05 versus C16+Ang-1 treated group; d, P < 0.05 versus levodopa-treated group. Inflammatory scores:0, without inflammation; 1, limited cellular infiltration around blood vessels and meninges; 2, mild infiltration (1–10 inflammatory cells/slide in parenchymal tissues); 3, moderate infiltration (11–100 inflammatory cells/slide in parenchymal tissues); 4, severe infiltration (> 100 inflammatory cells/slide in parenchymal tissues). |
The level of the microglia-specific marker Iba-1 increased in the corpus striatum of the PD mice (Figure 4L), while C16+Ang-1 treatment (Figure 4M), and especially C+A+L treatment (Figure 4O), suppressed the increase in microglia in the PD mice (Figure 4L), while levodopa only exhibited a partial effect (Figure 4N). Furthermore, quantitative analysis of activated astrocytes (indicated by GFAP positive staining) showed that C16+Ang-1 and C+A+L had more remarkable effects in suppressing astrocyte activation and inhibiting glial scar formation (arrow in Figure 4Q) compared to levodopa alone.
LRRK2 plays an important role in the regulation of inflammation.15 The immunostaining results of LRRK2 in the striatum indicated that LRRK2 was increased in the PD mice (Figure 4V) compared to the control group (Figure 4U). However, LRRK2 was inhibited by C16+Ang-1 (Figure 4W) and C+A+L (Figure 4Y) treatment, but not by levodopa (Figure 4X).
Western blot analysis indicated that CD3 (Figure 5A and B), Iba-1 (Figure 5C and D), Lrrk2 (Figure 5E and F), and GFAP (Figure 5G and H) expression were upregulated and that treatment with C16+Ang-1 or C+A+L reduced the expression of these markers in the PD mice. Levodopa alone did not affect CD3 and Iba-1 expression, and only partly reduced GFAP and Irrk2 expression.
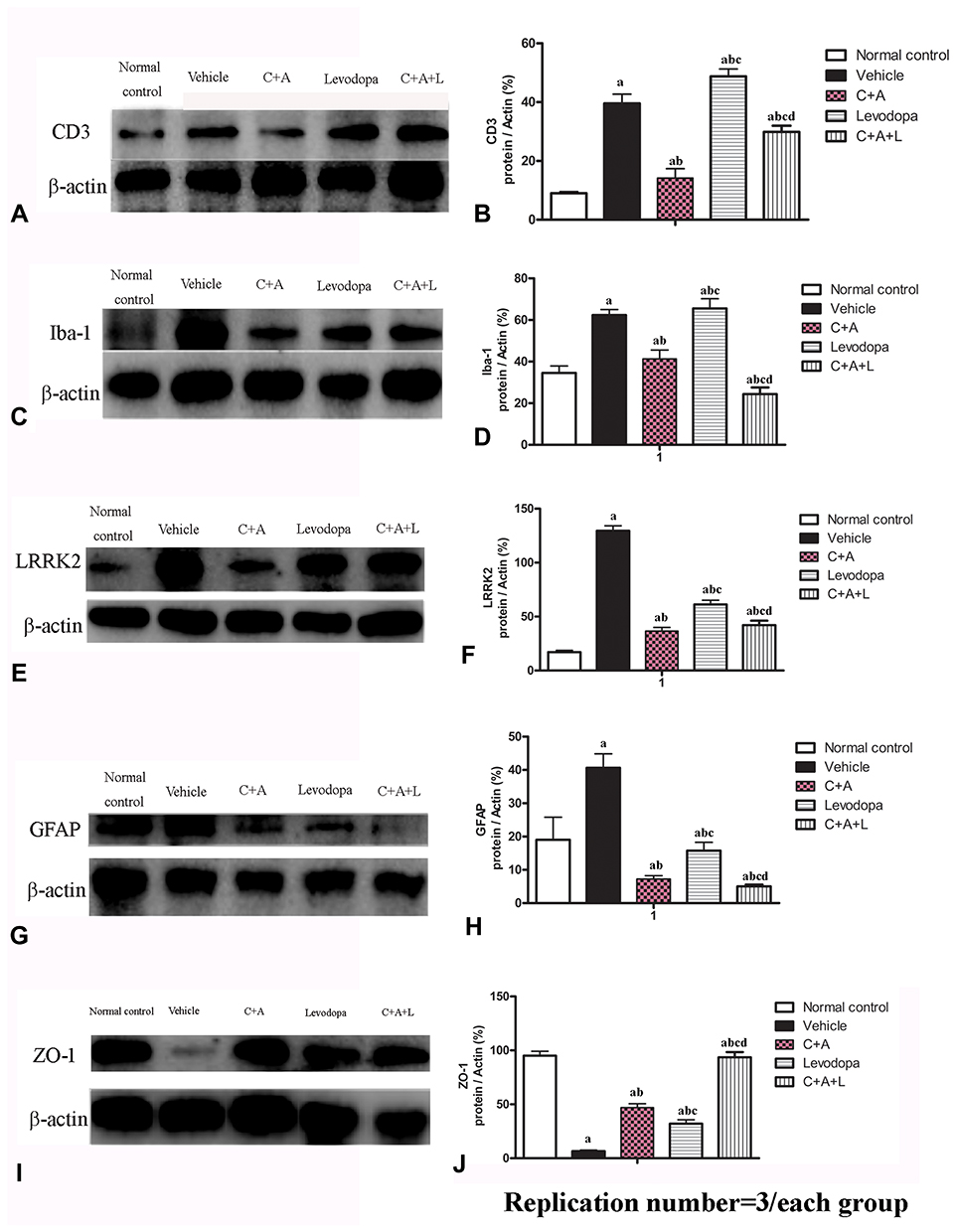 |
Figure 5 Expression of CD3 (A and B), Iba-1 (C and D), Lrrk2 (E and F), GFAP (G and H), and ZO-1 (I and J) by Western blot analysis. a, P < 0.05 versus control; b, P < 0.05 versus vehicle group; c, P < 0.05 versus C16+Ang-1 treated group; d, P < 0.05 versus levodopa-treated group. |
C16+Ang-1, but Not Levodopa, Reduces BBB Permeability and Blood Vessel Leakage
EB can be used as an indicator of edema and decreased blood vessel integrity in tissues. The vehicle group showed severe vasculature leakage (Figure 6B). C16+Ang-1 (Figure 6C) and C+A+L (Figure 6E) treatment, but not levodopa alone (Figure 6D), reduced the leakage (Figure 6K). Furthermore, the expression of ZO-1, an indicator of tight junctions in endothelial cells, was decreased in the PD mice (Figures 5I–J and 6G), but was recovered with C16+Ang-1 (Figure 6H) and C+A+L treatment (Figure 6J), but not levodopa alone (Figure 6I and L).
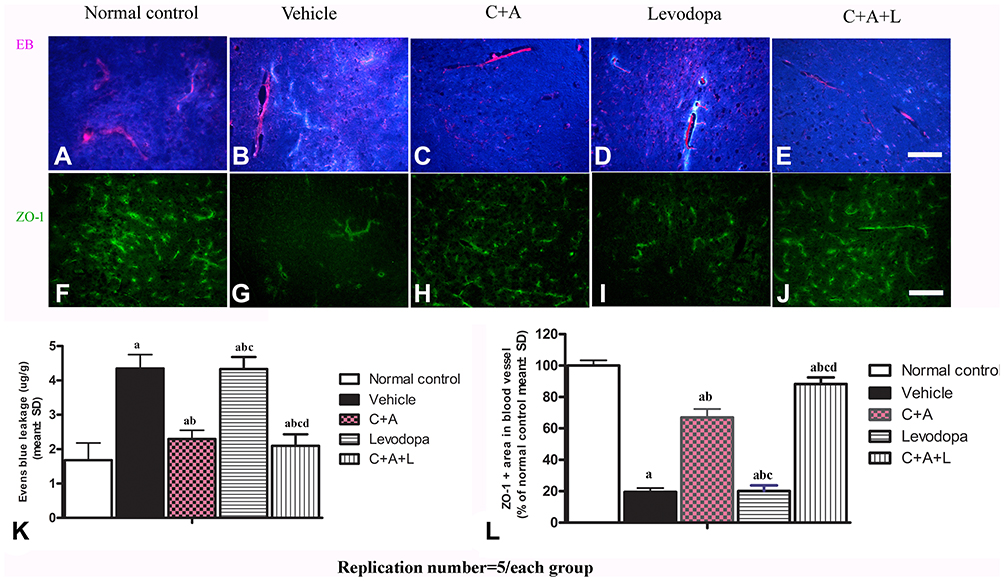 |
Figure 6 (A–E) C16+Ang-1, but not levodopa, reduced BBB permeability and blood vessel leakage. EB leakage showed by red colour. (F–J) Tight junction between microvascular endothelial cells, shown by immunofluorescence staining of ZO-1 (green), were reduced in the vehicle group, but maintained by C16+Ang-1 treatment. (K) Quantification of EB. (L) Quantification of ZO-1. Scale bar = 100 µm. a, P < 0.05 versus control; b, P < 0.05 versus vehicle group; c, P < 0.05 versus C16+Ang-1 treated group; d, P < 0.05 versus levodopa-treated group. |
C16+Ang-1 Improves the Effects of Levodopa in Reducing Neuronal Apoptosis, Restores Dopamine Neurons, and Alleviates Neuronal Death and Syn Loss in PD Mice
Nissl staining indicated that C16+Ang-1, levodopa, and especially C+A+L treatment reduced neuronal loss in the nigra substance (Figure 7A–E) and the striatum (Figure 7F–J) of PD mice. Moreover, the expression of caspase-3 in its active form, which is involved in cell apoptosis, was upregulated in the vehicle group (Figure 7L), but downregulated in the C16+Ang-1, levodopa, and C+A+L groups (Figure 7M–O).
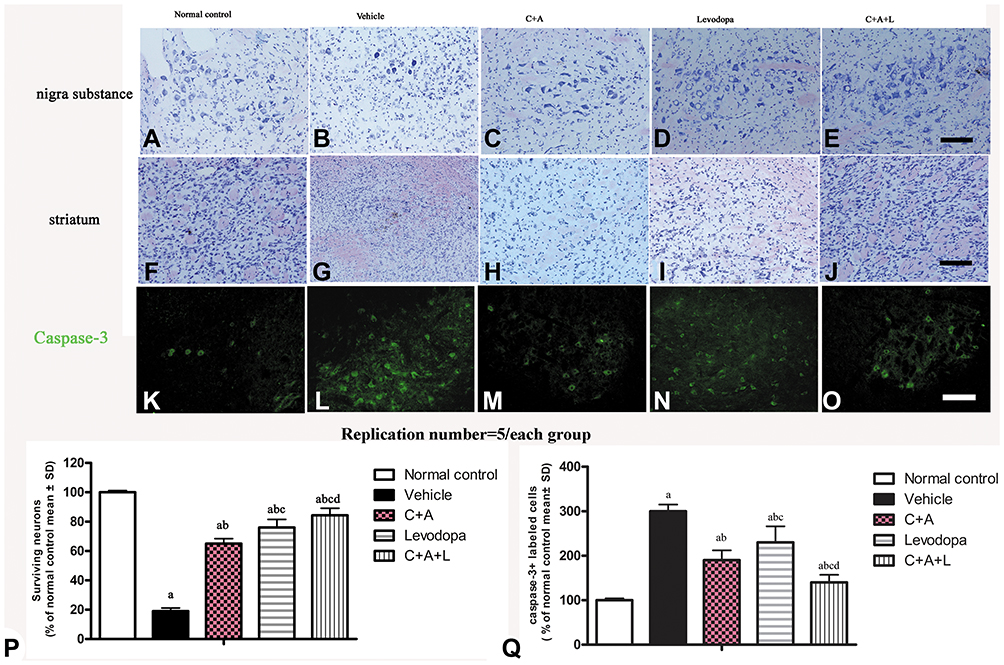 |
Figure 7 (A–J) Nissl staining of the SN and striatum. Neuronal loss is shown in the SN (A–E) and striatum (F–J). (K–O) Expression of active caspase-3 in (K) control; (L) MPTP; (M) MPTP and C16+Ang-1; (N) MPTP and levodopa; (O) MPTP and C+A+L; (P) The number of surviving neural cells was normalized to control. (Q) Quantification of caspase 3-labeled cells. Scale bar = 100 µm. a, P < 0.05 versus control; b, P < 0.05 versus vehicle group; c, P < 0.05 versus C16+Ang-1 treated group; d, P < 0.05 versus levodopa-treated group. |
The dopamine neurons, which are mainly located in the nigra substance, and the dopamine positive neuronal fibers, which mainly exist in the striatum region, were positive for TH immunostaining. In the control group, there were many TH positive neurons in the nigra substance (Figure 8A) and TH positive fibers in the striatum (Figure 8F). There was severe loss of both dopamine neurons and dopamine positive fibers in the vehicle group (Figure 8B and G). C16 +Ang-1 (Figure 8C and H), levodopa (Figure 8D and I), and C+A+L (Figure 8E and J) treatment reversed this phenomenon (Figure 8I and II).
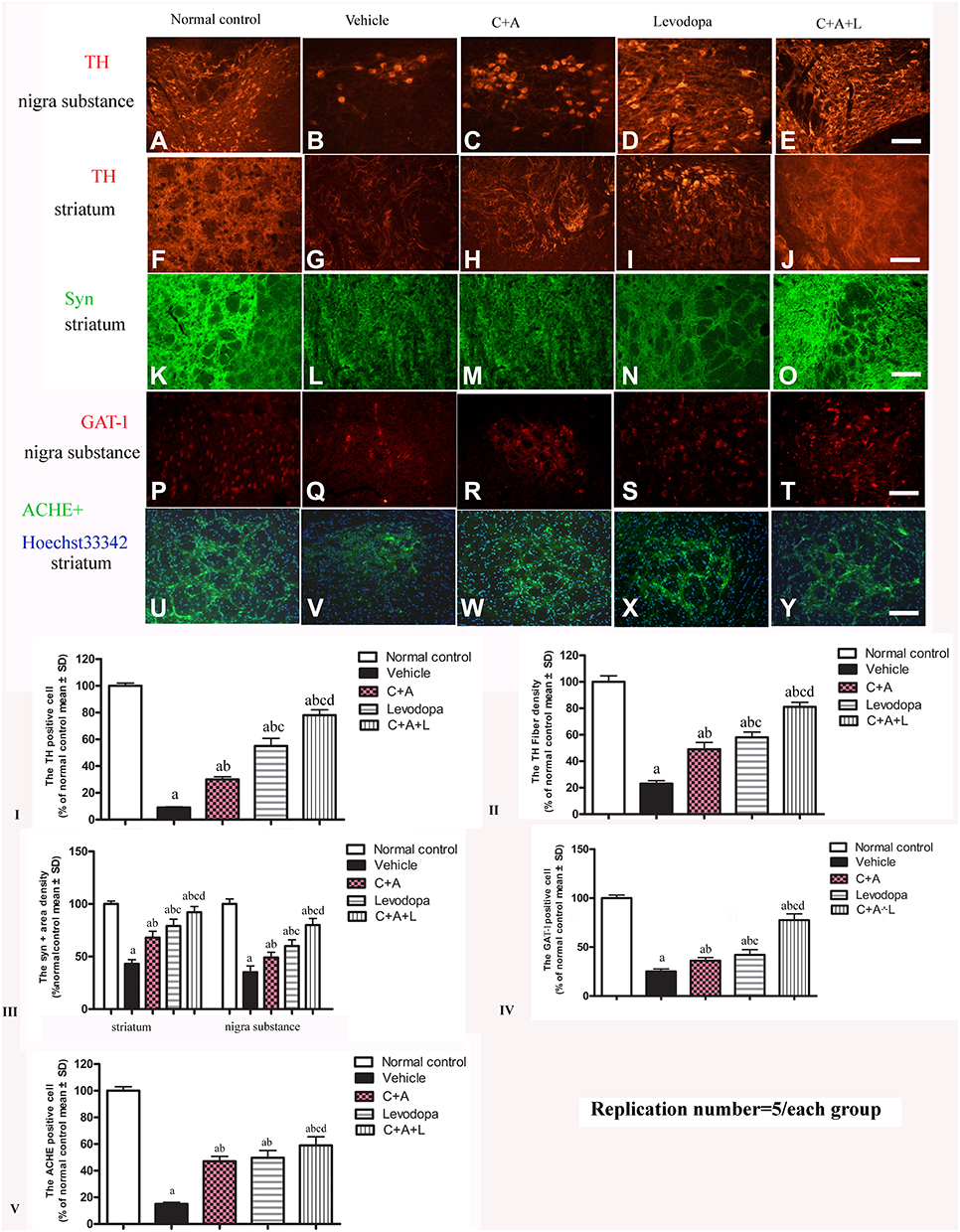 |
Figure 8 (A–J) Expression of TH in the SN and striatum. (A and F) control; (B and G) MPTP; (C and H) MPTP and C16+Ang-1; (D and I) MPTP and levodopa; (E and J) MPTP and C+A+L; (K–O) Expression of Syn in the striatum. Staining of Syn (green) in the striatum, (K) control; (L) MPTP; (M) MPTP and C16+Ang-1; (N) MPTP and levodopa; (O) MPTP and C+A+L; (P–T) Expression of GAT-1 in the nigra substance. (P) control; (Q) MPTP; (R) MPTP and C16+Ang-1; (S) MPTP and levodopa; (T) MPTP and C+A+L; (U–Y) Expression of ACHE in the striatum. (U) control; (V) MPTP; (W) MPTP and C16+Ang-1; (X) MPTP and levodopa; (Y) MPTP and C+A+L; Scale bar = 100 µm. (I–V) quantification of TH+ cells in the SN (I), TH+ fibers in the striatum (II), Quantification of Syn+ area (III), quantification of GAT-1+ cells (IV), and quantification of ACHE+ cells (V).a, P < 0.05 versus control; b, P < 0.05 versus vehicle group; c, P < 0.05 versus C16+Ang-1 treated group; d, P < 0.05 versus levodopa-treated group. |
Immunofluorescent staining of Syn, which is a synapse-associated protein that can promote synaptic plasticity (Figure 8L), was decreased in the PD mice, but C16+Ang-1 (Figure 8M), levodopa (Figure 8N), and C+A+L (Figure 8O) treatment restored its expressions (Figure 8III).
Expression of GAT-1 (Figure 8P–T) and ACHE (Figure 8U–Y) as well as serum GABA levels (Figure 3E) were all downregulated in the MPTP-treated mice. However, C16+Ang-1, levodopa, and C+A+L treatment all restored expression of these proteins (Fig 8IV and V). Western blot analysis also indicated that C16+Ang-1, levodopa, or C+A+L treatment restored the levels of TH (Figure 9C and D), Syn (Figure 9E and F), GAT-1 (Figure 9G and H), and ACHE (Figure 9I and J), and decreased active caspase-3 (Figure 9A and B).
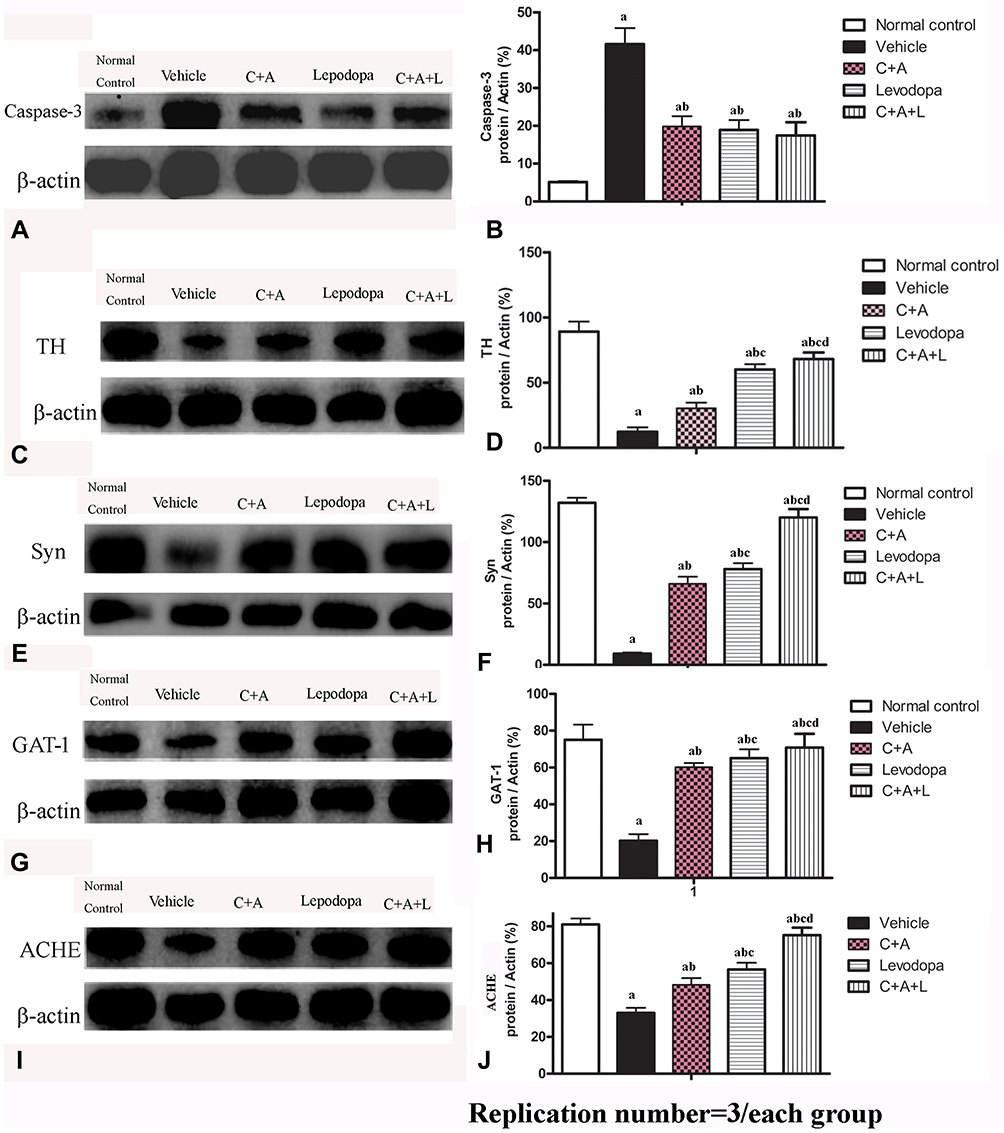 |
Figure 9 Expression of active caspase-3 (A and B), TH (C and D), Syn (E and F), GAT-1 (G and H), and ACHE (I and J) were measured using Western blot analysis. a, P < 0.05 versus control; b, P < 0.05 versus vehicle group; c, P < 0.05 versus C16+Ang-1 treated group; d, P < 0.05 versus levodopa-treated group. |
In normal neuronal nuclei, TEM ultrastructural morphology showed unconsolidated chromatin (Figure 10A). In myelinated axons in the control group, the myelin sheaths were dark and ring-shaped (Figure 10C). Mitochondria also demonstrated clear cristae (red arrow, Figure 10D). There was no tissue edema or blood vessel leakage (Figure 10B). In the PD mice, the neurons had shrunken nuclei, the nuclear chromatin was condensed, fragmented, and marginated (Figure 10E), and there were swollen cristae in the vacuolized mitochondria (blue arrow, Figure 10H). However, vascular vessel leakage was observed around the blood vessels, and there was severe tissue edema in the extracellular space (Figure 10F). Loose and fused changes were observed in the myelin sheath, in addition to splitting and vacuole changes (Figure 10G). However, in the C16+Ang-1 (Figure 10I–L) and levodopa (Figure 10M–P) groups, the nuclei morphology (Figure 10I and M), myelin, and axons (Figure 10K and O) were more normal. Furthermore, tissue edema and vascular vessel leakage in the C16+Ang-1 group (Figure 10J) were alleviated compared to the levodopa group (Figure 10N). The mitochondria morphology (Figure 10L and P) was also improved in both groups. In the C+A+L group, the nuclei morphology (Figure 10Q), myelin and axons (Figure 10S), mitochondria (Figure 10T), and blood vessels (Figure 10R) more closely approximated that of the normal group.
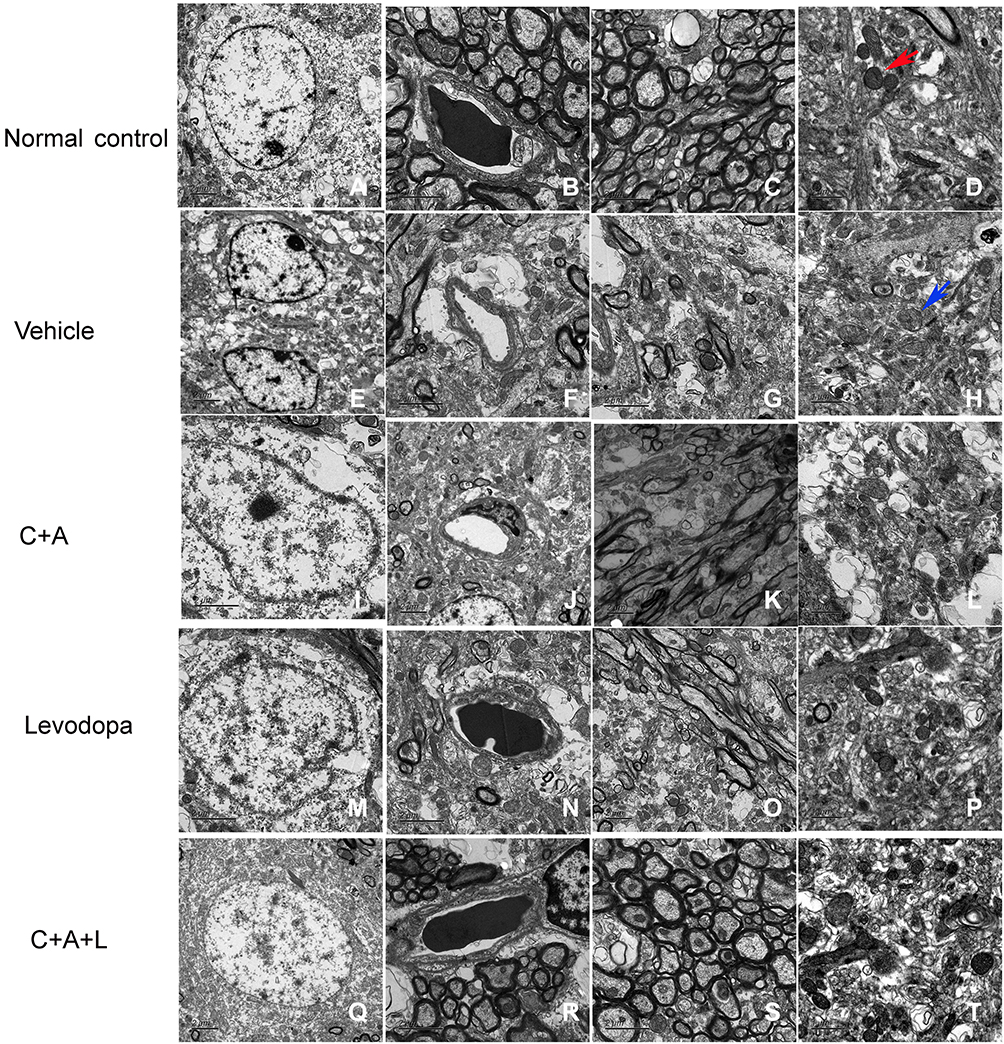 |
Figure 10 Ultrastructural morphology using TEM. (A–D) control; (E–H) MPTP; (I–L) MPTP and C16+Ang-1; (M–P) MPTP and levodopa; (Q–T) MPTP and C+A+L. Mitochondria with clear cristae are shown by the red arrow. Mitochondria vacuolization and swollen cristae are shown by the blue arrow. A-C, E-G, I-K, M-O, Q-S, Scale bar = 2 μm; D, H, L, P, T, Scale bar = 1 μm. |
Discussion
Inflammation related to aging can influence the degeneration of dopaminergic neurons, whereas activation of anti-inflammatory signaling pathways appears to protect against neuron degeneration in the SN.3 Levodopa is a gold-standard treatment for PD, application of the dopamine precursor levodopa can rescue dying neurons in the striatum and restore neuronal function related to motor symptoms, and alleviate the motor symptoms of PD. Unfortunately, pulsatile levodopa treatment can lead to a neuroinflammatory response in the striatum.17 Neuroinflammation has been shown to be deeply involved in PD initiation and progression in both humans and experimental models.16 Long-term administration of levodopa can irreversibly lead to abnormal involuntary movements (AIM), which dramatically affects a patient’s quality of life.1 Collectively, previous findings indicate that inflammatory mechanisms play a role in dyskinesia and impair the effects of levodopa in PD treatment. Thus, suppressing neuroinflammation in the CNS might improve the effects of levodopa.5
MPTP is commonly used as a PD inducer in non-human models, making it a reliable model to investigate novel therapies.2 MPTP can cross the BBB and transform into an active metabolite, a process that is mediated by monoamine oxidase B. Subsequently, MPTP is carried into the neurons in the SNpc by a dopamine transporter, where MPTP can inhibit mitochondrial complex I. MPTP can then increase oxidative stress and neuronal apoptosis, resulting in neuroinflammation in mouse models.18 Previous studies suggest that systemic inflammatory stimulation exacerbates AIM induced by levodopa treatment in MPTP lesioned mice and increases pro-inflammatory cytokines in the striatum of LID mice.1,2
In our MPTP-induced PD model, infiltration of inflammatory cells was found in the parenchymal striatum in the levodopa group, which is in accordance with previous studies showing that neuroinflammation could exacerbate levodopa-induced dyskinesia.19 Inflammation has a pathogenic role in neurodegenerative diseases. Both dystonia and LID belong to hyperkinetic movement disorders. Dystonia often occurs spontaneously, and LID is either dystonic or choreiform.20,21 Dystonia and LID can result in a circuit involving the cortex, basal ganglia, thalamus, and cerebellum. They also share striatal cholinergic signaling dysregulation and striatal synaptic plasticity abnormalities related to neuroinflammation. Recent studies indicate that neuroinflammation may also be involved in the pathogenesis of X-linked Dystonia-Parkinsonism.22 In our research, the PD mice in the vehicle and levodopa groups had dystonia, as revealed by electrophysiological testing, which was visibly mitigated by C16+Ang-1 or C+A+L treatment (Figure 2F), further demonstrating that alleviating inflammation can lessen PD symptoms and alleviate this functional disability.
A highly inflammatory environment can affect disease progression. Glia and immune cells are thought to play main roles in producing oxidants and ROS, which are deeply involved in CNS pathologies.23 Leukocyte infiltration (as indicated by CD3+ staining) into the brain is a key factor in increasing local inflammation. Blood vessel endothelial cells along with dopaminergic neurons die after MPTP exposure.11 Dopamine-induced oxidation and neurodegenerative changes are pivotal mechanisms in PD pathogenesis.11 Reducing inflammation may result in better outcomes for neurodegeneration in PD, especially in late stages of the disease.24 The perpetual cycle of microglial activation can further promote neuroinflammation and activation of microglia.11 Activated microglia (indicated in our study by Iba-1 upregulation) exacerbate inflammatory responses by increasing cytokine production that leads to the recruitment of immune cells. Following inflammation, astrocytes proliferated and promoted reactive astrogliosis in the CNS (indicated in our study by glial scar formation), which could inhibit axonal regeneration. Moreover, it has been reported that dysfunction of LRRK2 may influence microglial activation.25 Inhibition of LRRK2 can prevent dopaminergic neuron loss and motor deficits following LPS stimulation.26 In our study, inhibition of LRRK2 expression reduced production of pro-inflammatory mediators and decreased inflammatory responses, which is in accordance with results of previous research.27
The targets of C16 and Ang-1 are ανβ3 integrin and Tie2, respectively.28 C16 can competitively inhibit integrin αvβ3 signaling and inflammatory cell binding to the endothelium, preventing leukocyte transmigration.Furthermore, it can also act as an ανβ3 integrin agonist and promote endothelial cell angiogenesis.25 Ang-1 can maintain the integrity of endothelial cells, enhance the survival of endothelial cells, and alleviate blood vessel leakage by activating the Ang-1-Tie2 system. However, it can also decrease the expression of adhesion factors, thus thwarting inflammatory cell adhesion. Our previous studies indicated that C16+Ang1 treatment could up-regulate expression of Tie2 and p85, which is a subunit of PI3K. The activation of the PI3K/Akt pathway in microglia can promote anti-inflammatory effects, immune regulation, and tissue repair.7 The mechanisms for Ang-1 and C16 partly overlap; as such, combining Ang-1 and C16 might have a synergistic effect.
PD is a progressive neurodegenerative disorder characterized by the loss of dopaminergic and GABA neurons in the SN and striatum.29 The striatum plays a role in facilitating voluntary movement, and the cerebellum functions in the maintenance and coordination of voluntary movements. Motor loss can result from neuroinflammation and crosstalk with neurochemicals.30 GABA is the main inhibitory neurotransmitter in the brain and plays an important role in regulating neuronal excitability. GABA re-uptake from the synapse is dependent on specific transporters - mainly GAT-1, and its deficiency might responsible for abnormal motor behavior in parkinsonian symptoms.31 In previous study, LPS could activate Toll-like receptor 4 (TLR4) and promote the release of proinflammatory cytokines (IL-1β) in microglia, which can subsequently suppress postsynaptic GABA receptor activity and reduce presynaptic GABA synthesis.30 Moreover, in a mouse model of parkinsonism, downregulation of GAT-1 were found to lead to a tonic GABAergic inhibition of DA release.31 In the present study, the reduction in expression of GABA, GAT-1, synapse-associated proteins Syn (which can modulate the plasticity of the synapse),6 and decreasing expression of ACHE in the striatum (exhibited loss of cholinergic neurons) might all be involved in the neurodegeneration of PD.32 Fortunately, as an exogenous dopamine substitute, levodopa can restore GAT-1 and ACHE expression and increase the number of TH positive neurons and Syn expression area.
Conclusions
Levodopa can neither inhibit inflammation in the CNS nor protect the integrity of the BBB in PD mice, which may be the underlying cause of its side effects in the clinic. By improving the CNS microenvironment and reducing secondary tissue damage due to inflammation, the combination of C16 and Ang-1 could promote the functional recovery effects of levodopa, alleviate injury by inflammatory factors in the mitochondria, and prevent death of injured neurons, ultimately attenuating the side effects caused by inflammation. In conclusion, our study suggests that C16+Ang-1 might compensate for the shortcomings of levodopa and ameliorate the effects of levodopa. This treatment strategy might be developed as a combinatorial therapeutic in the future.
Abbreviations
PD, Parkinson disease; ACHE, Acetylcholinesterase; AIMs, abnormal involuntary movements; LID, termed levodopa-induced dyskinesia; COX2, cyclooxygenase-2; iNOS, inducible nitric oxide synthase; Ang-1, angiopoietin-1; BBB, blood-brain barrier; CNS, central nervous system; DARPP-32, dopamine- and cAMP-regulated phosphoprotein of 32 kDa; GAT, γ-aminobutyric acid transporter; ELISA, enzyme-linked immunosorbent assay; EC, endothelial cell; FITC, fluorescein isothiocyanate; GABA, γ-aminobutyric acid; IL-6, interleukin 6; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; SN, substantia nigra; SNpc, substantia nigra pars compacta; Syn, synaptophysin; PI3K, phosphoinositide 3-kinase; ROS, reactive oxygen species; TH, tyrosine hydroxylase; TRIFC, Trimolecular Fluorescence Complementation; ZO-1, zonal occludens-1; EAE, experimental autoimmune encephalomyelitis; MS, Multiple Sclerosis.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics Approval and Informed Consent
The experiments were performed abiding by the NIH guidelines and approved by the Ethics of Committee of Zhejiang University Medical College (SRRSH202102016, date of approval: 10-Feb-2021).
Acknowledgments
We thank Medjaden Inc. for editing and proofreading the manuscript.
Author Contributions
All authors took part in the study design, execution, acquisition of data, analysis, and interpretation. The corresponding author drafted the article, and all other authors substantially revised the article. During revision, all authors approved modification of the article before submission. All authors read and approved the final manuscript which has been submitted for review, as well as agree to take responsibility for the contents of the article.
Funding
This work was supported by the National Natural Science Foundation of China (project no. 81971069), Zhejiang Provincial Natural Science Foundation of China (project no. LY22H090017), and Science and Technology Planning Project of Jinhua City, P.R. China (project no. 2021-3-149).
Disclosure
The authors have declared that no conflicts of interest exists.
References
1. Radhakrishnan DM, Goyal V. Parkinson’s disease: a review. Neurol India. 2018;66:S26–s35. doi:10.4103/0028-3886.226451
2. Yan A, Song L, Zhang Y, Wang X, Liu Z. Systemic inflammation increases the susceptibility to levodopa-induced dyskinesia in 6-OHDA lesioned rats by targeting the NR2B-medicated PKC/MEK/ERK pathway. Front Aging Neurosci. 2020;12:625166. doi:10.3389/fnagi.2020.625166
3. Lanza K, Perkins AE, Deak T, Bishop C. Late aging-associated increases in L-DOPA-induced dyskinesia are accompanied by heightened neuroinflammation in the hemi-parkinsonian rat. Neurobiol Aging. 2019;81:190–199. doi:10.1016/j.neurobiolaging.2019.05.019
4. Marino BLB, de Souza LR, Sousa KPA, et al. Parkinson’s disease: a review from pathophysiology to treatment. Mini Rev Med Chem. 2020;20:754–767. doi:10.2174/1389557519666191104110908
5. Del-Bel E, Bortolanza M, Dos-Santos-Pereira M, Bariotto K, Raisman-Vozari R. l-DOPA-induced dyskinesia in Parkinson’s disease: are neuroinflammation and astrocytes key elements? Synapse. 2016;70:479–500. doi:10.1002/syn.21941
6. Liu Y, Zhang Y, Zheng X, et al. Galantamine improves cognition, hippocampal inflammation, and synaptic plasticity impairments induced by lipopolysaccharide in mice. J Neuroinflammation. 2018;15:112. doi:10.1186/s12974-018-1141-5
7. Fu X, Chen H, Han S. C16 peptide and angiopoietin-1 protect against LPS-induced BV-2 microglial cell inflammation. Life Sci. 2020;256:117894. doi:10.1016/j.lfs.2020.117894
8. Jiang H, Zhang F, Yang J, Han S. Angiopoietin-1 ameliorates inflammation-induced vascular leakage and improves functional impairment in a rat model of acute experimental autoimmune encephalomyelitis. Exp Neurol. 2014;261:245–257. doi:10.1016/j.expneurol.2014.05.013
9. Zhang F, Yang J, Jiang H, Han S. An ανβ3 integrin-binding peptide ameliorates symptoms of chronic progressive experimental autoimmune encephalomyelitis by alleviating neuroinflammatory responses in mice. J Neuroimmune Pharmacol. 2014;9:399–412. doi:10.1007/s11481-014-9532-6
10. Wang B, Tian KW, Zhang F, Jiang H, Han S. Angiopoietin-1 and C16 peptide attenuate vascular and inflammatory responses in experimental allergic encephalomyelitis. CNS Neurol Disord Drug Targets. 2016;15:496–513. doi:10.2174/1871527314666150821112546
11. Jackson-Lewis V, Przedborski S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat Protoc. 2007;2:141–151. doi:10.1038/nprot.2006.342
12. Zhang QS, Heng Y, Mou Z, Huang JY, Yuan YH, Chen NH. Reassessment of subacute MPTP-treated mice as animal model of Parkinson’s disease. Acta Pharmacol Sin. 2017;38:1317–1328. doi:10.1038/aps.2017.49
13. Cai HY, Fu XX, Jiang H, Han S. Adjusting vascular permeability, leukocyte infiltration, and microglial cell activation to rescue dopaminergic neurons in rodent models of Parkinson’s disease. NPJ Parkinsons Dis. 2021;7:91. doi:10.1038/s41531-021-00233-3
14. Su RJ, Zhen JL, Wang W, Zhang JL, Zheng Y, Wang XM. Time-course behavioral features are correlated with Parkinson’s disease‑associated pathology in a 6-hydroxydopamine hemiparkinsonian rat model. Mol Med Rep. 2018;17:3356–3363. doi:10.3892/mmr.2017.8277
15. Keller AV, Rees KM, Seibt EJ, et al. Electromyographic patterns of the rat hindlimb in response to muscle stretch after spinal cord injury. Spinal Cord. 2018;56:560–568. doi:10.1038/s41393-018-0069-z
16. Pajares M, Manda G, Boscá L, Cuadrado A, Cuadrado A. Inflammation in Parkinson’s disease: mechanisms and therapeutic implications. Cells. 2020;9:1687. doi:10.3390/cells9071687
17. Dou F, Chu X, Zhang B, et al. EriB targeted inhibition of microglia activity attenuates MPP(+) induced DA neuron injury through the NF-κB signaling pathway. Mol Brain. 2018;11:75. doi:10.1186/s13041-018-0418-z
18. Blandini F, Armentero MT. Animal models of Parkinson’s disease. Febs J. 2012;279:1156–1166. doi:10.1111/j.1742-4658.2012.08491.x
19. Bishop C. Neuroinflammation: fanning the fire of l-dopa-induced dyskinesia. Mov Disord. 2019;34:1758–1760. doi:10.1002/mds.27900
20. Calabresi P, Standaert DG. Dystonia and levodopa-induced dyskinesias in Parkinson’s disease: is there a connection? Neurobiol Dis. 2019;132:104579. doi:10.1016/j.nbd.2019.104579
21. Iderberg H, Francardo V, Pioli EY. Animal models of L-DOPA-induced dyskinesia: an update on the current options. Neuroscience. 2012;211:13–27. doi:10.1016/j.neuroscience.2012.03.023
22. Petrozziello T, Mills AN, Vaine CA, et al. Neuroinflammation and histone H3 citrullination are increased in X-linked Dystonia Parkinsonism post-mortem prefrontal cortex. Neurobiol Dis. 2020;144:105032. doi:10.1016/j.nbd.2020.105032
23. Mei M, Zhou Y, Liu M, et al. Antioxidant and anti-inflammatory effects of dexrazoxane on dopaminergic neuron degeneration in rodent models of Parkinson’s disease. Neuropharmacology. 2019;160:107758. doi:10.1016/j.neuropharm.2019.107758
24. Schwenkgrub J, Zaremba M, Joniec-Maciejak I, Cudna A, Mirowska-Guzel D, Kurkowska-Jastrzębska I. The phosphodiesterase inhibitor, ibudilast, attenuates neuroinflammation in the MPTP model of Parkinson’s disease. PLoS One. 2017;12:e0182019. doi:10.1371/journal.pone.0182019
25. Rui Q, Ni H, Li D, Gao R, Chen G. The role of LRRK2 in neurodegeneration of Parkinson disease. Curr Neuropharmacol. 2018;16:1348–1357. doi:10.2174/1570159x16666180222165418
26. Dwyer Z, Rudyk C, Thompson A, et al. Leucine-rich repeat kinase-2 (LRRK2) modulates microglial phenotype and dopaminergic neurodegeneration. Neurobiol Aging. 2020;91:45–55. doi:10.1016/j.neurobiolaging.2020.02.017
27. Gelders G, Baekelandt V, Van der Perren A. Linking neuroinflammation and neurodegeneration in Parkinson’s disease. J Immunol Res. 2018;2018:4784268. doi:10.1155/2018/4784268
28. Cai HY, Tian KW, Zhang YY, Jiang H, Han S. Angiopoietin-1 and ανβ3 integrin peptide promote the therapeutic effects of L-serine in an amyotrophic lateral sclerosis/Parkinsonism dementia complex model. Aging. 2018;10:3507–3527. doi:10.18632/aging.101661
29. Aryal S, Skinner T, Bridges B, Weber JT. The pathology of Parkinson’s disease and potential benefit of dietary polyphenols. Molecules. 2020;25:4382. doi:10.3390/molecules25194382
30. Abg Abd Wahab DY, Gau CH, Zakaria R, et al. Review on cross talk between neurotransmitters and neuroinflammation in striatum and cerebellum in the mediation of motor behaviour. Biomed Res Int. 2019;2019:1767203. doi:10.1155/2019/1767203
31. Roberts BM, Doig NM, Brimblecombe KR, et al. GABA uptake transporters support dopamine release in dorsal striatum with maladaptive downregulation in a parkinsonism model. Nat Commun. 2020;11:4958. doi:10.1038/s41467-020-18247-5
32. Kish SJ, Schut L, Simmons J, Gilbert J, Chang LJ, Rebbetoy M. Brain acetylcholinesterase activity is markedly reduced in dominantly-inherited olivopontocerebellar atrophy. J Neurol Neurosurg Psychiatry. 1988;51:544–548. doi:10.1136/jnnp.51.4.544
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.