")
Back to Journals » International Journal of Nanomedicine » Volume 16
Co-Administration of iRGD with Sorafenib-Loaded Iron-Based Metal-Organic Framework as a Targeted Ferroptosis Agent for Liver Cancer Therapy
Authors Liu X, Zhu X, Qi X, Meng X, Xu K
Received 17 November 2020
Accepted for publication 26 January 2021
Published 11 February 2021 Volume 2021:16 Pages 1037—1050
DOI https://doi.org/10.2147/IJN.S292528
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Yan Shen
Xianchuang Liu,1 Xinyang Zhu,1 Xun Qi,1 Xianwei Meng,2 Ke Xu1
1Department of Radiology and Key Laboratory of Diagnostic Imaging and Interventional Radiology of Liaoning Province, The First Affiliated Hospital of China Medical University, Shenyang, Liaoning, People’s Republic of China; 2Laboratory of Controllable Preparation and Application of Nanomaterials, Laboratory of Cryogenics, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing, People’s Republic of China
Correspondence: Ke Xu
Department of Interventional Radiology, The First Affiliated Hospital of China Medical University, No. 155 Nanjing North Street, Heping District, Shenyang, 110000, Liaoning, People’s Republic of China
Tel +86 24 8328 2999
Fax +86 24 8328 2997
Email [email protected]
Purpose: Hepatocellular carcinoma (HCC) is one of the most common fatal cancers, with no curative therapy available. The concept of ferroptosis is attracting increasing attention in cancer research. Herein, we describe the use of a nanodevice as an effective strategy for inducing ferroptosis to manage HCC.
Methods: To improve ferroptosis-induced treatment of HCC, we constructed sorafenib (sor)-loaded MIL-101(Fe) nanoparticles (NPs) [MIL-101(Fe)@sor] and evaluated the efficacy of ferroptosis-based HCC therapy after co-administration with the iRGD peptide both in vitro and in vivo.
Results: The prepared MIL-101(Fe) NPs have several promising characteristics including drug-loading, controllable release, peroxidase activity, biocompatibility, and T2 magnetic resonance imaging ability. MIL-101(Fe)@sor NPs significantly induced ferroptosis in HepG2 cells, increased the levels of lipid peroxidation and malondialdehyde, and reduced those of glutathione and glutathione peroxidase 4 (GPX-4). The in vivo results showed that the MIL-101(Fe)@sor NPs significantly inhibited tumor progression and decreased GPX-4 expression levels, with negligible long-term toxicity. Meanwhile, co-administration of MIL-101(Fe)@sor NPs with iRGD significantly accelerated ferroptosis.
Conclusion: Our findings suggest that MIL-101(Fe)@sor NPs co-administered with iRGD are a promising strategy for inducing HCC ferroptosis.
Keywords: iron-based metal-organic framework, iRGD, sorafenib, ferroptosis, HCC, nanoparticles
Introduction
Hepatocellular carcinoma (HCC) is a leading cause of cancer worldwide and accounts for about 782,000 deaths annually.1 Since early HCC is typically asymptomatic, more than 75% of patients are diagnosed at advanced stages. Current potential therapies include surgical resection, interventional therapy, radiotherapy, chemotherapy, targeted therapy, and immunotherapy. However, the treatment effects remain unsatisfactory,2,3 and novel therapies are urgently needed to improve patient outcomes.
Ferroptosis is a novel mechanism of regulated cell death (RCD) that was first proposed by Stockwell.4,5 It is distinct from apoptosis, necrosis, and autophagy at morphological, biochemical, and genetic levels.6 For example, cells that undergo ferroptotic cell death have normal nucleus size, reduced mitochondrial volume, increased mitochondrial membrane density, and reduced (or disappeared) mitochondrial cristae.7 Ferroptosis is mediated by the accumulation of fatal lipid peroxidation (LPO) in the presence of iron ions; massive LPO rupture cell membranes to induce ferroptotic cell death.8 This iron-dependent form of cell death is triggered by decreasing expression of glutathione peroxidase 4 (GPX-4), an LPO scavenger, which contributes to glutathione (GSH) depletion while reducing LPO.9 Evidence suggests that targeting ferroptosis is an effective strategy in the management of various types of cancer, especially HCC.10–12
Several small molecules, drugs, experimental compounds, and genes have been found to induce ferroptotic cell death in cancer.13 However, the efficacies of existing ferroptotic compounds are not sufficient. Nanotechnology has been used in the development of new ferroptosis inducers for cancer treatment, and satisfactory therapeutic effects have been reported in recent years.14,15 Among them, iron-based nanomaterials serve as a platform for directly loading ferroptosis inducers and can also release iron ions that participate in Fenton reactions and accelerate ferroptosis. Therefore, multifunctional theranostic nanoplatforms can be fabricated using iron-based nanomaterials for cancer ferroptosis management.16
In this study, an Fe-metal organic framework [MIL-101(Fe)] was designed to effectively induce tumor ferroptosis. Owing to its high drug-loading capacity, sorafenib (a ferroptosis inducer used to treat advanced HCC)17,18 was chosen and loaded onto MIL-101(Fe) nanoparticles (NPs) to synthesize MIL-101(Fe)@sor. Relying solely on passive transport does not achieve maximum nanomedicine accumulation or the maximum anti-tumor effect. To enhance the nanodrug tumor targeting and penetration abilities, an iRGD peptide (amino acid sequence: CRGDK/RGPD/EC) was introduced containing a tumor-homing motif (RGD) and a tissue-penetrating motif (CendR). The iRGD peptide can specifically bind to the neuropilin-1 (NRP-1) receptor, which is primarily located in tumor and vascular tissues, where it activates an endocytic transport pathway and promotes drug penetration into tumor tissues. Previous studies described favorable outcomes of iRGD treatment in liver cancer.19,20 Moreover, iRGD peptide co-administration can effectively augment drug efficacy.21,22
We prepared sorafenib-loaded MIL-101(Fe) NPs (MIL-101(Fe)@sor) and co-administered iRGD to induce liver cancer ferroptosis as a novel treatment strategy. When MIL-101(Fe)@sor is internalized by cancer cells, the released iron ions and sorafenib synergistically increase lipid peroxidation (LPO). Magnetic resonance (MR) was used to visualize NPs in the tumor sites, and the iRGD peptide was found to enhance NP delivery into the tumor parenchyma and accelerate ferroptosis induction.
Materials and Methods
Materials
FeCl3·6H2O, H2BDC, H2BDC-NH2, and N,N-dimethylformamide (DMF) were obtained from Aladdin Biochemical Co., Ltd. (Shanghai, China). Sorafenib, 4ʹ,6-diamidino-2-phenylindole (DAPI), and 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) were purchased from Dalian Meilun Biochemical Co., Ltd (Dalian, China). iRGD peptide was purchased from Shanghai Apeptide Co., Ltd (Shanghai, China). Ferrostain-1 (Fer-1) and deferoxamine mesylate (DFOM) were obtained from Macklin Biochemical Co., Ltd. (Shanghai, China). GSH assays, reactive oxygen species (ROS) and malondialdehyde (MDA) assays, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) preparations, and enhanced chemiluminescence (ECL) detection kits were obtained from Beyotime Biotechnology Co., Ltd (Shanghai, China). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), and trypsin were purchased from HyClone Biochemical Co., Ltd (Shanghai, China). BODIPY581/591-C11 was obtained from Thermo Fisher Scientific (Waltham, MA, USA). Anti-glutathione peroxidase 4 (anti-GPX-4) and goat anti-rabbit IgG H&L (fluorescein isothiocyanate) were purchased from Abcam (Cambridge, UK).
MIL-101(Fe) NP Preparation
The synthesis method of MIL-101(Fe) NPs referred to our previous work.23 First, H2BDC (0.011 g), H2BDC-NH2 (0.012 g), and FeCl3·6H2O (0.058 g) were dissolved in 120 mL DMF. After stirring for 20 min, the mixture was then transferred to a Teflon-lined, stainless steel autoclave and incubated at 120°C for 10 h. After natural cooling, the remaining solution was centrifuged for 30 min at 10,000 rpm and washed three times with ethanol. Finally, the precipitate was quantified and dispersed in alcohol.
NP Characterization
NP morphology was assessed with scanning electron microscopy (SEM; SU3500; Tokyo; Japan) and transmission electron microscopy (TEM; H-7650; Tokyo; Japan), and NP size was measured from the SEM images. NP elemental composition was analyzed using SEM mapping and SEM energy dispersive X-ray spectroscopy (EDS). For in vitro MRI examination, the T2*-weighted MR images of MIL-101(Fe) NPs at various concentrations (1.875, 3.75, 7.5, and 15 mg/mL) were scanned using an MR scanner (3.0 T, GE Healthcare, Chicago, IL, USA). Then, the reconstructed MR images were generated using a GE AW 4.6 Advantage Workstation.
Drug Loading
Initially, 5 mg of sorafenib was dissolved in 1 mL DMSO. Then, 10 mg of MIL-101(Fe) NPs were mixed with the sorafenib solution under vigorous stirring for 36 h. The precipitation was obtained after centrifuging at 10,000 rpm for 15 min and washed three times with ultrapure water to remove unloaded sorafenib. The supernatant solution obtained by centrifugation was collected to calculate the drug loading rate (DL%) and drug encapsulation rate (EE%) using a Multiskan Sky microplate reader (Thermo Fisher Scientific) at 265 nm. The EE% and DL% were calculated as follows: EE% = content of sor loaded in MIL-101(Fe)/initial sor content × 100%; DL% = content of sor loaded in MIL-101(Fe)/content of sor loaded in MIL-101(Fe) + weight of MIL-101(Fe) × 100%.
Drug Release Behavior
To investigate the effect of pH on sorafenib release, MIL-101(Fe)@sor NPs were dissolved in phosphate-buffered saline (PBS) containing 1% Tween 80 with different pH values (pH 5.5 and 7.4) with gentle shaking (200 rpm/min) at 37°C. The supernatant was collected after centrifugation (10,000 rpm, 15 mins) at different time points and added to an equal volume of fresh PBS. All collected supernatants were extracted using methanol and measured using a Multiskan Sky microplate reader at 265 nm to determine the amount of released sorafenib based on a standard curve.
Cell Culture
Human hepatoma HepG2 cells were obtained from the Chinese Academy of Sciences (CAS, Shanghai, China). Cell culture medium was DMEM supplemented with 10% (v/v) FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were cultured in a 37°C humidified incubator in an atmosphere of 5% CO2.
In vitro Cellular Toxicity
Briefly, HepG2 cells (5 × 103 cells/well) were uniformly seeded into 96-well plates and cultured in an atmosphere of 37°C and 5% CO2 overnight. Then, HepG2 cells were incubated with fresh culture medium containing different concentrations of MIL-101(Fe) NPs (1.5625, 3.125, 6.25, 12.5, 50, 100, and 200 μg/mL). After 24 h, 20 μL of MTT (5 mg/mL) reagent was added into the wells and incubated for another 4 h. Nest, 100 μL DMSO was added into each well. Finally, a Multiskan Sky microplate reader was used to measure absorbance at 490 nm.
Cellular Uptake Analysis
HepG2 cells were treated with MIL-101(Fe)@sor NPs and prepared in accordance with the experimental procedure for TEM imaging. Next, the HepG2 cells were incubated with DMEM, MIL-101(Fe)@sor NPs, and MIL-101(Fe)@sor NPs + iRGD for 6 h (using 10 μM Sor and 30 μmol/L iRGD).24 Then the cells were collected, washed with PBS three times, centrifuged, and uniformly dispersed in agarose gel. Finally, the cells were scanned with 3.0 T MR, and the T2* mapping image signals were reconstructed using a GE AW 4.6 Advantage Workstation.
Peroxidase (POD) Activity
H2O2 can undergo cleavage by MIL-100(Fe) to produce hydroxyl radicals (·OH) that belong to the ROS family; 3,3ʹ,5,5ʹ-tetramethylbenzidine (TMB) can be used as an indicator to detect ·OH.25,26 Therefore, TMB, TMB + H2O2, and TMB + H2O2 + MIL-101(Fe) were prepared. All samples were imaged and then measured using a microplate reader following a 30-min reaction. In addition, ·OH can be detected using the green fluorescence produced by the oxidation of DCFH-DA (2,7-dichlorofluorescein diacetate) by intracellular ROS.27 HepG2 cells were seeded into 6-well plates and allowed to adhere and grow. Then cells were treated with DMEM, H2O2, MIL-101(Fe), or MIL-101(Fe)+H2O2 and incubated for an additional 4 h. Fresh DMEM containing 10 μM DCFH-DA was added, and the incubation was continued for another 30 min. Finally, ·OH generation was observed using fluorescence microscopy.
In vitro Anti-Tumor Activity Evaluation
HepG2 cells were seeded into 96-well plates and incubated for 24 h before adding fresh culture medium containing 100 μM H2O2 and different concentrations of MIL-101(Fe) NPs (12.5, 50, 100, and 200 μg/mL) for another 24 h. Next, the cells were co-incubated for 24 h with culture medium containing sorafenib, MIL-101(Fe)@sor, or MIL-101(Fe)@sor + iRGD, with an equivalent dosage of 1.25, 2.5, 5, 10, or 20 μM sorafenib and 30 μM iRGD. The MTT method was used to quantitatively analyze in vitro anti-tumor activity. HepG2 cell morphological changes were observed using microscopy before and after MIL-101(Fe)@sor NP treatment.
Effect of Ferroptosis Inhibitors on Cell Viability
HepG2 cells were seeded into 96-well plates and incubated until attachment. Then the medium was replaced with fresh culture medium containing 200 μM DFOM (an iron-chelating agent),28 ferrostatin-1 (Fer-1, an inhibitor of ferroptosis; 1 μM),28 and DMEM and incubated for 1 h. Next, the cell culture medium was replaced with DMEM, MIL-101(Fe), sorafenib, MIL-101(Fe)@sor, or MIL-101(Fe)@sor/iRGD) for 24 h at an equivalent dosage of 10 μM sorafenib and 30 μmol/L iRGD. Finally, cell viability was quantitatively analyzed using MTT assays.
Intracellular Malondialdehyde (MDA) Assay
Intracellular MDA, a product of lipid peroxidation, was measured using an MDA assay kit.29 Briefly, HepG2 cells were seeded into 6-well plates for attachment. Then, DMEM, MIL-101(Fe), sorafenib, MIL-101(Fe)@sor, or MIL-101(Fe)@sor + iRGD was added, and cells were cultured for an additional 6 h (at an equivalent dosage of 10 μM sorafenib or 30 μmol/L iRGD). Subsequently, the relative MDA concentration in cell lysates was measured as indicated in the manufacturer’s instructions. Lastly, the percent content of MDA was determined relative to MDA content in the DMEM group, which was set at 100%.
Intracellular Glutathione (GSH) Assay
HepG2 cells were seeded into 6-well plates and cultured at 37°C overnight. Then, DMEM, MIL-101(Fe), sorafenib, MIL-101(Fe)@sor, or MIL-101(Fe)@sor + iRGD were added and cultured for an additional 6 h (at an equivalent dosage of 10 μM sorafenib or 30 μmol/L iRGD). Intracellular GSH levels were measured using a commercial GSH kit with a microplate reader at 412 nm. The percent content of GSH was determined relative to GSH content in the DMEM group, which was set at 100%.
Intracellular Lipid Peroxide (LPO) Assay
The fluorescent probe C11-BODIPY581/591 was used to determine intracellular LPO levels.30 HepG2 cells were seeded in 24-well plates until attachment. The medium was then replaced with fresh culture medium supplemented with DMEM, MIL-101(Fe), sorafenib, MIL-101(Fe)@sor, and MIL-101(Fe)@sor + iRGD (at an equivalent dosage of 10 μM sorafenib, 30 μmol/L iRGD) and incubated for an additional 4 h. Next, the cells were incubated in fresh DMEM containing 2 μM C11BODIPY581/591 and cultured for another 30 min. Finally, the cells were stained with DAPI to visualize nuclei, washed with PBS, and then imaged using fluorescence microscopy.
Western Blot (WB)
HepG2 cells were seeded in 6-well plates and grown in a cell incubator overnight. DMEM, MIL-101(Fe), sorafenib, MIL-101(Fe)@sor, or MIL-101(Fe)@sor + iRGD was added and cultured at an equivalent dosage of 10 μM sor or 30 μmol/L iRGD for an additional 24 h. Proteins were subjected to SDS-PAGE and later transferred to polyvinylidene fluoride membranes. After, the membranes were blocked with 5% BSA, they were incubated with anti-GPX4 antibody at 4°C overnight, followed by the secondary antibody for 1.5 h at room temperature. Anti-β-actin was used as an internal loading control. Membranes were visualized using an enhanced ultrasensitive ECL instrument. ImageJ software was used to quantify relative expression normalized to β-actin.
Animals and Tumor Models
The study was conducted according to the “Guide for the Care and Use of Laboratory Animals,” 8th edition (International Publication No: 978-0-309-15400-0), and the protocol was approved by the Institutional Animal Care and Use Committee of the China Medical University (CMU).
Five-week-old Kun Ming (KM) mice (approximate weight: 25 g) were purchased from Changsheng Organisms Co. Ltd. (Changchun, China). Mouse H22 hepatoma cells were prepared at a density of 5 × 106/mL in PBS. The liver cancer model was successfully established by subcutaneous injection of 200 μL H22 cells into the right hind leg of each mouse. At the end of the experiments, the mice were sacrificed under deep anesthesia with intraperitoneal administration of pentobarbital sodium (150 mg/kg body weight).
Hemolysis
This test was performed as previously described.24,31 Briefly, 5-mL blood samples were collected from New Zealand rabbits. Blood fibrinogen was removed by vigorous stirring, followed by three washes with approximately 10 times the volume of normal saline (NS). Next, a 2% erythrocyte suspension was made in NS, then the 2.5-mL erythrocyte suspension was mixed with MIL-101(Fe)@sor NPs into tubes 2–6 where the final concentrations of sorafenib were 6, 12, 18, 24 and 30 μg/mL, while deionized water (tube 7) and NS (tube 1) were used as the positive and negative controls, respectively. All tubes were reacted for 3 h at 37°C.
In vivo Tumor Permeability Assay
A total of 12 male KM mice were randomly divided into 4 groups. The liver cancer model was established, and the tumor volumes reached approximately 160 mm3. Next, 200 μL PBS, 4 mg/kg iRGD, 50 mg/kg Evans Blue, or 50 mg/kg Evans Blue + 4 mg/kg iRGD were injected into the tail vein of tumor-bearing mice.32 After 30 min, the left ventricle was cannulated, and the mouse hearts were perfused with 1% bovine serum albumin in PBS. After euthanasia, the primary organs (lung, liver, spleen, heart, kidney) were collected. Evans Blue was extracted from tissues after DMF was added at 10 μL/10 mg tissue and then incubated at 37°C for 20 h. Finally, the samples were centrifuged, and the amounts of Evans Blue were quantified using a Multiskan Sky microplate reader at 600 nm.
Toxicity Studies
An acute toxicity study was conducted prior to conducting animal experiments. A total of 20 male KM mice were randomly divided into 4 groups. MIL-101(Fe) NPs were dispersed in PBS and injected through the tail vein at doses of 0, 25, 50, and 100 mg/kg for groups 1–4, respectively. Mouse growth was evaluated daily, and body weight was recorded every 3 days. On day 15, blood samples were collected from the ophthalmic venous plexus for routine blood analysis and liver and kidney function tests. Specifically, we measured the biomarkers white blood cell (WBC), red blood cell (RBC), platelet (PLT), hemoglobin (HGB), alanine aminotransferase (ALT), glutamic oxaloacetic transaminase (AST), blood urea nitrogen (BUN), and creatinine (CR). After euthanasia, the primary organs (liver, heart, lungs, spleen, and kidneys) were removed, fixed in 4% neutral formaldehyde, and used for subsequent hematoxylin and eosin (H&E) staining.
In vivo Tumor Inhibition
After the H22 tumor-bearing liver cancer model was successfully established and the tumor volumes reached ~100 mm3, the mice were randomly divided into 5 groups (n = 6 per group). Mice were intravenously injected once every 3 days, for a total of 21 days (7 injections) with either PBS, MIL-101(Fe), sorafenib, MIL-101(Fe)@sor, or MIL-101(Fe)@sor + iRGD at an equivalent dosage of 5 mg/kg sorafenib.24 Body weight and tumor sizes were recorded every 3 days. The tumor volume was calculated according to the following formula: V = (A × B2)/2, where “A” represents the longest tumor diameter and “B” represents the shortest tumor diameter perpendicular to “A.” When the experiment was finished, liver and kidney function tests and routine blood tests were performed on samples collected from the ophthalmic venous plexus. After euthanasia, the major organs (spleen, lungs, heart, liver, and kidneys) and tumors were harvested, weighed, photographed, and fixed with 4% neutral formaldehyde prior to H&E and immunohistochemistry (IHC) staining. The samples were embedded in tissue-freezing medium at −80°C before immunofluorescence (IF) staining. During treatment, the mice were scanned using an MR scanner (GE, 3.0 T) at different time points (0, 1, 6, and 12 h) after intravenous injection. The MR signal intensities of tumor sites were quantified using the GE AW 4.6 Advantage Workstation.
Statistical Analysis
Results are presented as mean ± standard deviation (SD). Differences among groups were compared with one-way analysis of variance followed by LSD t-tests. SPSS version 22.0 software (IBM, Armonk, NY, USA) was used for all analyses, and p < 0.05 was considered statistically significant.
Results
NP Synthesis and Characterization
The synthesis of MIL-101(Fe)@sor is depicted in Figure 1. SEM and TEM results showed that the MIL-101(Fe) NPs had good dispersibility, uniform size, and smooth surfaces (Figure 2A and B). The EDS and elemental mapping results obtained by SEM indicated that the primary elements included Fe, Cl, O, and C (Figure 2C and D). MIL-101(Fe) NPs were ~200 nm as measured on SEM and TEM images (Figure 2E). Figure 2F shows that MIL-101(Fe) NPs had a good T2-weighted MRI properties. The bright T2 MRI signal was attenuated with increasing concentration of MIL-101(Fe) NPs, and the T2 relaxation rate at different concentrations was 8.3348. The DL% and EE% were 12.1% and 27.8%, respectively; the equation for the sorafenib curve was y = 0.1162x + 0.068, R2 = 0.999. As shown in Figure 2G, sorafenib release from MIL-101(Fe)@sor NPs occurred in a time- and pH-dependent manner. Sorafenib was gradually released from MIL-101(Fe)@sor NPs without an obvious burst-release effect. Drug release reached approximately 35% at pH 5.5 and only 10% at pH 7.4 after 60 h.
 |
Figure 1 Schematic illustration of the synthesis and application of MIL-101(Fe)@sor NPs. |
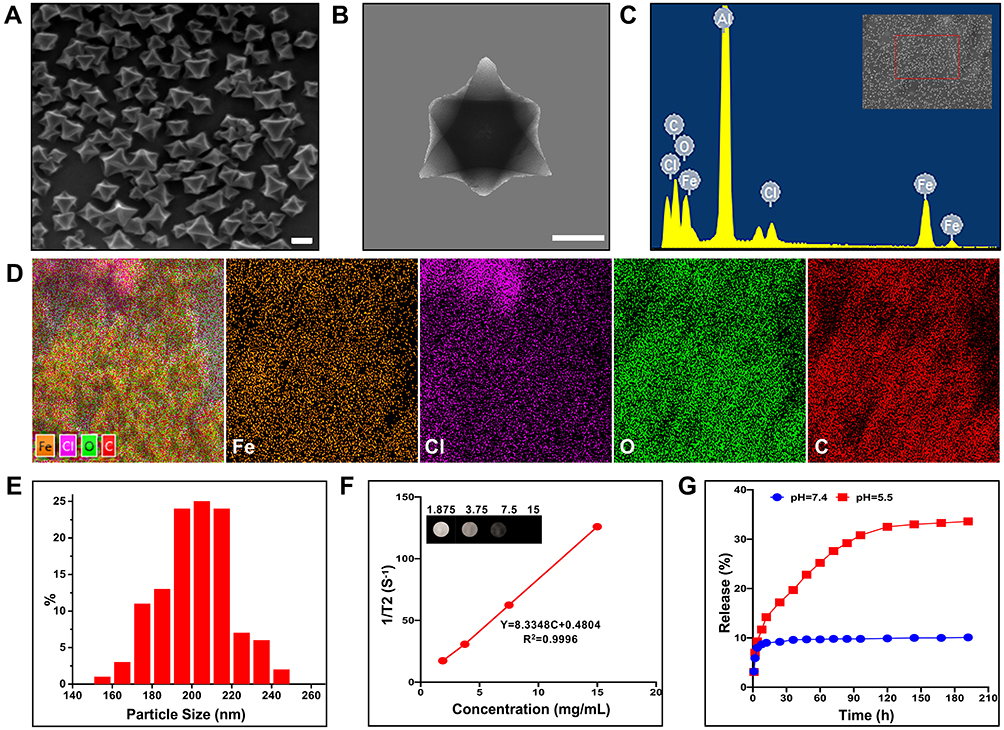 |
Figure 2 Characterization of MIL-101(Fe) NPs. (A) SEM and (B) TEM images of MIL-101(Fe) NPs (scale bar: 100 nm). (C) EDS spectrum and (D) element mappings of MIL-101(Fe)NPs. (E) Particle size distribution of MIL-101(Fe) NPs. (F) A plot of relaxation rate 1/T2 versus concentration of MIL-101(Fe) NPs using a MRI scanner; T2 MRI of diverse MIL-101(Fe) NPs concentrations in vitro (inset). (G) Sorafenib release profiles of MIL-101(Fe)@sor NPs at different pH values (5.5 and 7.4). |
POD Activity Assay
Upon combining MIL-101(Fe), H2O2, and TMB, the color changed from clear to navy blue and had an absorption peak at 652 nm that increased with time (Figure S1 A and 1B). When using DCFH-DA to evaluate ·OH generation, almost no green fluorescence was detected in HepG2 cells following treatment with DMEM, H2O2, and MIL-101(Fe) NPs. However, significant green fluorescence was observed after MIL-101(Fe) NPs were treated with H2O2 (Figure S1 C).
MIL-101(Fe) NP Toxicity and Biocompatibility
The in vitro cytotoxicity of MIL-101(Fe) NPs in HepG2 cells was assessed using MTT assays. Cells treated with MIL-101(Fe) NPs showed >85% viability at concentrations ranging from 1.56–200 μg/mL (Figure 3A). To further determine hemocompatibility, we performed in vitro hemolysis assays. As shown in Figure 3B, all red cells were precipitated in tubes 1–6, and the supernatant was colorless and transparent, indicating that the MIL-101(Fe)@sor NPs did not induce hemolysis; tube 7 was red, confirming that hemolysis occurred in the positive control sample.
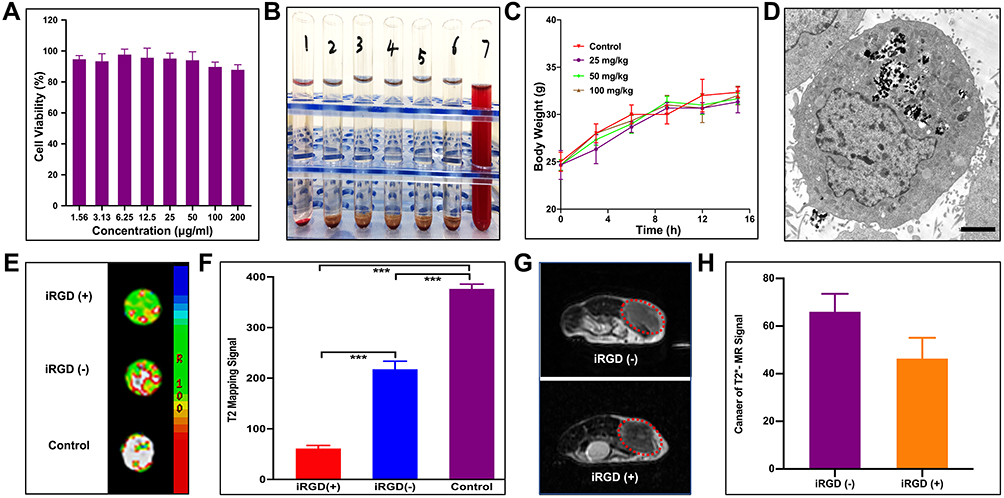 |
Figure 3 Toxicity and absorption of MIL-101(Fe) NPs in vitro and in vivo. (A) Viability of HepG2 cells treated with various concentrations of MIL-101(Fe) NPs for 24 h. (B) Photograph of hemolysis samples for MIL-101(Fe)@sor. Tube 1: negative control (NS), Tubes 2–6: five different concentrations MIL-101(Fe)@sor from low to high, Tube 7: positive control. (C) Change in body weights of mice injected with various concentrations of MIL-101(Fe) NPs for 15 days. (D) TEM images of HepG2 cells treated with MIL-101(Fe)@sor. (E) MR images and (F) quantized MR signal intensity of HepG2 cells treated with DMEM, MIL-101(Fe) @sor, or MIL-101(Fe)@sor + iRGD. (G) MR images and (H) quantized MR signal intensity of H22 liver tumors injected with MIL-101(Fe) NPs or MIL-101(Fe) NPs + iRGD. The red circle indicates the tumor. |
We then conducted in vivo acute toxicity experiments where different concentrations of MIL-101(Fe) NPs, were injected into mice. No variation in body weight was observed among the groups (Figure 3C). H&E staining did not reveal any obvious pathologic damage in the major organs (heart, liver, spleen, lung, and kidney) (Figure S2). Blood biochemical tests for WBC, RBC, PLT, HGB, ALT, AST, BUN, and CR were in the normal range for all groups (Figure S3).
Cellular Uptake and Tumor Targeting of iRGD
MIL-101(Fe) NP uptake and cellular distribution were detected by TEM. MIL-101(Fe) NPs were successfully absorbed by cells, accumulated in the cytoplasm, and did not enter the nuclei (Figure 3D). MR was conducted to evaluate the cellular uptake of MIL-101(Fe) NPs and targeting of iRGD. As shown in the reconstructed MR images in Figure 3E, HepG2 cells treated with NPs showed a lower signal intensity than the control group. Furthermore, the signal intensity of HepG2 cells treated with MIL-101(Fe)@sor + iRGD was significantly lower than that of the MIL-101(Fe)@sor group after quantitative analysis (Figure 3F, p < 0.05). To assess the in vivo tumor-penetration capacity of the iRGD peptide, we used Evans Blue dye33 and MR. The blue intensity indicates the quantity of accumulated Evans Blue. The tumor color in Evans Blue + iRGD group was the darkest among all the groups, while the other organs were clear (Figure S4 A). The Evans Blue content was highest in tumors from the Evans Blue + iRGD group (p < 0.001) compared with other groups (Figure S4 B). As shown in Figure 3G, after MIL-101(Fe)@sor + iRGD injection, a significant decrease in MRI was noted compared with MIL-101(Fe)@sor injection, as shown by a darkening of the tumor. The quantified MR signal intensity for the MIL-101(Fe)@sor + iRGD-injected group showed a significant decrease compared to the MIL-101(Fe)@sor-injected group (Figure 3H).
Mechanism of MIL-101(Fe)@sor-Induced Ferroptotic Cell Death
When 100 μM H2O2 was added to HepG2 cells, cell viability decreased with higher MIL-101(Fe) NPs concentration (Figure 4A). Nest, the anti-cancer effects of free sorafenib, MIL-101(Fe)@sor, and MIL-101(Fe)@sor + iRGD were evaluated in HepG2 cells; all treatment groups showed dose-dependent cytotoxicity with increased sorafenib concentration (Figure 4B). The MIL-101(Fe)@sor + iRGD group showed a stronger increase in cytotoxicity than the MIL-101(Fe)@sor group at an equivalent concentration range (1.25 μM to 20 μM). Compared with untreated cells, MIL-101(Fe)@sor NP-treated HepG2 cells showed cytopathic changes such as cellular volume swelling and nuclear pyknosis, followed by cell detachment compared (Figure S5).17
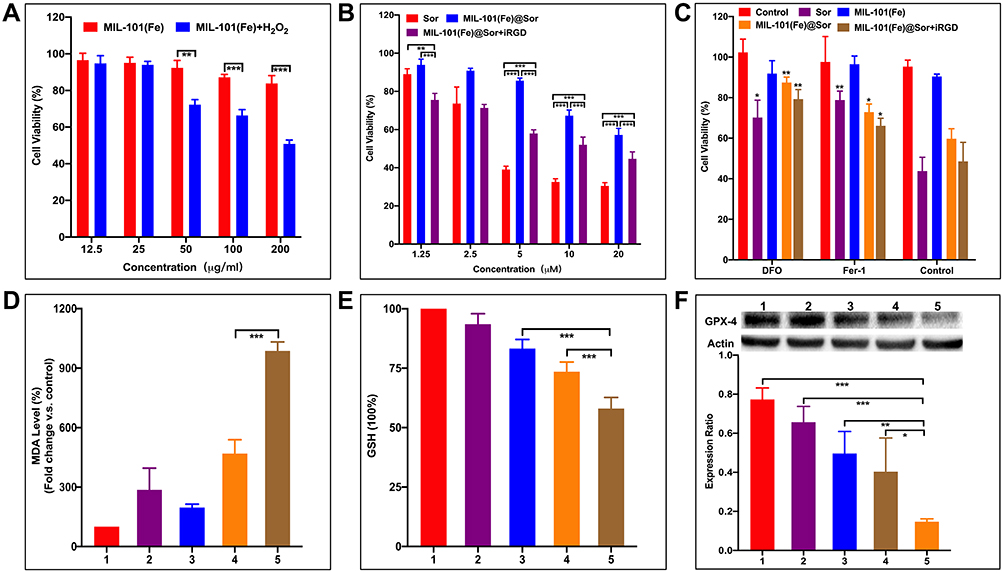 |
Figure 4 Mechanism of MIL-101(Fe)@sor-induced ferroptotic cell death. (A and B) MTT viability assays of HepG2 cells with different treatments. (C) MTT viability assays of HepG2 cells with different treatments and ferroptosis inhibitors. (D and E) MDA and GSH levels in HepG2 cells after different treatments. (F) WB analysis of GPX4 expression in HepG2 cells after different treatments. 1–5: control, MIL-101(Fe), sorafenib, MIL-101(Fe)@sor, and MIL-101(Fe)@sor + iRGD groups, respectively. *p < 0.05, **p < 0.01, ***p < 0.001. |
To determine the mechanism by which MIL-101(Fe)@sor induces cell death, HepG2 cells were pre-treated with ferroptosis inhibitors (Fer-1 and DFOM) prior to in vitro cytotoxicity assays. In the presence of either Fer-1 or DFOM, the toxicities of free sorafenib, MIL-101(Fe)@sor, and MIL-101(Fe)@sor + iRGD were significantly alleviated (p < 0.05, Figure 4C). In comparison, MIL-101(Fe) NPs induced negligible toxicity; therefore, the addition of Fer-1 and DFOM had an insignificant effect.
MDA is a product of LPO formed through free radical reactions mediated by hydroxyl ions and is a prominent cytotoxic product of the iron-catalyzed Fenton-like reaction.29 The MIL-101(Fe)@sor + iRGD group produced the highest MDA levels among the groups (Figure 4D), and the difference was statistically significant (p < 0.05). Furthermore, sorafenib, MIL-101(Fe)@sor, and MIL-101(Fe)@sor + iRGD depleted intracellular levels of GSH, which is an indicator of ferroptosis34 (Figure 4E). In contrast, GSH levels were significantly lower in HepG2 cells treated with MIL-101(Fe)@sor + iRGD compared to the other four groups (p < 0.05). Expression of GPX-4, another ferroptosis biomarker,9 was consistent with the change trend of GSH level, with the MIL-101(Fe)@sor + iRGD group having the strongest inhibitory effect (p < 0.05) as evidenced by WB (Figure 4F). These results indicate that MIL-101(Fe)@sor induces cell death through the ferroptotic pathway.
Intracellular LPO Generation
Intracellular LPO plays an important role in ferroptosis.8 Thus, we used the lipid peroxidation indicator C11-BODIPY581/591 to assess LPO levels. As shown in Figure 5, the nuclei and LPO signals were blue and green, respectively. MIL-101(Fe)@sor + iRGD-treated HepG2 cells had the strongest fluorescence signal among the groups, whereas the cells in the control and MMIL-101(Fe) groups showed very weak fluorescence.
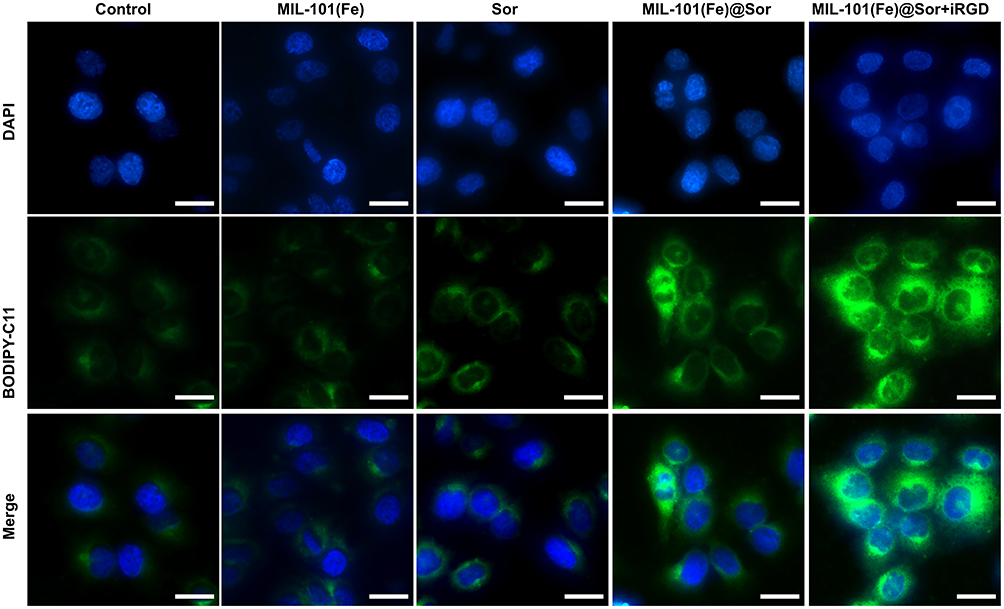 |
Figure 5 Representative image of LPO in HepG2 cells after different treatments. |
In vivo Tumor Suppression Evaluation
The in vivo tumor suppressive effect of the prepared NPs was evaluated using the H22 tumor-bearing mouse model. H22 tumors grew extremely rapidly in the control and MIL-101(Fe) NP groups, whereas they grew slowly in the other groups (Figure 6A). As shown in Figure 6B, harvested tumors were smallest in the MIL-101(Fe)@sor + iRGD group. MIL-101(Fe)@sor + iRGD-treated mice had the highest degree of tumor inhibition (p < 0.05) with significantly decreased tumor weights (p < 0.05) compared with other groups (Figure 6C). There was no significant decrease in body weight in the MIL-101(Fe)@sor + iRGD group during treatment (Figure 6D).
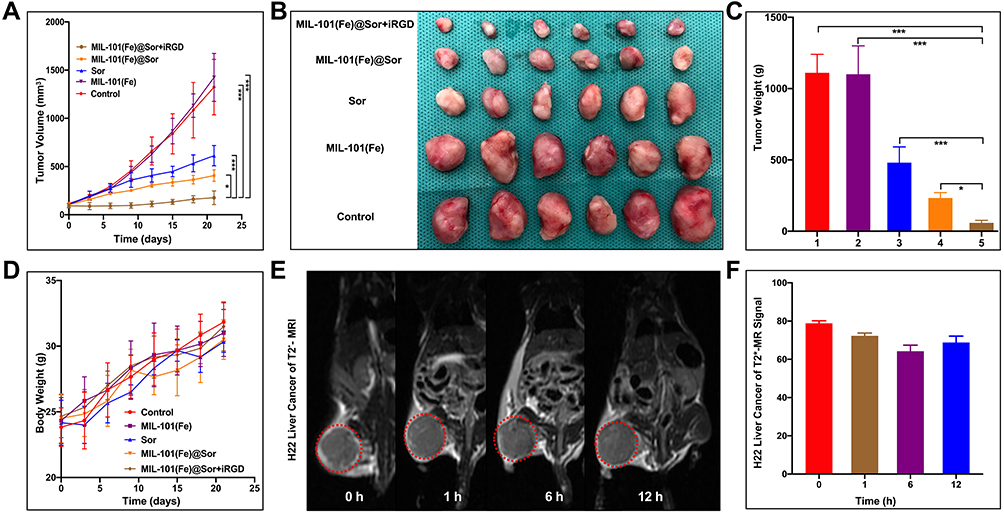 |
Figure 6 In vivo anti-tumor efficacy evaluation. (A) Tumor growth curves of different groups of tumor-bearing mice after various treatments indicated for 21 days (n=6). (B) Pictures of the harvested tumors (n=6). (C) Tumor weight of different groups (1–5: control, MIL-101(Fe), sorafenib, MIL-101(Fe)@sor, and MIL-101(Fe)@sor + iRGD groups, respectively). (D) Body weights of mice after different treatments. (E) MR images of mice at 0, 1, 6, and 12 h after treatment. The red circle indicates the tumor. (F) MR signal intensity of the tumors at the indicated time points. *p < 0.05, ***p < 0.001. |
T2* mapping images of H22 tumor-bearing KM mice in Figure 6E demonstrate significant contrast between the MR images of the liver and tumor. Following MR image reconstruction and quantization, the signal intensity showed a dynamic change in the tumor site, while the signal intensity of H22 liver cancer peaked at 6 h following MIL-101(Fe)@sor NP injection (Figure 6F).
The tumor slices were then stained and imaged. The MIL-101(Fe)@sor + iRGD group had the largest necrotic area in tumor tissues and the lowest number of GPX-4-positive cells compared with other groups, as evidenced by H&E and IHC (Figure 7). IF for GPX-4 in different tumor tissues demonstrated that MIL-101(Fe)@sor + iRGD significantly inhibited GPX-4 expression in tumor tissues, showing the strongest inhibitory effect compared with the other groups (Figure S6).
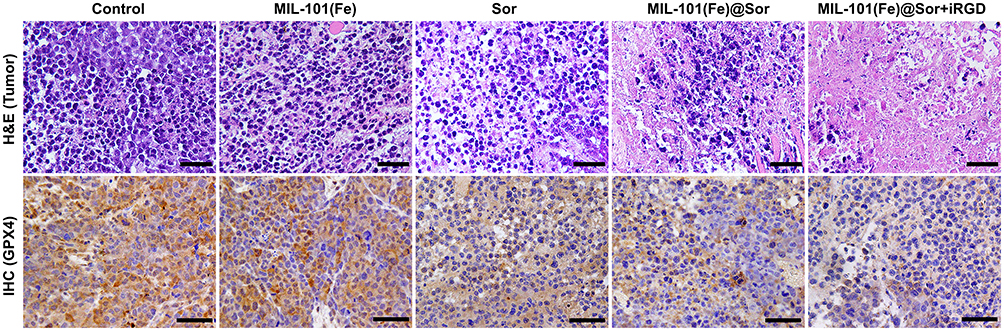 |
Figure 7 H&E and IHC staining of GPX4 in tumor sections collected from differently treated groups of mice (scale bar: 50 μm). |
Systemic Toxicity Evaluation
There was no significant decrease in the body weights of mice treated with MIL-101(Fe)@sor + iRGD during treatment. Although obvious lung inflammation and liver damage were observed in the sorafenib group (Figure 8), the organs of the remaining groups showed relatively normal tissue structure and morphology. In addition, blood test results revealed that the sorafenib group exhibited high AST levels compared with the control group, indicating liver damage (Figure S7). All other blood test results of the experimental groups were in the normal ranges.
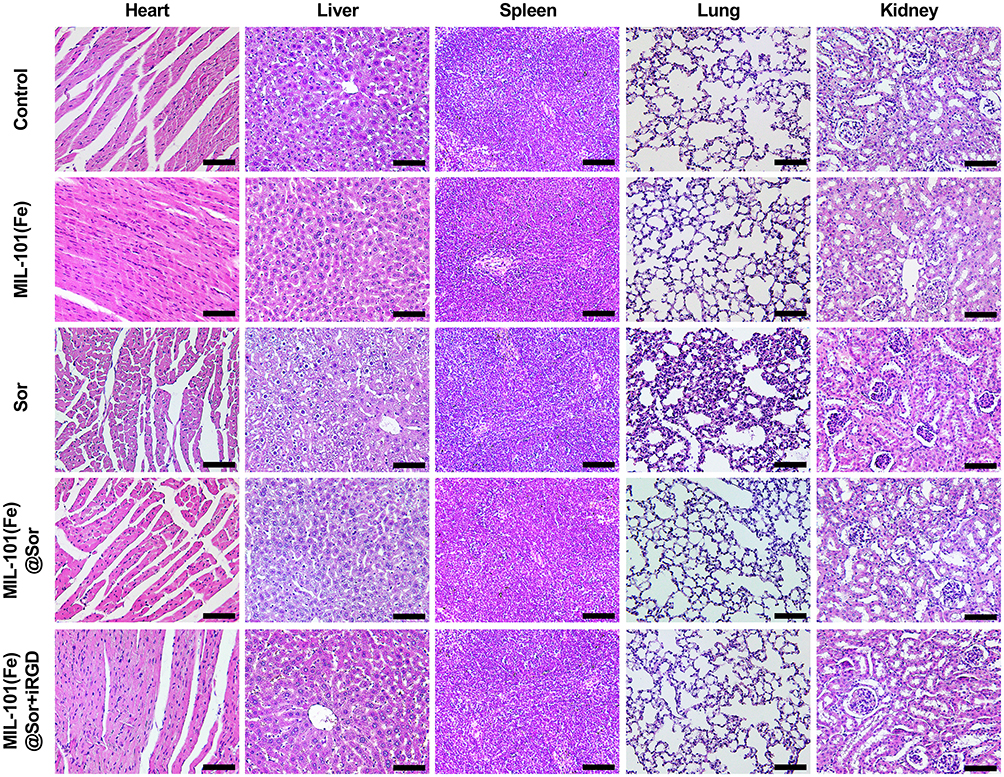 |
Figure 8 H&E staining of major organs from differently treated groups at day 21 (scale bar: 100 μm). |
Discussion
In this study, we introduced a novel therapeutic strategy that relies on ferroptosis and prepared MIL-101(Fe) as a carrier for loading the ferroptotic agent sorafenib. To increase active MIL-101(Fe)@sor delivery into tumors, the tumor-penetrating peptide iRGD was given with the nanodrugs. We found that MIL-101(Fe)@sor co-administration with iRGD significantly induced ferroptosis in liver cancer.
The enhanced permeability and retention (EPR) effect provided the theoretical basis for passive tumor targeting of nanoparticles in recent decades. However, because of unique tumor characteristics like heterogeneity, high interstitial fluid pressure, and dense tumor stroma, the therapeutic efficacy of anti-cancer nanomedicines remained limited to the EPR effect.35 The iRGD peptide can significantly promote drug penetration into tumor tissues by actively opening specific drug transport channels.20 Previous studies have demonstrated that iRGD co-administration improves the therapeutic effects of nanodrugs.21,22 In agreement with this, our results showed that iRGD significantly increased tumor drug accumulation in vitro and in vivo.
Most nanomaterials that can be used effectively to induce ferroptosis are iron-based, such as iron oxide nanoparticles (IONPs),36 iron-based up-conversion nanomaterials,37 iron-doped nanomaterials,38 iron-based metal-organic frameworks (MOF),39 iron-based metal-organic networks (MON),40 iron-based polymer micelles,41 and iron nanometallic glasses.42 MOFs have received considerable attention owing to their ultrahigh pore volume, enormous surface area, and biodegradability.43 We prepared MIL-101(Fe), which has a high drug-loading capacity, controllable release profile, excellent POD catalytic activity, and good biocompatibility; it can also be used as a MR contrast agent. Encapsulating sorafenib into MIL-101(Fe) resulted in satisfactory EE% and DL%, which improved its solubility and stability. The prepared MIL-101(Fe)@sor exhibited pH-responsive drug-controlled release properties that benefit from MIL-101(Fe) NP degradability.23 Through the Fenton reaction, POD converted H2O2 into ·OH, further contributing to ferroptotic cell death. Moreover, in vitro MRI examination of MIL-101(Fe) NPs proved that they were a good candidate for T2-weighted MRI. The MRI signal intensity decreased with increased cellular uptake of MIL-101(Fe) NPs. Indeed, their intracellular uptake could be quantified using MR image processing software. Additionally, we demonstrated that MIL-101(Fe) had negligible cytotoxicity, as evidenced by MTT assays, biochemical tests, and H&E staining. Inducing ferroptosis is a novel and promising therapeutic strategy against cancer; and developing ferroptosis-inducing nanodrugs for HCC therapy is essential. The mechanisms underlying ferroptosis are mostly associated with LPO accumulation and iron metabolism.7 To exploit the key mechanism of ferroptosis, we combined MIL-101(Fe) (iron-based nanomaterials) with the ferroptosis-inducing agent sorafenib. In HepG2 liver cancer cells, MIL-101(Fe)@sor NP-induced ferroptosis was characterized by increased levels of LPO, MDA, and GPX-4 protein, as well as reduced levels of GSH. The morphological changes of HepG2 cells observed following MIL-101(Fe)@sor NP treatment were consistent with the characteristics of ferroptosis.10 Moreover, the cytotoxicity induced by MIL-101(Fe)@sor NPs could be significantly alleviated using an iron-chelating agent (DFOM) and a ferroptosis inhibitor (Fer-1). Collectively, these results demonstrate that MIL-101(Fe)@sor NPs can significantly induce ferroptosis in liver cancer cells. Co-administration of iRGD increased MIL-101(Fe)@sor NP delivery and accelerated ferroptosis, thereby enhancing their efficacy.
The therapeutic effect was further investigated in vivo using an liver cancer xenograft mouse model. Encapsulation of MIL-101(Fe) NPs with sorafenib and co-administration with iRGD achieved the strongest suppressive effect on tumor growth in our study. GPX-4 plays an important role in inhibiting ferroptosis and suppressing LPO generation.9 Our IHC and IF results for GPX-4 demonstrated that MIL-101(Fe)@sor + iRGD had the strongest inhibitory effect on GPX-4 expression levels in all tumor tissues. These results demonstrate that when sorafenib, MIL-101(Fe) NPs, and iRGD peptide were combined, they successfully induced ferroptotic death in liver cancer cells. Moreover, the MR properties of MIL-101(Fe) NPs could help us monitor the process of ferroptosis in real time. Most importantly, MIL-101(Fe)@sor + iRGD showed no significant toxicity in vivo and did not serious side effects, indicating good biocompatibility and its possible clinical application. The low toxicity of MIL-101(Fe)@sor + iRGD benefited from nanodrug characteristics and the active targeting of iRGD peptide to tumors. We made full use of existing clinical anti-liver cancer drugs, which could expand the therapeutic value of clinical drugs. In summary, MIL-101(Fe)@sor was designed to eradicate cancer cells via the ferroptosis pathway; it accumulated in tumor tissues through the EPR effect and active targeting by iRGD.
Conclusion
We developed a sorafenib-loaded MIL-101(Fe) nanodrug to be co-administered with iRGD as a ferroptosis-associated therapy for liver cancer. In vitro studies showed excellent efficacy in eradicating cancer cells. More importantly, the cell death induction mechanism was thoroughly investigated. This system can consume GSH, decrease GPX-4 levels, enhance LPO generation, and simultaneously supply iron ions. In vivo studies in an H22-bearing liver cancer xenograft mouse model demonstrated that this nanodrug can effectively inhibit tumor growth with good biological safety. Our study provides a promising strategy to construct highly effective ferroptosis-inducing nanomedicines. Such treatments may have significant therapeutic advantages and warrant further investigation.
Abbreviations
HCC, hepatocellular carcinoma; sor, sorafenib; NPs, nanoparticles; POD, peroxidase; MR, magnetic resonance; RCD, regulated cell death; LPO, lipid peroxidation; GPX-4, glutathione peroxidase 4; GSH, glutathione; DMF, N,N-dimethylformamide; DAPI, 4,6-diamidino-2-phenylindole; MTT, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide; Fer-1, ferrostain-1; DFOM, deferoxamine mesylate; DMEM, Dulbecco’s modified Eagle’s medium; FBS, fetal bovine serum; SEM, scanning electron microscopy; TEM, transmission electron microscopy; DL %, drug loading rate; EE %, drug encapsulation rate; ·OH, hydroxyl radicals; TMB, 3,3ʹ,5,5ʹ-tetramethylbenzidine; MDA, malondialdehyde; WB, Western blot; PVDF, polyvinylidene fluoride; CMU, China Medical University; NS, normal saline; H&E, hematoxylin and eosin; IHC, immunohistochemistry; IF, immunofluorescence; SD, standard deviation; WBC, white blood cell; RBC, red blood cell; PLT, platelet; HGB, hemoglobin; ALT, alanine aminotransferase; AST, glutamic oxaloacetic transaminase; BUN, blood urea nitrogen; CR, creatinine; EPR, enhanced permeability and retention; IO NPs, iron oxide nanoparticles; MOF, metal-organic frameworks; MON, metal-organic networks.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Beijing, China) [grant number 81630053].
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed on the journal to which the article will be submitted; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Baig B, Halim SA, Farrukh A, et al. Current status of nanomaterial-based treatment for hepatocellular.carcinoma. Biomed Pharmacother. 2019;116:108852. doi:10.1016/j.biopha.2019.108852
3. Hamza AA, Heeba GH, Elwy HM, et al. Molecular characterization of the grape seeds extract’s effect against chemically induced liver cancer: in vivo and in vitro analyses. Sci Rep. 2018;8(1):1270. doi:10.1038/s41598-018-19492-x
4. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
5. Stockwell BR, Friedmann AJP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. doi:10.1016/j.cell.2017.09.021
6. Angeli JPF, Shah R, Pratt DA, Conrad M. Ferroptosis inhibition: mechanisms and opportunities. Trends Pharmacol Sci. 2017;38(5):489–498. doi:10.1016/j.tips.2017.02.005
7. Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–379. doi:10.1038/cdd.2015.158
8. Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26(3):165–176. doi:10.1016/j.tcb.2015.10.014
9. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331. doi:10.1016/j.cell.2013.12.010
10. Louandre C, Marcq I, Bouhlal H, et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015;356(2):971–977. doi:10.1016/j.canlet.2014.11.014
11. Yu H, Guo P, Xie X, Wang Y, Chen G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med. 2017;21(4):648–657. doi:10.1111/jcmm.13008
12. Nie J, Lin B, Zhou M, Wu L, Zheng T. Role of ferroptosis in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2018;144(12):2329–2337. doi:10.1007/s00432-018-2740-3
13. Liang C, Zhang X, Yang M, Dong X. Recent progress in ferroptosis inducers for cancer therapy. Adv Mater. 2019;31(51):e1904197. doi:10.1002/adma.201904197
14. Liu M, Liu B, Liu Q, Du K, Wang Z, He NJCCR. Nanomaterial-induced ferroptosis for cancer specific therapy. Coord Chem Rev. 2019;382:160–180. doi:10.1016/j.ccr.2018.12.015
15. Yi J, Minikes AM, Jiang X. Aiming at cancer in vivo: ferroptosis-inducer delivered by nanoparticles. Cell Chem Biol. 2019;26(5):621–622. doi:10.1016/j.chembiol.2019.05.002
16. Shan X, Li S, Sun B, et al. Ferroptosis-driven nanotherapeutics for cancer treatment. J Control Release. 2020;319:322–332. doi:10.1016/j.jconrel.2020.01.008
17. Louandre C, Ezzoukhry Z, Godin C, et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer. 2013;133(7):1732–1742. doi:10.1002/ijc.28159
18. Lachaier E, Louandre C, Godin C, et al. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014;34(11):6417–6422.
19. Schmithals C, Köberle V, Korkusuz H, et al. Improving drug penetrability with iRGD leverages the therapeutic response to sorafenib and doxorubicin in hepatocellular carcinoma. Cancer Res. 2015;75(15):3147–3154. doi:10.1158/0008-5472.CAN-15-0395
20. Zuo H. iRGD: a promising peptide for cancer imaging and a potential therapeutic agent for various cancers. J Oncol. 2019;2019:9367845. doi:10.1155/2019/9367845
21. Liu Y, Ji M, Wong MK, Joo KI, Wang P. Enhanced therapeutic efficacy of iRGD-conjugated crosslinked multilayer liposomes for drug delivery. Biomed Res Int. 2013;2013:378380.
22. Li X, Wu M, Pan L, Shi J. Tumor vascular-targeted co-delivery of anti-angiogenesis and chemotherapeutic agents by mesoporous silica nanoparticle-based drug delivery system for synergetic therapy of tumor. Int J Nanomed. 2015;11:93–105. doi:10.2147/IJN.S81156
23. Ma X, Ren X, Guo X, et al. Multifunctional iron-based metal-organic framework as biodegradable nanozyme for microwave enhancing dynamic therapy. Biomaterials. 2019;214:119223. doi:10.1016/j.biomaterials.2019.119223
24. Zhong Y, Su T, Shi Q, et al. Co-administration of iRGD enhances tumor-targeted delivery and anti-tumor effects of paclitaxel-loaded PLGA nanoparticles for colorectal cancer treatment. Int J Nanomed. 2019;14:8543–8560. doi:10.2147/IJN.S219820
25. Zhang JW, Zhang HT, Du ZY, Wang X, Yu SH, Jiang HL. Water-stable metal-organic frameworks with intrinsic peroxidase-like catalytic activity as a colorimetric biosensing platform. Chem Commun (Camb). 2014;50(9):1092–1094. doi:10.1039/C3CC48398C
26. Yang Q, Xu Q, Jiang HL. Metal-organic frameworks meet metal nanoparticles: synergistic effect for enhanced catalysis. Chem Soc Rev. 2017;46(15):4774–4808. doi:10.1039/C6CS00724D
27. Xue T, Xu C, Wang Y, Wang Y, Tian H, Zhang Y. Doxorubicin-loaded nanoscale metal-organic framework for tumor-targeting combined chemotherapy and chemodynamic therapy. Biomater Sci. 2019;7(11):4615–4623. doi:10.1039/C9BM01044K
28. Sang M, Luo R, Bai Y, et al. BHQ-cyanine-based “Off-On” long-circulating assembly as a ferroptosis amplifier for cancer treatment: a lipid-peroxidation burst device. ACS Appl Mater Interfaces. 2019;11(46):42873–42884. doi:10.1021/acsami.9b12469
29. Sun X, Ou Z, Chen R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63(1):173–184. doi:10.1002/hep.28251
30. Yang B, Liu Q, Yao X, et al. FePt@MnO-based nanotheranostic platform with acidity-triggered dual-ions release for enhanced MR imaging-guided ferroptosis chemodynamic therapy. ACS Appl Mater Interfaces. 2019;11(42):38395–38404. doi:10.1021/acsami.9b11353
31. Zhang J, Wang T, Mu S, Olerile LD, Yu X, Zhang N. Biomacromolecule/lipid hybrid nanoparticles for controlled delivery of sorafenib in targeting hepatocellular carcinoma therapy. Nanomedicine (Lond). 2017;12(8):911–925. doi:10.2217/nnm-2016-0402
32. Zhang Q, Zhang Y, Li K, Wang H, Li H, Zheng J. A novel strategy to improve the therapeutic efficacy of gemcitabine for non-small cell lung cancer by the tumor-penetrating peptide iRGD. PLoS One. 2015;10(6):e0129865. doi:10.1371/journal.pone.0129865
33. Sugahara KN, Teesalu T, Karmali PP, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16(6):510–520. doi:10.1016/j.ccr.2009.10.013
34. Tang H, Chen D, Li C, et al. Dual GSH-exhausting sorafenib loaded manganese-silica nanodrugs for inducing the ferroptosis of hepatocellular carcinoma cells. Int J Pharm. 2019;572:118782. doi:10.1016/j.ijpharm.2019.118782
35. El-Kharrag R, Amin A, Hisaindee S, Greish Y, Karam SM. Development of a therapeutic model of precancerous liver using crocin-coated magnetite nanoparticles. Int J Oncol. 2017;50(1):212–222. doi:10.3892/ijo.2016.3769
36. Sang M, Luo R, Bai Y, et al. Mitochondrial membrane anchored photosensitive nano-device for lipid hydroperoxides burst and inducing ferroptosis to surmount therapy-resistant cancer. Theranostics. 2019;9(21):6209–6223. doi:10.7150/thno.36283
37. Hu P, Wu T, Fan W, et al. Near infrared-assisted Fenton reaction for tumor-specific and mitochondrial DNA-targeted photochemotherapy. Biomaterials. 2017;141:86–95. doi:10.1016/j.biomaterials.2017.06.035
38. Klein S, Wegmann M, Distel LVR, Neuhuber W, Kryschi C. APTES-Terminated ultrasmall and iron-doped silicon nanoparticles as X-Ray dose enhancer for radiation therapy. Biochem Biophys Res Commun. 2018;498(4):855–861. doi:10.1016/j.bbrc.2018.03.070
39. Wang D, Zhou J, Chen R, et al. Core-shell metal-organic frameworks as Fe2+ suppliers for Fe2+-mediated cancer therapy under multimodality imaging. Chem Mater. 2017;29(8):3477–3489. doi:10.1021/acs.chemmater.6b05215
40. Zheng DW, Lei Q, Zhu JY, et al. Switching apoptosis to ferroptosis: metal-organic network for high-efficiency anticancer therapy. Nano Lett. 2017;17(1):284–291. doi:10.1021/acs.nanolett.6b04060
41. Kwon B, Han E, Yang W, et al. Nano-Fenton reactors as a new class of oxidative stress amplifying anticancer therapeutic agents. ACS Appl Mater Interfaces. 2016;8(9):5887–5897. doi:10.1021/acsami.5b12523
42. Zhang C, Bu W, Ni D, et al. Synthesis of iron nanometallic glasses and their application in cancer therapy by a localized Fenton reaction. Angew Chem Int Ed Engl. 2016;55(6):2101–2106. doi:10.1002/anie.201510031
43. He C, Liu D, Lin W. Nanomedicine applications of hybrid nanomaterials built from metal-ligand coordination bonds: nanoscale metal-organic frameworks and nanoscale coordination polymers. Chem Rev. 2015;115(19):11079–11108. doi:10.1021/acs.chemrev.5b00125
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.