")
Back to Journals » International Journal of Nanomedicine » Volume 14
Co-Administration Of iRGD Enhances Tumor-Targeted Delivery And Anti-Tumor Effects Of Paclitaxel-Loaded PLGA Nanoparticles For Colorectal Cancer Treatment
Authors Zhong Y , Su T , Shi Q, Feng Y, Tao Z, Huang Q, Li L, Hu L, Li S, Tan H, Liu S, Yang H
Received 17 June 2019
Accepted for publication 5 October 2019
Published 1 November 2019 Volume 2019:14 Pages 8543—8560
DOI https://doi.org/10.2147/IJN.S219820
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Yi Zhong,1,* Tao Su,1,* Qiuxiao Shi,1 Yanru Feng,1 Ze Tao,1 Qiuxia Huang,1 Lan Li,1 Liqiang Hu,1 Shengfu Li,1 Hong Tan,2 Shan Liu,3 Hao Yang1
1Key Lab of Transplant Engineering and Immunology, MOH, West China-Washington Mitochondria and Metabolism Research Center, West China Hospital, Sichuan University, Chengdu 610041, People’s Republic of China; 2Department of General Surgery, Chengdu Integrated TCM & Western Medicine Hospital (Chengdu First People’s Hospital), Chengdu 610041, People’s Republic of China; 3Department of Laboratory Medicine, Sichuan Academy of Medical Sciences & Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China, Chengdu 610072, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shan Liu
Department of Laboratory Medicine, Sichuan Academy of Medical Sciences & Sichuan Provincial People’s Hospital, No. 32, West Second Section First Ring Road, Chengdu, Sichuan 610072, People’s Republic of China
Tel +86 28 6138 2518
Email [email protected]
Hao Yang
Key Lab of Transplant Engineering and Immunology, MOH, West China Hospital, No. 1, Keyuan 4th Road, Hi-tech Zone, Chengdu 610041, People’s Republic of China
Tel +86 28 85164031
Email [email protected]
Background: Nanoparticles exhibit great promise for improving the solubility and tissue-specific distribution of chemotherapeutic agents; however, the passive and highly variable enhanced permeability and retention (EPR) effects observed in tumors frequently leads to insufficient delivery of nanodrugs into tumors. The tumor-penetrating peptide iRGD can actively enhance tumor-selective delivery of nanoparticles into tumors by binding to integrin and interacting with tissue-penetrating receptor neuropilin-1.
Materials and methods: To improve colorectal cancer treatment, in this study, we prepared a paclitaxel (PTX)-loaded PLGA nanoparticle (PLGA-PTX) and evaluated its tumor-targeting and antitumor activity by co-administration with iRGD.
Results: Compared to free PTX, encapsulated PTX retained preferential cytotoxicity toward various colorectal cancer cells while effectively sparing healthy cells. PLGA-PTX treatment resulted in cell cycle arrest at the G2/M phase and apoptosis, leading to inhibition of cancer cell migration and invasion. PLGA-PTX combined with iRGD displayed little enhancement of cytotoxicity in vitro. Despite this, iRGD receptors integrin and neuropilin-1 were found to be primarily overexpressed on abundant tumor vessels in mice bearing colorectal tumors. Consequently, co-administration of nanoparticles with iRGD promoted the selective delivery of nanoparticles into tumor tissues in vivo. Additionally, the combined regimen enhanced the antitumor effects compared to those of each individual reagent.
Conclusion: Our findings suggest that PLGA nanoparticles combined with the iRGD peptide provide a promising drug delivery strategy for facilitating active drug accumulation into tumors, given that iRGD receptors are overexpressed on tumor vessels. This co-administration system lacking covalent conjugation provides a more convenient means to combine various therapeutic agents with iRGD to achieve personalized nanotherapy.
Keywords: tumor penetrating peptide, nanoparticle, iRGD, paclitaxel, tumor vasculature, colorectal cancer
Introduction
Colorectal cancer is the third most common cancer in the world and is responsible for more than 880,000 deaths in 2018.1 The rapid growth of colorectal cancer is characterized by the abundant development of blood vessels, a characteristic that contributes to its malignancy and aggression.2,3 Although surgery can be used to treat colorectal cancer when it is diagnosed at its earlier stages, in most cases, it is difficult to completely remove primary or metastatic tumors. Thus, adjuvant chemotherapy is typically used to suppress tumor growth and prolong survival in patients, especially those harboring unresectable tumors. The drug cocktails FOLFOX (leucovorin, fluorouracil, and oxaliplatin) or CAPOX (capecitabine and oxaliplatin) that act as DNA-damaging therapeutic agents have been commonly combined with surgery, radiotherapy, or targeted blockade regents to treat colorectal cancer depending upon cancer stage and molecular subtype.4–6 Use of these chemotherapy regimens as the treatment backbone provides notable activity but still requires improvement for prolonging disease-free survival and ultimately providing a long-term cure.
Paclitaxel (PTX), isolated from the bark of Taxus brevifolia, is one of the most successful first-line antitumor drugs that possesses confirmed cytotoxicity against a broad range of tumors. PTX targets microtubules and reduces their dynamicity, as a result arresting mitotic action and inducing proliferating cell apoptosis.7 PTX combined with certain pathway inhibitors exhibited remarkable cytotoxicity in colorectal cancer cells.8,9 Intriguingly, the addition of PTX to FOLFOX (POF regimen) as a first-line therapy has successfully improved patient response rate and survival in a randomized phase II clinical trial for advanced gastric cancer treatment when compared to FOLFOX alone.10,11 These inspiring outcomes of preclinical and clinical studies in colorectal and gastric cancer treatment appear to show great promise for the use of this application of PTX in colorectal cancer therapy. Due to its poor aqueous solubility, PTX (brand name, Taxol) is commonly formulated in a vehicle composed of a 1:1 blend of Cremophor EL and ethanol prior to administration. The toxic effects of Cremophor EL, however, have been noted in both animal models and patients. Additionally, PTX can kill actively dividing normal cells, consequently resulting in toxic side effects.12–15
Nanoparticles have been demonstrated to significantly promote solubility and to mitigate the systematic toxicity of conventional antitumor agents, ultimately enhancing the therapeutic index. Importantly, nanodrugs are capable of passively targeting solid tumors by an enhanced permeability and retention (EPR) effect.16,17 Attesting to the advantages in cancer therapy, a number of nanodrugs, including the albumin-bound PTX nanoparticle Abraxane and liposome-delivered doxorubicin Doxil, have been approved in the US and Europe for primary and metastatic cancer treatment.18 Solid tumors, however, are typically densely heterogeneous and their blood vessels can vary with according to tumor type and growth status, and this, in turn causes highly variable EPR effects in tumors. The frequent occurrence of low EPR, especially in clinical tumors, compromises EPR-dependent nanoparticle delivery and often leads to insufficient accumulation of nanodrugs within solid tumors.19,20
To achieve more desirable therapeutic outcomes, novel diagnostic tools can be designed to identify the extent of the ERP effect in tumors and to select patients who will benefit from nanotherapy. Alternatively, nanoparticles have been extensively modified by the addition of tumor-homing ligands to actively enhance particle delivery into tumors.21,22 Of these ligands, the tumor-penetrating peptide iRGD is a bifunctional ligand containing a tumor-homing motif (RGD) and a tissue-penetrating motif (CendR). Covalent conjugation of iRGD to nanoparticles can significantly promote the delivery of encapsulated drugs into tumors, resulting in improved tumor-targeting and tissue-penetrating properties of the payloads.23–25 More interestingly, the enhanced delivery of nanodrugs into tumors can also be achieved through co-administration with iRGD in addition to conjugation with the peptide. Once bound to tumor sites via the RGD motif, the tissue-penetrating motif CendR can stimulate vascular permeability in a VEGF-like manner and facilitate the transport of peptides and co-administered payloads across the endothelial cell barrier from the tumor vessels into the tumor parenchyma. Enhanced antitumor activity has been observed for several nanodrugs when they were co-administered with iRGD.26–28
Poly lactic-co-glycolic acid (PLGA) is a US Food and Drug Administration (FDA)-approved biodegradable materials for drug delivery and the PLGA-based nanoparticles have been extensively used for delivery of both therapeutic small compounds and biomacromolecules.29 In the present study, we prepared and characterized a PTX-loaded PLGA nanoparticle (PLGA-PTX) for use in colorectal cancer treatment. PLGA-PTX was co-administered with iRGD to promote tumor-targeting drug delivery. Although this combination treatment exhibited few synergistic effects directly against tumor cell growth in vitro, iRGD improved the delivery of PLGA nanoparticles and the encapsulated PTX from tumor blood vessels into tumor parenchyma, and it enhanced the antitumor effects of PLGA-PTX in vivo. This co-administration drug delivery system exhibits great promise as a convenient method to combine various therapeutic agents for colorectal cancer treatment.
Materials And Methods
Materials
Monomethoxy-poly(ethylene glycol)-poly lactic-co-glycolic acid (PEG-PLGA) was obtained from Jinan Dai Gang Biomaterial Co., Ltd. (Jinan, China). PTX was purchased from Dalian Melone Biomart Co., Ltd, and iRGD (CRGDRGPDC) was provided by Genscript Co., Ltd. (Guilin, China). Coumarin-6 and polyvinylalcohol (PVA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Cell counting kit-8 (CCK-8), Annexin V apoptosis detection kits, and PI/RNase cell cycle assay kits were obtained from Dojindo (Kumamoto, Japan). Caspase-Glo® 3/7 assay kits were purchased from Promega (Madison, WI, USA), and 4,6-diamidino-2-phenylindole (DAPI) was obtained from Molecular Probes (Eugene, OR, USA). Alexa Fluor 647 anti-mouse CD31 antibody was purchased from Biolegend (San Diego, CA, USA), and 1,1ʹ-dioctadecyl-3,3,3ʹ,3ʹ- tetramethylindodicarbocyanine (DID) was provided by Key GEN Bio TECH Co., Ltd. Anti-integrin and neuropilin-1 antibodies were purchased from Abcam (Cambridge, MA, USA).
Peptide Design And Characterization
The iRGD peptide targets to tumors through the action of its integrin-binding RGD motif. After this initial binding, its CendR motif exposed by proteolysis can bind to neuropilin-1 and then trigger tissue penetration. Therefore, this cyclic peptide possessing an amino acid sequence of CRGDRGPDC is a composite of the integrin-binding RGD motif and the cryptic CendR motif. The iRGD peptide was synthesized using standard solid-phase fluorenylmethoxycarbonyl (Fmoc) chemistry techniques and was custom-made by Genescript Inc. (Nanjing, China). The purity of the peptide (>95%) was analyzed by reversed-phase high-performance liquid chromatography (HPLC), and the mass of the peptide was determined and characterized using high resolution mass spectrometry in the presence and absence of DTT. The peptide was separated into aliquots, lyophilized, and stored at 20°C for further use.
Cell Culture
Human colorectal cancer cells (LS174T, COLO205, HCT116, and SW620) and mouse normal liver cells (BNL CL.2) were obtained from American Type Culture Collection (ATCC), and they were cultured in RPMI-1640 or DMEM containing 10% fetal bovine serum, 2 mmol/L L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a 5% CO2 humidified atmosphere.
Preparation Of Nanoparticles
Nanoparticles were prepared by the emulsion/solvent evaporation technique according to a previously reported method.30 Briefly, 10 mg of PEG-PLGA and 1 mg of PTX were dissolved into 1 mL of dichloromethane. Then, the solution was added to 4 mL of 1% PVA and emulsified by sonication at 100 W for 70 s on ice using a probe sonicator (Ningbo Scientz Biotechnology Co. Ltd., China). Finally, the emulsion was evaporated under a vacuum to remove the dichloromethane. The obtained solution was centrifuged (25,000 ×g) at 4°C for 40 min and washed three times with deionized water. The sediment was resuspended into phosphate buffered saline (PBS, pH 7.4) containing 137 mM NaCl, 2.68 mM KCl, 10 mM Na2HPO4, and 2 mM KH2PO4 or lyophilized for further use.
Characterization Of Nanoparticles
Particle Size, Zeta Potential, And Appearance
The mean particle size and zeta potential of nanoparticles were both measured by a particle size analyzer (Zetasizer Nano-ZS, Malvern, UK) following dilution of an appropriate amount of nanoparticles into 2 mL of deionized water. The morphology of the nanoparticles was detected by transmission electronic microscopy (TEM) (Tecai G2 F20, FEI Co., Ltd. Hong Kong, China) after negative staining with a 2% phosphotungstic acid solution. To determine stability, the freshly prepared nanoparticles were stored in 1% PVA or PBS, pH 7.4, at 4°C for 28 days. At designated time intervals, changes in particle size were determined as described above.
Entrapment Efficiency And Drug Loading Capacity
The entrapment efficiency (EE%) and drug loading capacity (DL%) of PTX-loaded PLGA were both determined using an Agilent 1260 HPLC system with a C18 column (150 × 4.6 mm, 3 μm, Thermo Fisher, USA) at 30°C. The lyophilized nanoparticles (1 mg) were dissolved into 1 mL of dichloromethane under vortex to extract PTX from the nanoparticles. After solvent evaporation, the residue was re-dissolved into 1 mL of deionized water and acetonitrile (50:50 ratio). PTX content was measured by HPLC at a flow rate of 0.6 mL/min using an ultraviolet detector at 227 nm.31 The EE% and DL% were calculated as follows:
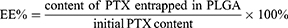

In Vitro Release
The release behavior of PTX in PLGA was monitored in PBS, pH 7.4 or pH 6.4, containing 0.1% Tween 80 as previously described with a slight modification.32 Briefly, an aliquot of nanoparticles equivalent to 30 μg of PTX was diluted into 1 mL of PBS containing 0.1% Tween 80. The suspension was shaken at 37°C at a rate of 100 rpm. At the designated time points (0, 0.5, 1, 2, 4, 8, 12, 24, 48, or 72 h), the supernatant was replaced with 1 mL of fresh PBS buffer following centrifugation. Subsequently, the PTX released into the supernatant was immediately determined by HPLC as described above. The accumulative release of PTX from nanoparticles was calculated and plotted versus time.
Hemolysis
The use of human blood was approved by the Medical Ethics Committee of Sichuan Provincial People’s Hospital and authorized by the written informed consent from donors. Human erythrocytes (RBCs) obtained from healthy donors were isolated by centrifugation at 1200 g for 5 min, washed with normal saline (NS) for three times, and then suspended in NS at a 2% concentration (volume ratio). For the hemolysis experiment, the 2.5 mL erythrocyte suspension was treated with PTX or PLGA-PTX at 37°C for 3 h. Deionized (DI) water and NS were used as the positive and negative control, respectively. The supernatant was collected following centrifugation, and then the absorbance was measured at 540 nm using an ultraviolet-visible spectrophotometer (Shimadzu UV-1601PC, Japan). The percentage of sample-induced hemolysis was calculated as follows:

In Vitro Cytotoxicity
In vitro cytotoxicity was measured by the CCK-8 assay according to the manufacturer’s specifications. Briefly, cells were seeded into 96-well plates at a density of 1×104 cells/well and incubated for 12–18 h to allow for attachment to the plate. Free PTX or PLGA-PTX was diluted into culture medium at a concentration of 0.5, 1, 2.5, 5, 10, 20, 100, or 1000 ng/mL. Diluted drugs (100 μL) were added to each well in the presence or absence of 30 μmol/L iRGD and then further incubated at 37°C for the indicated time. Next, 10 μL of CCK-8 solution was added to each well, and the absorbance was detected at 450 nm after 1–4 h of incubation. Cells incubated with culture medium alone were regarded as the 100% viability control to calculate the survival ratio.
Cell Cycle And Apoptosis Assay
Cell apoptosis and cell cycle were analyzed following PLGA-PTX treatment over a time course. Cells seeded into 96-well plates were treated with 100 μL of 100 ng/mL PLGA-PTX, and the changes in cell morphology were observed under a microscope. Simultaneously, the activation of caspase-3 was detected by adding 100 μL of Caspase-Glo 3/7 reagent to each well and then incubating for 1 h at room temperature. Alternatively, cells were stained with the cell-permeable dye Hoechst 33342 at the indicated time point following PLGA-PTX treatment and observed under a fluorescence microscope. For flow cytometry analysis, cells were cultured in 6-well plates and treated with 2.5 mL of 100 ng/mL PLGA-PTX over time. At the designated time points, cells were detached and washed with PBS twice. For the cell cycle assay, cells were fixed with 70% cold ethanol overnight and then stained with 500 μL of PI/RNase working solution for 30 min according to the kit instructions. For the cell apoptosis assay, cells were dual-stained with FITC-Annexin V and PI for 15 min, and this was followed by flow cytometry analysis.
Tumor Spheroid-Based Migration And Invasion Assays
To establish tumor spheroids, tumor cells were dispensed into an ULA (Ultra-Low Attachment) 96-well round-bottomed microplate (Corning) as described by Vinci et al.33 Cells were further cultured for 3–4 days to allow for formation of multicellular tumor spheroids possessing a three-dimensional compact structure. For the tumor spheroid migration assay, tumor spheroids were transferred into a 96-well flat-bottomed microplate pre-coated with 50 μg/mL of fibronectin (EMD Millipore). Tumor spheroids were further cultured in the presence or absence of 2.5–10 ng/mL PLGA-PTX. The migration of tumor spheroids was imaged using a phase contrast microscope. The quantification of migration inhibition was analyzed by measuring the areas covered by migrating cells. The fold change in migration area was normalized to the initial size of each tumor spheroid. For the tumor invasion assay, tumor spheroid medium was gently replaced by an equivalent volume of matrigel (Corning). Following matrigel solidification, culture medium was added to the top of the matrigel. PLGA-PTX or PBS (10 ng/mL) was diluted into both matrigel and the overlying medium. The invasion of tumor cells from tumor spheroids into the matrigel was imaged using an inverted microscope.
In Vivo Experiments
Tumor Vasculature And Receptor Expression
All animal experiments were approved by the Medical Ethics Committee of Sichuan Provincial People’s Hospital. The animal welfare and use were in agreement with the guidelines approved by the Medical Ethics Committee of Sichuan Provincial People’s Hospital. LS174T cells (1×106) dispersed in 100 μL of PBS were injected subcutaneously into the right flank of female BALB/c (nu/nu) mice. When the tumor nodules reached the expected size (about 200–300 mm3), tumor sections were excised from the mice. Then, the tumors were immunostained with anti-CD31 to reveal the blood vessels and anti-integrin and neuropilin-1 antibodies to reveal the iRGD receptors.
In Vivo Imaging
BALB/c nude mice were established with LS174T xenografts as described above. When the tumor nodules reached a size of approximately 150–200 mm3, six mice were divided into two groups and injected with PLGA-DID or PLGA-DID + iRGD. The doses of DID and iRGD were maintained at 0.2 mg/kg and 10 mg/kg, respectively. The mice were anaesthetized and imaged at 4, 8, and 24 h post-administration using the SPECTRAL Lago and Lago X Imaging Systems (Spectral, AZ, UAS) with an emission bandpass filter at 640 nm and excitation at 690 nm. Next, the mice were sacrificed, and tumors and other organs (the brain, heart, liver, spleen, lung, and kidney) were collected and imaged.
In Vivo Distribution
BALB/c nude mice were established with the LS174T xenografts as described above. The, the mice were divided into two groups and injected intravenously with coumarin-6-loaded-PLGA and coumarin-6-loaded-PLGA + iRGD, respectively, at a dose of 10 mg/kg iRGD and 0.4 mg/kg coumarin-6. Three hours later, the mice were anesthetized, and the tumor was excised into thick frozen sections (100 μm). For immunostaining, the slides were fixed and incubated with Alexa Fluor 647 anti-mouse CD31 antibody (1:200 dilution) for 1.5 h at 37°C. Nuclei were stained by DAPI.
In Vivo Antitumor Assessment
Antitumor activity was investigated in a subcutaneous LS174T tumor-bearing mouse model as described above. At the onset of a palpable tumor (about 50 mm3), mice were divided into 5 groups that were intravenously injected with free PTX, PTX + iRGD, PLGA-PTX, PLGA-PTX + iRGD (PTX and iRGD at a dose of 10 mg/kg), or an equivalent volume of normal saline (NS) every 2 days for a total of three injections. The tumor volume (mm3) was calculated every day as length×width2×0.5. The body weight of each mouse was recorded throughout the entire experimental period. All animals were sacrificed at the conclusion of the observation, and the tumor weights were then measured.
Statistical Analysis
Results are presented as the mean ± SD of at least three independent experiments. Analysis of variance was used to analyze the differences. A P value <0.05 was considered statistically significant. *P<0.05; **P<0.01; ***P<0.001.
Results
Characterization Of Nanoparticles And The iRGD Peptide
The prepared colloidal solutions of PLGA and PTX-loaded PLGA presented with an opalescent appearance with a strong Tyndall Effect (Figure 1A). The detailed characteristics of the PLGA nanoparticles were further evaluated, and these included particle size, polydispersity index (PDI), zeta potential, entrapment efficiency, and drug loading capacity. It is believed that particle size is one of the pivotal factors influencing the tissue distribution and tumor targeting of the nanoparticles in vivo.34 As shown in Figure 1B and in Table 1, the size of PLGA and PLGA-PTX were respectively 138.9 ± 10.8 and 147.5 ± 9.5 nm with a narrow size distribution (PDI < 0.1), indicating a suitable particle size and distribution for drug delivery into tumors. The zeta potential of PLGA and PLGA-PTX were −2.26 and −1.84 mV, respectively (Table 1), which were close to neutrality, confirming the presence of PEG chains shielding the negative charges that existed at the surface of the nanoparticles. TEM images verified that the nanoparticles were spherical in shape and possessed smooth surfaces (Figure 1C).
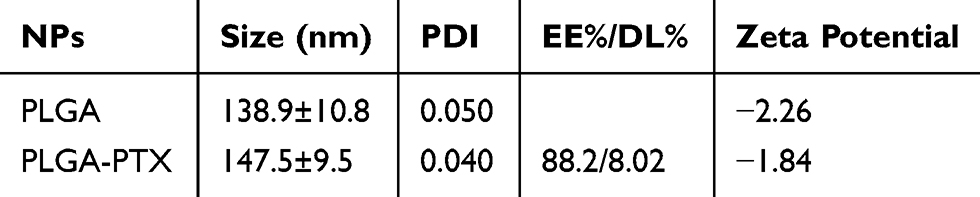 |
Table 1 Characterization Of Nanoparticles (n=3, Mean±SD) |
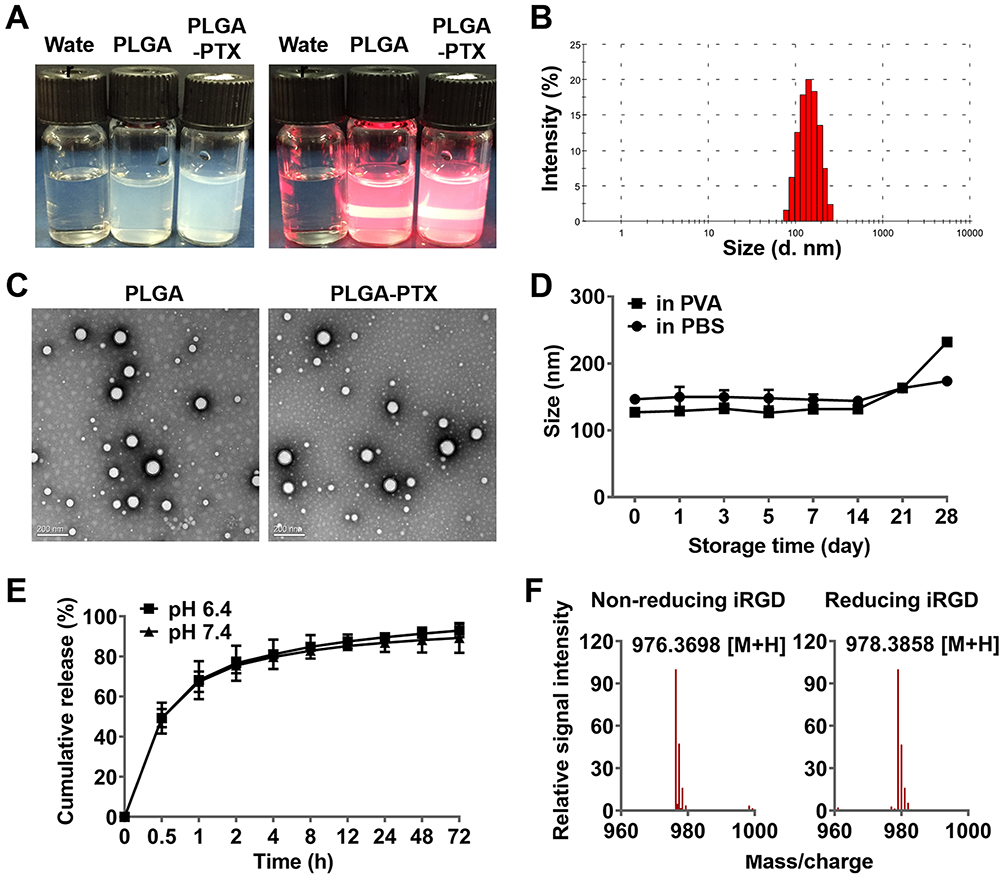 |
Figure 1 Characterization of nanoparticles and the iRGD peptide. Notes: (A) Nanoparticles exhibited opalescence and Tyndall effect compared to that observed for deionized water. (B) PLGA-PTX exhibited an average particle size of 147.5±9.5 nm with a narrow size distribution (PDI<0.1). (C) Nanoparticles were spherical in shape and possessed smooth surfaces as shown in the transmission electron microscopy image (scale bar = 200 nm). (D) Changes in PLGA-PTX particle size at different storage times in PBS and PVA at 4°C. (E) PLGA-PTX exhibited a sustained release in pH 6.4 and pH 7.4 PBS containing 0.1% Tween-80. (F) The disulfide bond of iRGD was determined in the absence (non-reducing) or presence (reducing) of DTT by mass spectrometry. Abbreviations: PLGA, poly lactic-co-glycolic acid; PTX, paclitaxel; PLGA-PTX, paclitaxel-loaded PLGA; PDI, polydispersity index; PBS, phosphate buffer saline. DTT, dithiothreitol. |
HPLC analysis revealed that PLGA-PTX exhibited 88.2% EE% and 8.02% DL% (Table 1). Additionally, the stability investigation suggested that PLGA-PTX remained stable without a significant change in particle size for at least 2 weeks in both PVA and PBS solutions at 4°C, indicating that the nanoparticles possessed adequate storage stability (Figure 1D). Further storage at 4 weeks led to an increase in the particle size of PLGA-PTX. The drug release profile of the nanoparticles in vitro was assayed in PBS and determined by HPLC. As shown in Figure 1E, PTX exhibited sustained release from PLGA without an obvious burst-release effect from 0.5 to 72 h in both pH 6.4 and 7.4 PBS containing 0.1% tween 80. Accordingly, these results suggest that we have successfully constructed well-characterized nanoparticles that exhibit good storage stability and controlled-release properties.
The cyclic peptide formed via a disulfide bond is a crucial factor that retains the high binding activity of iRGD to the integrin receptor. The disulfide bonds of iRGD were determined by high-resolution mass spectrometry. As shown in Figures 1F and S1, the accurate mass (975.3698 Da) of an isotopic species was almost identical to the theoretical mass (975.3651 Da) of the cyclic iRGD peptide. A reduction of the disulfide bond with DTT resulted in an approximate 2 Da mass increase (977.3858 Da), verifying the intrinsic disulfide-based cyclic structure in iRGD.
In Vitro Cytotoxicity
To determine the cytotoxicity of PLGA-PTX, colorectal cancer COLO205 cells were treated with increasing concentrations of nanodrugs over times ranging from 24 h to 96 h. As shown in Figure 2A, an enhanced loss of cell viability was observed in conjunction with an extension of treatment time from 24 h to 72 h. A further increase of treatment time up to 96 h showed little promotion of cytotoxicity in the context of tumor cells. To compare the cytotoxicity in the presence of iRGD with that in the absence of the peptide, COLO205 cells were treated with free PTX or PLGA-PTX combined with or without iRGD for 72 h. As shown in Figure 2B, all treatment groups showed a dose-dependent cytotoxicity in COLO205 cells. The loss of cell viability reached 70–80% at a concentration of 2.5 ng/mL PTX. These results suggest that PTX at a low concentration displayed superior cytotoxicity in COLO205 cells. Additionally, PLGA encapsulation retained the cell killing effects of PTX against COLO205 cells. The combination of iRGD with PLGA-PTX or free PTX, however, showed no obvious improvement of cytotoxicity compared to that of these drugs alone. To validate the cell killing activities of different PTX formulations and their combinations with iRGD, the colorectal cancer cell line LS174T, HCT116, and SW620 were treated under the same conditions. Similarly, both PLGA-PTX and PTX alone at 2.5 ng/mL inhibited 30%–60% of tumor cell growth following 72 h treatment. Despite this, no substantial enhancement of cytotoxicity in these cancer cells was observed when these drugs were combined with iRGD (Figure 2C). Additionally, encapsulation of PTX into PLGA retained the preferential cytotoxicity of PTX on cancer cells. PLGA-PTX at high concentrations of up to 1000 ng/mL induced less than 40% normal liver cell death in the presence or absence of iRGD (Figure S2). PLGA-PTX treatment also induced no obvious hemolysis, while free PTX that was dissolved into the solvent consisting of Cremophor-EL and ethanol induced 13.43% lysis in erythrocytes (Figure 2D). These data demonstrate that PLGA-PTX is a desirable controlled-release formulation with highly efficient activities against colorectal cancer cells. The combination of iRGD with PLGA-PTX in vitro displays little improvement of the cytotoxicity directly on tumor cells.
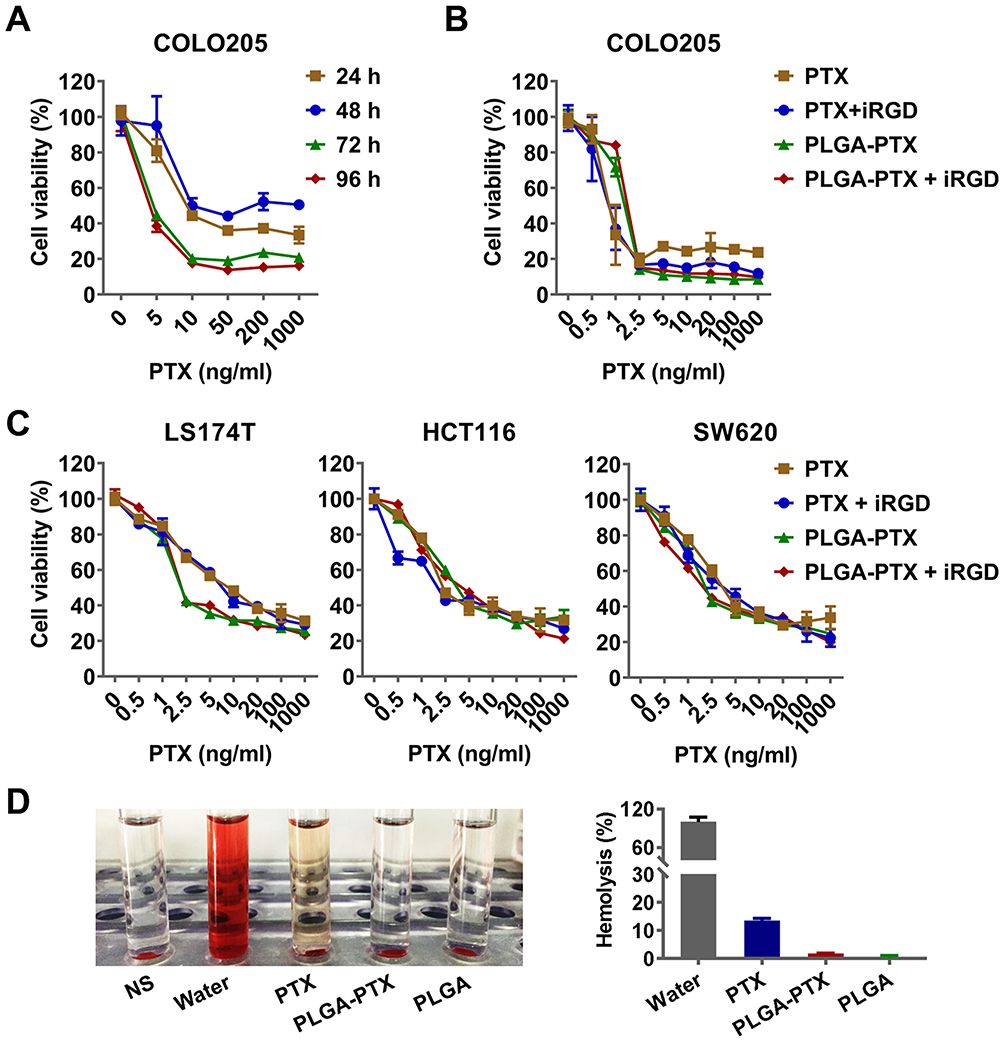 |
Figure 2 In vitro cytotoxicity and hemolysis assay of nanoparticles. Notes: (A) COLO205 colorectal cancer cells were treated with PLGA-PTX over a time course, and this was followed by cell viability measurement using the CCK-8 assay. (B) COLO205 cells were treated with free PTX or encapsulated PLGA-PTX combined with or without iRGD for 72 h. (C) Cytotoxicity of PTX or PLGA-PTX against various colorectal cancer cells. (D) Hemolysis assay. DI water and NS were used as the positive (100% hemolysis) and negative control, respectively. The appearance of hemolysis (left panel) and percentage of hemolysis (right panel) were assessed for each group. Abbreviations: PLGA, poly lactic-co-glycolic acid; PTX, paclitaxel; PLGA-PTX, paclitaxel-loaded PLGA; DI water, deionized water; NS, normal saline. |
Cell Cycle Arrest And Tumor Cell Apoptosis
To explore the mechanism of tumor cell death induced by nanodrugs, LS174T tumor cells were treated with 100 ng/mL PLGA-PTX. As shown in Figure 3A, remarkable cell shrinkage and cell membrane blebbing were observed concomitantly with the loss of cell number as treatment times were increased from 24 h to 72 h. Additionally, PLGA-PTX induced nuclear condensation in most tumor cells after 24 h of treatment and gradually led to nuclear fragmentation and DNA loss after treatment for 48 h to 72 h. Simultaneously, flow cytometry analysis demonstrated that the percentages of early (Annexin V+/PI-) and late (Annexin V+/PI+) apoptotic cells were 29.6% and 23.7%, respectively, following PLGA-PTX treatment for 24 h. Extension of treatment time resulted in a decrease in the percentage of early apoptotic cells to less than 10% with a concomitant increase in the percentage of late apoptotic cells to approximately 80% after 72 h of treatment. Additionally, the significant activation of apoptotic executioner caspase 3 was detected from 8 h to 24 h after treatment, indicating PLGA-PTX-mediated induction of the initiation of apoptosis at early time points prior to the alteration of cell morphology and loss of DNA (Figure 3B). Within 24 h of treatment, flow cytometry revealed that PLGA-PTX induced a significant accumulation of DNA at the G2/M phase with a concomitant decrease of DNA content in G0/G1 phase, indicating a time-dependent tumor cell cycle arrest (Figure 3C). These results suggest that the encapsulated PTX in PLGA gives rise to cell cycle arrest of the fast-proliferating tumor cells and hence induces cell apoptosis following activation of caspase.
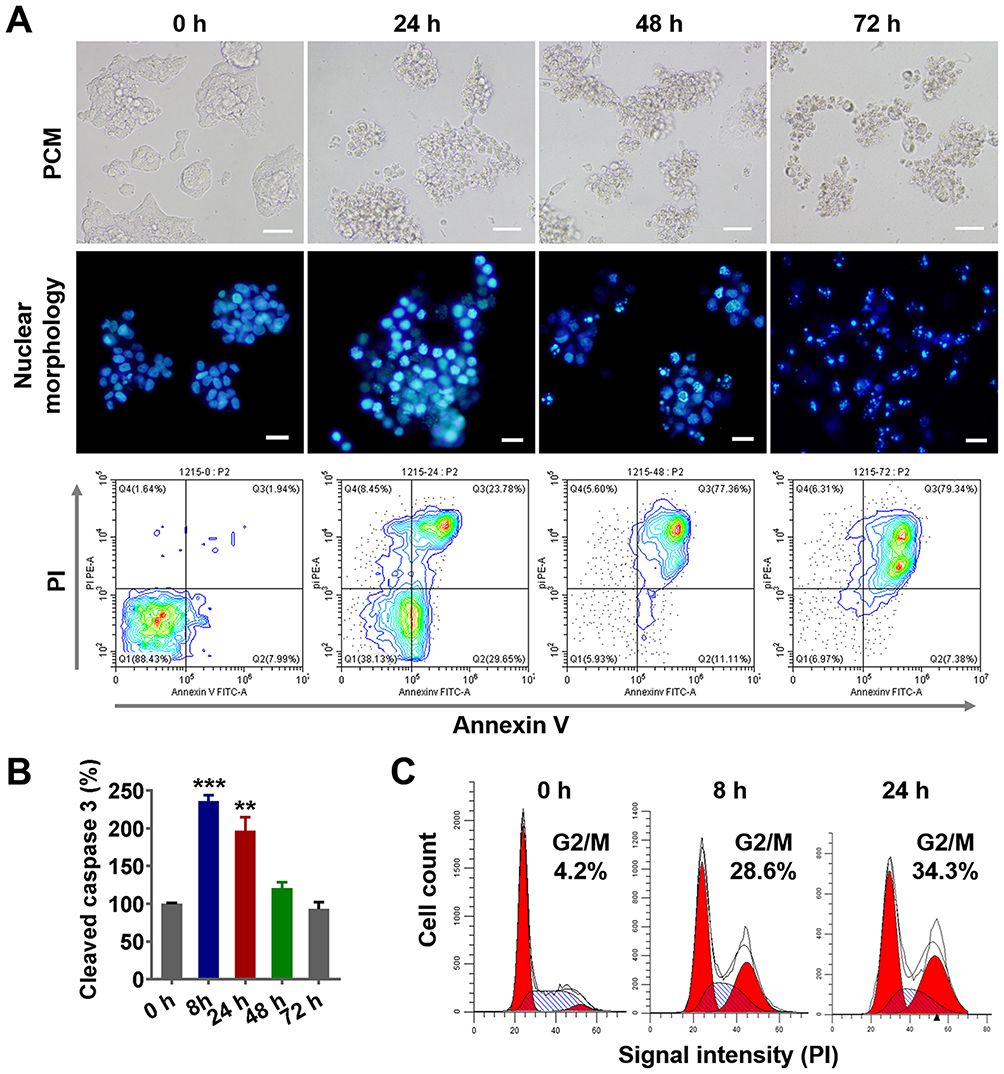 |
Figure 3 PLGA-PTX-induced cell cycle arrest and tumor cell apoptosis. Notes: Colorectal cancer cells (LS174T) were treated with PLGA-PTX over a time course. (A) Cell morphology was recorded by PCM, and cell nuclei were stained via Hoechst dye, and this was followed by fluorescence microscope imaging. Cell apoptosis was assayed by flow cytometry after dual staining with FITC-labeled Annexin V and PI. Scale bar, 50 μm. (B) Caspase 3 activation was analyzed by quantification of cleaved caspase 3 relative to a reference at 0 h. (C) Flow cytometry analysis revealed PLGA-PTX treatment induced accumulation of DNA from G0/G1 phase into G2/M phase over time following PI staining. **P<0.01; ***P<0.001. Abbreviations: PLGA, poly lactic-co-glycolic acid; PTX, paclitaxel; PLGA-PTX, paclitaxel-loaded PLGA; PCM, phase contrast microscope; FITC, fluorescein; isothiocyanate; PI, propidium iodide. |
Inhibition Of Tumor Cell Migration And Invasion From Tumor Spheroids
The migration of tumor cells assay was based on a cultured three-dimensional tumor spheroid by employing HCT116 as a cell model, as this cell line was able to form a compact spheroid structure useful for further functional assays.33 The tumor spheroid was formed in an ultra-low attachment round-bottomed plate and then transferred into a fibronectin-coated flat-bottomed 96-well plate, and this was followed by treatment with a low concentration (2.5 to 10 ng/mL) of PLGA-PTX over time. As shown in Figure 4A, the multicellular tumor spheroids attached to the plate within 8 h in the PBS group. Subsequently, tumor cells exhibited a rapid migration along the coated plate away from tumor spheroids after 24 h, and the leading edge of the migrating cells was almost beyond the microscope field of view after 48 h. In contrast, the migration of tumor cells was dramatically inhibited after tumor spheroids were treated with PLGA-PTX (Figures 4A and S3). The migration areas in the PLGA-PTX group decreased to at least half that of the PBS group after 48 h of treatment (Figure 4B). To detect tumor invasion, tumor spheroids were cultured in round-bottomed plates, and this was followed by the addition of Matrigel. Tumor spheroids were allowed to grow further in the semisolid gel-like matrix in the presence or absence of PLGA-PTX. After incubation for 48 h, numerous tumor cells disseminated from spheroids and invaded into the matrix in the PBS group. In contrast, treatment with a low concentration (10 ng/mL) of PLGA-PTX markedly inhibited the invasion of tumor cells from cultured spheroids. Tumor cells remained closely attached to tumor spheroids, and only several tumor cells invaded into the matrix far from spheroids (Figure 4C). Additionally, the combination of PLGA-PTX and iRGD resulted in comparable with PLGA-PTX treatment alone levels of inhibition of the migration and invasion of HCT116 tumor spheroids (Figures S4 and S5). These data indicate that PLGA-PTX nanodrugs given at a low concentration exhibit significant effects in the context of inhibition of tumor cell migration and invasion.
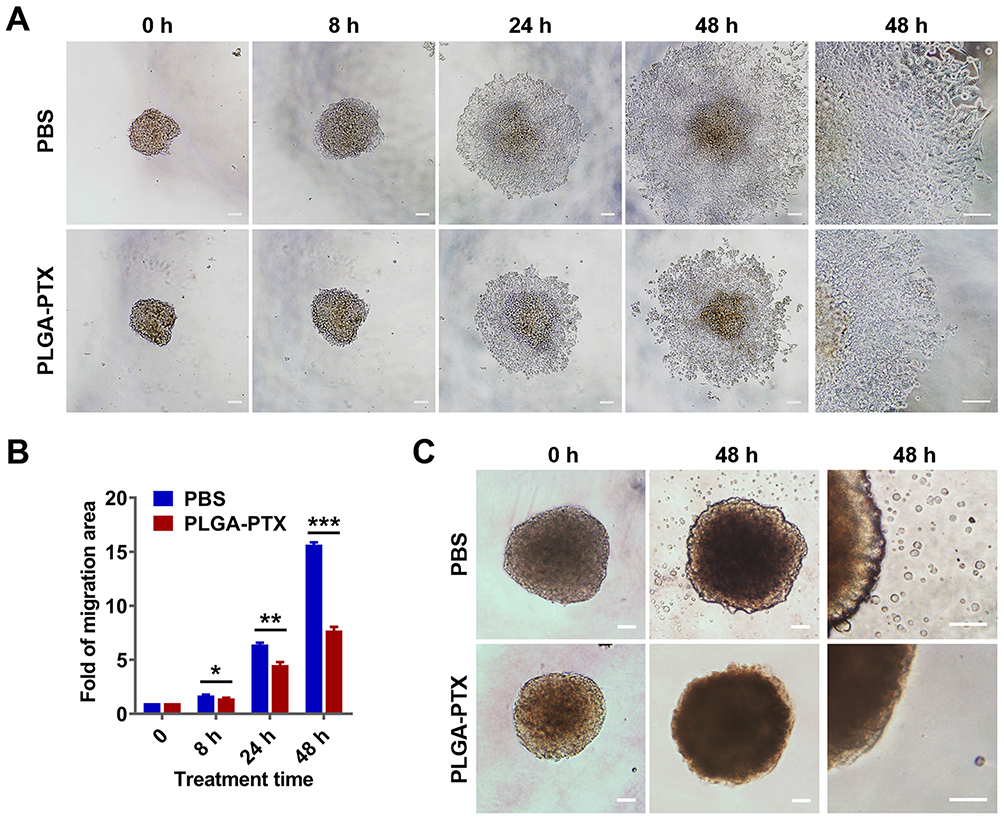 |
Figure 4 PLGA-PTX inhibits tumor spheroid cell migration and invasion. Notes: Colorectal cancer cells (HCT116) were cultured in an ultra-low attachment plate to form tumor spheroids, and this was followed by treatment with low concentration of PLGA-PTX in culture medium over time. (A) The tumor cell migration away from tumor spheroids in the absence or presence of 10 ng/mL of PLGA-PTX was recorded using an inverted microscope. Scale bar, 100 μm. (B) The migration areas were measured and presented as the fold change relative to the areas at 0 h. (C) The invasion of tumor cells that disseminated from spheroids into the matrigel matrix was imaged using an inverted microscope. Scale bar, 100 μm. *P<0.05; **P<0.01; ***P<0.001.Abbreviations: PLGA, poly lactic-co-glycolic acid; PTX, paclitaxel; PLGA-PTX, paclitaxel-loaded PLGA; PBS, phosphate buffer saline. |
Tumor Vasculature Distribution And Receptor Expression
Drug delivery into tumor sites in vivo primarily depends upon the distribution of tumor blood vessels within the tumor tissues. The architecture of the tumor vasculature was investigated in LS174T tumors by immunofluorescence staining of blood vessel endothelial cells. Red fluorescence of CD31 in Figure 5A demonstrates that blood vessels were particularly abundant in LS174T tumors, which provided a fundamental condition for efficient drug transport into tumors. To achieve tumor-specific drug delivery via iRGD, the expression of the iRGD receptors integrin and neuropilin-1 was further assessed in tumors. Figure 5B shows that the iRGD-binding integrin α5, but not integrin αv, was highly expressed and enriched on tumor blood vessels. Similarly, the tissue-penetrating receptor neuropilin-1 was also highly expressed and predominantly located on tumor vessels. Only a minor amount of integrin α5 and neuropilin-1 expression was observed on tumor cells. The abundant tumor vessels and high expression of integrin and neuropilin-1 suggest that iRGD may possess the potential for improving the active delivery of nanodrugs into LS174T tumors.
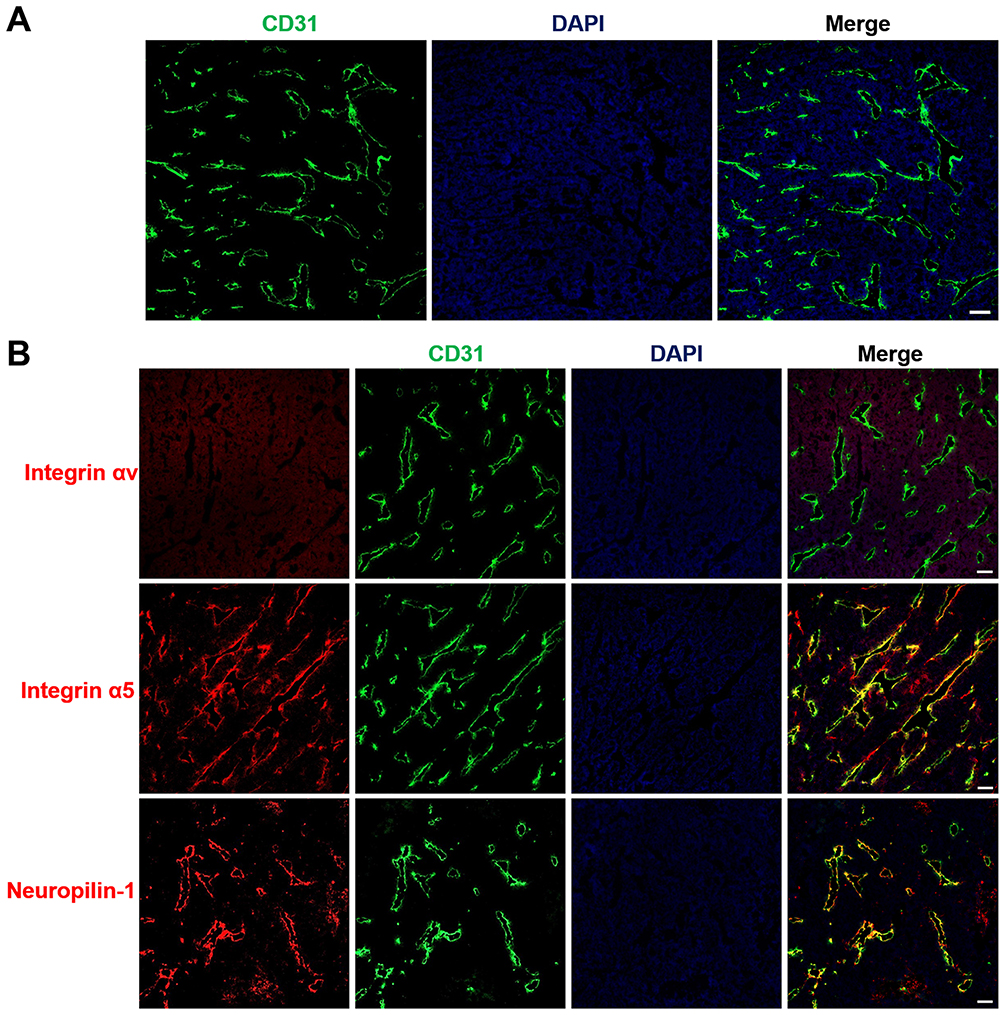 |
Figure 5 Tumor vasculature distribution and expression of the iRGD receptor. Notes: LS174T tumor frozen sections were prepared for immunofluorescence staining and imaging. (A) Blood vessel distributions were stained with anti-CD31 antibody (red). (B) The expression of iRGD receptors, including integrin αv, integrin α5, and neuropilin-1, were assayed by staining with anti-receptor antibodies (red). Tumor blood vessels (green) and nuclei (blue) were co-stained with anti-CD31 antibody and DAPI. Scale bar, 100 µm. Abbreviations: CD31, cluster of differentiation 31; DAPI, 4,6-diamidino-2-phenylindole. |
In Vivo Imaging And Tumor Penetration
To evaluate the tumor-targeting enrichment of nanoparticles, live optical imaging was performed on LS174T tumor-bearing mice injected with NIR fluorescent probe DID-loaded PLGA. Co-injection of PLGA-DID with iRGD appeared to enhance the accumulation of PLGA nanoparticles in tumors at 4, 8, and 24 h post-injection compared to that of PLGA-DID alone (Figure 6A). The ex vivo tissues were also imaged and quantified after the mice were sacrificed at 24 h. In agreement with the live imaging results, co-administration of PLGA-DID with iRGD enhanced accumulation by approximately 2-fold more than that of the payload in tumors without an obvious increase of payload delivery into normal tissues, and these tissues included the brain, heart, liver, spleen, lung, and kidneys (Figure 6B). The high accumulation of nanoparticles was also observed in the liver and spleen due to the well-established arrestment and clearance of nanoscale materials by cells of the mononuclear phagocyte system (MPS). Consequently, our results suggest that co-administration with iRGD can effectively and specifically improve the delivery of nanoparticles into the tumor site.
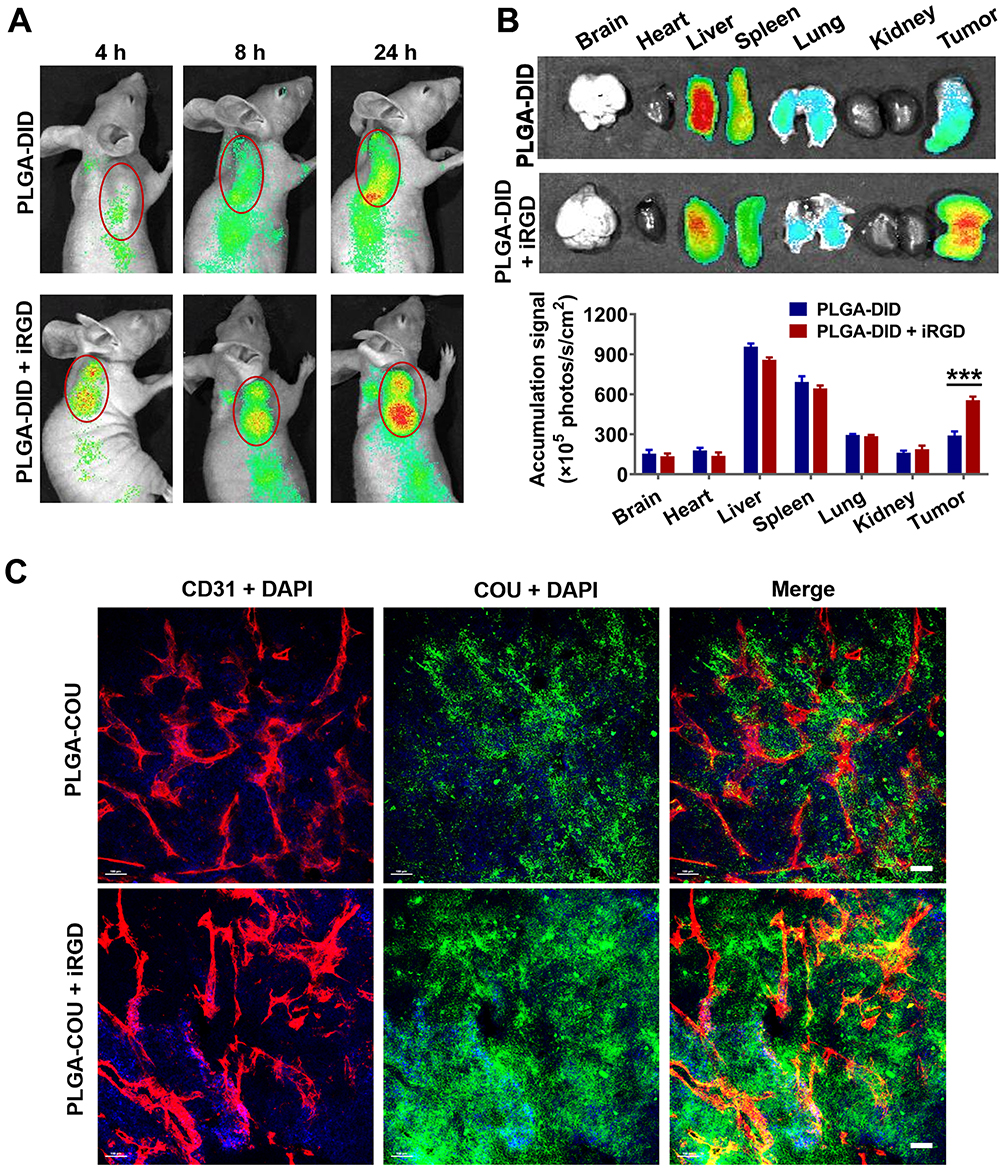 |
Figure 6 Distribution of nanoparticles in tumor-bearing mice and tumor xenografts. Notes: Mice bearing LS174T tumors (150–200 mm3) were given a single intravenous injection of PLGA-DID alone or combined with iRGD. (A) Mice were imaged using the SPECTRAL Lago NIR imaging system over time. The tumor location was marked with red circles. (B) At 24 h post injection, mice were sacrificed, and tumors and major organs were harvested for ex vivo imaging. Fluorescence intensity in tumors and organs was quantified. n=3. (C) LS174T tumors were sectioned after the injection of mice with PLGA-COU alone or in combination with iRGD. The distribution of PLGA-COU (green) within tumors was imaged under a confocal microscope. Tumor vessels were labeled with anti-CD31 antibody (red), and nuclei were visualized with DAPI (blue). Scale bar, 100 µm. ***P<0.001.Abbreviations: PLGA, poly lactic-co-glycolic acid; DID, 1,1ʹ-dioctadecyl-3,3,3ʹ,3ʹ- tetramethylindodicarbocyanine; COU, coumarin-6; CD31, cluster of differentiation 31; DAPI, 4,6-diamidino-2-phenylindole. |
To further determine if iRGD-mediated nanoparticles could efficiently distribute into tumor parenchyma, coumarin 6-loaded PLGA was co-administered with iRGD (PLGA-COU + iRGD), and tumor tissues were sectioned at 3 h post injection. The payload distribution in the tumor was detected by the green fluorescence signal of coumarin-6. Confocal microscopy revealed that PLGA-COU alone principally distributed around tumor vessels, while PLGA-COU co-injected with iRGD significantly promoted the deep penetration and distribution of the payload into the tumor parenchyma away from tumor vessels (Figure 6C). These results suggest that co-administration with iRGD can enhance tumor-targeting accumulation and penetration of PLGA particles into tumors.
In Vivo Antitumor Assessment
The in vivo antitumor effect was further evaluated using mice bearing LS174T tumors. When the tumor volume reached approximately 50 mm3, free PTX or PLGA-PTX were intravenously administered at 10 mg/kg every other day for a total of three treatments that were combined with or without 10 mg/kg iRGD. Alternatively, mice were administered with an equivalent volume of normal saline (NS).
As shown in Figure 7A, LS174T tumors grew quickly in the control NS group, and the tumor volume increased to over 900 mm3 at 12-days post-inoculation. All groups treated with each formulation of PTX showed significant suppression of tumor growth, suggesting highly efficient antitumor effects of PTX in vivo. Encapsulating PTX in PLGA resulted in a stronger suppressive effect on tumor growth than that observed after treatment with free PTX. Additionally, it was clear that co-administration of PLGA-PTX with iRGD dramatically enhanced tumor growth suppression efficacy compared to that observed with PLGA-PTX alone (P<0.05). Similarly, PTX combined with iRGD displayed stronger antitumor effects than free PTX (P<0.01). The iRGD peptide promoted the delivery of PLGA-PTX into tumors and thereby further enhanced the antitumor efficacy of the nanodrugs.
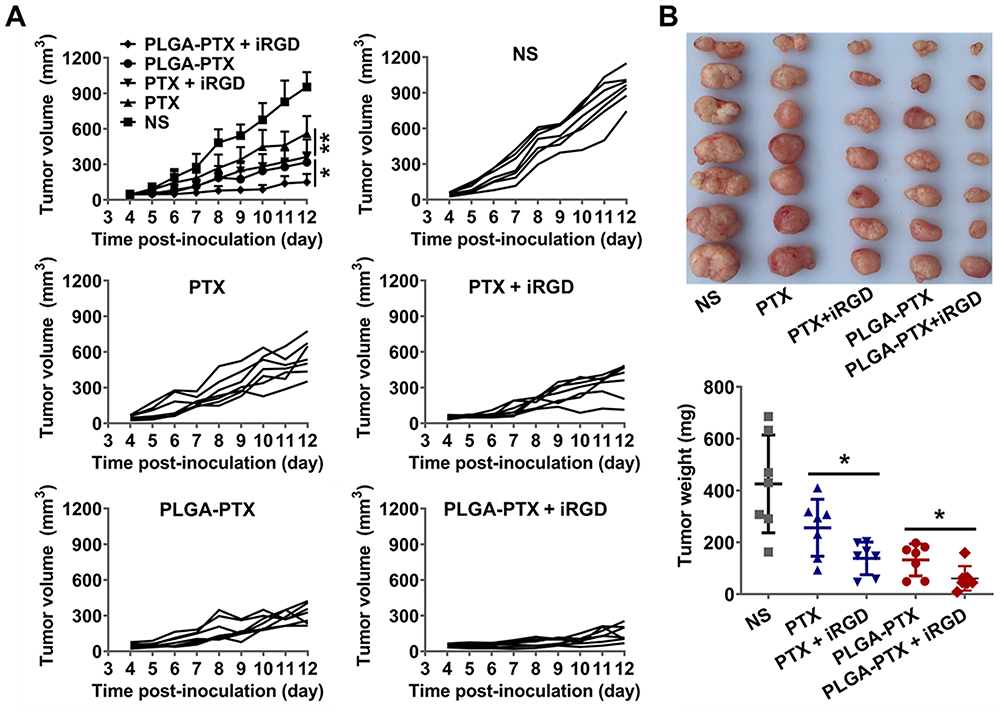 |
Figure 7 In vivo antitumor efficacy evaluation. Notes: (A) At the onset of a palpable tumor (about 50 mm3), BALB/c nude mice bearing LS174T tumors were intravenously injected with free PTX, PTX + iRGD, PLGA-PTX, PLGA-PTX + iRGD, or NS every 2 days for a total of three injections (PTX and iRGD at dose of 10 mg/kg). Tumor volume was calculated as length×width2×0.5. The mean tumor volume of each group and the tumor volume of each mouse in each group were presented over time. (B) Tumors obtained from the sacrificed mice were imaged, and tumor weight was measured for each group. *P<0.05; **P<0.01. Abbreviations: PLGA, poly lactic-co-glycolic acid; PTX, paclitaxel; PLGA-PTX, paclitaxel-loaded PLGA; NS, normal saline. |
At the conclusion of the observation, the mean tumor sizes in the free PTX, PTX + iRGD, PLGA-PTX, and PLGA-PTX+ iRGD groups were 561±55.5 mm3, 363±55.8 mm3, 317±31.5 mm3, and 150±26.2 mm3, respectively, compared to 953±47.3 mm3 in the control group. The corresponding tumor weights were reduced by 39.9%, 67.6%, 69%, and 85.7% after treatment with free PTX (256 ± 42 mg), PTX + iRGD (138 ± 24 mg), PLGA-PTX (132 ± 23 mg), and PLGA-PTX + iRGD (61 ± 18 mg), respectively, compared to that of the NS control group (426 ± 71 mg) (Figure 7B). Additionally, treatment with all formulation of PTX resulted in little change in mice body weight (Figure S6) compared to that of the NS group. These data suggest that encapsulation of PTX in PLGA and co-administration with tumor-penetrating iRGD peptide can substantially enhance the therapeutic effects of PTX against colorectal cancer in vivo.
Discussion
The clinical application of conventional chemotherapeutic agents such as PTX is limited by their poor water solubility and a lack of specific tissue distribution. Nanoparticles can improve solubility, bioavailability, and passive targeted distribution.35 In this study, we prepared a controlled-release formulation of PTX via encapsulation of this compound into PLGA. To increase the active delivery of PLGA-PTX into tumors, iRGD as the tumor-penetrating peptide was co-administered with the nanodrugs. We found that co-administration with iRGD significantly promoted the accumulation of nanoparticles preferentially into colorectal tumor xenografts and enhanced their therapeutic effects in vivo.
The EPR effect has been widely used as the rationale for employing nanoparticles to treat cancer; however, it has become increasingly clear that this passive tumor-targeting strategy through EPR effects frequently results in low delivery efficiency due to the porosity of tumor vessels and the degree of tumor vascularization that are highly variable according to tumor type and status.19,20,36 Contrary to passive drug delivery, active targeting relies on ligand-receptor interactions that induce specific binding to overexpressed receptors at the tumor site, and thus they can enhance drug retention compared to that of the EPR effect alone.37
The iRGD peptide is a tumor-targeting and tissue-penetrating peptide that can enhance vascular and tissue penetration in a tumor-specific manner. In addition to modification of iRGD on the surface of nanoparticles,38,39 it has been demonstrated that co-administration of drugs and free iRGD without creating a new chemical entity can also improve the activity of anticancer drugs.25,40 Additionally, co-administration of iRGD is more convenient and can bypass the conjugation technique. The combination and proportion between iRGD and drugs can also be prepared according to clinical acquirements for personalized medicine. Hence, in this study, we co-administered nanoparticles with the iRGD peptide and evaluated the synergistic effect of drug delivery both in vitro and in vivo.
Here, the PLGA nanoparticle was chosen as the drug carrier due to its merits that include physical stability, sustained drug release, and FDA approval for safe use in humans. Encapsulating PTX into PLGA resulted in sufficient EE% and DL%. The prepared PLGA-PTX was stable without changes in particle size for at least 2 weeks, and it exhibited the sustained-release characteristic of PTX. Additionally, PLGA-PTX demonstrated remarkable cytotoxicity on a variety of colorectal cancer cells a in time- and concentration-dependent manner while displaying relatively low cytotoxicity against normal liver cells (Figures 2A and S2). PLGA-PTX induced cell cycle arrest in the G2/M phase and resulted in caspase activation and cell apoptosis (Figure 3). At low concentrations, PLGA-PTX inhibited tumor cell migration and invasion (Figures 4 and S3), indicating a potential ability to reduce tumor metastasis. The combination of PLGA-PTX with iRGD, however, showed no substantial enhancement of cytotoxicity of PTX in vitro (Figures 2A and S2)
Successful drug delivery mediated by iRGD primarily depends upon the activity of the iRGD peptide, the density of tumor vessels, and the expression level of iRGD receptors.41 Based on selective accumulation in tumor vessels via the integrin receptor, iRGD is then able to be activated and interact with the neuropilin-1 receptor to trigger tissue penetration and drug delivery (Figure 8). Therefore, when employing iRGD as a drug carrier, the tumor vessel distribution and iRGD receptor expression should be assessed to ensure the satisfying conditions for drug transport and penetration into tumors are present. Colorectal cancer has been characterized by the abundant development of blood vessels.2,3 In this study, the extensive distribution of tumor vasculature in LS174T xenografts was revealed by immunofluorescence staining of blood endothelial cells (Figure 5A). Additionally, the tumor-targeting receptor integrin and the tumor-penetrating receptor neuropilin-1 were found to be highly expressed on tumor vessels (Figure 5B), suggesting a suitable tumor microenvironment for employing iRGD as the drug delivery vehicle. The RGD motif has great affinity to integrin αvβ3, αvβ5 and α5β1.42 The high expression of integrin subunit α5 but not αv in LS174T tumors indicates α5β1 but not αvβ3 or αvβ5 is the primary integrin on LS174T tumor vessels. This result ties well with previous studies wherein α5β1 is robustly and uniformly expressed on blood vessels of many tumor types and is a promising target for tumor theranostics.42–44
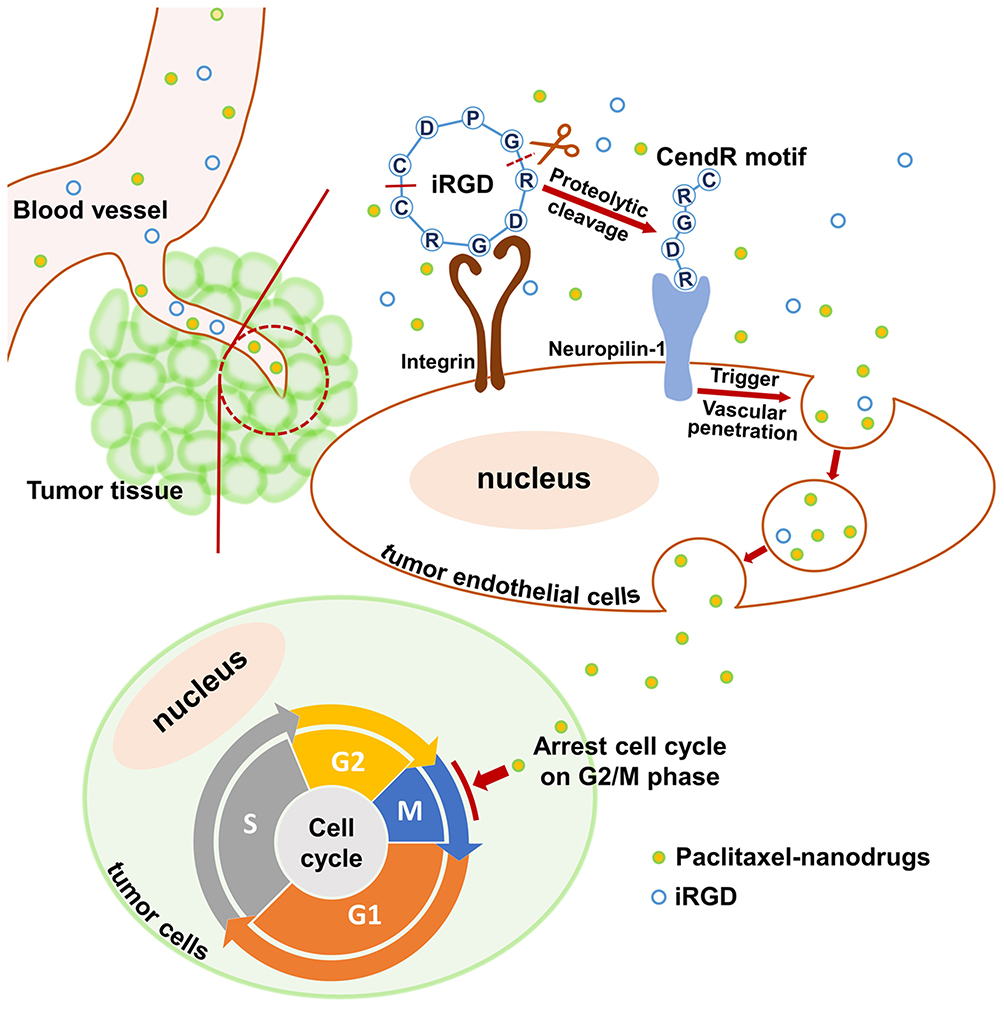 |
Figure 8 Schematic diagram of co-administration of iRGD with nanodrugs. Notes: When the co-administered nanodrugs and iRGD peptide reach the tumor tissue, iRGD binds to the integrin receptor and is proteolytically cleaved. The exposed tissue-penetrating motif CendR interacts with the neuropilin-1 receptor and triggers nanodrug penetration into tumor tissues. Ultimately, paclitaxel nanodrugs induce tumor cell cycle arrest and sequential apoptosis. |
Consequently, intravenous co-administration of nanoparticles with iRGD promoted tumor-preferential accumulation of the payload within the tumor site without apparent alteration of the particle distribution in normal tissues (Figures 6A and 6B). Additionally, iRGD enhanced the penetration of cargos away from tumor vessels into the tumor parenchyma (Figure 6C). It has been demonstrated that the enhanced drug penetration via the co-administration of iRGD relies on iRGD-mediated vascular permeability and trans-cell transportation of drugs deep into the tumor parenchyma.45,46 The relative low expression of integrin and neuropilin-1 receptors on LS174T tumor cells indicated that the improved drug delivery observed in this study could be predominantly attributed to enlarged transport of cargoes across tumor vessels (Figure 8). Eventually, a much stronger tumor growth suppression was observed in mice bearing LS174T tumors following treatment with PLGA-PTX + iRGD than that observed after treatment with PTX alone or PLGA-PTX (Figure 7). Thus, co-administration of iRGD is a competent strategy for the enhancement of nanoparticle delivery selectively into tumors.
Conclusion
In this study, a PTX nanodrug co-administered with iRGD was developed for targeted therapy of colorectal cancer. The iRGD-co-administration delivery system was found to possess obvious advantages over iRGD-free nanoparticles, including enhancing tumor accumulation, tumor penetration, and antitumor activity. All these findings suggest that co-administration with iRGD may provide a favorable drug delivery strategy for the treatment of colorectal tumors that possess abundant blood vessels with highly expressed integrin and neuropilin-1 receptors. Co-administration of iRGD with nanodrugs is an alternative manner that may also provide a convenient preparation process compared to that of the conjugation technique. Additionally, the combination and proportion between iRGD and drugs can also feasibly be optimized for use in precision medicine.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81603018 and 81871475) and the Central Universities Foundation of University of Electronic Science and Technology of China (No. ZYGX2019J109).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Coutelle O, Schiffmann LM, Liwschitz M, et al. Dual targeting of Angiopoetin-2 and VEGF potentiates effective vascular normalisation without inducing empty basement membrane sleeves in xenograft tumours. Br J Cancer. 2015;112(3):495–503. doi:10.1038/bjc.2014.629
3. Ruggiero A, Villa CH, Holland JP, et al. Imaging and treating tumor vasculature with targeted radiolabeled carbon nanotubes. Int J Nanomedicine. 2010;5:783–802. doi:10.2147/IJN.S13300
4. Loree JM, Sha A, Soleimani M, et al. Survival impact of CAPOX versus FOLFOX in the adjuvant treatment of stage III colon cancer. Clin Colorectal Cancer. 2018;17(2):156–163. doi:10.1016/j.clcc.2018.01.010
5. Martini G, Troiani T, Cardone C, et al. Present and future of metastatic colorectal cancer treatment: a review of new candidate targets. World J Gastroenterol. 2017;23(26):4675–4688. doi:10.3748/wjg.v23.i26.4675
6. Dienstmann R, Vermeulen L, Guinney J, Kopetz S, Tejpar S, Tabernero J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat Rev Cancer. 2017;17(2):79–92. doi:10.1038/nrc.2016.126
7. Alushin GM, Lander GC, Kellogg EH, Zhang R, Baker D, Nogales E. High-resolution microtubule structures reveal the structural transitions in alphabeta-tubulin upon GTP hydrolysis. Cell. 2014;157(5):1117–1129. doi:10.1016/j.cell.2014.03.053
8. Wang Y, Zhang C, Zhang S, et al. Kanglaite sensitizes colorectal cancer cells to Taxol via NF-kappaBeta inhibition and connexin 43 upregulation. Sci Rep. 2017;7(1):1280. doi:10.1038/s41598-017-01480-2
9. Zou H, Li L, Garcia Carcedo I, Xu ZP, Monteiro M, Gu W. Synergistic inhibition of colon cancer cell growth with nanoemulsion-loaded paclitaxel and PI3K/mTOR dual inhibitor BEZ235 through apoptosis. Int J Nanomedicine. 2016;11:1947–1958. doi:10.2147/IJN.S100744
10. Lin R, Li H, Chen Y, et al. FOLFOX versus POF (paclitaxel plus FOLFOX) versus IP PAC (intraperitoneal paclitaxel plus FOLFOX) as a first-line treatment in advanced gastric cancer (AGC): a multicenter, randomized phase II trial, FNF-004 trial. J Clin Oncol. 2019;37(4_suppl):6. doi:10.1200/JCO.2019.37.4_suppl.6
11. Lin R, Zhao S, Li H, Liu J, Fan N. A phase II Study of FOLFOX versus POF (paclitaxel plus FOLFOX) versus IP PAC (intraperitoneal paclitaxel) plus FOLFOX as a first-line treatment in advanced gastric cancer (AGC): a feasibility analysis. J Clin Oncol. 2017;35(4_suppl):56. doi:10.1200/JCO.2016.69.1378
12. Hassan MS, Awasthi N, Li J, et al. Superior therapeutic efficacy of nanoparticle albumin bound paclitaxel over cremophor-bound paclitaxel in experimental esophageal adenocarcinoma. Transl Oncol. 2018;11(2):426–435. doi:10.1016/j.tranon.2018.01.022
13. Alves RC, Fernandes RP, Eloy JO, Salgado HRN, Chorilli M. Characteristics, properties and analytical methods of paclitaxel: a review. Crit Rev Anal Chem. 2018;48(2):110–118. doi:10.1080/10408347.2017.1416283
14. Choudhury H, Gorain B, Tekade RK, Pandey M, Karmakar S, Pal TK. Safety against nephrotoxicity in paclitaxel treatment: oral nanocarrier as an effective tool in preclinical evaluation with marked in vivo antitumor activity. Regul Toxicol Pharmacol. 2017;91:179–189. doi:10.1016/j.yrtph.2017.10.023
15. Wang F, Porter M, Konstantopoulos A, Zhang P, Cui H. Preclinical development of drug delivery systems for paclitaxel-based cancer chemotherapy. J Control Release. 2017;267:100–118. doi:10.1016/j.jconrel.2017.09.026
16. Cui X, Fan Q, Shi S, et al. A Novel Near-infrared Nanomaterial with high quantum efficiency and its applications in Real Time in-Vivo imaging. Nanotechnology. 2018;29:205705. doi:10.1088/1361-6528/aab2fa
17. Ma J, Wu H, Li Y, et al. Novel core-interlayer-shell DOX/ZnPc co-loaded MSNs@ pH-sensitive CaP@PEGylated liposome for enhanced synergetic chemo-photodynamic therapy. Pharm Res. 2018;35(3):57. doi:10.1007/s11095-017-2295-z
18. Wu D, Si M, Xue HY, Wong HL. Nanomedicine applications in the treatment of breast cancer: current state of the art. Int J Nanomedicine. 2017;12:5879–5892. doi:10.2147/IJN.S123437
19. Tietjen GT, Saltzman WM. Nanomedicine gets personal. Sci Transl Med. 2015;7(314):314fs347. doi:10.1126/scitranslmed.aad3106
20. Danhier F. To exploit the tumor microenvironment: since the EPR effect fails in the clinic, what is the future of nanomedicine? J Control Release. 2016;244(Pt A):108–121. doi:10.1016/j.jconrel.2016.11.015
21. Zhu L, Zhao H, Zhou Z, et al. Peptide-functionalized phase-transformation nanoparticles for low intensity focused ultrasound-assisted tumor imaging and therapy. Nano Lett. 2018;18:1831–1841.
22. Sharma S, Mann AP, Molder T, et al. Vascular changes in tumors resistant to a vascular disrupting nanoparticle treatment. J Control Release. 2017;268:49–56. doi:10.1016/j.jconrel.2017.10.006
23. Wang T, Wang D, Liu J, et al. Acidity-triggered ligand-presenting nanoparticles to overcome sequential drug delivery barriers to tumors. Nano Lett. 2017;17(9):5429–5436. doi:10.1021/acs.nanolett.7b02031
24. Ma L, Chen Q, Ma P, et al. iRGD-functionalized PEGylated nanoparticles for enhanced colon tumor accumulation and targeted drug delivery. Nanomedicine. 2017;12(16):1991–2006. doi:10.2217/nnm-2017-0107
25. Liu C, Yao S, Li X, Wang F, Jiang Y. iRGD-mediated core-shell nanoparticles loading carmustine and O(6)-benzylguanine for glioma therapy. J Drug Target. 2017;25(3):235–246. doi:10.1080/1061186X.2016.1238091
26. Yu H, Tang Z, Song W, Zhang D, Zhang Y, Chen X. Co-administration of iRGD enhancing the anticancer efficacy of cisplatin-loaded polypeptide nanoparticles. J Control Release. 2015;213:e145–e146. doi:10.1016/j.jconrel.2015.05.246
27. Zhang WQ, Yu KF, Zhong T, et al. Does ligand-receptor mediated competitive effect or penetrating effect of iRGD peptide when co-administration with iRGD-modified SSL? J Drug Target. 2015;23(10):897–909. doi:10.3109/1061186X.2015.1034279
28. Wang K, Zhang X, Liu Y, Liu C, Jiang B, Jiang Y. Tumor penetrability and anti-angiogenesis using iRGD-mediated delivery of doxorubicin-polymer conjugates. Biomaterials. 2014;35(30):8735–8747. doi:10.1016/j.biomaterials.2014.06.042
29. Li X, Jiang X. Microfluidics for producing poly (lactic-co-glycolic acid)-based pharmaceutical nanoparticles. Adv Drug Deliv Rev. 2018;128:101–114. doi:10.1016/j.addr.2017.12.015
30. Xu B, Jin Q, Zeng J, et al. Combined tumor- and neovascular-”dual targeting” gene/chemo-therapy suppresses tumor growth and angiogenesis. ACS Appl Mater Interfaces. 2016;8(39):25753–25769. doi:10.1021/acsami.6b08603
31. Zeng X, Tao W, Mei L, Huang L, Tan C, Feng SS. Cholic acid-functionalized nanoparticles of star-shaped PLGA-vitamin E TPGS copolymer for docetaxel delivery to cervical cancer. Biomaterials. 2013;34(25):6058–6067. doi:10.1016/j.biomaterials.2013.04.052
32. Abouelmagd SA, Sun B, Chang AC, Ku YJ, Yeo Y. Release kinetics study of poorly water-soluble drugs from nanoparticles: are we doing it right? Mol Pharm. 2015;12(3):997–1003. doi:10.1021/mp500817h
33. Vinci M, Gowan S, Boxall F, et al. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol. 2012;10:29. doi:10.1186/1741-7007-10-29
34. Gaumet M, Vargas A, Gurny R, Delie F. Nanoparticles for drug delivery: the need for precision in reporting particle size parameters. Eur J Pharm Biopharm. 2008;69(1):1–9. doi:10.1016/j.ejpb.2007.08.001
35. Aftab S, Shah A, Nadhman A, et al. Nanomedicine: an effective tool in cancer therapy. Int J Pharm. 2018;540(1–2):132–149. doi:10.1016/j.ijpharm.2018.02.007
36. Kreuter J. Drug delivery to the central nervous system by polymeric nanoparticles: what do we know? Adv Drug Deliv Rev. 2014;71:2–14. doi:10.1016/j.addr.2013.08.008
37. Huang Y, Fan CQ, Dong H, Wang SM, Yang XC, Yang SM. Current applications and future prospects of nanomaterials in tumor therapy. Int J Nanomedicine. 2017;12:1815–1825. doi:10.2147/IJN.S127349
38. Simon-Gracia L, Hunt H, Scodeller P, et al. iRGD peptide conjugation potentiates intraperitoneal tumor delivery of paclitaxel with polymersomes. Biomaterials. 2016;104:247–257. doi:10.1016/j.biomaterials.2016.07.023
39. Yan F, Wu H, Liu H, et al. Molecular imaging-guided photothermal/photodynamic therapy against tumor by iRGD-modified indocyanine green nanoparticles. J Control Release. 2016;224:217–228. doi:10.1016/j.jconrel.2015.12.050
40. Gu G, Gao X, Hu Q, et al. The influence of the penetrating peptide iRGD on the effect of paclitaxel-loaded MT1-AF7p-conjugated nanoparticles on glioma cells. Biomaterials. 2013;34(21):5138–5148. doi:10.1016/j.biomaterials.2013.03.036
41. Kaiser J. Mixed results from cancer replications unsettle field. Science (New York, NY). 2017;355(6322):234–235. doi:10.1126/science.355.6322.234
42. Yao VJ, Ozawa MG, Varner AS, et al. Antiangiogenic therapy decreases integrin expression in normalized tumor blood vessels. Cancer Res. 2006;66(5):2639–2649. doi:10.1158/0008-5472.CAN-05-1824
43. Parsons-Wingerter P, Kasman IM, Norberg S, et al. Uniform overexpression and rapid accessibility of alpha5beta1 integrin on blood vessels in tumors. Am J Pathol. 2005;167(1):193–211. doi:10.1016/s0002-9440(10)62965-3
44. Majumder P, Bhunia S, Bhattacharyya J, Chaudhuri A. Inhibiting tumor growth by targeting liposomally encapsulated CDC20siRNA to tumor vasculature: therapeutic RNA interference. J Control Release. 2014;180:100–108. doi:10.1016/j.jconrel.2014.02.012
45. Pang HB, Braun GB, Friman T, et al. An endocytosis pathway initiated through neuropilin-1 and regulated by nutrient availability. Nat Commun. 2014;5:4904. doi:10.1038/ncomms5972
46. Liu X, Lin P, Perrett I, et al. Tumor-penetrating peptide enhances transcytosis of silicasome-based chemotherapy for pancreatic cancer. J Clin Invest. 2017;127(5):2007–2018. doi:10.1172/JCI92284
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.