")
Back to Journals » Cancer Management and Research » Volume 10
Clofoctol and sorafenib inhibit prostate cancer growth via synergistic induction of endoplasmic reticulum stress and UPR pathways
Authors Fan L, He Z, Head SA, Zhou Y, Lu T, Feng X, Zhang X, Zhang M, Dang Y, Jiang X, Wang M
Received 24 May 2018
Accepted for publication 31 July 2018
Published 23 October 2018 Volume 2018:10 Pages 4817—4829
DOI https://doi.org/10.2147/CMAR.S175256
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Antonella D'Anneo
Lixia Fan,1,* Zhenglei He,1,* Sarah A Head,2 Yinghui Zhou,1 Ting Lu,1 Xulong Feng,1 Xueqing Zhang,1 Meng Zhang,1 Yongjun Dang,3 Xinghong Jiang,4 Minghua Wang1
1Department of Biochemistry and Molecular Biology, Medical College, Soochow University, Suzhou, Jiangsu, China; 2Department of Pharmacology and Molecular Sciences, Johns Hopkins School of Medicine, Baltimore, MD, USA; 3Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Fudan University Shanghai Medical College, Shanghai, China; 4Department of Neurobiology, Medical College, Soochow University, Suzhou, China
*These authors contributed equally to this work
Background/Purpose: Prostate cancer is a major burden on public health and a major cause of morbidity and mortality among men worldwide. Drug combination therapy is known as a powerful tool for the treatment of cancer. The aim of this study is to evaluate the synergistic inhibitory mechanisms of clofoctol and sorafenib in the treatment of prostate cancer. However, the molecular mechanisms of this phenomenon have not been illuminated clearly. In this study, we investigated the anti-tumor effects of clofoctol in combination with sorafenib in vitro and in vivo.
Methods: The activity and mechanism of clofoctol in combination with sorafenib were examined in PC-3cells. mRNA and protein expression of key players in the ER stress pathway were detected with RT-PCR and Western blotting. Cell viability was estimated by CCK-8 assay or Alamar blue assay, and apoptosis and cell cycle were monitored and measured by flow cytometry. PC-3 cells were inoculated subcutaneously in male BALB/c nude mice. The therapeutic regimen was initiated when the tumor began showing signs of growth and treatment continued for 5 weeks.
Results: Our data indicate that clofototol and sorafenib induce cell death through synergistic induction of endoplasmic reticulum (ER) stress, resulting in activation of the unfolded protein response (UPR). Combination therapy with clofoctol and sorafenib induced an upregulation of markers of all three ER stress pathways: PERK, IRE1 and ATF6. In addition, combination therapy with clofoctol and sorafenib markedly inhibited the growth of prostate cancer xenograft tumors, compared with clofoctol or sorafenib alone.
Conclusion: The combination of clofoctol and sorafenib can serve as a novel clinical treatment regimen, potentially enhancing antitumor efficacy in prostate cancer and decreasing the dose and adverse effects of either clofoctol or sorafenib alone. These results lay the foundation for subsequent research on this novel therapeutic regimen in human prostate cancer.
Keywords: drug combination, endoplasmic reticulum stress, unfolded protein response, prostate cancer, clofoctol
Introduction
Prostate cancer remains the second leading cause of cancer-related deaths among men in the US, with an estimated death toll exceeding 26,120 in 2015.1 Although early treatment can be performed by radical prostatectomy or radiation, most patients will have local recurrence and distant metastases later.2,3 Drug combinations are widely used to treat the most difficult diseases such as cancer.4,5 The main purpose of the combination therapy is to achieve a synergistic effect, reduce toxic doses and reduce or delay the induction of drug resistance.6 The effective drug combination results in a reduction in the concentration of each drug required to produce the desired effect compared to the dose of each drug to produce the same effect as a single agent. Thus, the good literature described in the use of combination therapy in cancer has the advantage of producing synergistic antitumor effects through the use of anticancer drugs with different mechanisms and/or reducing the possibility of developing resistant cancer cell clones.7
In previous studies, we screened the Johns Hopkins Drug Library for the inhibition of prostate cancer cell proliferation to identify new anti-prostate cancer activity in known drugs. We identified an antibacterial drug clofoctol as a novel inhibitor of prostate cancer cell proliferation and systematically investigated the mechanism underlying its anticancer activity in vitro and in vivo. The study also showed that clofoctol exerts its anticancer effect through activation of endoplasmic reticulum (ER) stress and all three unfolded protein response (UPR) pathways.8 Sorafenib is a multi-kinase inhibitor and tyrosine kinase receptor of the Raf/MEK/ERK pathway and has been shown to induce apoptosis and inhibit proliferation and angiogenesis in a variety of tumor cells.9
The ER plays an important role in the quality control of proteins, determining whether the protein is correctly folded before it reaches its final destination.10 When proteins are misfolded, they are retained in the ER and delivered to the cytosol for proteasomal degradation, a process known as ER-related degradation.11 If the misfolded protein accumulates beyond the limit of the ER-related degradation ability, it will lead to the activation of the UPR process.12 UPR is a cytoprotective mechanism that responds to the misfolded protein excess in ER. The UPR pathway includes three specific branches: IRE1, ATF6 and PERK.13 Both PERK and IRE1 contain a cytoplasmic kinase domain, which is known to be activated by homodimerization and autophosphorylation in the presence of ER stressors.14 The UPR increases protein folding capacity via activation of IRE1 and ATF6 that leads to increased production of ER chaperones and also decreases the unfolded protein burden via PERK-dependent inhibition of translation initiation.15 In the case of ATF6, accumulation of unfolded proteins induces ATF6 transport from the ER to the Golgi apparatus, where it is cleaved by two transmembrane protein site 1 (S1P) and site 2 (S2P) proteases.16 Cleaved ATF6 produces a cytoplasmic protein that is an active transcription factor. Short-term ER stress events lead to prosurvival transcriptional activities through UPR pathways. When cells undergo irreversible ER stress, the UPR pathway eliminates damaged cells by apoptosis.17–19 Significantly, other reports have demonstrated that sorafenib is a potent inducer of ER stress, manifested by a rapid mobilization of cytoplasmic calcium, PERK activation, induction of IRE1, XBP1 splicing, phosphorylation of eIF2 and induction of the CHOP (also known as DDIT-3) and the GADD34 (also known as PPP1R15A).20 Thus, UPR is an important sensor for cells to make a life or death decision under conditions of ER stress.
In this study, we identified clofoctol and sorafenib as a synergistic drug combination for inhibition of PC-3 cell growth through high-throughput screening. We further investigated this synergistic mechanism and found that treatment of sorafenib with clofoctol enhanced PC-3 cell death by inducing ER stress and activating the UPR pathway. In addition, we found that clofoctol induces the accumulation of ubiquitinated modified proteins, which is a general indicator of the inhibition of ubiquitin proteasome system (UPS) function. Sorafenib alone does not cause accumulation of ubiquitinated modified proteins, but sorafenib enhances clofoctol’s action when used in combination. Finally, we used mouse xenografts of PC-3 cells to demonstrate synergistic effects of clofoctol and sorafenib in vivo. Our results indicate that these two compounds have a synergistic effect on PC-3 cell death induced by the ER stress signaling pathway and demonstrate that this combination is an effective therapeutic potential in prostate cancer.
Materials and methods
Cell cultures and reagents
PC-3, DU145 and LNCaP were obtained from the Shanghai Institute for Biological Sciences and grown in the Roswell Park Memorial Institute-1640 (RPMI-1640) culture medium supplemented with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA), penicillin (100 U/mL) and streptomycin (100 µg/mL). Cultured cells were grown at 37°C in a humidified atmosphere of 5% CO2 and were passaged using pancreatic enzymes two or three times a week. Sorafenib tosylate (BAY 43-9006), clofoctol, MG132 and cycloheximide (CHX) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA).
Cell viability assays
PC-3 cells were seeded on 96-well plates (2,000 cells/well in 100 µL), with three replicates used for each concentration. The cells were treated with various concentrations of sorafenib or clofoctol before incubation with 10 µL per well of Cell Counting Kit-8 (CCK-8) reagent (Dojindo Molecular Technologies Inc., Shanghai, China) or Alamar blue (Sigma-Aldrich, MO, USA) solution for 3 hours. Cell viability was estimated by CCK-8 assay or Alamar blue assay.21 The OD was measured using a microplate reader (Thermo Fisher Scientific). The IC50 values were calculated using the GraphPad Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA). The combined effect can be compared to the expected additive effect given by the common formula Eab = Ea + (1-Ea) Eb, where Eab represents the theoretical effect of the two drug combinations and Ea and Eb represent the two drugs.22 To determine the potential mechanism of drug–drug interactions, we used the CompuSyn software (CompuSyn, Cambridge, UK) to calculate the combination index (CI). A CI of <1.0 was considered to be indicative of synergism.
Cell apoptosis and cell-cycle analysis
Cells were treated with dimethyl sulfoxide (DMSO), sorafenib (6 µM), clofoctol (10 µM) and in combination for 24 hours, before being harvested and centrifuged. Cell apoptosis and cell cycle were examined by flow cytometry using Annexin V-Fluorescein-isothiocyanate (FITC)/propidium iodide (PI) double staining and PI staining of DNA, respectively.
Western blot analysis
Cells were treated with sorafenib (6 µM) and clofoctol (10 µM) alone or in combination for the indicated periods of time. Cells were harvested and subjected to Western blot analysis as described previously.23 The primary antibodies used in the experiments were as follows: GAPDH (1:1,000; Cell Signaling Technology, Danvers, MA, USA), eIF2α (1:1,000; Cell Signaling Technology), phospho-eIF2α (1:1,000; Cell Signaling Technology), transcription factor CHOP (1:500; Cell Signaling Technology), ATF4 (1:1,000; Cell Signaling Technology), PERK (1:1,000; Cell Signaling Technology) and ubiquitin (1:1,000; Santa Cruz Biotechnology Inc., Dallas, TX, USA).
XBP1 splicing assay and quantitative real-time PCR
Total RNA from cells was isolated by TRIzol reagent (Thermo Fisher Scientific), and cDNAs were synthesized using cDNA Synthesis Kit (Takara Bio, Otsu, Japan) with oligo-(dT) primers. The cDNAs were PCR amplified using specific primers for XBP1 designed by Roche program. Primers of XBP1 were as follows: hXBP1.3S: AAA CAG AGTAGCAGCTCAGACTGC and mXBP1.12AS: TCCTTCTGGGTAGACCTCTGGGAG. The PCR products were resolved on 2% agarose gel. To distinguish the unspliced XBP1 mRNA from the spliced form, the PCR products of XBP1 mRNA were digested with Pst I prior to gel electrophoresis. Quantitative real-time PCR (qPCR) was carried out using Roche LightCycler 96 instrument (Hoffman-La Roche Ltd., Basel, Switzerland). Sequences of primers for GRP78/BiP (commonly known as BiP), CHOP, ATF6, ATF4, PERK and GADD34 are summarized in Table S1.
Xenograft tumor mouse model
The animal experiment was approved by the ethics committee of Soochow University (Approval No. ECSU-201800066) and followed the guideline for Guide for the Care and Use of Laboratory Animals. Male BALB/c nude mice (4 weeks old) were purchased from the Soochow University Experimental Animal Center. Mice were allowed to get accustomed to their new environment for 1 week before commencement of the experiment.
Mice were inoculated subcutaneously with 2×106 PC-3 cells suspended in 200 µL PBS on the right back. When xenograft tumors reached a volume of ~100 mm3, mice were randomly assigned to four groups (n=7 each group) and treated intraperitoneally. Therapeutic schedule based on our in vitro results, preliminary experiments and other researchers’ studies was as follows: 1) control group: solvent; 2) clofoctol-treated group: clofoctol 100 mg/kg; 3) sorafenib-treated group: sorafenib 18 mg/kg; and 4) combination treatment group: clofoctol 100 mg/kg and sorafenib 18 mg/kg. Treatment cycle was 2 days, and the whole treatment process lasted for 5 weeks. Mice were weighed every 4 days. Tumor sizes were monitored every 4 days using a caliper, and tumor volume was calculated according to the formula L×S2×0.5,24 in which L represents the longest diameter and S represents the shortest diameter of the tumor.
Statistical analyses
The quantitative data are presented as mean±SD values and plotted using GraphPad Prism 5. Statistically significant differences were determined by an unpaired Student’s t-test. Values of P≤0.05, P≤0.01 and P≤0.001 were denoted by asterisks *, ** and ***, respectively, by comparing the experimental (treated) vs vehicle group. The significance of the differences between the experimental conditions was determined using Student’s t-test for unpaired observations.
Drug synergistic effect: The software Calcusyn (BioSoft, Ferguson, MO, USA and Cambridge, UK) was used for calculating drug combination effect. CI was used as the indicator of the drug combination dose effect.
Results
Screening of the inhibitors and verification of the combination effect
For synergistic screening, we selected 40 inhibitors of Johns Hopkins Drug Library (JHDL) drugs to inhibit prostate cancer cells. A total of 19 different drug classes were selected, including different classes of antiviral drugs, vasodilators and antihistamines drugs (Table 1). According to the Chou–Talalay formula and the dose–response method, different drugs were combined with clofoctol for the treatment of PC-3 cells. In our previous study, we found that clofoctol is active on all six prostate cancer cell lines with IC50 values ranging from 10 to 15 µM.8 To determine IC50 values, we initially treated PC-3 cells with increasing concentrations of sorafenib, ranging from 1.95 to 15 µM (Table 2). The IC50 concentration of sorafenib in PC-3 cells was 5.98 µM (5.458–6.559 µM). The chemical structures of clofoctol and sorafenib are as follows (Figure 1A and B). We found that clofoctol and sorafenib can inhibit cell proliferation in prostate cancer cells (PC-3, DU145 and LNCaP). The Alamar blue assay was carried out at 72 hours with the cells treated with a range of doses; 2, 4 and 6 µM sorafenib and 2, 4, 6, 8, 10 and 12 µM clofoctol. Treatment of PC-3, DU145 and LNCaP cells with sorafenib and/or clofoctol showed that the inhibitory effect of the combination of the two drugs was far greater than the inhibitory effect of the single drug (Figure S1A–C). The concept of synergy or antagonism has a well-defined definition: they represent a greater or lesser effect on the combination drug, respectively, rather than a simple additive effect expected by the action of each drug alone. If the actual efficiency is greater than the theoretical frequency, it indicates that the two drugs are synergistic. We have found that the effect of the two drugs in combination with multiple doses is greater than the additive effect in PC-3, DU145 and LNCaP cells. The effect of the combination of the two drugs was found to be more pronounced in PC-3 cells (Table S2).
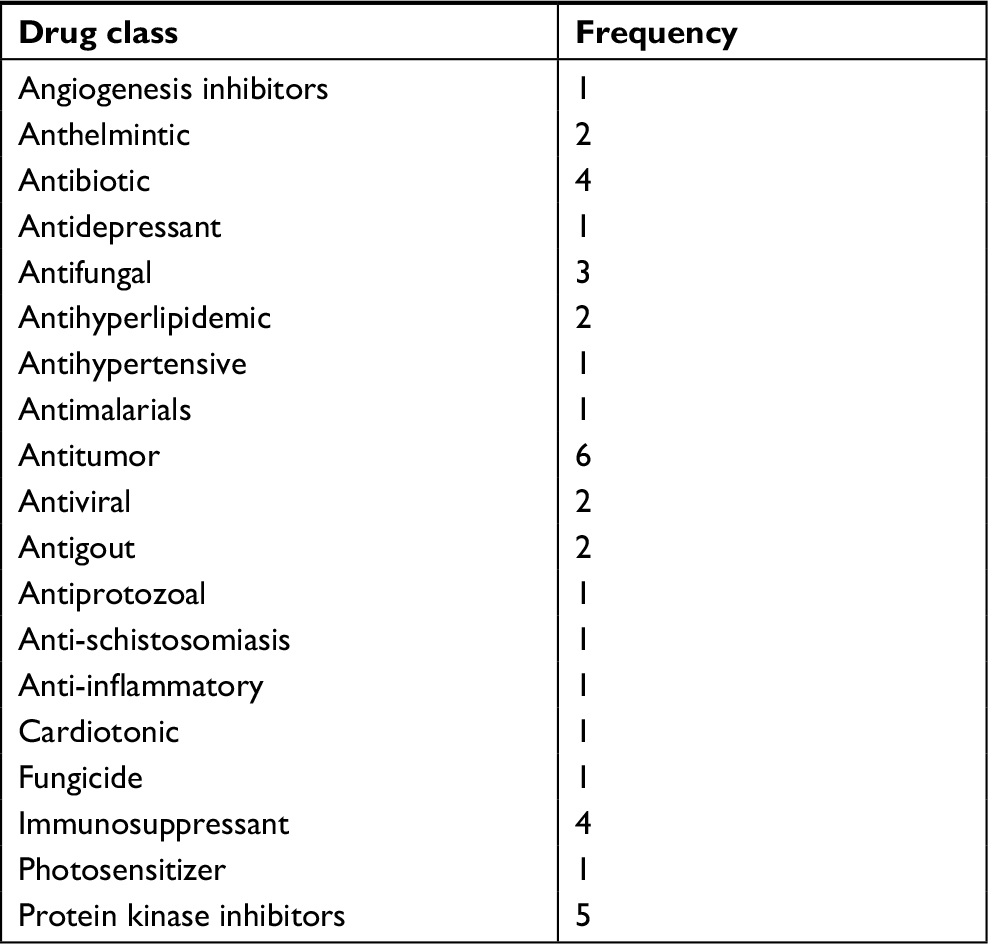 | Table 1 Distribution of drug classes |
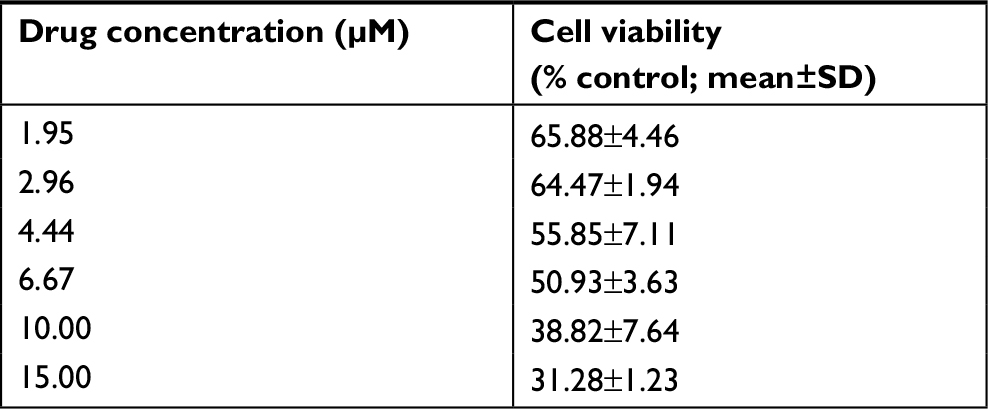 | Table 2 The dose-dependent cell proliferation assay of sorafenib on PC-3 cells |
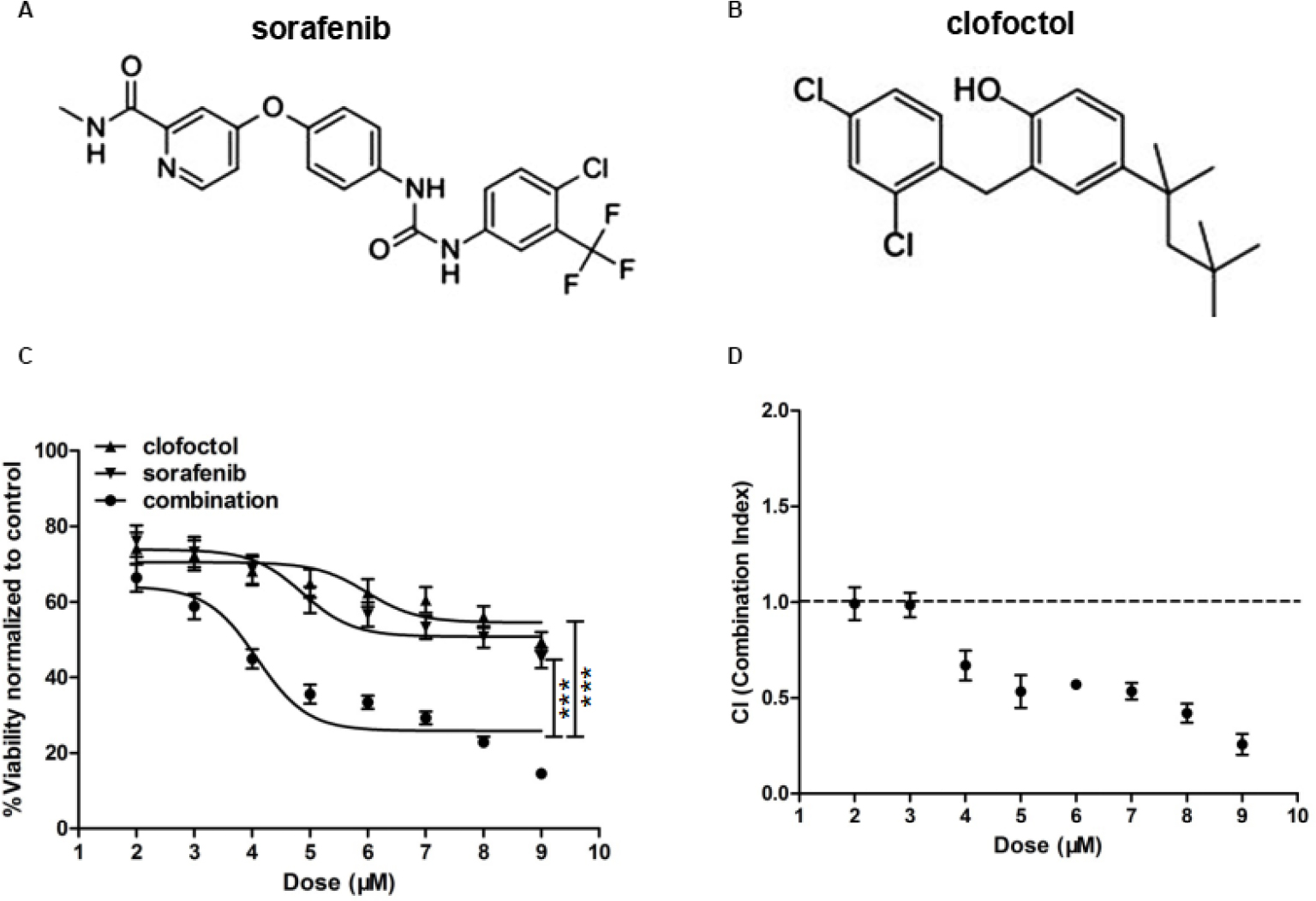 | Figure 1 Clofoctol and sorafenib synergistically inhibit PC-3 cell proliferation. Notes: (A) and (B) Chemical structures of clofoctol and sorafenib. (C) PC-3 cells were treated with a series of concentrations of clofoctol alone, sorafenib alone and a combination of clofoctol and sorafenib. Cell viability was determined by CCK-8 assay. Cells untreated with sorafenib or clofoctol were used as negative control. X-axis values represent each drug concentration. Y-axis represents the ratio of the value at 48 hours divided by the value at 1 hour for each concentration and normalized to the negative control, which was set to 100%. Graph bars represent mean±SD. ***P<0.001 indicate statistical significance as determined by using the unpaired t-test. (D) The key parameter returned by the Chou–Talalay method is the CI. A CI of 1 indicates pure additivity, a CI of >1 indicates antagonism and a CI between 0 and 1 indicates synergy. Abbreviations: CCK-8, Cell Counting Kit-8; CI, combination index. |
To further determine that clofoctol and sorafenib synergistically inhibit the growth of prostate cancer cells. This protocol is shown in Figure 1C and D and utilizes the cell viability assay of PC-3 cells. Cell viability was assessed by CCK-8 assay at 48 hours, and cells were treated with a series of doses of 2–9 µM clofoctol and 2–9 µM sorafenib. Treatment of PC-3 cells with clofoctol or sorafenib for 48 hours increased the antiproliferative effect in a dose-dependent manner. Cell viability (%) represents the ratio of the value at 48 hours divided by the value at 1 hour for each concentration and normalized to the negative control (untreated cells). Combination therapy resulted in a significant increase in antiproliferative effect when compared to treatment with clofoctol or sorafenib as a single treatment (***P<0.001; Figure 1C). CI is the natural law-based general expression of pharmacologic drug interactions. The CI theorem offers a quantitative definition for additive effect (CI=1), synergism (CI<1) and antagonism (CI>1) in drug combinations. When calculated for clofoctol and sorafenib, the CI was <1 in PC-3 cells with increasing dose, indicating synergistic effects (Figure 1D).
Clofoctol and sorafenib synergistically inhibit PC-3 cell proliferation
Based on our results, we selected IC50 doses of clofoctol (10 µM) and sorafenib (6 µM) for further combination studies. Studies of PC-3 cell apoptosis and cell cycle were performed. Clofoctol and sorafenib were minimally toxic by Annexin V analysis when administered alone at concentrations as high as 10 and 6 µM, respectively. The percentage of apoptotic cells is expressed as the percentage of cells double positive for PI and Annexin V. When cells were exposed to the combination of sorafenib and clofoctol, 78.84% cells were arrested in the G1 phase (Figure 2A and B) and the apoptosis rate increased up to 21.94% (Figure 2C and D). These results suggest that clofoctol in combination with sorafenib induces cell-cycle arrest and enhances the apoptotic effect.
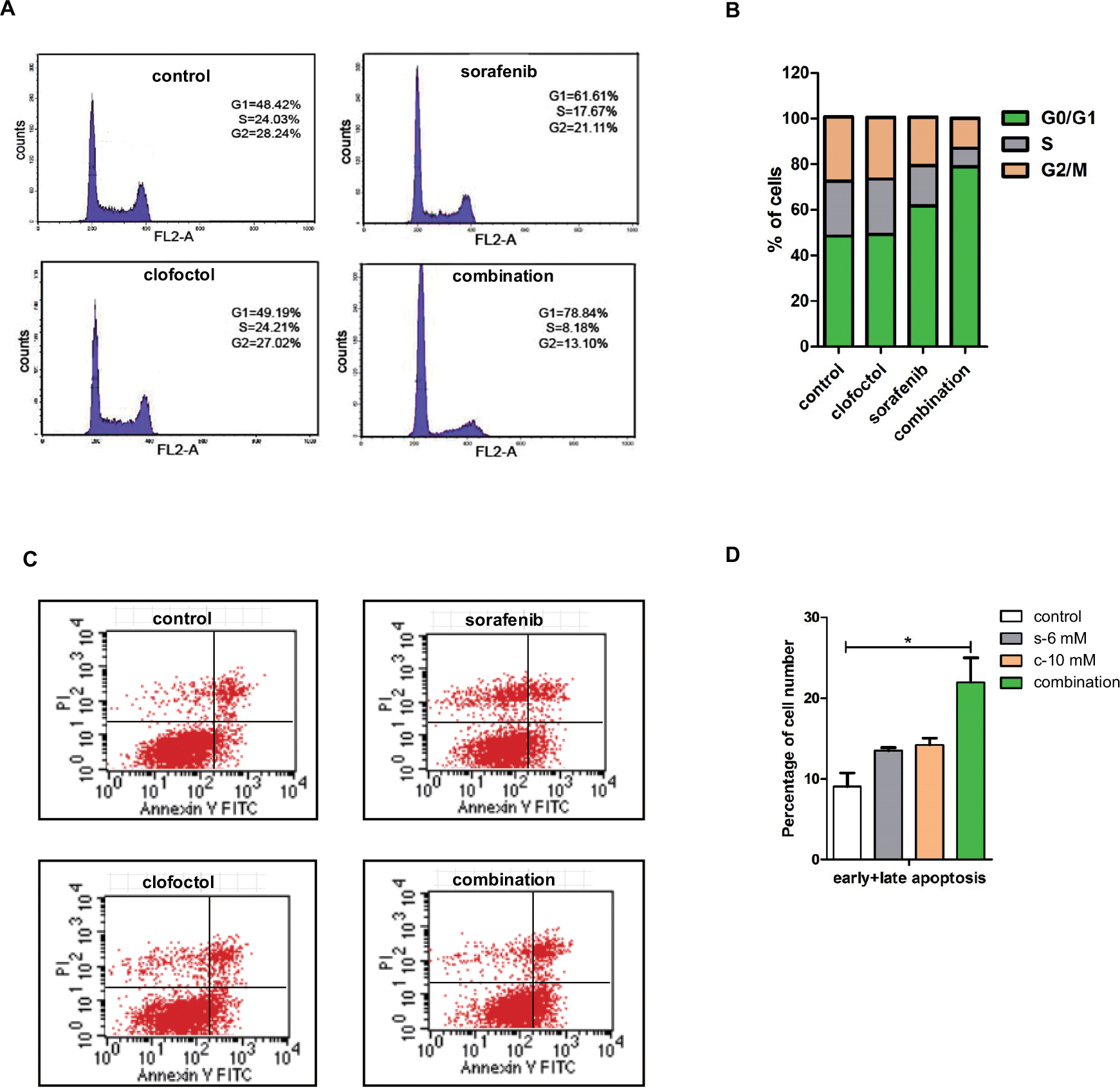 | Figure 2 Clofoctol and sorafenib induce cell-cycle arrest and apoptosis. Notes: (A and B) PC-3 cells show increased accumulation of cells in G1 with combination of clofoctol and sorafenib. Cell-cycle distribution was analyzed by flow cytometry with PI-stained PC-3 cells treated with DMSO, clofoctol, sorafenib and combination for 24 hours. Difference in the distribution is shown in histograms on the right. (C and D) The percentage of Annexin V-positive PC-3 cells treated with DMSO, clofoctol, sorafenib and combination for 24 hours was determined through Annexin V/PI assay. The difference percentage of early and late apoptosis is displayed in the right histogram. *P<0.05 indicates statistical significance as determined by using the unpaired t-test t-test. s and c are the acronyms for sorafenib and clofoctol, respectively. Abbreviations: PI, propidium iodide; DMSO, dimethyl sulfoxide. |
Clofoctol combined with sorafenib inhibits PC-3 cell growth by activating UPR pathways
In our previous studies, we demonstrated PC-3 cells treated with clofoctol-induced massive vacuolization and activated ER stress. Indeed, all three UPR pathways including IRE1, PERK and ATF6 were activated by clofoctol. Activation of UPR pathways by clofoctol led to the inhibition of protein translation in cells and activation of the JNK signaling pathway, resulting in autophagic cell death. Hence, PC-3 cells treated with clofoctol and sorafenib could not only induce ER stress separately but might also have a synergistic effect on some pathways in ER stress. We thus investigated the effects of sorafenib and clofoctol on UPR pathways, including IRE1, PERK and ATF6. Real-time PCR analysis was performed to determine the expression of mRNAs involved in the UPR pathways in PC-3 cells after 12 hours of clofoctol and sorafenib (Figure 3A–C). We observed that the Pst I digestion products from XBP1 mRNA were decreased after treatment with combination of sorafenib with clofoctol for 24 hours (Figure 3D). These results were validated by qPCR and Western blot, which confirmed that ATF4 and CHOP pathways were activated and could induce cell death even though there was no obvious difference in phosphorylation of eIF2α (Figure 3E–G).
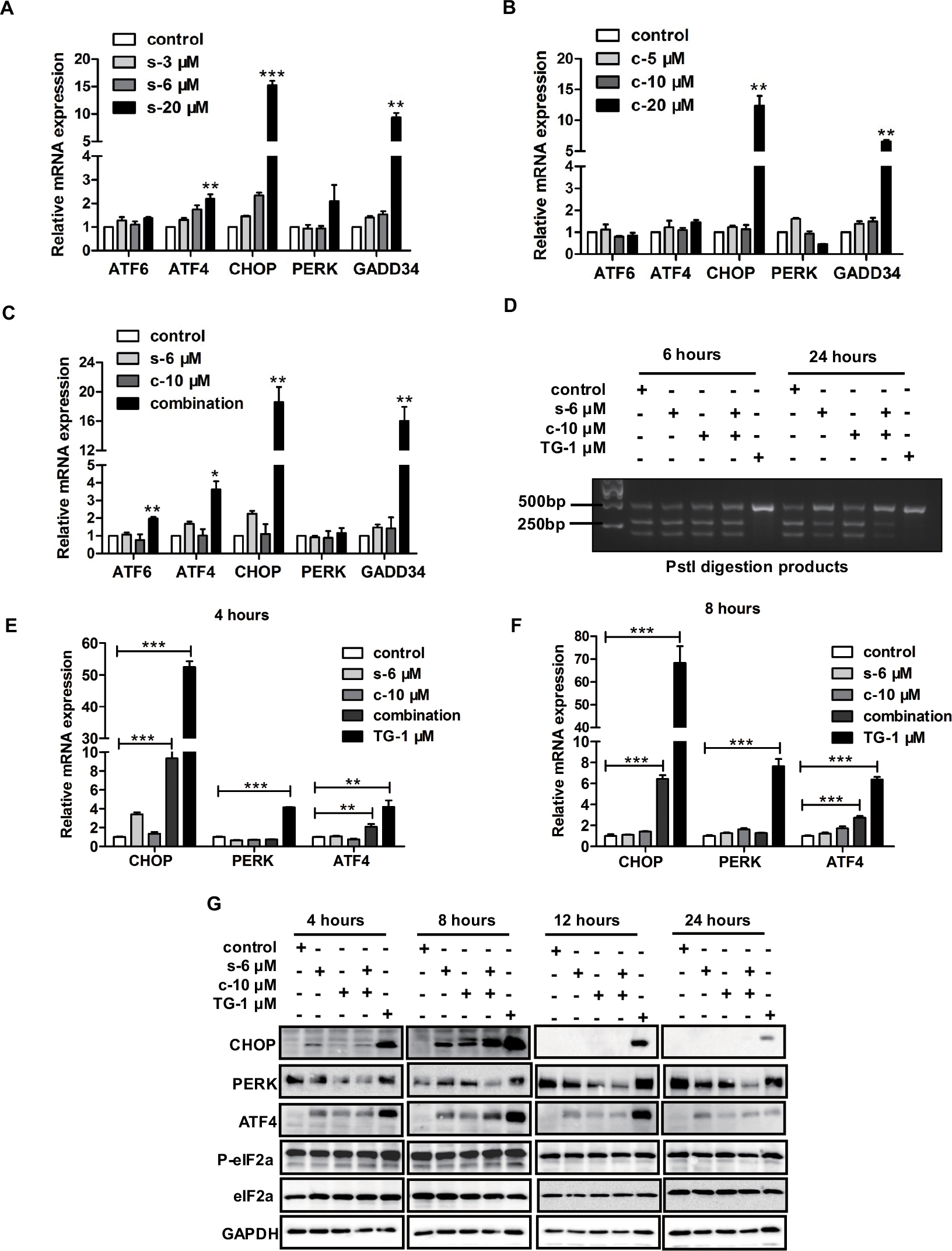 | Figure 3 Combination of sorafenib and clofoctol induces ER stress in PC-3 cells. Notes: (A–C) The hallmarks of ER stress were measured by real-time PCR levels after treatment with compounds for 12 hours. Values were normalized to DMSO treatment. Graph bars represent mean±SD from three independent experiments. *P<0.05, **P<0.01 and ***P<0.001 indicate the statistical significance by using the unpaired t-test t-test. (D) XBP1 splicing after PC-3 cells treatment with compounds (sorafenib, clofoctol and TG used as a positive control) for 6 hours and 24 hours. The two bands at the bottom represent the products from digestion of XBP1u, and the top band in gel represents XBP1s, which cannot be digested by the restriction endonuclease Pst I. (E-F) We detected mRNA expression levels of UPR pathway-activated markers by qPCR, such as CHOP, PERK and ATF4. (G) We detected protein expression levels of UPR pathway-activated markers by Western blot, such as CHOP, PERK, ATF4 and eIF2α. The protein molecular weights of CHOP, PERK, ATF4, phospho-eIF2α, eIF2α and GAPDH were 27, 140, 49, 38, 38, 37 kDa, respectively. and c are the acronyms for sorafenib and clofoctol, respectively. Abbreviations: ER, endoplasmic reticulum; DMSO, dimethyl sulfoxide; TG, thapsigargin; UPR, unfolded protein response. |
Clofoctol could induce the accumulation of ubiquitinated proteins
The ATP-driven chaperone VCP, also known as p97, is an AAA-ATPase central to UPS-dependent protein turnover. (VCP)/p97 governs critical steps in ubiquitin-dependent protein quality control and intracellular signaling pathways.25,26 The VCP-centered ER-associated protein degradation pathway serves to reduce excessive levels of unfolded polypeptides in the ER, thereby mitigating ER stress. Once ER stress is activated, ER stress-associated apoptotic signaling is initiated, as exhibited by cellular vacuolization.27 In this study, we sought to determine whether clofoctol affects the UPS pathway. PC-3 cells were exposed to DMSO, 6 µM sorafenib, 10 µM clofoctol or the combination of these two compounds for 6 and 24 hours, and 5 µM MG132 was used as a positive control. Equal amounts of protein from whole cell lysates were subjected to Western blot for the VCP proteins (a general indicator of inhibited UPS function). Clofoctol causes the accumulation of ubiquitinated proteins by inhibiting the ubiquitination signaling pathway, while sorafenib can attenuate the effects of clofoctol. We speculated that sorafenib may play an important role in the ubiquitination signaling pathway to attenuate the inhibition of clofoctol at 6 and 24 hours. In particular, this effect is more pronounced within 24 hours (Figure 4A). We further showed that clofoctol could cause a dose-dependent vacuolization of the cytoplasm and sorafenib alone does not cause vacuolization of the cytoplasm, but in combination, sorafenib enhances the effect of clofoctol (Figure 4B).
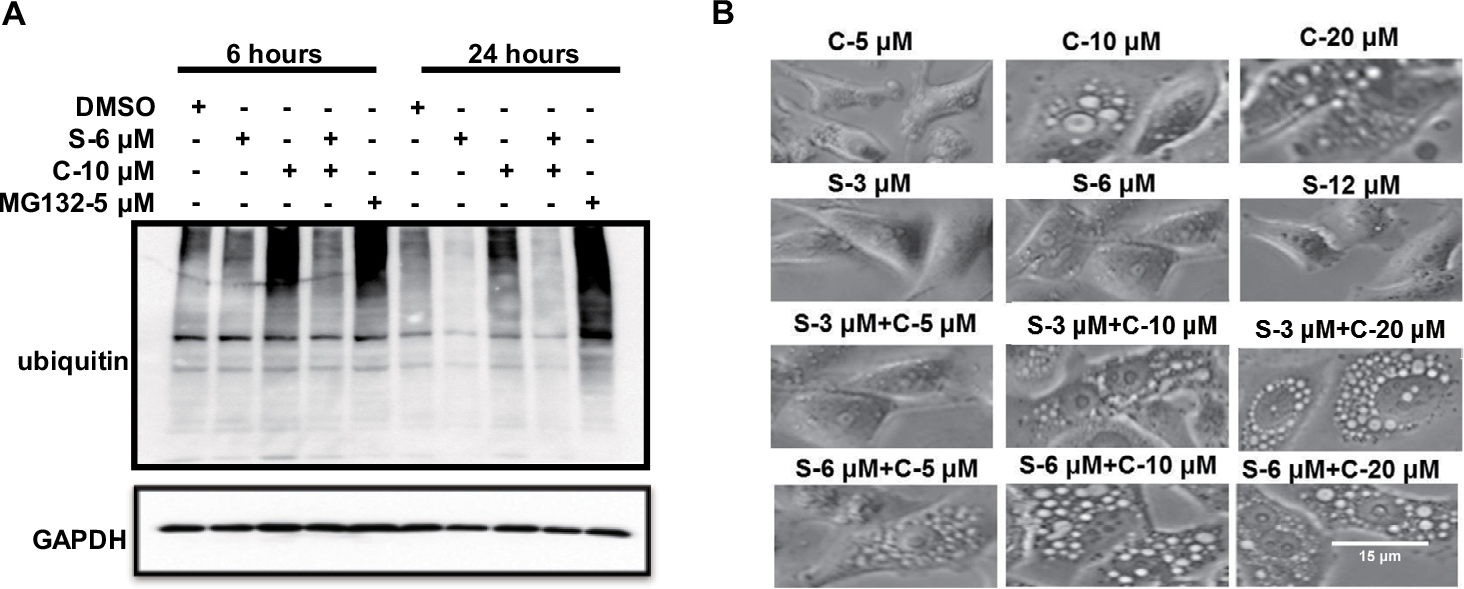 | Figure 4 Clofoctol and sorafenib induced vacuolization and ubiquitin accumulation in PC-3 cells. Notes: (A) PC-3 cells expressing ubiquitin with HA-tag were exposed to compounds as described earlier for 6 hours and 24 hours and immunoblotted for ubiquitin. The molecular weight of ubiquitin was 8 kDa. (B) Clofoctol could induce vacuolization of the cytoplasm, although less prominent vacuolization was sometimes observed in cells treated only with sorafenib. Cells treated with clofoctol plus sorafenib showed a significantly greater vacuolization than did cells with either treatment alone. The vacuolization becomes more apparent upon increasing the concentration of clofoctol but not that of sorafenib. s and c are the acronyms for sorafenib and clofoctol, respectively. |
Antitumor activity of combination of clofoctol with sorafenib in human prostate cancer xenografts
Male BALB/c nude mice with PC-3 cell xenograft tumors were treated with vehicle, clofoctol, sorafenib or the combination for 5 weeks. When tumors were taken out (Figure 5A), it was obvious that the tumors in the clofoctol and sorafenib treatment groups were smaller than those in the control group, and the inhibition was even more remarkable in the clofoctol–sorafenib co-treatment group (Figure 5B and C). During the whole intervention process, clofoctol, sorafenib and their combination were well tolerated by the mice with appropriate dose according to the weight of the mice, and the weight in all the three treatment groups was not significantly different from that of the control group (Figure 5D). There was no difference in daily food and water consumption between groups. Then, qPCR was performed on RNA extracted from the tumors of these four groups. qPCR results showed that the expression levels of CHOP, GADD34, ATF6, ATF4, PERK and GRP78 mRNA increased significantly. The results further validated that clofoctol and sorafenib could lead to synergistic PC-3 cell death via induction of ER stress by activating UPR pathways (Figure 5E).
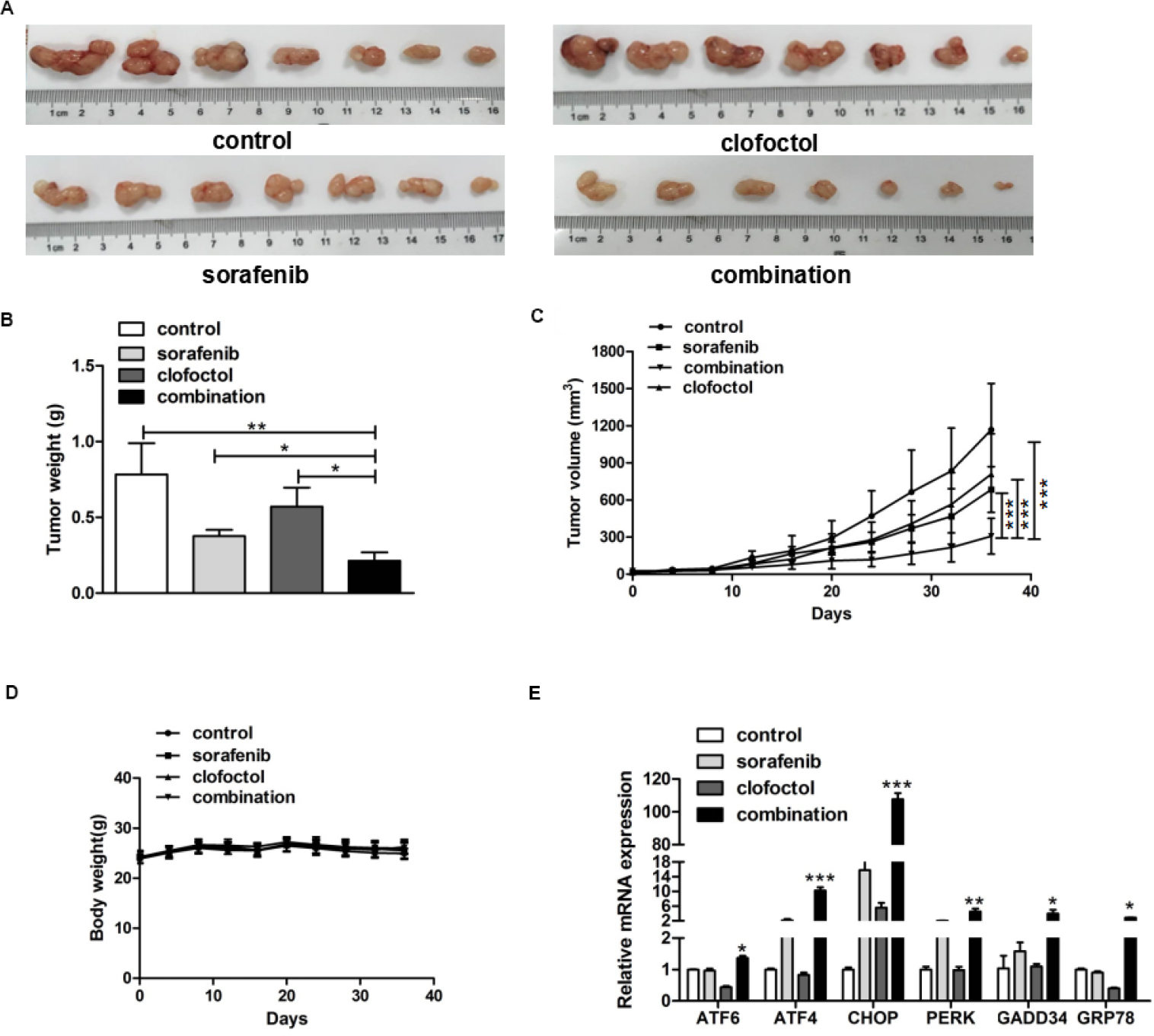 | Figure 5 Clofoctol plus sorafenib enhanced inhibition of PC-3 xenograft tumor growth. Notes: (A) PC-3 xenograft tumors were smaller in the clofoctol and sorafenib treatment groups than vehicle control, and the inhibition was more remarkable in the clofoctol–sorafenib co-treatment group. (B and C) Tumor weight and tumor volume were reduced significantly by the combination therapy. Graph bars represent mean±SD from three independent experiments. *P<0.05, **P<0.01 and ***P<0.001 indicate the statistical significance by using the unpaired t-test t-test. (D) There was no significant difference in mouse weight between groups. (E) Extracted RNA from the isolated mice tumors was used to measure mRNA levels of genes related to ER stress. qPCR data are represented as mean±SD. *P<0.05, **P<0.05 and ***P<0.001 denote a significant difference by using the unpaired t-test t-test between combination treatment group and control group. Abbreviations: ER, endoplasmic reticulum; qPCR, quantitative real-time PCR. |
Discussion
With the increase in morbidity and mortality of prostate cancer and the occurrence of adverse reactions, combination therapy has attracted attention due to its advantages such as enhanced anticancer effect, less medication and reduced adverse reactions.28 Given that monotherapy has more adverse effects, we tested the combination of clofoctol and PC-3 cells for 40 compounds in prostate cancer patients according to the Chou–Talalay formula and dose–response method. The use of Chou–Talalay analysis determines the presence of synergy, which requires parallel production of individual drug dose–response curves and combined proliferation data.29 The Chou–Talalay method uses the median effect equation to determine if the combination of the two drugs produces more effects than simply adding individual drug effects.30
This study investigated whether ER stress and the UPR pathway are co-activated by clofoctol and sorafenib in vitro and in vivo in PC-3 cells. We found consistent evidence that clofoctol and sorafenib induce cell death via synergistic induction of ER stress by activating UPR pathways, which was characterized by increasing protein and mRNA expression of UPR pathways markers, eg, CHOP, GADD34, ATF6 and ATF4. Studies show that ATF4 promotes many adaptive responses, restores ER function and maintains cell survival.31 ATF4 can also promote apoptosis by regulating CHOP.32 In cells with irreversible damage, ATF4 also participates in the cell death pathway by inducing the transcription factor CHOP, a member of the ROS and the apoptosis regulator BCL-2 family.33 CHOP is a transcription factor induced by cellular stress and has been thought to play a role in signal transduction from ER stress to apoptosis.34 It has been shown that accumulation of CHOP enhances apoptosis in response to ER stress in various systems.35,36 GADD34 expression is induced by ER stress because its coding gene is activated by CHOP.37 In the present study, treatment with clofoctol and sorafenib resulted in a rapid increase in CHOP and GADD34 accumulation. These events represent prototypical markers of the ER stress-dependent UPR signaling pathway. The results showed that treatment with clofoctol and sorafenib increased the ATF4 mRNA expression, implying that PERK pathway is one of mechanisms of clofoctol- and sorafenib-induced ER stress.
In addition to PERK, IRE1 and ATF6 represent two other ER transmembrane proteins that serve as post-sensors of ER stress and mediate UPR.38 In response to the accumulation of unfolded protein in the ER, IRE1 undergoes activation and initiates XBP1 mRNA splicing, resulting in translation of the transcriptionally active form of XBP1. XBP1 is a transcription factor that regulates expression of important genes in ER stress response.39 When UPR is activated, IRE1 oligomerizes and activates its ribonuclease domain, which catalyzes the splicing of the expressed form of XBP1 mRNA (XBP1u). The unspliced form of XBP1 mRNA contains a Pst I digestion site that is removed upon splicing. This process will generate a spliced isoform of XBP1 mRNA (XBP1s) as a marker for activation of IRE1.40 ATF6 is transported from the ER to the Golgi apparatus, where it is cleaved into a cytoplasmic fraction and migrates to the nucleus to activate transcription of the UPR.41 In the current study, we found that the two compounds combined to induce PC-3 cell death through the synergistic induction of ER stress. An increase in ATF6 mRNA expression indicates that clofoctol and sorafenib activate the ATF6 pathway. In the XBP1 splicing assay, we observed a decrease in the Pst I digestion product of XBP1 mRNA after treatment with sorafenib combination with clofoctol for 24 hours. At the same time, we confirmed the results of synergistically induced ER stress by the combination of sorafenib and clofoctol by qPCR and Western blot.
In addition, clofoctol induces cytoplasmic vacuolization, although less pronounced vacuolization is sometimes observed in cells treated with sorafenib alone. Cells treated with clofoctol and sorafenib showed a significantly greater vacuolization than either treated cells. As the concentration of clofoctol increases, vacuolation becomes more pronounced than sorafenib. Thus, the underlying mechanisms of induction of ER stress between sorafenib and clofoctol are different, but they have a synergistic effect in ER stress. We further combined clofoctol and sorafenib in a PC-3 xenograft model and found that the combination therapy was more effective than the control in inhibiting the growth of xenograft tumors than either clofoctol alone or sorafenib alone. Then, qPCR was performed on RNA extracted from these four groups of tumors. We monitored the three UPR pathways: PERK pathway, IRE1 pathway and ATF6 pathway. We also measured the glucose-regulated protein GRP78/Bip mRNA expression, which is a marker of ER stress.42 The chaperone proteins, such as GRP78/BiP, GRP94, are major regulators of all three pathways.43 The results further validated that clofoctol and sorafenib could lead to synergistic PC-3 cell death via induction of ER stress by activating UPR pathways.
Conclusion
Our data confirms the synergy between clofoctol and sorafenib by their effect on the ER-stress pathway to enhance antiproliferation and pro-apoptotic events of prostate cancer cells in vivo and in vitro. Our results suggest that the combination of clofoctol and sorafenib may be a viable strategy for the treatment of prostate cancer.
Acknowledgment
This study was supported by a grant from the National Natural Science Foundation of China (grant number 81572923, 81071957, 81872417), the Jiangsu Province Postdoctoral Research Funding (grant number 7131708615), the Suzhou City Science and Technology Program (grant number SYS201419) and the Priority Academic Program Development of Jiangsu Higher Education Institutions of China (PAPD).
Disclosure
The authors report no conflicts of interest in this work.
References
Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66(4):271–289. | ||
Falchook AD, Chen RC. Adjuvant vs.salvage radiotherapy for patients at high risk for recurrence after radical prostatectomy. Urol Oncol. 2015;33(11):451–455. | ||
Chung BH. The role of radical prostatectomy in high-risk prostate cancer. Prostate Int. 2013;1(3):95–101. | ||
Borges AH, Dubrow R, Silverberg MJ. Factors contributing to risk for cancer among HIV-infected individuals, and evidence that earlier combination antiretroviral therapy will alter this risk. Curr Opin HIV AIDS. 2014;9(1):34–40. | ||
de Witt P, Maartens DJ, Uldrick TS, Sissolak G. Treatment outcomes in AIDS-related diffuse large B-cell lymphoma in the setting roll out of combination antiretroviral therapy in South Africa. J Acquir Immune Defic Syndr. 2013;64(1):66–73. | ||
Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–446. | ||
Zhou Y, Yang J, Zhang R, Kopeček J. Combination therapy of prostate cancer with HPMA copolymer conjugates containing PI3K/mTOR inhibitor and docetaxel. Eur J Pharm Biopharm. 2015;89:107–115. | ||
Wang M, Shim JS, Li RJ, Rj L, et al. Identification of an old antibiotic clofoctol as a novel activator of unfolded protein response pathways and an inhibitor of prostate cancer. Br J Pharmacol. 2014; 171(19):4478–4489. | ||
Liu L, Cao Y, Chen C, et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006; 66(24):11851–11858. | ||
Delaunay-Moisan A, Appenzeller-Herzog C. The antioxidant machinery of the endoplasmic reticulum: Protection and signaling. Free Radic Biol Med. 2015;83:341–351. | ||
Vashistha N, Neal SE, Singh A, Carroll SM, Hampton RY. Direct and essential function for Hrd3 in ER-associated degradation. Proc Natl Acad Sci U S A. 2016;113(21):5934–5939. | ||
Bernasconi R, Galli C, Kokame K, Molinari M. Autoadaptive ER-associated degradation defines a preemptive unfolded protein response pathway. Mol Cell. 2013;52(6):783–793. | ||
Rashid HO, Yadav RK, Kim HR, Chae HJ. ER stress: Autophagy induction, inhibition and selection. Autophagy. 2015;11(11):1956–1977. | ||
Gupta A, Hossain MM, Read DE, et al. PERK regulated miR-424(322)-503 cluster fine-tunes activation of IRE1 and ATF6 during Unfolded Protein Response. Sci Rep. 2015;5:18304. | ||
Gupta A, Read DE, Gupta S. Assays for induction of the unfolded protein response and selective activation of the three major pathways. Methods Mol Biol. 2015;1292:19–38. | ||
Ye J, Rawson RB, Komuro R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6(6):1355–1364. | ||
Weatherbee JL, Kraus JL, Ross AH. ER stress in temozolomide-treated glioblastomas interferes with DNA repair and induces apoptosis. Oncotarget. 2016;7(28):43820–43834. | ||
Haywood J, Yammani RR. Free fatty acid palmitate activates unfolded protein response pathway and promotes apoptosis in meniscus cells. Osteoarthritis Cartilage. 2016;24(5):942–945. | ||
Bustany S, Cahu J, Guardiola P, Sola B. Cyclin D1 sensitizes myeloma cells to endoplasmic reticulum stress-mediated apoptosis by activating the unfolded protein response pathway. BMC Cancer. 2015; 15:262. | ||
Rahmani M, Davis EM, Crabtree TR, et al. The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol Cell Biol. 2007;27(15):5499–5513. | ||
Li N, Sun C, Zhou B, et al. Low concentration of quercetin antagonizes the cytotoxic effects of anti-neoplastic drugs in ovarian cancer. PLoS One. 2014;9(7):e100314. | ||
Foucquier J, Guedj M. Analysis of drug combinations: current methodological landscape. Pharmacol Res Perspect. 2015;3(3):e00149. | ||
Ortholan C, Durivault J, Hannoun-Levi JM, et al. Bevacizumab/docetaxel association is more efficient than docetaxel alone in reducing breast and prostate cancer cell growth: a new paradigm for understanding the therapeutic effect of combined treatment. Eur J Cancer. 2010;46(16):3022–3036. | ||
Wang C, Tao H, Cheng L, Liu Z. Near-infrared light induced in vivo photodynamic therapy of cancer based on upconversion nanoparticles. Biomaterials. 2011;32(26):6145–6154. | ||
Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol. 2012;14(2):117–123. | ||
Nalbandian A, Ghimbovschi S, Wang Z, et al. Global gene expression profiling in R155H knock-in murine model of VCP disease. Clin Transl Sci. 2015;8(1):8–16. | ||
Lin YF, Lee YF, Liang PH. Targeting β-tubulin:CCT-β complexes incurs Hsp90- and VCP-related protein degradation and induces ER stress-associated apoptosis by triggering capacitative Ca2+ entry, mitochondrial perturbation and caspase overactivation. Cell Death Dis. 2012;3:e434. | ||
Buys SS, Partridge E, Black A, et al. Effect of screening on ovarian cancer mortality: the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Randomized Controlled Trial. JAMA. 2011;305(22):2295–2303. | ||
Segovia-Mendoza M, Díaz L, González-González ME, et al. Calcitriol and its analogues enhance the antiproliferative activity of gefitinib in breast cancer cells. J Steroid Biochem Mol Biol. 2015;148:122–131. | ||
Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. | ||
Yamaguchi S, Ishihara H, Yamada T, et al. ATF4-mediated induction of 4E-BP1 contributes to pancreatic beta cell survival under endoplasmic reticulum stress. Cell Metab. 2008;7(3):269–276. | ||
Ma J, Yang YR, Chen W, et al. Fluoxetine synergizes with temozolomide to induce the CHOP-dependent endoplasmic reticulum stress-related apoptosis pathway in glioma cells. Oncol Rep. 2016;36(2):676–684. | ||
Carpio MA, Michaud M, Zhou W, et al. BCL-2 family member BOK promotes apoptosis in response to endoplasmic reticulum stress. Proc Natl Acad Sci U S A. 2015;112(23):7201–7206. | ||
Liu W, Zhu X, Wang Q, Wang L. Hyperglycemia induces endoplasmic reticulum stress-dependent CHOP expression in osteoblasts. Exp Ther Med. 2013;5(5):1289–1292. | ||
Song Q, Gou WL, Zhang R. FAM3A attenuates ER stress-induced mitochondrial dysfunction and apoptosis via CHOP-Wnt pathway. Neurochem Int. 2016;94:82–89. | ||
Gurlo T, Rivera JF, Butler AE, et al. CHOP Contributes to, But Is Not the Only Mediator of, IAPP Induced β-Cell Apoptosis. Mol Endocrinol. 2016;30(4):446–454. | ||
Dalton LE, Healey E, Irving J, Marciniak SJ. Phosphoproteins in stress-induced disease. Prog Mol Biol Transl Sci. 2012;106:189–221. | ||
Badiola N, Penas C, Miñano-Molina A, et al. Induction of ER stress in response to oxygen-glucose deprivation of cortical cultures involves the activation of the PERK and IRE-1 pathways and of caspase-12. Cell Death Dis. 2011;2:e149. | ||
Lee GH, Kim HK, Chae SW, et al. Bax inhibitor-1 regulates endoplasmic reticulum stress-associated reactive oxygen species and heme oxygenase-1 expression. J Biol Chem. 2007;282(30):21618–21628. | ||
Homma T, Fujii J. Heat stress promotes the down-regulation of IRE1α in cells: An atypical modulation of the UPR pathway. Exp Cell Res. 2016;349(1):128–138. | ||
Galindo I, Hernáez B, Muñoz-Moreno R, et al. The ATF6 branch of unfolded protein response and apoptosis are activated to promote African swine fever virus infection. Cell Death Dis. 2012;3:e341. | ||
Zhang H, Lv M, Jia J, et al. Expression of the 78 kD glucose-regulated protein is induced by endoplasmic reticulum stress in the development of hepatopulmonary syndrome. Gene. 2014;537(1):115–119. | ||
Fros JJ, Major LD, Scholte FEM, et al. Chikungunya virus non-structural protein 2-mediated host shut-off disables the unfolded protein response. J Gen Virol. 2015;96(Pt 3):580–589. |
Supplementary materials
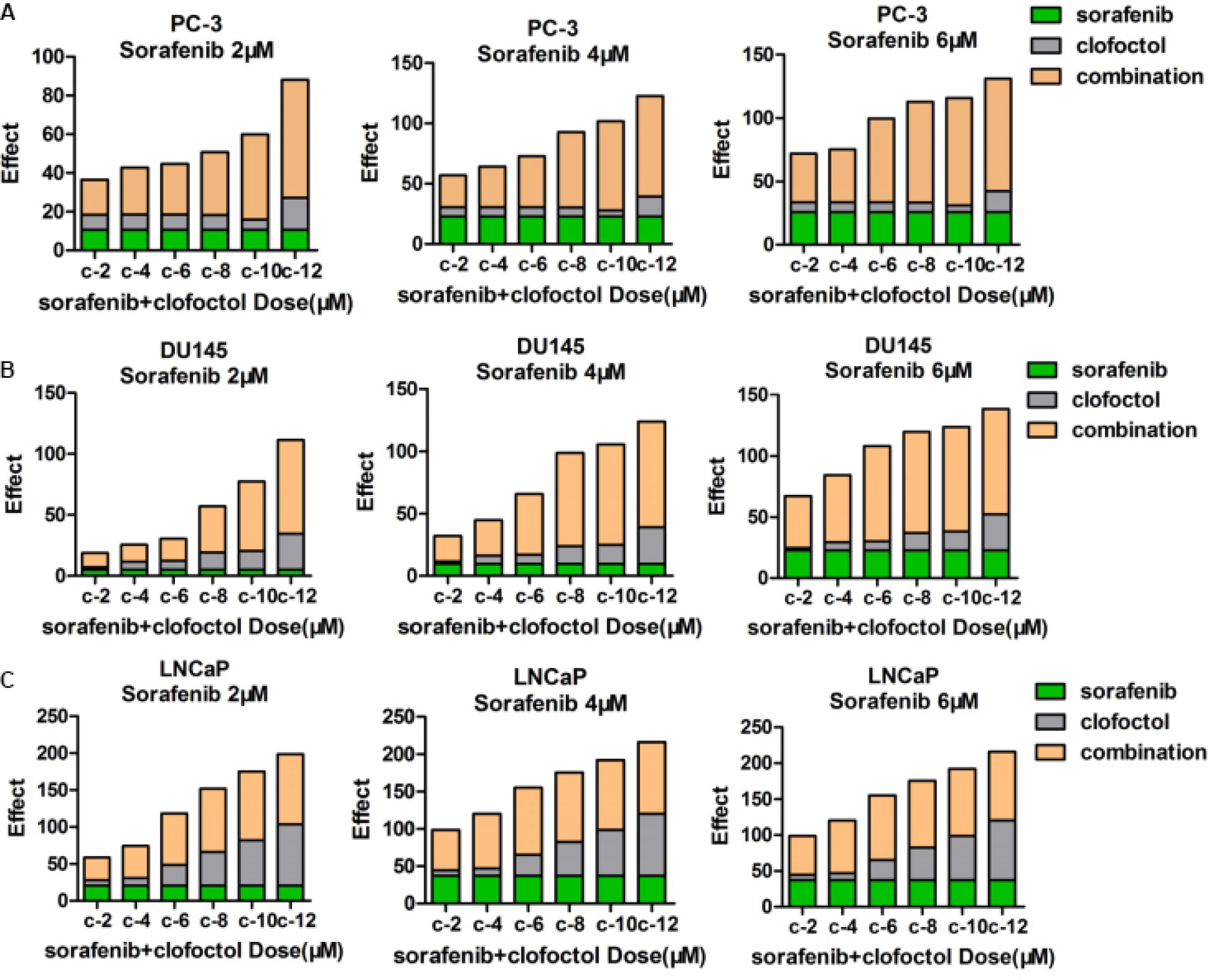 | Figure S1 Clofoctol and sorafenib synergistically inhibit PC-3, DU145 and LNCaP cells proliferation. Notes: (A–C) Alamar blue assays were performed 72 hours after treatment of PC-3, DU145 and LNCaP cells with a range of doses, 2–6 µM sorafenib and 2–12 µM clofoctol. It was verified by cell proliferation experiments whether the two drugs, clofoctol and sorafenib, synergistically inhibit prostate cancer. Repeat three times for each experiment, and Calculate the average of the plot. s and c are the acronyms for sorafenib and clofoctol, respectively. |
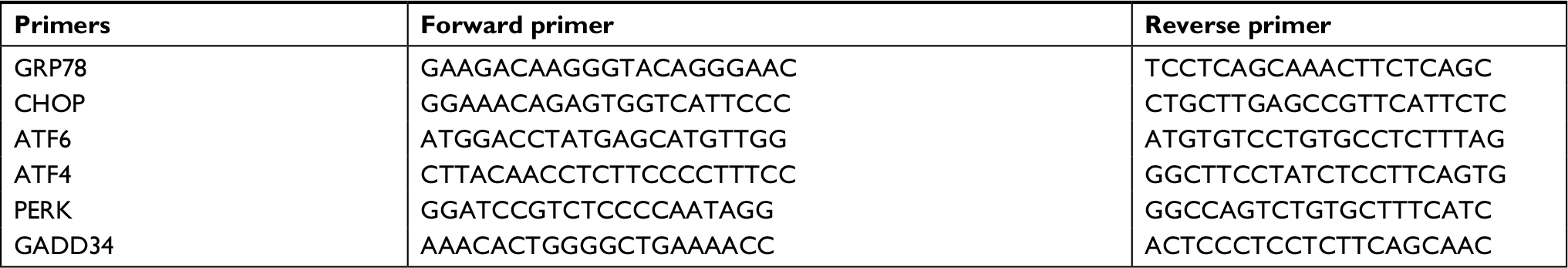 | Table S1 Primers of quantitative PCR |
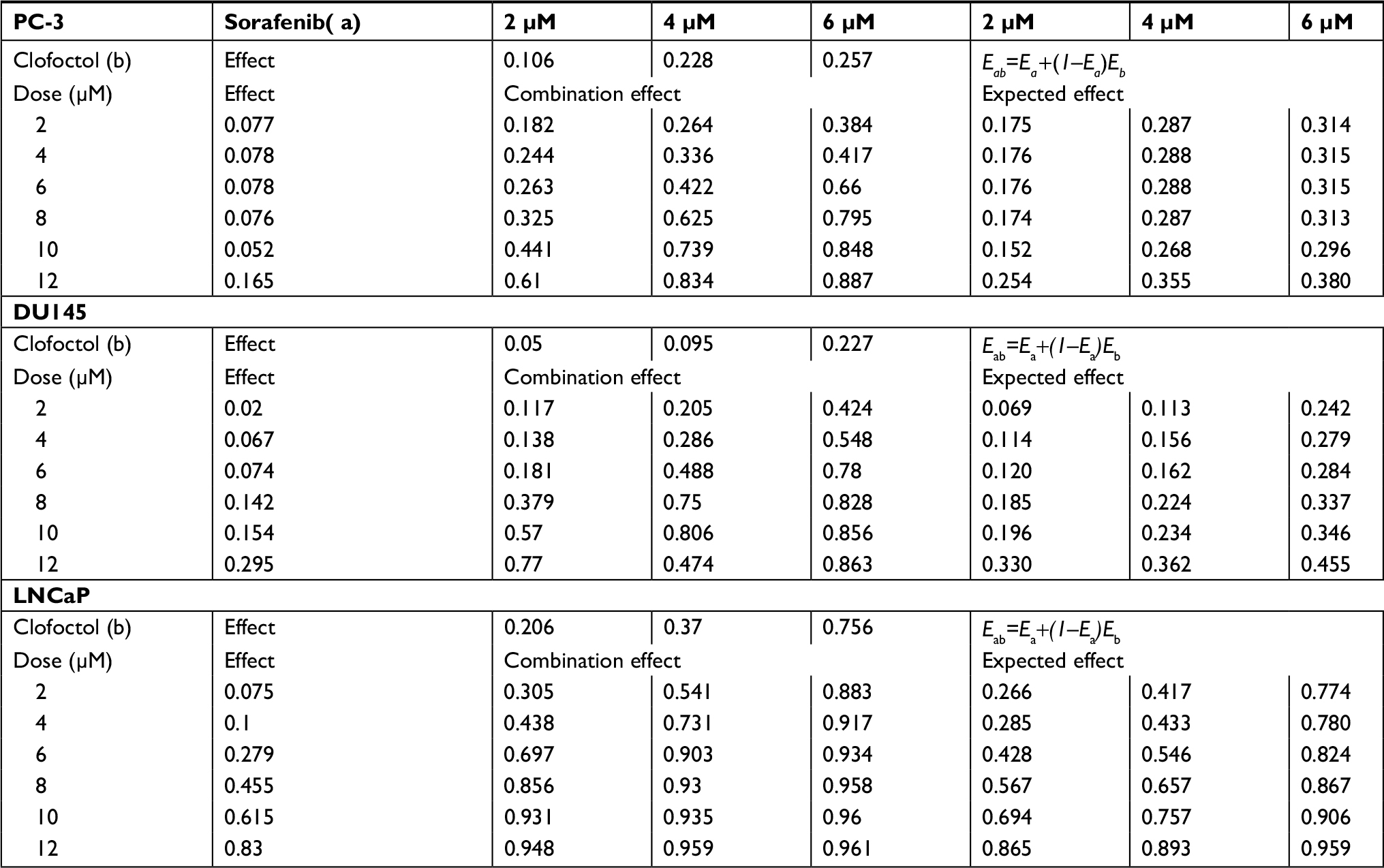 | Table S2 The combination effect and the expected additive effect |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.