")
Back to Journals » Cancer Management and Research » Volume 12
Clinicopathological Characteristics and Mutational Profiling of Adult T-Cell Lymphoblastic Lymphoma in a Chinese Population
Authors Chen F, Pang D, Guo H, Jiang X, Liu S, Huang L, Wei X, Liang Z, Wang X, Li W
Received 19 December 2019
Accepted for publication 9 April 2020
Published 30 April 2020 Volume 2020:12 Pages 3003—3012
DOI https://doi.org/10.2147/CMAR.S242903
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Kenan Onel
Feili Chen,1 Diwen Pang,1 Hanguo Guo,1 Xinmiao Jiang,1 Sichu Liu,1 Ling Huang,1 Xiaojuan Wei,1 Zhanli Liang,1 Xiaoxia Wang,2 Wenyu Li1
1Lymphoma Division, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, School of Medicine, South China University of Technology Guangzhou, Guangdong, People’s Republic of China; 2Nanjing Geneseeq Technology Inc, Nanjing, Jiangsu, People’s Republic of China
Correspondence: Wenyu Li
Lymphoma Division, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, School of Medicine, South China University of Technology, No. 123, West of Huifu Road, Guangzhou, Guangdong, People’s Republic of China
Tel/Fax +86-20-81884713-80631
Email [email protected]
Purpose: The purpose of this study is to perform a retrospective analysis of disease outcomes and mutational profiles in patients with adult T-cell lymphoblastic lymphoma (T-LBL).
Patients and Methods: A total of 43 patients were treated over a 9-year period at a single institution. The study examined treatment outcomes, clinical characteristics, and the use of circulating tumor DNA (ctDNA) and mutational profiling for patient diagnosis.
Results: The estimated overall survival (OS) and progression-free survival (PFS) time for all patients was 37.0 (95% CI: 17.7– 56.2) and 28.1 (95% CI: 0.9– 55.4) months, respectively. Chidamide maintenance was used in five patients exhibiting unfavorable genetic alterations, with no evidence of relapse. Next-generation sequencing of pretreatment tumor tissue was undertaken for 15 patients. NOTCH1 mutations were the most frequent genetic alterations, followed by mutations in PHF6, TP53, JAK1, JAK3, PTEN, and DNM2. The genetic profile of the blood was similar to that of the tumor. Kappa coefficient analysis (14 patients, 56 time points, kappa = 1.0, p = 0.00) indicated a 92.6% agreement between ctDNA response and tumor volume measurements at post treatment when compared with baseline. Detection of ctDNA predicted disease relapse in two patients.
Conclusion: The prognosis of patients with adult T-LBL remains very poor. Detection of tumor-associated sequences in ctDNA may be an effective method for diagnosing T-LBL and measuring treatment efficacy. Incorporation of new drugs such as histone deacetylase inhibitors (HDACi)has the potential to improve outcomes in these patients.
Keywords: lymphoblastic lymphoma, gene mutations, HDACi, circulating tumor DNA
Introduction
T-cell lymphoblastic lymphoma (T-LBL) is a rare and aggressive form of non-Hodgkin’s lymphoma, typically presenting as a bulky mass in the anterior mediastinum.1 It primarily affects children and young adults, with males disproportionately affected relative to females. Pleural effusion is among the most common symptoms, along with bone marrow infiltration and a high risk of central nervous system involvement.2
At present, management of patients with T-LBL remains difficult owing to the aggressiveness of the disease. The prognosis of patients treated using conventional non-Hodgkin’s lymphoma protocols remains unsatisfactory. Although the use of acute lymphoblastic leukemia (ALL) protocols improves long-term survival, approximately half of all patients experience relapse or refractory disease even with intensive chemotherapy.3 Thus, it is of great importance to identify high-risk patients at the time of diagnosis. The GRAALL/LYSA study developed an oncogenetic prognostic model (favourable: NOTCH1/FBXW7 mutation and/or no RAS/PTEN mutation/deletion) with an independent prognostic value for survival, indicative of the importance of mutation detection at the time of diagnosis.4 Monitoring of minimal residual disease (MRD) is highly predictive of treatment outcome in adult ALL.5,6 At present, the use of circulating tumor DNA (ctDNA) as a liquid biopsy has emerged as a feasible, non-invasive tool for disease monitoring in malignant lymphomas,7,8 while next-generation sequencing (NGS) technology is a promising approach for mutation profiling of ctDNA owing to its high-throughput capacity, as well as its sensitivity and specificity;8–10 however, the utility of using ctDNA as a biomarker for T-LBL diagnosis has not been investigated.
Herein, we report our experience treating 43 T-LBL patients in a tertiary hospital over a 9-year period, from 2009 to 2018. We examined treatment outcomes, clinical characteristics, and the use of ctDNA and mutational profiling for patient diagnosis.
Patients and Methods
Patient Selection
From July 2009 to April 2018, 43 eligible patients from Guangdong Provincial People’s Hospital, China, were identified for inclusion in this study. The inclusion criteria were as follows: (a) patients with pathologically confirmed T-LBL; (b) patients who were ≥16 years of age; (c) patients who were newly diagnosed; (d) patients who underwent radiological tests for clinical staging; and (e) patients receiving at least one cycle of chemotherapy.
The present study was approved by the Clinical and Research Ethics Committee of Guangdong Provincial People’s Hospital, China. All procedures that involved human participants were performed in accordance with the Declaration of Helsinki. All patients provided written informed consent to participate in this study.
Treatment Protocol
Most patients (31/43, 72.1%) were treated using the modified BFM90 regimen (Supplementary Table S1), and five of these patients also received chidamide (10 mg, twice every week) during the maintenance phase and for the first 6 months after completion of chemotherapy. Twenty-one patients completed all the cycles of the BFM-90 regimen. The other 10 patients did not complete the regimen due to disease progression. Four patients received 6 cycles of the CHOPE regimen (cyclophosphamide [750 mg/m2, i.v., qd, day 1], epirubicin [75 mg/m2, i.v., qd, day 1], vincristine [2 mg, i.v., qd, day 1], prednisone [90 mg, p.o., qd, days 1–5], and etoposide [100 mg/m2, i.v., days 1–3]) and no maintenance. Eight patients received 6 cycles of the hyper CVAD/MA regimen (hyperfractionated cyclophosphamide [300 mg/m2, i.v., q12h, d1–3], vincristine [2 mg, i.v., days 4 and 11], doxorubicin [50 mg/m2, i.v., day 4], and dexamethasone [40 mg, i.v., d1–4, days 11–14], alternating with high doses of methotrexate [1000 mg/m2, i.v., day 1] and cytarabine [3000 mg/m2, i.v., q12h, days 2–3]). Intrathecal chemotherapy with cytarabine (30 mg) and methotrexate (15 mg) was given on day 1 of every cycle. Two patients with hyper CVAD/MA received allogeneic stem cell transplantation, and one was treated using autologous stem cell transplantation.
Evaluation and Follow-Up
All patients underwent enhanced computed tomography (CT), or positron emission tomography (PET)/CT for evaluation. Bone marrow biopsy was performed for patients with bone marrow involvement at baseline. The treatment response was evaluated according to the revised efficacy evaluation criteria set forth by the malignant lymphoma International Working Group (IWG).11
Following completion of therapy, patients were followed up every 3 months for the first 2 years, every 6 months from year 3 to year 5, and once every year thereafter. The examined parameters included blood cell count and serum lactate dehydrogenase (LDH) levels.
Sample Collection and Processing
Tumor biopsy samples (n = 15) were obtained from formalin-fixed paraffin-embedded (FFPE) tissues. Peripheral blood samples (n = 14) were collected over the course of the disease, stored at −80 °C, and then shipped to the central testing laboratory (Nanjing Geneseq Technology Inc., Nanjing, China). Sequencing of tumor and plasma samples was performed using a panel specifically targeting 102 lymphoma-associated genes (Supplementary Table S2), as previously reported.8
Statistical Analysis
SPSS 21.0 statistical software (IBM, Chicago, IL, USA) was used for data analysis. The Kaplan–Meier method was used to calculate all survival end points, which were compared by the Log rank test. Cox regression analysis was used for multivariate analyses. In all the figures, error bars represent standard error of the mean. Paired samples were analyzed by a Wilcoxon signed-rank test, while unpaired values were analyzed by a Mann–Whitney test to obtain p-values. A Kappa coefficient test was used to assess for agreement between plasma ctDNA and enhanced CT evaluation of tumor response to therapy. Two-sided p-values <0.05 were considered significant. Overall survival (OS) time was calculated from the date of diagnosis to the time of death from any cause. Progression-free survival (PFS) time was calculated from the start of treatment to the time of disease progression or death due to T-LBL.
Results
Patient Demographics and Clinical Characteristics
A total of 43 adult patients with T-LBL were identified over a 9-year period in Guangdong Provincial People’s Hospital, China. Baseline characteristics are shown in Table 1. The median age of the patients was 24 years (range, 16–68 years). Most patients were male (31/43, 72.1%) and presented evidence of a bulky mass in the mediastinum (defined as ≥7 cm, 26/43, 60.5%). The Eastern Cooperative Oncology Group12 performance status of patients was ≥2 in 60.5% of patients at the time of diagnosis. Most of the patients (88.4%) were classified as Ann Arbor stage I–II. The median LDH level was 233 U/L (range, 78–6560 U/litter, upper limit of normal range: 245 U/litter). Twelve (27.9%) patients had B symptoms. More than half of the patients (24/43, 55.8%) had bone marrow involvement, 7 (16.3%) had central nervous system involvement at the time of diagnosis or progression, and 24 (55.8%) had pleural effusion.
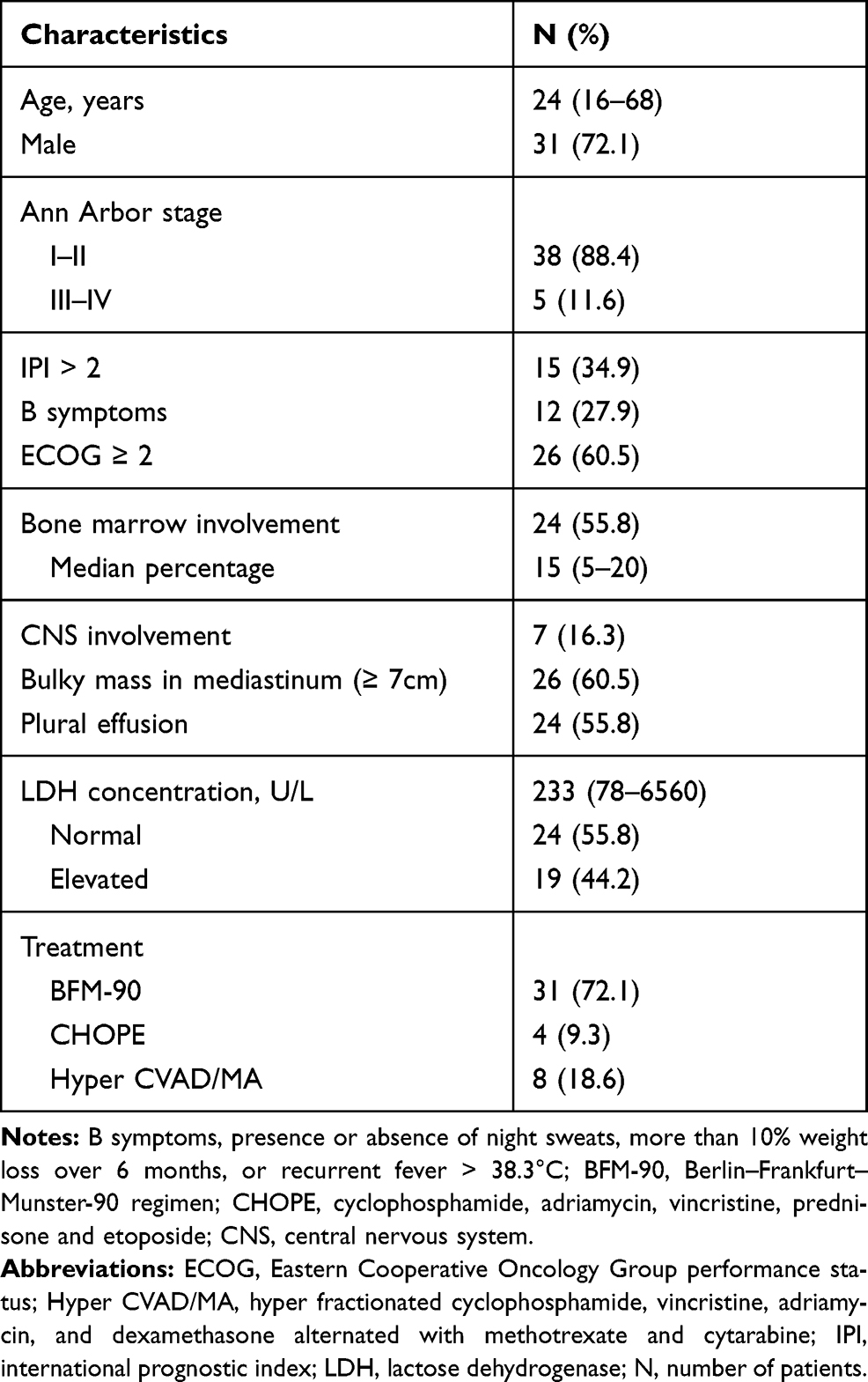 |
Table 1 Patient Characteristics |
Response to Therapy and Patient Survival
The median follow-up time for eligible living patients was 29.0 months (range: 0.9–122.0 months). Twenty patients were alive at the time of the last follow up. One patient achieved complete remission after receiving a modified BFM-90 regimen but died of myeloid leukemia 3 years after completion of chemotherapy. Most of the patients died as a result of disease progression. The estimated OS and PFS time for all the patients were 37.0 (95% CI: 17.7–56.2) and 28.1 (95% CI: 0.9–55.4) months, respectively (Figure 1). The estimated 5-year OS and PFS rates for all the patients were 42.9% (95% CI: 42.0–43.8%) and 40.5% (95% CI: 39.7–41.4%), respectively (Figure 1A and B).
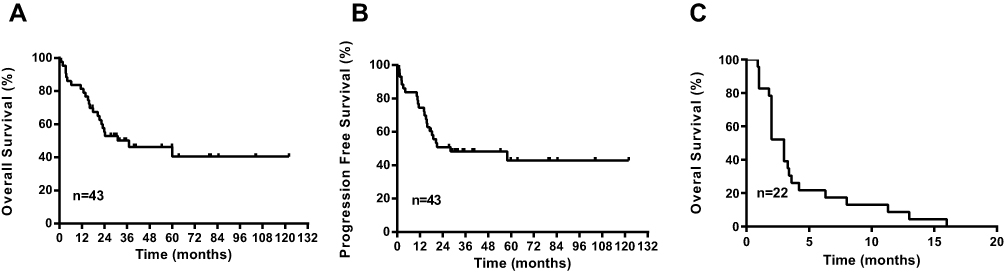 |
Figure 1 Kaplan–Meier curves for progression-free and overall survival. (A) Overall survival of the whole study population. (B) Progression-free survival of the whole study population. (C) Overall survival of patients with relapsed/refractory disease. |
More than half of the patients (22/43, 51.2%) had relapsed or refractory disease. Seven of these patients exhibited no response to initial treatment, while the remaining 15 relapsed after complete remission. The median relapse time was 16.5 months, with most patients relapsing within 18 months after diagnosis. For patients receiving the modified BFM-90 regimen, relapse usually occurred during the maintenance phase, with none of the relapsed or refractory patients surviving (Figure 1C).
Next, we examined the relationship between clinical characteristics and survival. Younger age and the absence of central nervous system lymphoma were both associated with longer survival time (Supplementary Table S3). Multivariate analysis was performed with age, central nervous system involvement, and LDH level as cofactors. Elevated LDH level (PFS: hazard ratio [HR] 4.8, 95% CI 1.6–14.9, p = 0.01; OS: HR 3.2, 95% CI 1.1–9.1, p = 0.03) was independent prognostic factors for OS and PFS time (Supplementary Table S4).
In the GRAAL-LYSA LL03 trial, a NOTCH1/FBXW7/RAS/PTEN oncogene classifier was identified as an independent prognostic factor for adult T-LBL.3 To improve clinical outcomes, patients harboring NOTCH1 and RAS/PTEN mutations received oral chidamide (10 mg, twice every week) in addition to routine maintenance treatment during the maintenance phase and for the first 6 months after completion of chemotherapy. In total, five patients received chidamide. At the last follow up, none of these patients had relapsed (Supplementary Table S5).
Noninvasive Mutational Profiling of Tumor Genetic Heterogeneity in T-LBL
To evaluate the technical performance of the targeted sequencing approach and the clinical utility of ctDNA for identifying T-LBL tumor genotypes, we analyzed 15 diagnostic T-LBL tumor biopsies and 82 longitudinal plasma samples, 14 of which were obtained before chemotherapy (Supplementary Table S6). Twelve patients had paired tumor and plasma samples. All the patients received the modified BFM-90 regimen. We identified somatic alterations in 100% of tumors, with a total of 92 mutations (Figure 2A and B). When applied to the prechemotherapy plasma, our assay detected ctDNA in 100% of the patients (Figure 2A). In addition, 89.1% of tumor-confirmed mutations could be noninvasively genotyped directly from prechemotherapy plasma (Figure 2A and B). At least one tumor-confirmed variant was identified by noninvasive genotyping in 100% of prechemotherapy plasma samples (Figure 2A). However, the mutant allele frequencies were significantly lower in plasma ctDNA than in tumor biopsies (Figure 2C). Plasma and tumor tissue were obtained simultaneously in 3 patients. For the other 9 patients, biopsy was performed before the collection of blood for plasma ctDNA sequencing (median: 4 days; range: 3–10 days). They also received steroids for symptom relief, which may have affected the release of ctDNA into the blood.
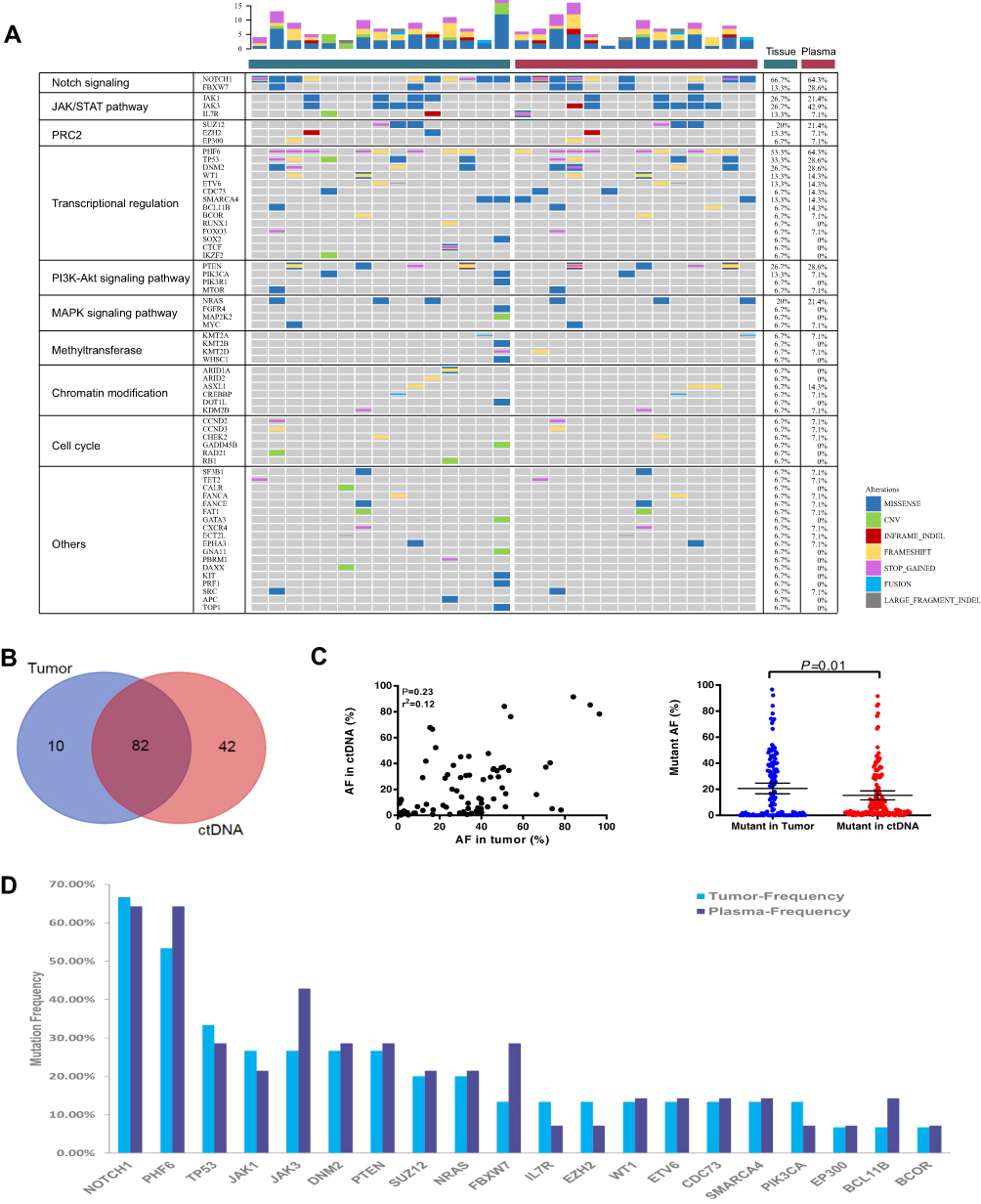 |
Figure 2 Non-invasive genotyping of T-LBL is feasible. (A) Case-level mutational profiles of 15 tumor and 14 plasma samples from patients with T-LBL genotyped by our sequencing panel. Each column represents one sample, each row represents one gene. (B) A total of 92 and 124 mutations were detected in tumor and plasma samples respectively. Eighty-two identified mutations were shared between the archival tumor tissue (blue) and plasma (red) ctDNA. (C) Correlation assessment of mutant allele frequencies between SNVs of matching tumor/pre-chemotherapy plasma pairs, shown for all tumor/pre-chemotherapy plasma pairs analyzed in this work (n=12). Mutant allele frequencies were significantly higher in tumor samples. (D) The 20 most recurrently mutated genes in our cohort are depicted. |
The results of the mutational profiling are shown in Figure 2A and D. The mutational profiles of T-LBL were similar to those of T-ALL. Oncogenic NOTCH signaling is activated in more than 65% of T-ALL patients and increasing evidence has indicated that it is a major regulator of leukemia cell growth and metabolism.13 In our cohort, mutations in NOTCH1 were the most frequently identified genetic alterations in tumor tissues (10/15, 66.7%), followed by mutations in PHF6 (8/15, 53.3%), TP53 (5/15, 33.3%), JAK1 (4/15, 26.7%), JAK3 (4/15, 26.7%), PTEN (4/15, 26.7%), and DNM2 (4/15, 26.7%) (Figure 2D). Additional mutations were also observed in several histone modification-associated genes such as SUZ12 (3/15, 20%), EZH2 (2/15, 13.3%), and EP300 (1/15, 6.7%).
Prognostic Value of ctDNA in T-LBL
We next evaluated whether ctDNA analysis could facilitate early identification of clinically relevant risk groups in T-LBL by comparing the total ctDNA burden at diagnosis with clinical indices (Figure 3). We did not observe a correlation between the amount of ctDNA and serum LDH levels or tumor volume (Figure 3A and B), which may have been due to the small sample size. However, we found that ctDNA concentrations at initial diagnosis were significantly correlated with the International Prognostic Index (IPI) (p = 0.01, Figure 3C). Patients with elevated LDH levels also had higher ctDNA concentrations (p = 0.01, Figure 3D). Notably, whereas 100% of prechemotherapy samples had detectable ctDNA, only 50% of samples had abnormally high concentrations of LDH, demonstrating the superior sensitivity of ctDNA.
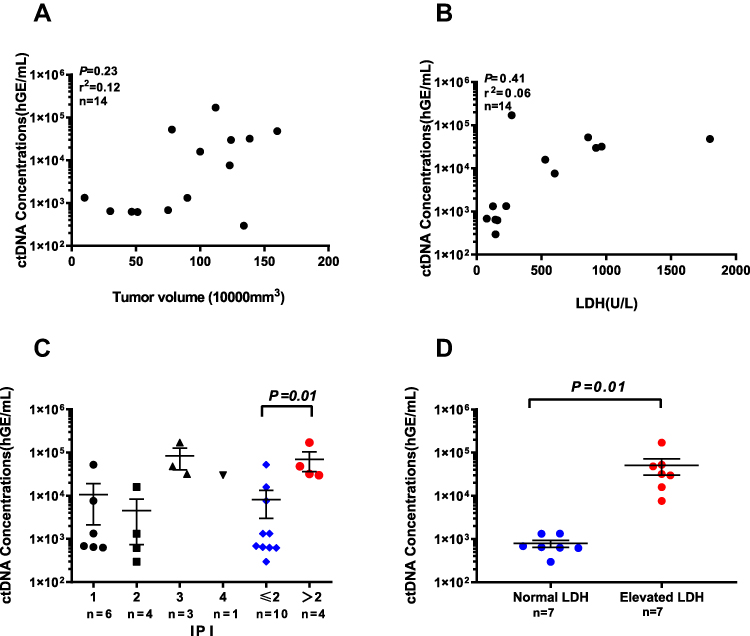 |
Figure 3 Quantification of ctDNA in relation to T-LBL clinic indices. (A) Correlation between tumor volume measured from enhanced CT imaging and ctDNA concentrations from pre-chemotherapy plasma. (B) Relationship between LDH and ctDNA concentration from pre-chemotherapy plasma time points. Pre-chemotherapy LDH and MTV values in (A) and (B) were obtained as close in time as possible to blood draws used for plasma ctDNA sequencing (median, 3 days for LDH and 4 days for tumor volume). r, Pearson correlation coefficient. (C) Association between ctDNA concentration at diagnosis and IPI. Statistical comparison between low IPI (1+2) and high IPI (3+4) patients was performed using Mann–Whitney U-test. Means and SEMs are indicated. (D) Comparison of ctDNA concentration at diagnosis between patients with LDH above or below the upper limit of normal (ULN; 245 U/liter) was performed using Mann–Whitney U-test. Means and SEMs are indicated. |
Treatment Response Assessment Using ctDNA Quantification
To assess if ctDNA can be employed as a surrogate biomarker of treatment response, we analyzed the longitudinal plasma samples of 14 patients. Patients received enhanced CT at baseline, preconsolidation therapy, prereinduction therapy, premaintenance therapy and 1 month after maintenance. They also received enhanced CT when disease progressed. Blood collection times for plasma ctDNA sequencing were as close to each other as possible (median: 3 days). Overall, we found that ctDNA levels were decreased following chemotherapy, similar to the findings for tumor volume measurements, where a significant decrease in tumor burden was also observed after chemotherapy (Figure 4A). We report an agreement of 92.6% between ctDNA response and tumor volume measurements at post treatment when compared with baseline using Kappa coefficient analysis (14 patients, 56 time points, kappa = 1.0, p = 0.00). These findings demonstrate that the clinical utility of using plasma ctDNA levels for monitoring response is comparable to that of enhanced CT evaluation. Patient 4 developed a residual mass in the mediastinal with ctDNA clearance in plasma after therapy. However, the patient refused a PET/CT scan and biopsy. He was alive until the last follow up but has ended all therapy for more than 6 months.
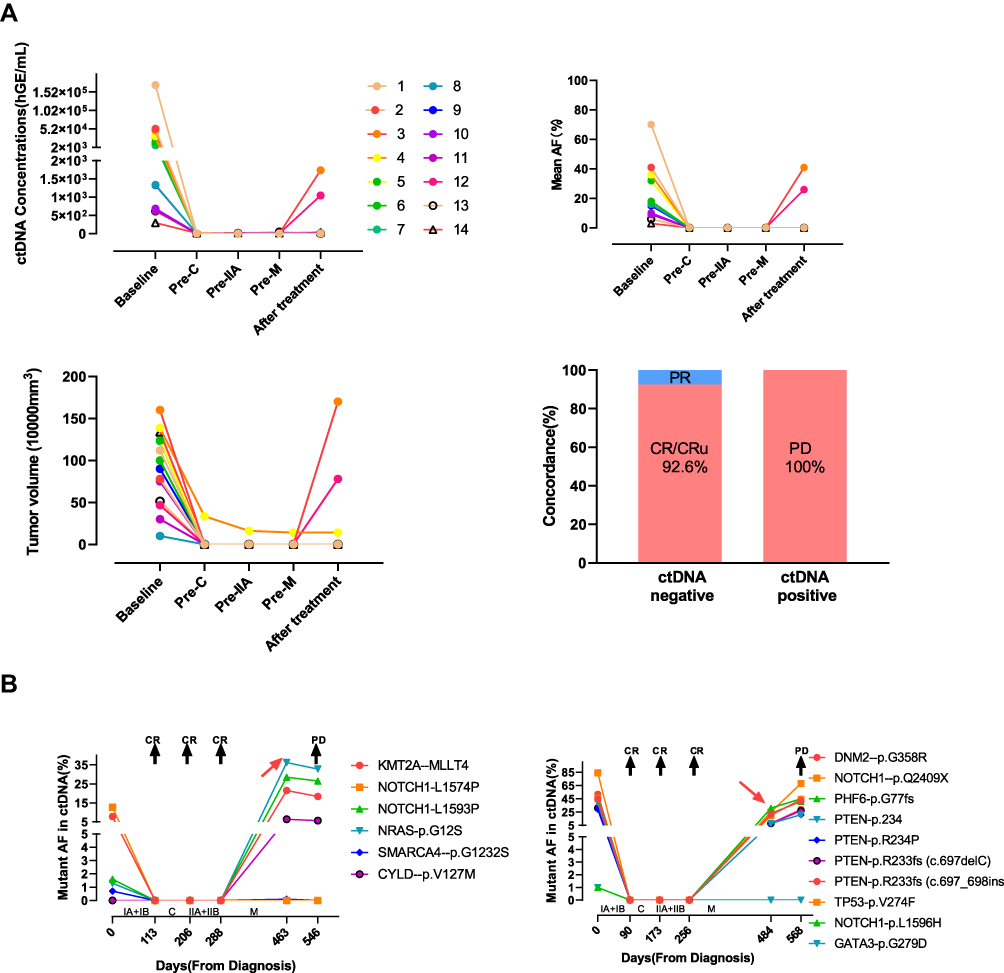 |
Figure 4 Temporal analysis of plasma ctDNA agrees with response to therapy. (A) Serial plasma ctDNA analysis was done throughout course of treatment in patients receiving modified BFM-90 regimen. Significant decrease in plasma ctDNA and tumor volumes were observed in response to therapy. These findings were comparable to changes in tumor volume measurements. (B) Two patients got disease progression during maintenance. They got complete remission with the clearance of plasma ctDNA after the re-induction phase. Plasma ctDNA arose during the maintenance phase. Then the patients complained of cough and pleural effusion. Progression was confirmed by enhanced CT. |
Patients 3 and 12 exhibited disease progression during the maintenance phase. Although ctDNA was identified at the time of clinical relapse (Figure 4B), it was also detectable as MRD before relapse in both patients. The elapsed time between the first ctDNA-positive time point and clinical relapse was 83 and 84 days, respectively. These results highlight the potential of using ctDNA profiling by targeted sequencing to improve MRD assessment and early relapse detection. However, only 2 patients in our cohort relapsed. The sample size was small, and a larger cohort is required to further evaluate this result.
Discussion
Here, we report the results for all the adult T-LBL patients treated in a tertiary hospital over a 9-year period, from 2009 to 2018. The clinical characteristics of the patients in our cohort were broadly consistent with previous descriptions. The prognosis of our patients remains unsatisfactory, with an average PFS time of 28.1 months (95% CI: 0.9–55.4; Figure 1). The estimated 5-year OS and PFS rates for all the patients were 42.9% (95% CI: 42.0–43.8%) and 40.5% (95% CI: 39.7–41.4%), respectively (Figure 1). These survival rates were slightly lower than those reported in other studies using ALL-like intensive chemotherapy regimens in similar patients, which have reported 5-year progression-free survival rates ranging from 62% to 77%.14–16 Moreover, none of the relapsed or refractory patients in our study survived.
In an attempt to improve survival, we focused on new drugs. We found that over 50% of the patients in our cohort had mutations that resulted in the activation of Notch-1 signaling. HDACi drugs have shown significant therapeutic potential against T-ALL via epigenetic targeting of Notch-1-driven transcription.17 Chidamide is a HDACi drug used for the treatment of relapsed or refractory peripheral T-cell lymphoma,18 and was also shown to be capable of killing lymphoid leukemia cells in a preclinical study.19 Thus, chidamide (10 mg, twice every week) was added to the maintenance chemotherapy for 5 patients with NOTCH1 and RAS/PTEN mutations. At the point of the last follow up, none of these patients had relapsed. However, the sample size in this study was small, and a larger prospective study is required to further clarify the effect of chidamide in T-LBL.
Recent advancements in genomic profiling have provided new opportunities for novel therapeutic interventions for patients with lymphoma.7,20,21 However, tumor tissue is not always available, and malignant lymphoma is a genetically heterogeneous, systemic disease, involving multiple organs at initial diagnosis. In contrast, NGS-based ctDNA testing can be easily deployed when tumor tissue is unavailable, while also providing insight into the heterogeneity of the tumor. In our study, a total of 134 genetic alterations were detected, consisting of 82 (61.2%) mutations that were shared between the archival tumor tissue and blood ctDNA. Moreover, more mutations were seen in ctDNA than in archival tumor tissue, highlighting the ability of ctDNA to reveal tumor heterogeneity. We also observed a correlation between the amount of ctDNA and clinical indices. Patients with high LDH and IPI had higher amounts of ctDNA. Thus, plasma ctDNA might complement traditional clinical indices and serve as an independent prognostic biomarker in the future.
Our results also indicated the unique strength of ctDNA detection for longitudinal assessment of tumor response to therapy, which is complementary to imaging in T-LBL. Despite a small sample size, we found a significant reduction in ctDNA concentrations following chemotherapy. Similar patterns in temporal changes in ctDNA concentrations and enhanced CT assessments of tumor response indicated that our analysis of plasma was highly translational. Moreover, detection of ctDNA might be more sensitive and specific than imaging. T-LBL patients usually present with a bulky mass in the anterior mediastinum; however, it can be difficult to discriminate between thymus hyperplasia and residual disease in imaging. In our study, one patient showing residual disease in enhanced CT and clearance of ctDNA also survived after completion of chemotherapy. We also observed that patients had detectable ctDNA levels before clinical progression.
Our study had some limitations. First, this was a retrospective study with a relatively small sample size. Moreover, patients were treated during a 9-year period, and methods for disease diagnosis and monitoring can change over time. Only 2 patients with progressive disease had NGS-based ctDNA analysis. Thus, it’s hard to clarify the correlation of mutation profile and ctDNA mutation with treatment outcome. It’s also hard to clarify the correlation of early clearance of ctDNA with survival time. Second, NGS-based ctDNA analysis was performed for 14 patients, whereas only a few received a PET/CT scan for disease surveillance. Consequently, it was difficult to evaluate the correlation between ctDNA detection and PET/CT in our study. Third, we did not test bone marrow in patients with bone marrow infiltration, or cerebrospinal fluid (CSF). Detecting ctDNA in CSF has been shown to be a more sensitive method for the detection of central nervous system metastasis than cytology.22 In the future, we will prospectively combine detection of ctDNA in CSF, bone marrow, and plasma for molecular characterization and surveillance of T-LBL.
Conclusion
In conclusion, we reported the clinicopathological characteristics of adult T-LBL in a Chinese population. The overall prognosis of these patients was poor. Incorporation of chidamide might improve outcomes in patients with unfavorable genetic mutations. We also observed that plasma ctDNA analysis is a feasible and promising method to use for the detection of mutational load. Detecting ctDNA in plasma may provide an additional method for molecular disease characterization and might also serve as a supplementary means for evaluating tumor response to T-LBL treatment. However, given the small sample size and retrospective nature of the study, more research will be necessary to assess the effects on disease outcomes.
Abbreviations
ALL, acute lymphoblastic leukemia; CI, confidence interval; CR, complete response; CT, computed tomography; ctDNA, circulating tumor DNA; ECOG, Eastern Cooperative Oncology Group; FFPE, formalin-fixed paraffin-embedded; HDACi, histone deacetylase inhibitor; IPI, International Prognostic Index; LDH, lactate dehydrogenase; MRD, minimal residual disease; ORR, overall response; OS, overall survival; PD, progressive disease; PET, positron emission tomography; PFS, progression-free survival; PR, partial remission; SD, stable disease; T-LBL, T-cell lymphoblastic lymphoma.
Ethics and Consent Statement
The present study was approved by the Clinical and Research Ethics Committee of Guangdong Provincial People’s Hospital, China. All procedures that involved human participants were performed in accordance with the Declaration of Helsinki. All patients provided written informed consent to participate in this study.
Author Contributions
Feili Chen, Diwen Pang and Hanguo Guo contributed equally to the work, Feili Chen contributed to the conception, design, the interpretation of the data and drafting the manuscript. Hanguo Guo and Diwen Pang contributed to the acquisition, analysis of data. Xinmiao Jiang, Ling Huang, Sichu Liu, Zhanli Liang and Xiaojuan Wei contributed to the conception, the acquisition of the data. Xiaoxia Wang contributed to the interpretation and analysis of the data. In addition, Wenyu Li contributed to the final manuscript approval for submission and publication. All in all, all authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This article was supported by the Guangdong Provincial Department of Science and Technology Fund Project (to W.L., grant number 2016A050502028).
Disclosure
Dr Xiaoxia Wang is affiliated with Nanjing Geneseeq Technology Inc. The authors report no other conflicts of interest in this work.
References
1. Portell CA, Sweetenham JW. Adult lymphoblastic lymphoma. Cancer J (Sudbury, Mass). 2012;18(5):432–438. doi:10.1097/PPO.0b013e31826b1232
2. Hoelzer D, Gokbuget N. Treatment of lymphoblastic lymphoma in adults. Best Pract Res Clin Haematol. 2002;15(4):713–728. doi:10.1053/beha.2002.0230
3. Lepretre S, Touzart A, Vermeulin T, et al. Pediatric-like acute lymphoblastic leukemia therapy in adults with lymphoblastic lymphoma: the GRAALL-LYSA LL03 study. J clin oncol. 2016;34(6):572–580. doi:10.1200/JCO.2015.61.5385
4. Huguet F, Leguay T, Raffoux E, et al. Pediatric-inspired therapy in adults with Philadelphia chromosome-negative acute lymphoblastic leukemia: the GRAALL-2003 study. J clin oncol. 2009;27(6):911–918. doi:10.1200/JCO.2008.18.6916
5. Bassan R, Spinelli O, Oldani E, et al. Improved risk classification for risk-specific therapy based on the molecular study of minimal residual disease (MRD) in adult acute lymphoblastic leukemia (ALL). Blood. 2009;113(18):4153–4162. doi:10.1182/blood-2008-11-185132
6. Bruggemann M, Schrauder A, Raff T, et al. Standardized MRD quantification in European ALL trials: proceedings of the Second International Symposium on MRD assessment in Kiel, Germany, 18–20 September 2008. Leukemia.;24(3):521–535. doi:10.1038/leu.2009.268
7. Scherer F, Kurtz DM, Newman AM, et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci Transl Med. 2016;8(364):364ra155. doi:10.1126/scitranslmed.aai8545
8. Sun P, Chen C, Xia Y, et al. Mutation profiling of malignant lymphoma by next-generation sequencing of circulating cell-free DNA. J Cancer. 2019;10(2):323–331. doi:10.7150/jca.27615
9. Dubois S, Viailly PJ, Mareschal S, et al. Next-generation sequencing in diffuse large B-cell lymphoma highlights molecular divergence and therapeutic opportunities: a LYSA study. Clin Cancer Res. 2016;22(12):2919–2928. doi:10.1158/1078-0432.CCR-15-2305
10. Dubois S, Jardin F. The role of next-generation sequencing in understanding the genomic basis of diffuse large B cell lymphoma and advancing targeted therapies. Expert Rev Hematol. 2016;9(3):255–269. doi:10.1586/17474086.2016.1130616
11. Cheson BD, Horning SJ, Coiffier B, et al. Report of an international workshop to standardize response criteria for non-Hodgkin’s lymphomas. NCI Sponsored International Working Group. J clin oncol. 1999;17(4):1244. doi:10.1200/JCO.1999.17.4.1244
12. Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern cooperative oncology group. Am J Clin Oncol. 1982;5(6):649–655. doi:10.1097/00000421-198212000-00014
13. Balbach ST, Makarova O, Bonn BR, et al. Proposal of a genetic classifier for risk group stratification in pediatric T-cell lymphoblastic lymphoma reveals differences from adult T-cell lymphoblastic leukemia. Leukemia. 2016;30(4):970–973. doi:10.1038/leu.2015.203
14. Hoelzer D, Gokbuget N, Digel W, et al. Outcome of adult patients with T-lymphoblastic lymphoma treated according to protocols for acute lymphoblastic leukemia. Blood. 2002;99(12):4379–4385. doi:10.1182/blood-2002-01-0110
15. Thomas DA, O’Brien S, Cortes J, et al. Outcome with the hyper-CVAD regimens in lymphoblastic lymphoma. Blood. 2004;104(6):1624–1630. doi:10.1182/blood-2003-12-4428
16. Cortelazzo S, Intermesoli T, Oldani E, et al. Results of a lymphoblastic leukemia-like chemotherapy program with risk-adapted mediastinal irradiation and stem cell transplantation for adult patients with lymphoblastic lymphoma. Ann Hematol. 2012;91(1):73–82. doi:10.1007/s00277-011-1252-x
17. Waibel M, Vervoort SJ, Kong IY, et al. Epigenetic targeting of Notch1-driven transcription using the HDACi panobinostat is a potential therapy against T-cell acute lymphoblastic leukemia. Leukemia. 2018;32(1):237–241. doi:10.1038/leu.2017.282
18. Shi Y, Jia B, Xu W, et al. Chidamide in relapsed or refractory peripheral T cell lymphoma: a multicenter real-world study in China. J Hematol Oncol. 2017;10(1):69. doi:10.1186/s13045-017-0439-6
19. Lu X, Ning Z, Li Z, Cao H, Wang X. Development of chidamide for peripheral T-cell lymphoma, the first orphan drug approved in China. Intractable Rare Dis Res. 2016;5(3):185–191. doi:10.5582/irdr.2016.01024
20. Schmitz R, Wright GW, Huang DW, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. 2018;378(15):1396–1407. doi:10.1056/NEJMoa1801445
21. Hickmann A-K, Frick M, Hadaschik D, et al. Molecular tumor analysis and liquid biopsy: a feasibility investigation analyzing circulating tumor DNA in patients with central nervous system lymphomas. BMC Cancer. 2019;19(1):192. doi:10.1186/s12885-019-5394-x
22. Ballester LY, Glitza Oliva IC, Douse DY, et al. Evaluating circulating tumor DNA from the cerebrospinal fluid of patients with melanoma and leptomeningeal disease. J Neuropathol Exp Neurol. 2018;77(7):628–635. doi:10.1093/jnen/nly046
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.