")
Back to Journals » Journal of Inflammation Research » Volume 11
Clinical utility of ustekinumab in Crohn’s disease
Authors Kotze PG, Ma C , Almutairdi A, Panaccione R
Received 17 November 2017
Accepted for publication 9 January 2018
Published 8 February 2018 Volume 2018:11 Pages 35—47
DOI https://doi.org/10.2147/JIR.S157358
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Paulo Gustavo Kotze,1,2 Christopher Ma,1 Abdulelah Almutairdi,1,3 Remo Panaccione1
1Inflammatory Bowel Disease Unit, Division of Gastroenterology and Hepatology, Cumming School of Medicine, University of Calgary, Calgary, AB, Canada; 2Inflammatory Bowel Disease Outpatient Clinics, Colorectal Surgery Unit, Catholic University of Paraná, Curitiba, Brazil; 3Department of Medicine, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia
Abstract: The introduction of anti-tumor necrosis factor (TNF) therapy marked an important milestone in the management of moderate-to-severe Crohn’s disease (CD). However, there remains a pressing demand for alternative therapeutic options for patients with primary nonresponse, secondary loss of response, or intolerable side effects to conventional treatment and TNF antagonists. Ustekinumab (UST) is a fully human IgG1κ monoclonal antibody that inhibits the p40 subunit shared by the proinflammatory cytokines, the interleukin (IL)-12 and -23. This blockade leads to dampening of the inflammatory cascade and differentiation of inflammatory T cells. The clinical development program for UST in CD includes dose finding Phase II (Crohn’s Evaluation of Response to Ustekinumab Anti-Interleukin-12/23 for Induction [CERTIFI]) and the pivotal Phase III (UNITI) trials that demonstrated both the clinical efficacy and safety in anti-TNF-naive and anti-TNF-exposed patients. Real-world evidence has further defined the role of UST in CD management. In this review, we discuss the mechanism of action of UST, describe the results of the randomized controlled trials with this agent, and review the real-world efficacy and safety data from observational cohorts. Finally, we identify areas of future research in the IL-12/23 inflammatory pathway and discuss the positioning of this novel therapeutic option in CD treatment algorithms.
Keywords: ustekinumab, Crohn’s disease, interleukin
Introduction
Crohn’s disease (CD) is a progressive, pan-intestinal, systemic form of inflammatory bowel disease (IBD). Its precise etiology is not fully defined. The pathophysiology of CD is multifactorial and is influenced by genetic predisposition, environmental triggers, and increased intestinal permeability, allowing luminal antigens to enter the lamina propria to trigger an uncontrolled inflammatory cascade.1 This results in transmural inflammation, mucosal ulceration, and complications, which include fibrostenosis, free perforation, abscess formation, and fistulae.2
The medical management of CD has traditionally been based on the use of nonspecific agents such as antibiotics and mesalamine, which have limited to no utility, corticosteroids, and immunomodulators (azathioprine [AZA], 6-mercaptopurine [6-MP], and methotrexate [MTX]). Patients with failure to conventional medical therapy are usually treated with biological agents.3 However, more recently, there has been a move toward earlier introduction of biological therapy. The first class of biological agents approved for the management of CD were tumor necrosis factor (TNF) alpha inhibitors, including infliximab (IFX), adalimumab (ADA), and certolizumab pegol (CZP).4–8 The introduction of anti-TNF agents was revolutionary for the management of CD due to their remarkable efficacy, rapidity of onset, capacity to induce and maintain mucosal healing, and ultimately, ability to reduce or delay the need for surgery and hospitalization and alter the natural progressive course of the disease.9,10
Despite the significant response and remission rates achieved with anti-TNF agents, ~30–40% of patients are primary nonresponders and ~20–30% of patients per year experience secondary loss of response.11 Alternative targets for treatment are required for these patients with refractory disease. In the past decade, several new classes of therapy have been introduced, including anti-integrins12–14 and anti-interleukin (IL) molecules.15 Ustekinumab (UST) is a novel monoclonal antibody that inhibits IL-12 and IL-23 by blocking the common p40 subunit of these proinflammatory cytokines.16 Its efficacy has been demonstrated in landmark clinical trials for induction and maintenance of response and remission in CD patients, independent of their previous exposure to anti-TNF agents.17–19
The aim of this descriptive, nonsystematic review was to discuss the mechanism of action of UST, summarize the main findings from the UST clinical trial programs, review the real-world efficacy and safety data with UST, and examine the positioning of this novel therapeutic option in the CD management algorithm.
Why anti-IL-12/23? The mechanism of action of UST
The most widely accepted hypothesis for IBD pathogenesis is that environmental triggers in genetically predisposed individuals induce abnormalities in the innate and adaptive immune response, modulated by the presence of gut microbiota.20–22 IL-12 and IL-23 are major players in activating adaptive immunity. Targeting this proinflammatory cytokine pathway has become an area of intense therapeutic exploration in autoimmune diseases, including psoriasis, psoriatic arthritis, and CD.23
Traditionally, CD was thought to be predominantly mediated by a classical T helper cell 1 (Th1) immune response, while ulcerative colitis (UC) was thought to be primarily an atypical T helper cell 2 (Th2)-driven process.20 Recently, a new set of T helper cells producing IL-17 (Th17) have been described, challenging the Th1/Th2 paradigm. Th17 responses are implicated in the pathogenesis of CD, psoriasis, psoriatic arthritis, and other inflammatory conditions.24,25 The description of effector Th17 cells by Harrington et al26 came after the initial description of the novel cytokine IL-23 by Oppmann et al.27 This cytokine plays a major role in the expansion and stabilization of committed Th17.25,28
Interestingly, IL-23 was discovered in the process of preclinical development of UST through the discovery that it shares the same p40 subunit with IL-12.16 IL-12, formerly termed cytotoxic lymphocyte maturation factor (CLMF), was initially described by Stern et al and Guber et al.29,30 IL-12 is important for the development of Th1 cells.31 It is a heterodimeric molecule composed of two subunits (p40 and p35 chains), while IL-23 is a heterodimer that consists of covalently linked p40 and p19 protein subunits.27 The p40 protein subunit is shared between the two cytokines and is the target molecule for UST, an anti-IL-12/23p40 fully human monoclonal antibody.
Both IL-12 and -23 bind to the shared IL-12 receptor (IL-12R) β1 subunit: IL-12 binds to a heterodimeric receptor complex consisting of IL-12Rβ1 and IL-12Rβ2 chains32 expressed on the surface of T cells and natural killer (NK) cells16 and IL-23 binds to IL-12Rβ1 and IL-23 receptor (IL-23R).33 Figure 1 demonstrates the relationship between subunits and receptors of both IL-12 and IL-23 and the mechanism of inhibition of the p40 subunit by UST.
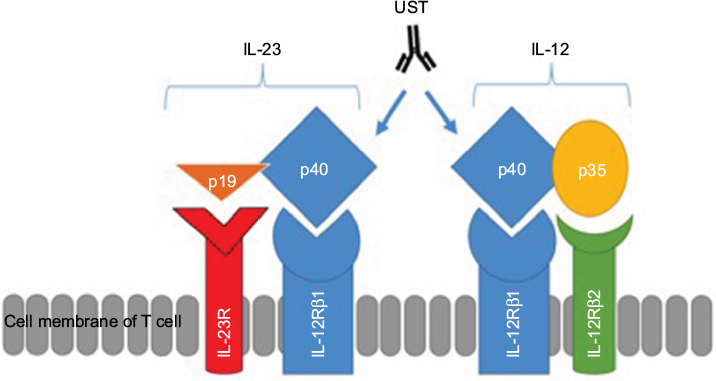 | Figure 1 Structures of IL-12 and IL-23, their receptors and the site of action of UST. Notes: IL-12 is composed of both p40 and p35 subunits, while IL-23 is composed of p40 and p19 subunits. IL-12 receptor is composed of two subunits, such as IL-12Rβ1 and IL-12Rβ2. IL-23 receptor is composed of two subunits, such as IL-12Rβ1 and IL-23R. Abbreviations: IL, interleukin; IL-23R, IL-23 receptor; UST, ustekinumab. |
The rationale behind targeting IL-12 in inflammatory conditions was derived from two observations. First, it was identified that animal models lacking the IL-12p40 subunit were resistant to experimentally induced autoimmune disorders, including multiple gut disease models. However, this hypothesis was refined following the discovery of IL-23: animal models deficient in IL-23 but not those deficient in IL-12 were resistant to developing autoimmune diseases.24,34,35 The second observation leading to the targeting of IL-23 was derived from genome-wide association studies (GWAS), which showed a protective role of the IL-23R variant (rs11209026) in CD.36
Once dendritic cells and macrophages are activated by microbial antigens via surface toll-like receptors (TLRs), different cytokines are released including IL-12, IL-23, IL-1β, transforming growth factor-β (TGF-β), and interferon γ (IFN-γ).28,37 IL-12 plays a major role in differentiating a naive T cell to the Th1 subtype16,31 that consequently secretes TNF-α, IL-2, and IFN-γ. In contrast, IL-23 activates the secretion of IL-17A, IL-17F, and IL-22 from a previously committed Th17 cell.38 In addition, IL-23 stabilizes Th17 and prolongs its survival while suppressing T regulatory (Treg) cell subtype differentiation from a naive T cell.39,40 By inhibiting Treg differentiation, IL-23 further potentiates the inflammatory cascade.
UST is a fully human IgG1 kappa (κ) monoclonal antibody that blocks the IL-12/23 p40 subunit. This prevents the common p40 subunit of IL-12 and IL-23 from interacting with their common receptor IL-12Rβ1 (Figure 1), leading to subsequent neutralization of human IL-12- and IL-23-mediated cell signaling, cell activation, and cytokine production, with a consequent reduction in an important step of the CD inflammatory process.
Clinical development program of UST in CD
The UST development programs comprised several important clinical trials17–19 with different designs and methodology, evaluating both clinical and objective endpoints in patient populations stratified by previous exposure to anti-TNF agents.
Phase IIa trial
The first randomized clinical trial evaluating the efficacy of UST in CD was published in 2008 by Sandborn et al.18 This was a double-blind cross-over study that included two different populations. Population 1 was composed of 104 patients with moderate-to-severe CD (defined by CD activity index [CDAI] between 220 and 450) and randomized in a 1:1:1:1 ratio to one of the following four groups: 1) subcutaneous (SC) placebo at weeks 0, 1, 2, and 3 and then SC 90 mg UST at weeks 8, 9, 10, and 11; 2) SC 90 mg UST at weeks 0, 1, 2, and 3 and then placebo at weeks 8, 9, 10, and 11; 3) intravenous (IV) placebo at week 0 and then 4.5 mg/kg UST at week 8; and 4) IV 4.5 mg/kg UST at week 0 and then placebo at week 8. Population 2 was an open-label study with 27 patients who were primary nonresponders or had secondary loss of response to IFX. These patients were randomized in a 1:1 ratio to receive either SC 90 mg UST at weeks 0, 1, 2, and 3 or IV 4.5 mg/kg UST at week 0. No additional treatment was administered at week 8.
The primary endpoint of the study was clinical response in population 1 at week 8, defined as a decrease in the CDAI of 25% or a CDAI of <150. Although there was a difference in clinical response rates at weeks 4 and 6 (53% UST vs 30% placebo, P=0.02), there was no statistically significant difference between patients treated with UST and placebo at week 8 in population 1 (49 vs 40%, respectively, P=0.337). These findings are illustrated in Figure 2. However, there was suggestion of effect modification by previous drug exposure: in population 1, patients with previous exposure to IFX (n=49) had more significant response rates than placebo in all visits up to week 8 (P<0.05), and in population 2, clinical response rates for patients in the combined UST group were 22 and 41% at weeks 2 and 4, respectively. At weeks 6 and 8, 48% of patients in the combined UST group demonstrated a clinical response.
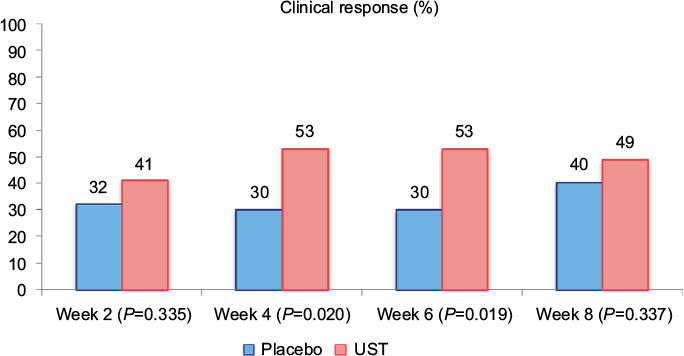 | Figure 2 Clinical response in Phase IIa trial of UST in population 1 (n=104 randomized patients). Notes: The primary endpoint (clinical response at week 8) was not met, and the trial was considered negative. However, a significant difference in clinical response was observed at weeks 4 and 6. Data from Sandborn et al.18 Abbreviation: UST, ustekinumab. |
Two potential explanations for the failure of this Phase IIa trial to meet the primary week 8 clinical response endpoint have been proposed: first, trial power was limited by the high placebo rate. Second, the administered UST induction dose may have been insufficient.
Phase IIb trial (Crohn’s Evaluation of Response to Ustekinumab Anti-Interleukin-12/23 for Induction [CERTIFI])
The CERTIFI study was a 36-week randomized double-blind placebo-controlled Phase IIb trial that evaluated the efficacy and safety of UST in CD for induction and maintenance.19 Included patients had moderate-to-severe CD (CDAI between 220 and 450) and prior nonresponse, secondary loss of response, or unacceptable adverse events to previous anti-TNF therapy.
The induction phase (weeks 0–8) included 526 patients, who were randomized into four groups (IV UST 1 mg/kg, 3 mg/kg, 6 mg/kg, and IV placebo at week 0). In the maintenance phase, patients with a response to induction with UST (defined by decrease in CDAI of 100 points) were re-randomized to receive SC 90 mg UST or placebo at weeks 8 and 16. Patients who had a response to placebo received placebo SC injections at weeks 8 and 16, and those who did not have a response to placebo received 270 mg of SC UST at week 8 followed by a 90 mg injection at week 16. The primary efficacy analysis was performed at week 22, and patients were followed to week 36 for the safety analysis.
The primary outcome of the CERTIFI study was clinical response (reduction in the CDAI of 100 points) at week 6, based on initial results of the aforementioned Phase IIa trial regarding optimal timing of separation from placebo.18 Secondary endpoints were clinical remission (CDAI <150) at week 6, clinical response at week 4, and clinical remission at week 22 among responders to UST at week 6.
The results of the primary endpoint of the CERTIFI study are illustrated in Figure 3. At week 6, clinical response was more prevalent in patients receiving 6 mg/kg of UST than in patients receiving placebo (39.7 vs 23.5%, Δ16.2% [95% CI, 5.1%, 27.3%], P=0.005). With respect to secondary induction outcomes, clinical remission at week 6 was observed in 10.6% of the cases in the placebo group, 16% in the UST 1 mg/kg (P=0.20) group, 15.9% in the UST 3 mg/kg (P=0.21) group, and 12.2% in the UST 6 mg/kg (P=0.68) group. Clinical response at week 4 was observed in 16.7% in the placebo group, 27.5% in the UST 1 mg/kg (P=0.04) group, 37.1% in the UST 3 mg/kg (P<0.001) group, and 30.5% in the UST 6 mg/kg (P=0.008) group.
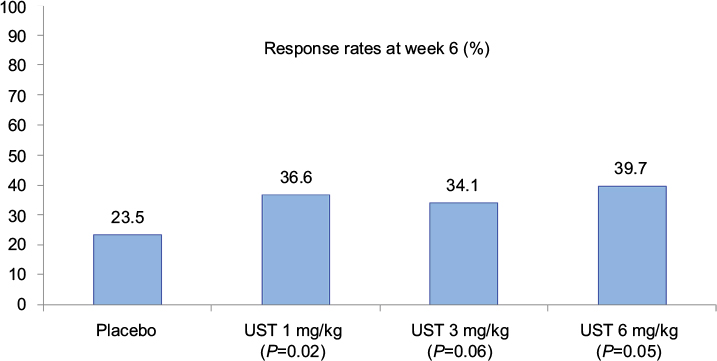 | Figure 3 Clinical response rates at week 6 (induction) in the CERTIFI trial (primary outcome). Notes: Statistical significance reached in the UST 1 and 6 mg/kg groups. Data from Sandborn et al.19 Abbreviation: UST, ustekinumab. |
Among responders to induction with UST, 41.7% of patients receiving 90 mg of UST in the maintenance phase were in clinical remission at week 22, as compared to 27.4% of those receiving placebo (Δ14.3% [95% CI: 2.0%, 27.1%], P=0.03).
In the induction phase, no major differences in adverse events between the four groups were observed. Infusion reactions at the first dose were similar between patients with IV UST and placebo (4.3 vs 4.5%, respectively). In the maintenance phase, the rates of adverse events, serious adverse events, and duration of follow-up were similar in the UST group as compared to placebo. There were no deaths, serious opportunistic infections, major cardiovascular events, and tuberculosis in the study. One patient from the UST 1 mg/kg group receiving 90 mg SC UST at weeks 8 and 16 developed a basal-cell carcinoma.
Phase III trials (UNITI)
The landmark study leading to the approval of UST for the indication of CD by regulatory agencies worldwide was the UNITI trial (A Study to Evaluate the Safety and Efficacy of Ustekinumab Induction Therapy in Patients with Moderately-to-Severely Active Crohn’s Disease), published in 2016.17 It was composed of two identical induction studies (UNITI-1, enrolling patients with primary nonresponse, secondary loss of response, or unacceptable adverse events to previous anti-TNF therapy, and UNITI-2, enrolling patients who had failure or unacceptable adverse events to conventional therapy with immunomodulators or corticosteroids). The maintenance phase of the study (IM-UNITI) included patients who responded to the induction studies. All three studies were randomized, double-blind, placebo-controlled trials.
Included patients had a diagnosis of moderate-to-severe CD (CDAI 220-450) for >3 months. Inclusion was restricted to patients with positive objective inflammatory parameters (C-reactive protein [CRP] >3.0 mg/L; fecal calprotectin > 250 mg/kg; ulcerations at the ileum, colon, or both).
In the induction trials, patients were randomized at week 0 in a 1:1:1 ratio to receive 1) single dose of 130 mg of IV UST, 2) ~6 mg/kg of IV UST (patients ≤55 kg received 260 mg, those >55 and ≤85 kg received 390 mg, and those >85 kg received 520 mg), or 3) placebo. The designs of the two induction trials were identical with the only difference being the inclusion population. For the maintenance phase, patients who responded to induction were re-randomized in a 1:1:1 ratio to receive 1) 90 mg of UST SC every 8 weeks, 2) 90 mg of UST SC every 12 weeks, or 3) placebo for 40 weeks. They were then followed up to week 44 of the maintenance study for a total study duration of 52 weeks (8 weeks induction with 44 weeks maintenance).
The primary endpoint for the induction trials (UNITI-1 and -2) was clinical response (defined by reduction in the CDAI of 100 points) at week 6. Major secondary endpoints were clinical response and remission (CDAI <150) at week 8 and a decrease in baseline CDAI of 70 points at weeks 3 and 6. In the maintenance trial, the primary endpoint was clinical remission (CDAI <150) at week 44. Major secondary endpoints were clinical response (decrease in CDAI >100 points), maintenance of remission among those in remission at initiation of the maintenance trial, steroid-free remission, and remission exclusively in the UNITI-1 population, all assessed at week 44.
In the UNITI-1 trial, a total of 714 patients were included (247 in the placebo group, 245 in the UST 130 mg group, and 249 in the UST 6 mg/kg group). In the UNITI-2 induction study, a total of 628 patients were analyzed (210 in the placebo, 209 in the UST 130 mg group, and 209 in the UST 6 mg/kg group). There was a low percentage of discontinuation in both studies, and the baseline characteristics were similar among all study groups.
The primary endpoint results of both induction studies are described in detail in Figure 4. The response rates at week 6 were higher in all UST groups as compared with placebo in both trials. These rates were nominally higher in UNITI-2, demonstrating that previous failure to anti-TNF therapy can negatively impact the induction effect of UST. Regarding the main secondary outcome of the induction trials, clinical remission at week 8 was more prevalent in the UST groups as compared to placebo. In UNITI-1, remission rates were 7.3% for placebo, 15.9% for UST 130 mg (P=0.003), and 20.9% for UST 6 mg/kg (P<0.001). In UNITI-2, the clinical remission rates were 19.6% for placebo, 30.6% for UST 130 mg (P=0.009), and 40.2% for UST 6 mg/kg (P<0.001). As observed, remission rates were higher in the UNITI-2 population, without previous failure to anti-TNF therapy, in a similar pattern as observed for the primary outcome of clinical response at week 6.
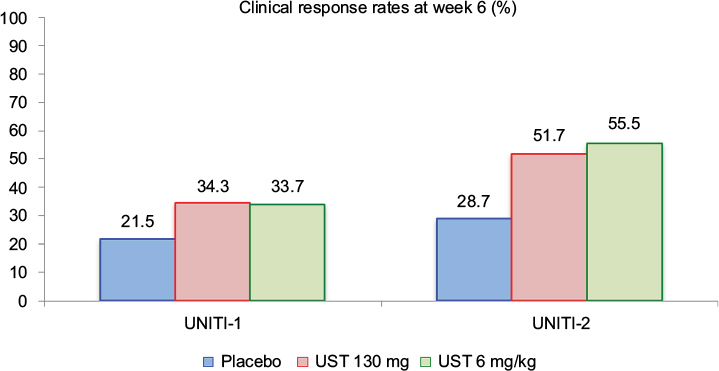 | Figure 4 Primary endpoint results of the UNITI-1 and UNITI-2 studies. Notes: Clinical response at week 6 was more prevalent in the UST-treated groups. In UNITI-1, P-values as compared to placebo were 0.002 (UST 130 mg) and 0.003 (UST 6 mg/kg). In UNITI-2, P-values as compared to placebo were <0.001 for both UST 130 mg and UST 6 mg/kg. Data from Feagan et al.17 Abbreviation: UST, ustekinumab. |
A total of 397 patients with induction response were re-randomized for the IM-UNITI maintenance phase. Among this population, 133 patients were randomized to the placebo group, 132 patients were randomized to the UST q12 weeks group, and 132 patients were randomized to the UST q8 weeks group. Figure 5 demonstrates the remission rates at week 44 of the maintenance trial in all patients and then stratified by induction trial in separate analysis. Higher remission rates were achieved in patients receiving 90 mg SC UST q8 weeks compared to placebo in all analyses. Additionally, steroid-free remission was more prevalent in the UST-treated groups as compared to placebo (29.8% placebo vs 42.6% UST q12 weeks vs 46.9% UST q8 weeks; P=0.04 and 0.004, respectively). Durable reductions in the CDAI, CRP, and fecal calprotectin levels were also more prevalent in the UST groups as compared to patients receiving maintenance with placebo.
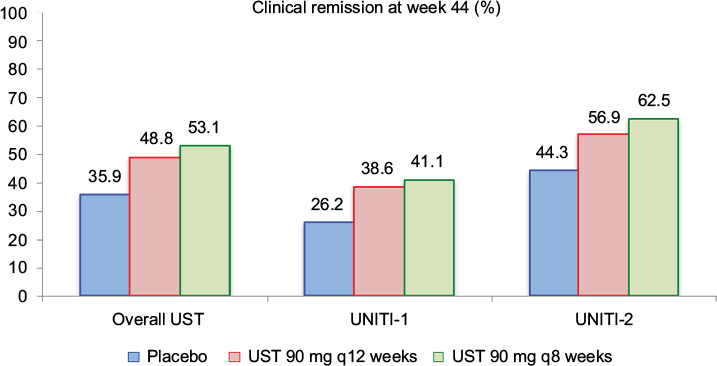 | Figure 5 Clinical remission rates at week 44 of the IM-UNITI maintenance study. Notes: In the pooled overall UST group, P=0.040 (q12 weeks) and P=0.005 (q8 weeks) for UST as compared to placebo. Among patients derived from the UNITI-1 induction trial, P=0.14 (q12 weeks) and P=0.10 (q8 weeks). Among patients derived from UNITI-2 trial, P=0.15 (q12 weeks) and P=0.02 (q8 weeks). Data from Feagan et al.17 Abbreviation: UST, ustekinumab. |
In the safety analysis, adverse events were observed in ~2/3 of patients during the UNITI-1 trial, with no important differences between groups. Serious adverse events occurred in 4.9–7.2% of patients. In the UNITI-2 trial, adverse events were observed in ~50% of patients, with serious adverse events being observed in 2.9–5.8% of the patients. At week 44 in the maintenance IM-UNITI study, at least one adverse event was observed in 81.7% of patients receiving UST 90 mg q8 weeks, 80.3% of patients receiving UST 90 mg q12 weeks, and 83.5% of patients receiving placebo. Serious adverse events were observed in 9.9, 12.1, and 15% in the same groups, respectively. Serious infections were observed in 13 patients overall, representing 2.3% in the UST 90 mg q8 weeks, 5.3% in the UST 90 mg q12 weeks, and 2.3% in the placebo groups.
One patient from the UNITI-1 trial receiving 6 mg/kg of UST developed multiple myeloma, despite not participating in the maintenance trial. In the UNITI-2 trial, one patient in the placebo group developed a basal cell carcinoma. In the IM-UNITI maintenance trial, two patients presented with basal cell carcinomas (one in the placebo group and one in the UST q8 weeks group). One patient from the UST q12 weeks trial had a diagnosis of metastatic adenocarcinoma in the small bowel after resection. The following three opportunistic infections were observed: one listeria meningitis in a patient from UNITI-1 trial receiving UST 6 mg/kg who was also on 30 mg/day of prednisone and two cases of uncomplicated esophageal candidiasis (one patient from UNITI-2 with concomitant steroids and MTX and another patient receiving UST q8 weeks in the maintenance trial). One case of tuberculosis was reported in a patient who had a single dose of 130 mg UST IV and had placebo maintenance 10 months after the UST infusion. There were no deaths in the UNITI program at publication.
In terms of objective markers of response, a significantly higher proportion of patients in the UST groups from the IM-UNITI trial achieved fecal calprotectin levels <250 mg/kg as compared to placebo at week 44 (27.5% in the UST q12 weeks, 30.1% in the UST q8 weeks, and 10.8% in the placebo groups).17
Endoscopic outcomes data from the IM-UNITI trial have been reported in abstract form.41 The primary endpoint of this substudy was a change in the CD Simple Endoscopic Score (SES-CD) at week 8 in patients with baseline colonoscopy. Secondary endpoints included a decrease of >3 points in the SES-CD, a reduction of 50% in the SES-CD, and the absence of ulcers (mucosal healing). Reduction in SES-CD was significantly higher in patients with UST (mean: -2.8) as compared to placebo (mean: -0.7) at week 8 (P=0.01). Other endoscopic data also favored UST over placebo, with more patients achieving >3 points SES-CD reduction (47.7 vs 29.9%, P<0.01).
The efficacy of UST in perianal fistulizing CD has also recently been reported in subgroup analysis of patients with active fistula at baseline in the Phase IIb (CERTIFI) and Phase III (UNITI) programs.42 Although ~40% of the included patients in the UNITI-1, UNITI-2, and CERTIFI studies had a history of perianal fistulizing CD, a smaller proportion (10.8 and 15.5%) had active fistulas at baseline. In the CERTIFI maintenance phase, fistula response occurred in 47% (9/19) in the UST group as compared to 30% (6/20) in the placebo group at week 22. In patients from IM-UNITI study, fistula response was observed in 80% of patients in the combined UST group and 45.5% (5/11) of patients in the placebo arm (P=0.64). The authors believe that a prospective study with UST in perianal fistulas is warranted.
Observational UST experiences
Multiple open-label observational cohort studies have assessed the efficacy and safety of UST for CD.43–51 Primarily, these reflect retrospective treatment experiences with UST prior to regulatory approval and evaluate patients with disease refractory to conventional therapy. Open-label studies have some advantages over clinical trials. First, they reflect “real-life” data; experience with anti-TNF agents has shown that response and remission rates in clinical practice can exceed those reported in randomized controlled trials.52 Second, open-label studies allow the evaluation of therapy in populations who would otherwise be excluded from clinical trials, including patients with comorbidities and those patients who are unable or unwilling to enroll in rigorous clinical trial regimens.
However, evidence from open-label studies must be interpreted recognizing the limitations of this study design. Importantly in CD, these studies often use nonstandardized dosing regimens and nonvalidated definitions of response and remission. Second, these studies are limited by smaller sample sizes and increased loss to follow-up compared to formalized clinical trial protocols. Third, these observations are uncontrolled and there may be a tendency for both investigators and patients to overstate subjective responses to therapy. This is difficult to constrain without strict outcome definition criteria, and endpoint assessment in retrospective studies is particularly prone to recall bias.53 Nonetheless, open-label studies provide an important context in the literature for novel therapies.
Clinical response and remission with UST
Four multicenter cohort studies have been conducted evaluating CD patients treated with UST. Wils et al51 summarized an observational cohort of 122 consecutive CD patients refractory to anti-TNF therapy, from 20 tertiary care centers in Europe as part of the Groupe d’Etude Therapeutique des Affections Inflammatoires du Tube Digestif [Therapeutic Study Group for Inflammatory Disorders of the Digestive Tube] (GETAID) cohort. They demonstrated that 65% of patients had clinical benefit within 3 months of receiving UST, defined as a significant improvement in CD-related symptoms and laboratory tests assessed by the patient’s physician with a decision to continue treatment, weaning from corticosteroids, and without surgery or rescue immunosuppression. Thirteen different UST induction regimens were used in this study, with a mean cumulative dose administered in the first month of therapy of 149 mg (±64 mg).
A second multicenter experience by Khorrami et al47 reported on 116 CD patients treated at 42 Spanish centers as a part of the Spanish Ustekinumab Study group (USTEK). Clinical outcomes in this cohort were defined by the Harvey-Bradshaw Index (HBI):54 response was characterized by the reduction of ≥3 points from baseline vs remission was defined by an HBI score of ≤4. Again, multiple dosing regimens were employed, with 48% of patients receiving a cumulative induction dose >360 mg. In this cohort, 83.6% (97/116) of patients achieved a clinical benefit (response or remission) after induction therapy and 76.4% of patients had achieved a clinical benefit by 6 months of treatment.
The largest open-label UST experience, including 167 CD patients, was reported by Ma et al from two tertiary care centers in Alberta, Canada.49,50 Using more strict definitions of clinical response (decrease in HBI ≥3 points compared to baseline with complete corticosteroid tapering), 38.9 and 60.3% of patients had achieved a clinical response at 3 and 6 months, respectively. Response was stratified by induction dosing and route of drug administration (IV vs SC), with no statistically significant differences at 3 or 6 months between the 3 vs 6 mg/kg induction groups and IV vs SC induction therapy.
Finally, Harris et al46 reported efficacy outcomes of 45 patients with complicated, refractory CD treated with UST at Vanderbilt University and University of Maryland School of Medicine. All patients received a novel SC dosing regimen consisting of UST 90 mg at weeks 0, 4, and 12 with a 270 mg booster dose given at week 8 if there was no or limited clinical response. Interestingly, 87% (39/45) of patients received the 270 mg booster dose at week 8. A total of 46% of patients demonstrated a reduction in the HBI score of ≥3 points and 35% of patients achieved clinical remission.
Three single-center open-label cohorts of CD patients treated with UST have also been reported. The first experience was described in 2014 by Batista et al43 from the Mayo Clinic. Eighteen patients with CD, 89% of whom had failed at least two anti-TNF agents, were treated with SC induction UST. Cumulative probability of clinical response was 11.1% at 1 month and 44.4% at 6 months, with 26.7% able to successfully taper off corticosteroids. Subsequently, Kopylov et al48 presented a cohort of 38 CD patients from McGill University, Montreal, QC, Canada, treated with UST. A total of 95% of these patients had failed multiple biological agents, but 73.6% of them achieved an initial clinical response by the first visit after induction. Clinical response in this study was defined by improvement in patient symptoms and a decision to continue UST therapy. Follow-up results were available for 31 patients in the cohort at 6 months; 80% of initial responders had maintained response and 64.5% of patients overall were considered responding to therapy. The McGill group has subsequently reported a mixed prospective and cross-sectional cohort of UST-treated CD patients, with a primary focus on evaluating the effect of drug levels on clinical outcomes.44 Finally, in a cohort of 79 patients treated at the University of British Columbia (UBC), Vancouver, BC, Canada, Greenup et al45 demonstrated symptomatic benefit, defined by physician assessment and improvement in subjective quality of life, in 56% of patients at 3 months. A total of 47% (9/19) of patients could withdraw corticosteroids at 3 months.
Long-term maintenance in open-label cohorts
In open-label cohorts, 12-month clinical response rates varied from 45 to 72%. In the three multicenter cohort studies describing 12-month clinical outcomes, the reported response rate was 68% in the GETAID cohort,51 72% in the Alberta cohort,50 and 64% in the USTEK cohort.47 Response rates based on the proportion of patients remaining on therapy in long-term follow-up are inflated by conditional censoring by drug discontinuation in nonresponders; therefore, the Alberta cohort and the USTEK cohort described the cumulative probability of maintained UST response at 12 months among all treated patients; this was similar in both cohorts (72 and 74%, respectively).47,50
Objective response to UST therapy in open-label cohorts
Endoscopic, radiographic, and serologic outcomes have been assessed in several open-label studies. In the GETAID cohort, CRP response was evaluated in 58 patients with initial clinical benefit, demonstrating CRP reductions in 95% of patients, normalization in 41% of patients, and a median reduction of 18 mg/L (IQR 8–32 mg/L).51 Similarly, Kopylov et al48 reported CRP response in all clinical responders and normalization in 61.5% of patients treated with UST. Endoscopic and radiographic outcomes were most extensively assessed in the Alberta cohort, where 141 patients had objective evaluation of disease with ileocolonoscopy, contrast-enhanced ultrasound, CT enterography, or MR enterography.49 Objective response, defined by improvement in radiographic inflammatory parameters or absence of deep mucosal ulcerations, was demonstrated in 54.5 and 55.8% of UST-treated patients at 6 and 12 months, respectively. Objective remission, defined by normalization of radiographic appearance or endoscopic mucosal healing, was achieved in 24.4 and 26.7% of UST-treated patients at 6 and 12 months, respectively. In other cohorts, endoscopic response rates range from 64 to 76%.45,46
Safety of UST in open-label studies
Overall, the safety profile of UST in open-label studies has been favorable with very few reported serious adverse events. The overall adverse event rate has been demonstrated to range from 9 to 56%, although there were no placebo groups against which these rates are compared, definition for adverse events is mixed, and direct association with therapy is often unclear. For example, the most commonly reported complications include myalgias and arthralgias, infections, and headache. Whether joint-related symptoms on UST are related to withdrawal of anti-TNF therapy and subsequent flare of extraintestinal articular manifestations or whether they are directly related to UST therapy is unclear. Serious adverse events are rare, and requirement for drug discontinuation due to adverse events in open-label cohorts has been reported to be <7%.47,51 There does not appear to be an increased risk of malignancy, and in all cohorts, only a single death on UST treatment has been reported,49 although this was in a patient with significant pretreatment cardiopulmonary comorbidity and the cause of death was not thought to be related to therapy.
Other issues
In the open-label environment, dose adjustment of UST has been attempted for patients with inadequate primary response and secondary loss of response to therapy. Most frequently, patients are advanced from q8-week maintenance to q4-week maintenance therapies. Alternative strategies to consider also include reloading with high-dose SC therapy or reloading with IV induction. Reported rates of escalation are mixed, primarily due to varying indications for dose adjustment. In the McGill cohort, almost half of patients underwent dose intensification and this was successful in 11/18 (61%) patients.48 In contrast, only 3/16 (19%) patients responded to escalated maintenance dosing in the UBC cohort.45 These discrepancies may reflect differences in indication for dose escalation, use of concomitant therapy, or decision for concurrent re-induction prior to dose intensification.
Efficacy of UST for the treatment of perianal disease has been evaluated in several cohorts with promising results. In the Alberta cohort, 14/45 (31%) patients with active perianal disease at induction achieved complete radiographic healing.49 Response rates in smaller samples have also been encouraging, with 8/12 (67%) patients responding in the GETAID cohort,51 11/18 (61%) patients responding in the USTEK cohort,47 and 9/13 (69%) patients responding in the McGill cohort.48 Open-label evidence for the efficacy of UST for other extraintestinal manifestations of CD remains sparse.
Potential predictors of UST response in univariate and multivariate models derived from observational cohorts have been inconsistent, and regression model power has been generally limited by small sample size. Concurrent immunomodulation was identified as a positive predictor of induction response in the GETAID cohort and of maintenance response in both the Alberta and USTEK cohorts.47,50,51 This was not replicated in subgroup analysis of the UNITI program, and the potential biological mechanisms are unclear, as the immunogenicity to UST is low17 and serum UST levels have not been shown to significantly vary with immunomodulator use.44 Battat et al evaluated serum trough UST concentrations associated with clinical and endoscopic responses: they demonstrated that 26-week threshold UST level of 4.5 µg/mL was associated with endoscopic response.44 Other potential negative predictors of response include previous surgical resection47 and prior primary anti-TNF nonresponse.45 A summary of significant relevance and weaknesses of the included open-label studies with UST is presented in Table 1.
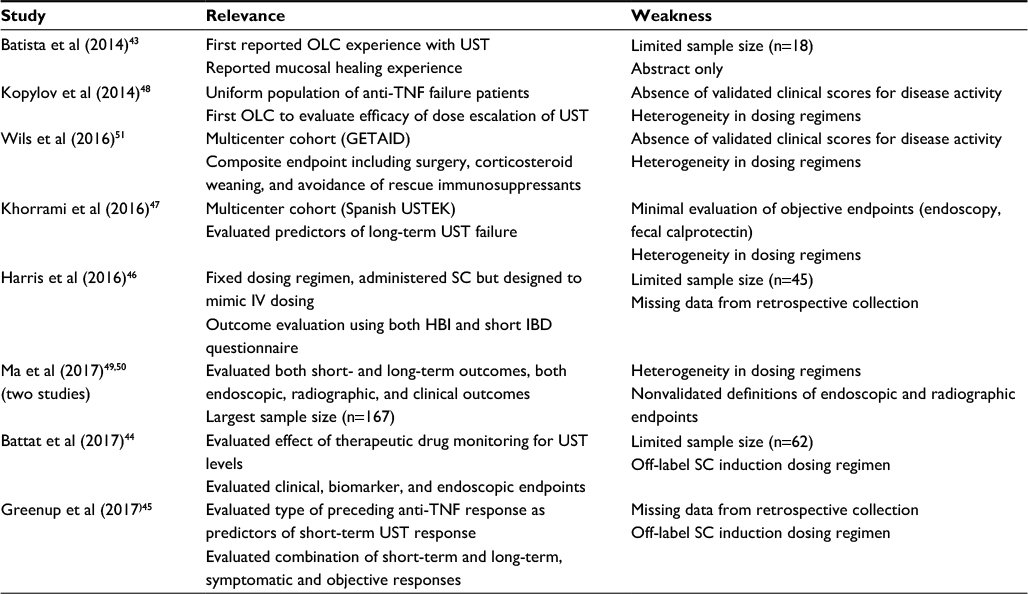 | Table 1 Summary of open-label cohorts of UST for Crohn’s disease Abbreviations: GETAID, [Therapeutic Study Group for Inflammatory Disorders of the Digestive Tube]; HBI, Harvey-Bradshaw Index; IBD, inflammatory bowel disease; IV, intravenous; SC, subcutaneous; TNF, tumor necrosis factor; UST, ustekinumab; USTEK, Spanish Ustekinumab Study group; OLC, open label cohort; IQR, interquartile range; CT, computed tomography; MR,magnetic resonance. |
Future directions
Use of UST today and tomorrow
With the mounting evidence for both safety and efficacy from randomized controlled trials and open-label real-world cohorts, we anticipate that UST will become an increasingly important part of the therapeutic armamentarium for the management of moderate-to-severe CD. In particular, as the number of patients with loss of response to anti-TNF therapy increases over time, second-line biological therapies such as UST will be needed to manage this difficult-to-treat population with refractory disease. However, there are still many unanswered questions regarding the optimal use of UST that need to be addressed.
First, the positioning of UST in the therapeutic algorithm for CD is not defined.55 It has been extensively studied after the failure of conventional therapy and anti-TNF agents, and whether using UST as a first-line biologic achieves better long-term outcomes in moderate-to-severe CD is unclear. Previous anti-TNF failure is associated with nominally lower response rates in the UNITI trial program and has been associated with poorer response in open-label studies. However, there are currently no head-to-head comparisons of UST vs either anti-TNF or anti-integrin as first-line therapy. This specific question is currently being evaluated in a prospective open-label cohort study (Clinicaltrials.gov: NCT03108326).
Furthermore, the positioning of UST as a second-line agent after anti-TNF failure is debatable. There are no data to support the use of UST over anti-integrin molecules such as vedolizumab or over oral small molecule therapy. Factors such as patient preference for ease of SC administration and presence of extraintestinal manifestations may play a role in deciding on second-line treatment options. However, the clinical “gestalt” that UST is superior to vedolizumab with respect to overall efficacy, rapidity of onset, efficacy in small bowel disease, and achievement of endoscopic or histologic healing is largely unfounded. As more agents are introduced as treatment options in CD, the positioning of UST in the treatment algorithm may change.
Third, there remain unanswered questions regarding the optimal conditions for UST administration. Future research should consider whether 1) UST induction should be bridged with corticosteroids or other therapy, 2) concomitant immunomodulator therapy is beneficial or specific subgroups of patients benefit from concomitant immunomodulators, 3) combination therapy with other classes of biological agents is safe and effective for severe disease, and 4) prospective therapeutic drug monitoring improves long-term outcomes in UST-treated patients. Finally, as with all novel therapies, postmarketing studies are needed to carefully monitor for safety signals of rare adverse events not captured in finite clinical trial programs.
Beyond UST: selective IL-23 blockade
Although the efficacy of blocking both IL-12 and IL-23 has been well demonstrated, an exciting horizon in the targeting of this inflammatory pathway is selective IL-23 blockade.56 The rationale for IL-23 selective blockade is twofold: first, IL-23 appears to be the critical step in disease pathogenesis in murine models. Second, targeting the unique p19 subunit of IL-23 would allow normal IL-12-mediated Th1 responses that mediate host protection against mycobacterial disease. Thus, the aim of IL-23-specific blockers would be to reduce inflammatory disease burden while preserving host immunity and reducing off-target adverse events.57
Several IL-23-specific monoclonal antibodies are in clinical trial development programs, including brazikumab, risankizumab, tildrakizumab, and guselkumab.57 Phase II trials of risankizumab and brazikumab in CD patients have been completed with promising results.58,59 In a randomized controlled trial of 121 CD patients (93% with previous anti-TNF exposure), Feagan et al58 reported that 31% of risankizumab-treated patients achieved week 12 clinical remission compared to 15% of placebo-treated patients (P=0.0489). Similarly, in the Phase IIa trial of 121 patients with moderate-to-severe CD failing or intolerant to anti-TNF therapy, 49.2% of brazikumab-treated patients achieved clinical response or remission compared to 26.7% of patients receiving placebo (P=0.010).59
Finally, perhaps the most intriguing evidence for IL-23 selective inhibition has recently been published in the psoriasis literature. In a head-to-head Phase II randomized trial of risankizumab vs UST in 166 patients with psoriasis, achievement of the primary endpoint of 90% reduction in baseline Psoriasis Area and Severity Index (PASI) score at week 12 occurred in 77% (64/83) of risankizumab-treated patients compared to only 40% (16/40) of UST-treated patients (P<0.001).60 Although translation of these results to the CD population is limited by differences in disease pathogenesis and burden of systemic inflammation, the story of IL-23 selective inhibitors in CD will certainly continue to evolve over the next decade.
Conclusion
The inhibition of the IL-12 and -23 pathways has been demonstrated to be an effective strategy in the management of CD. In pivotal randomized controlled trials and multiple real-world observational cohorts, UST is safe and effective for the induction and maintenance of response and remission, in both anti-TNF-naive and anti-TNF-exposed CD patients. However, the optimal treatment strategy and positioning of UST in the CD management algorithm remain undefined and these decisions should be individualized and discussed with patients. Future studies are needed to delineate the efficacy of UST for UC, determine the effect of concomitant immunomodulator use, and optimize implementation of therapeutic drug monitoring with UST. These pathways will continue to be paved with increased experience and expertise with UST over time.
Disclosure
CM is supported in part by the Canadian Institutes of Health Research and the Canadian Association of Gastroenterology. PGK has received consulting fees from AbbVie, Takeda, and Pfizer and speaker’s bureau fees from AbbVie, Takeda, Ferring, Pfizer, UCB, and Janssen. RP has received scientific advisory board fees from Abbott/AbbVie, Amgen, Janssen, Merck, Pfizer, Prometheus Laboratories, Salix Pharma, Shire, Takeda, and Warner Chilcott, consulting fees from Abbott/AbbVie, Amgen, Aptalis, Astra Zeneca, Baxter, BMS, Centocor, Elan/Biogen, Eisai, Ferring, GSK, Janssen, Merck, Millennium, Pfizer, Proctor & Gamble, Prometheus Therapeutics and Diagnostics, Schering-Plough, Shire, Takeda, UCB Pharma, and Warner Chilcott, research grants from Abbott/AbbVie, Amgen, Aptalis, Astra Zeneca, Baxter, BMS, Centocor, Eisai, Elan/Biogen, Ferring, GSK, Janssen, Merck, Millennium, Pfizer, Proctor & Gamble, Prometheus, Shire, Schering-Plough, Takeda, UCB Pharma, and Warner Chilcott, and speaker’s bureau fees from Abbott/AbbVie, Amgen, Aptalis, Astra Zeneca, Baxter, BMS, Centocor, Eisai, Elan/Biogen, Ferring, GSK, Janssen, Merck, Millennium, Pfizer, Proctor & Gamble, Prometheus, Schering-Plough, Shire, Takeda, UCB Pharma, and Warner Chilcott. The authors report no other conflicts of interest in this work.
References
Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet. 2012; 380(9853):1590–1605. | ||
Freeman HJ. Natural history and long-term clinical course of Crohn’s disease. World J Gastroenterol. 2014;20(1):31–36. | ||
Gomollon F, Dignass A, Annese V, et al; ECCO. 3rd European evidence-based consensus on the diagnosis and management of Crohn’s disease 2016: part 1: diagnosis and medical management. J Crohns Colitis. 2017;11(1):3–25. | ||
Hanauer SB, Feagan BG, Lichtenstein GR, et al; ACCENT I Study Group. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet. 2002;359(9317):1541–1549. | ||
Sands BE, Anderson FH, Bernstein CN, et al. Infliximab maintenance therapy for fistulizing Crohn’s disease. N Engl J Med. 2004;350(9):876–885. | ||
Colombel JF, Sandborn WJ, Rutgeerts P, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s disease: the CHARM trial. Gastroenterology. 2007;132(1):52–65. | ||
Schreiber S, Khaliq-Kareemi M, Lawrance IC, et al; PRECISE 2 Study Investigators. Maintenance therapy with certolizumab pegol for Crohn’s disease. N Engl J Med. 2007;357(3):239–250. | ||
Panaccione R, Colombel JF, Sandborn WJ, et al. Adalimumab maintains remission of Crohn’s disease after up to 4 years of treatment: data from CHARM and ADHERE. Aliment Pharmacol Ther. 2013;38(10):1236–1247. | ||
Lichtenstein GR, Yan S, Bala M, Blank M, Sands BE. Infliximab maintenance treatment reduces hospitalizations, surgeries, and procedures in fistulizing Crohn’s disease. Gastroenterology. 2005;128(4):862–869. | ||
Rutgeerts P, Van Assche G, Sandborn WJ, et al. Adalimumab induces and maintains mucosal healing in patients with Crohn’s disease: data from the EXTEND trial. Gastroenterology. 2012;142(5):1102–1111.e2. | ||
Qiu Y, Chen BL, Mao R, et al. Systematic review with meta-analysis: loss of response and requirement of anti-TNFalpha dose intensification in Crohn’s disease. J Gastroenterol. 2017;52(5):535–554. | ||
Targan SR, Feagan BG, Fedorak RN, et al. Natalizumab for the treatment of active Crohn’s disease: results of the ENCORE Trial. Gastroenterology. 2007;132(5):1672–1683. | ||
Sands BE, Feagan BG, Rutgeerts P, et al. Effects of vedolizumab induction therapy for patients with Crohn’s disease in whom tumor necrosis factor antagonist treatment failed. Gastroenterology. 2014;147(3):618–627.e3. | ||
Sandborn WJ, Feagan BG, Rutgeerts P, et al; GEMINI 2 Study Group. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2013;369(8):711–721. | ||
Panaccione R, Sandborn WJ, Gordon GL, et al. Briakinumab for treatment of Crohn’s disease: results of a randomized trial. Inflamm Bowel Dis. 2015;21(6):1329–1340. | ||
Benson JM, Peritt D, Scallon BJ, et al. Discovery and mechanism of ustekinumab: a human monoclonal antibody targeting interleukin-12 and interleukin-23 for treatment of immune-mediated disorders. MAbs. 2011;3(6):535–545. | ||
Feagan BG, Sandborn WJ, Gasink C, et al; UNITI–IM-UNITI Study Group. Ustekinumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2016;375(20):1946–1960. | ||
Sandborn WJ, Feagan BG, Fedorak RN, et al; Ustekinumab Crohn’s Disease Study Group. A randomized trial of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn’s disease. Gastroenterology. 2008;135(4):1130–1141. | ||
Sandborn WJ, Gasink C, Gao LL, et al; CERTIFI Study Group. Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N Engl J Med. 2012;367(16):1519–1528. | ||
Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3(7):390–407. | ||
Colombel JF, Panaccione R, Bossuyt P, et al. Superior endoscopic and deep remission outcomes in adults with moderate to severe Crohn’s disease managed with treat to target approach versus clinical symptoms: data from calm. Gastroenterology. 2017;152(5):S155. | ||
de Souza HS, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol. 2016;13(1):13–27. | ||
Deepak P, Loftus EV Jr. Ustekinumab in treatment of Crohn’s disease: design, development, and potential place in therapy. Drug Des Devel Ther. 2016;10:3685–3698. | ||
Damsker JM, Hansen AM, Caspi RR. Th1 and Th17 cells: adversaries and collaborators. Ann N Y Acad Sci. 2010;1183:211–221. | ||
Patel DD, Kuchroo VK. Th17 cell pathway in human immunity: lessons from genetics and therapeutic interventions. Immunity. 2015;43(6):1040–1051. | ||
Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6(11):1123–1132. | ||
Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13(5):715–725. | ||
Verstockt B, Van Assche G, Vermeire S, Ferrante M. Biological therapy targeting the IL-23/IL-17 axis in inflammatory bowel disease. Expert Opin Biol Ther. 2017;17(1):31–47. | ||
Stern AS, Podlaski FJ, Hulmes JD, et al. Purification to homogeneity and partial characterization of cytotoxic lymphocyte maturation factor from human B-lymphoblastoid cells. Proc Natl Acad Sci U S A. 1990;87(17):6808–6812. | ||
Gubler U, Chua AO, Schoenhaut DS, et al. Coexpression of two distinct genes is required to generate secreted bioactive cytotoxic lymphocyte maturation factor. Proc Natl Acad Sci U S A. 1991;88(10):4143–4147. | ||
Dave M, Papadakis KA, Faubion WA Jr. Immunology of inflammatory bowel disease and molecular targets for biologics. Gastroenterol Clin North Am. 2014;43(3):405–424. | ||
Presky DH, Yang H, Minetti LJ, et al. A functional interleukin 12 receptor complex is composed of two beta-type cytokine receptor subunits. Proc Natl Acad Sci U S A. 1996;93(24):14002–14007. | ||
Parham C, Chirica M, Timans J, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168(11):5699–5708. | ||
Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201(2):233–240. | ||
Teng MW, Bowman EP, McElwee JJ, et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med. 2015;21(7):719–729. | ||
Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314(5804):1461–1463. | ||
Blanco P, Palucka AK, Pascual V, Banchereau J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth Factor Rev. 2008;19(1):41–52. | ||
Sarra M, Pallone F, Macdonald TT, Monteleone G. IL-23/IL-17 axis in IBD. Inflamm Bowel Dis. 2010;16(10):1808–1813. | ||
Izcue A, Hue S, Buonocore S, et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity. 2008;28(4):559–570. | ||
Shen W, Durum SK. Synergy of IL-23 and Th17 cytokines: new light on inflammatory bowel disease. Neurochem Res. 2010;35(6):940–946. | ||
Sandborn W, Gasink C, Chan D, et al. PD-012 endoscopic healing in the ustekinumab phase 3 UNITI/IMUNITI Crohn’s disease program and relationship of clinical outcomes to baseline ulceration status. Inflamm Bowel Dis. 2017;23:S9. | ||
Sands BE, Gasink C, Jacobstein D, et al. Fistula healing in pivotal studies of ustekinumab in Crohn’s disease. Gastroenterology. 2017;152:S185. | ||
Batista DD, Yadav S, Harmsen WS, et al. Su1420 ustekinumab treatment for Crohn’s disease in clinical practice: experience at a tertiary medical center. Gastroenterology. 2014;146:S464–S465. | ||
Battat R, Kopylov U, Bessissow T, et al. Association between ustekinumab trough concentrations and clinical, biomarker, and endoscopic outcomes in patients with Crohn’s disease. Clin Gastroenterol Hepatol. 2017;15(9):1427–1434.e2. | ||
Greenup AJ, Rosenfeld G, Bressler B. Ustekinumab use in Crohn’s disease: a Canadian tertiary care centre experience. Scand J Gastroenterol. 2017;52(12):1354–1359. | ||
Harris KA, Horst S, Gadani A, et al. Patients with refractory Crohn’s disease successfully treated with ustekinumab. Inflamm Bowel Dis. 2016;22(2):397–401. | ||
Khorrami S, Ginard D, Marin-Jimenez I, et al. Ustekinumab for the treatment of refractory Crohn’s disease: the Spanish experience in a large multicentre open-label cohort. Inflamm Bowel Dis. 2016;22(7):1662–1669. | ||
Kopylov U, Afif W, Cohen A, et al. Subcutaneous ustekinumab for the treatment of anti-TNF resistant Crohn’s disease – the McGill experience. J Crohns Colitis. 2014;8(11):1516–1522. | ||
Ma C, Fedorak RN, Kaplan GG, et al. Clinical, endoscopic and radiographic outcomes with ustekinumab in medically-refractory Crohn’s disease: real world experience from a multicentre cohort. Aliment Pharmacol Ther. 2017;45(9):1232–1243. | ||
Ma C, Fedorak RN, Kaplan GG, et al. Long-term maintenance of clinical, endoscopic, and radiographic response to ustekinumab in moderate-to-severe Crohn’s disease: real-world experience from a multicenter cohort study. Inflamm Bowel Dis. 2017;23(5):833–839. | ||
Wils P, Bouhnik Y, Michetti P, et al; Groupe d’Etude Thérapeutique des Affections Inflammatoires du Tube Digestif. Subcutaneous ustekinumab provides clinical benefit for two-thirds of patients with Crohn’s disease refractory to anti-tumor necrosis factor agents. Clin Gastroenterol Hepatol. 2016;14(2):242–250.e1–e2. | ||
Hindryckx P, Baert F, Hart A, et al; Clinical Trial Committee Clincom of the European Crohn’s and Colitis Organisation (ECCO). Clinical trials in luminal Crohn’s disease: a historical perspective. J Crohns Colitis. 2014;8(11):1339–1350. | ||
Concato J, Shah N, Horwitz RI. Randomized, controlled trials, observational studies, and the hierarchy of research designs. N Engl J Med. 2000;342(25):1887–1892. | ||
Harvey RF, Bradshaw JM. A simple index of Crohn’s-disease activity. Lancet. 1980;1(8167):514. | ||
Danese S, Bonovas S, Peyrin-Biroulet L. Positioning ustekinumab in Crohn’s disease: from clinical evidence to clinical practice. J Crohns Colitis. 2017;11(10):1258–1266. | ||
Abraham C, Dulai PS, Vermeire S, et al. Lessons learned from trials targeting cytokine pathways in patients with inflammatory bowel diseases. Gastroenterology. 2017;152(2):374–388.e4. | ||
Deepak P, Sandborn WJ. Ustekinumab and anti-interleukin-23 agents in Crohn’s disease. Gastroenterol Clin North Am. 2017;46(3):603–626. | ||
Feagan BG, Sandborn WJ, D’Haens G, et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe Crohn’s disease: a randomised, double-blind, placebo-controlled phase 2 study. Lancet. 2017;389(10080):1699–1709. | ||
Sands BE, Chen J, Feagan BG, et al. Efficacy and safety of MEDI2070, an antibody against interleukin 23, in patients with moderate to severe Crohn’s disease: a phase 2a study. Gastroenterology. 2017;153(1):77–86.e6. | ||
Papp KA, Blauvelt A, Bukhalo M, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376:1551–1560. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.