")
Back to Journals » OncoTargets and Therapy » Volume 7
Clinical utility of trabectedin for the treatment of ovarian cancer: current evidence
Authors Mascilini F, Amadio G, Di Stefano MG, Ludovisi M, Di Legge A, Conte C, De Vincenzo R, Ricci C, Masciullo V, Salutari V, Scambia G, Ferrandina G
Received 10 April 2014
Accepted for publication 9 June 2014
Published 12 July 2014 Volume 2014:7 Pages 1273—1284
DOI https://doi.org/10.2147/OTT.S51550
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Floriana Mascilini,* Giulia Amadio,* Maria Grazia Di Stefano, Manuela Ludovisi, Alessia Di Legge, Carmine Conte, Rosa De Vincenzo, Caterina Ricci, Valeria Masciullo, Vanda Salutari, Giovanni Scambia, Gabriella Ferrandina
Gynecologic Oncology Unit, Department of Oncology, Catholic University of Rome, Italy
*These authors contributed equally to this work
Abstract: Among the pharmaceutical options available for treatment of ovarian cancer, attention has been increasingly focused on trabectedin (ET-743), a drug which displays a unique mechanism of action and has been shown to be active in several human malignancies. Currently, single agent trabectedin is approved for treatment of patients with advanced soft tissue sarcoma after failure of anthracyclines and ifosfamide, and in association with pegylated liposomal doxorubicin for treatment of patients with relapsed partially platinum-sensitive ovarian cancer. This review aims at summarizing the available evidence about the clinical role of trabectedin in the management of patients with epithelial ovarian cancer. Novel perspectives coming from a better understanding of trabectedin mechanisms of action and definition of patients subgroups likely susceptible to benefit of trabectedin treatment are also presented.
Keywords: ET-743, ovarian cancer, clinical trials
Introduction
Epithelial ovarian carcinoma is one of the most common gynecological malignancies, and the fifth most frequent cause of cancer death in women.1 The estimated numbers of new cases of ovarian cancer per year in the United States is 21,980, with an estimated number of deaths of 14,270.1,2
The standard of care for the management of ovarian cancer patients includes optimal cytoreduction (ie, residual tumor <1 cm) followed by adjuvant chemotherapy with a platinum/taxane combination associated and followed by bevacizumab in advanced stage, high-risk patients.3–5 However, despite the advances in surgical efforts and the achievement of high response rates (70%–80%) to front-line treatment, ovarian cancer remains the most lethal gynecological malignancy, with 50%–75% of cases experiencing progression/recurrence of disease; indeed, the 5-year overall survival (OS) is around 25%–30% in advanced stage disease.1,2
The major determinants of clinical outcome have been represented for years by the extent of residual tumor at primary surgery and sensitivity to platinum-based therapy, the latter parameter being defined according to the duration of the platinum-free interval (PFI).6 In the clinical setting, patients are considered platinum resistant if progression/recurrence of disease occurs during chemotherapy or within 6 months from its completion; salvage single-agent chemotherapy with nonplatinum drugs has always resulted in short-lived response rates in approximately 10%–25% of patients,7 until recent evidence showed that the addition of bevacizumab provides higher response rates and even prolongation of OS.8
On the other hand, patients defined as platinum sensitive (those recurring/progressing after 6 months from the end of primary treatment) have been usually triaged to platinum-based combinations,6 although this therapeutic paradigm has been modified by the increasingly relevant evidence from the OCEANS trial showing the advantage of combining the platinum-based regimen with bevacizumab as compared to the platinum-taxane regimen.9 In the context of the so-called platinum-sensitivity, relapse/progression within 6–12 months after the administration of primary chemotherapy represents a sort of gray zone in terms of platinum resistance/responsiveness, and this is strongly supported by the clinical evidences that in this subset of patients, response rates range between 27% and 33% in patients administered either platinum-based rechallenge versus nonplatinum drugs.10
In this context, the optimal management of partially platinum-sensitive recurrent ovarian cancer patients still remains to be firmly defined; indeed, based on preclinical and retrospective studies, it has been proposed that prolongation of PFI through the administration of platinum agents could result in a higher response rate to subsequent platinum rechallenge.11,12 This hypothesis represents the rationale sustaining the setup of prospective Phase III randomized trials aimed at verifying whether prolongation of PFI through the use of nonplatinum agents could result in a higher response rate and better clinical outcome compared to the immediate rechallenge with platinum-based regimens in partially platinum-sensitive recurrent ovarian cancer patients.
Overall, these results highlight the need to clarify the therapeutic scenario for each clinical setting in order to optimize any drug or drug combination therapeutic performances.
In this context, among the pharmaceutical options currently available in the armamentarium of medical treatment of ovarian cancer, much emphasis has been progressively placed on trabectedin (ET-743, Yondelis®; Zeltia, Madrid, Spain, and Johnson & Johnson, New Brunswick, New Jersey, USA), which had gained much attention because of its unique mechanism of action and the demonstration of clinical activity in ovarian cancer as well as other solid malignancies.13–16
Since 2007, trabectedin represents the first anticancer marine-derived drug that has obtained marketing authorization from the European Medicines Agency (EMA), and in many other countries worldwide for treatment of patients with advanced soft tissue sarcoma after failure of anthracyclines and ifosfamide, or for those patients who are unsuitable to receive these agents. Moreover, based on the reported results of OVA-301 Phase III randomized study,16 in 2009 EMEA granted marketing authorization for trabectedin combined with pegylated liposomal doxorubicin (PLD) for treatment of patients with relapsed partially platinum-sensitive ovarian cancer.17 This review will focus on the clinical role of trabectedin in the management of patients with epithelial ovarian cancer. A brief overview of the process of trabectedin development, as well as the new perspectives of trabectedin employment, will also be provided.
Trabectedin structure and mechanisms of action
Trabectedin is a marine-derived tetrahydroisoquinoline alkaloid with antitumor activity, originally isolated from the tunicate Ecteinaiscidia turbinata and currently synthetically produced.18 As shown in Figure 1, trabectedin is constituted by a pentacyclic backbone composed of two fused tetrahydroisochinoline rings linked to a ten-membered lactone bridge through a benzylic sulfide linkage and attached to an additional ring system.
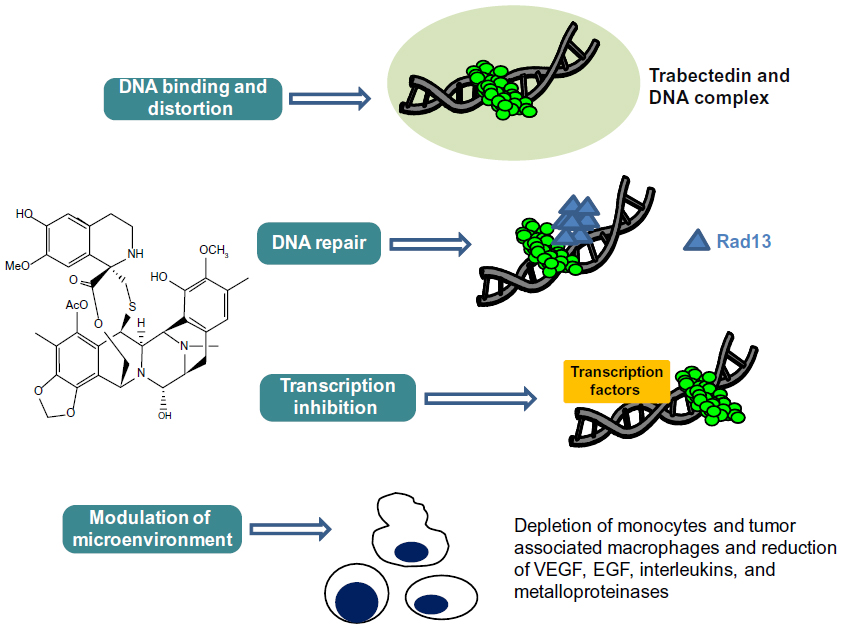 | Figure 1 Schematic picture of the unique and complex mechanism of action of trabectedin. |
Several studies have proposed that the mechanism of action of this compound is more complex than those exerted by conventional cytotoxic agents: in particular, its peculiar structure might allow the drug to interact with DNA through a covalent binding at the N2-guanine at the minor DNA groove, and also to protrude outside of the DNA helix, thus being available to bind several DNA-binding molecules such as transcriptional factors and DNA repair proteins.19,20 Binding to the minor DNA groove induces a bend of the helix toward the major groove and a DNA damage; this is recognized by the nucleotide excision repair (NER) system, and results in the accumulation of ternary DNA-trabectedin–protein repair complexes which lead, after collision with the replication fork, to the formation of double-strand DNA breaks, block of cell cycle, and induction of p53-independent apoptosis.21–24 These findings support the demonstration that, contrary to what is known for other DNA-binding agents, a functional NER system seems required for efficient trabectedin cytotoxicity; in fact, it has been shown that Chinese hamster and human ovarian cell lines deficient for genes essential for NER system activity, such as xeroderma pigmentosum, complementation group A, B, D, F and G (XPA, XPB, XPD, XPF, XPG), and excision repair cross-complementation 1 (ERCC1), are resistant to trabectedin, and this resistance is reverted by complementation of the cells with the corresponding gene.21,25
On the other hand, trabectedin is expected to be more effective in cells lacking functional homologous recombinant repair (HRR) mechanisms, such as those endowed with BReast CAncer (BRCA) gene mutation or BRCAness phenotype: preclinical data confirm that, besides the requirement of a functional transcription coupled NER machinery, a deficient HRR system is also needed for in vitro sensitivity to trabectedin.22
A retrospective analysis conducted in a cohort of paraffin-embedded tumor tissues from sarcoma patients treated with trabectedin as rescue therapy showed a correlation between low tumor BRCA1 messenger ribonucleic acid (mRNA) expression levels and both a higher percentage of patients free of disease progression at 6 months (33% versus 11%, P=0.02), and a longer median OS (15 months versus 5 months, P=0.0003) compared to patients with high tumor BRCA1 mRNA expression levels.26 Taken together, all these observations support the hypothesis that sensitivity to trabectedin correlates with functional NER and deficient HRR systems, features that can be exploited for optimal management of specific settings of patients.
Besides these direct effects on the DNA helix, trabectedin is able to interfere with transcription regulatory pathways in a promoter- and gene-dependent manner, as well as in a cell-dependent fashion; in particular, trabectedin has been shown to inhibit binding of transcription factors to DNA, thus blocking their transactivating effects. This has been reported in myxoid/round cell liposarcoma xenograft models in which therapeutic doses of trabectedin led to a detachment of the abnormal fusion protein FUS-CHOP from its promoters; this phenomenon translates into an improved drug sensitivity which becomes higher as far as the block of the transactivating activity of FUS-CHOP is prolonged.27 This area of research is getting more and more attention not only because it can provide novel information for a deeper understanding of the pleiotropic activity of trabectedin, but also because it might help defining the clinical settings most suitable to benefit of this peculiar drug.
Besides disrupting DNA function and interfering with transcription regulatory pathways, very recent studies have also highlighted that trabectedin could exert its antitumor activity by targeting some normal host cells. In particular, trabectedin has been shown to selectively deplete blood monocytes and tumor-associated macrophages in tumor-bearing mice as well as in tissue biopsies from soft tissue sarcoma and ovarian cancer patients.28–30 This selective effect seems to be ascribed to the ability of trabectedin to rapidly trigger the activation of Caspase 8 downstream of the TNF-related apoptosis-inducing ligand (TRAIL) membrane receptor, which is highly expressed in monocytes and macrophages compared to lymphocytes and neutrophils.29,30 Indeed, tumor-associated macrophages play a major role in the production of growth factors (ie, epidermal growth factor, chemokines, interleukins, metalloproteinases, and vascular endothelial growth factor), which promote tumor cell survival and metastatic phenotype. The demonstration that trabectedin was effective in a trabectedin-resistant in vivo model, while being not effective in the same cancer cell lines in vitro, clearly showed that the specific trabectedin-induced tumor cytotoxicity on circulating monocytes and macrophages could be involved in antitumor activity, and that part of antitumor activity of the drug could be ascribed to its ability to act as a tumor microenvironment modifier.
Overall, trabectedin can be considered more than a cytotoxic drug on the basis of the combination of multiple mechanisms of action, which provide the chance for a more multifaceted activity than initially formulated. Through these mechanisms trabectedin is likely to impact on relevant biological pathways involved in cancer biology, which may influence disease outcome.
Trabectedin pharmacokinetic features and Phase I studies
Trabectedin shows a high plasma protein binding (94%–98%), and a longer terminal half-life; in a population pharmacokinetic review on single agent trabectedin administered according to different schedules, the terminal half-life resulted at around 180 hours.31 Moreover, the distribution volume at steady state was generally reported to be large (up to 4,000 L) in the weekly administration as well as in the 3-weekly schedules.32–39
Trabectedin is metabolized in the liver mainly by the CYP3A4 enzyme, but also through the CYPC29, CYP2C19, and CYP2D6 isoenzymes and glutathione transferases.40 The major involvement of CYP3A4 isoenzyme in trabectedin metabolism in vivo may have some clinical implications. The interaction between trabectedin and pharmaceutical agents which share the same metabolic pathway cannot be excluded;41 moreover, the consequences of the higher levels and faster conversion performances of CYP3A4 in female human microsomes have to be taken into account.42 At the end, almost 60% of the drug is eliminated in the feces and only 6% in the urine.43,44
Table 1 summarizes the studies investigating the safety and assessing the maximum tolerated dose (MTD) for trabectedin used as single agent;32–39 different dose escalation and schedules have been explored, including 3-weekly schedules (1 day, 1-hour to 72-hour infusion; 5 consecutive days, 1-hour infusion), or every 4 weeks schedule (1 day, 1-hour to 3-hour infusion) (Table 1).
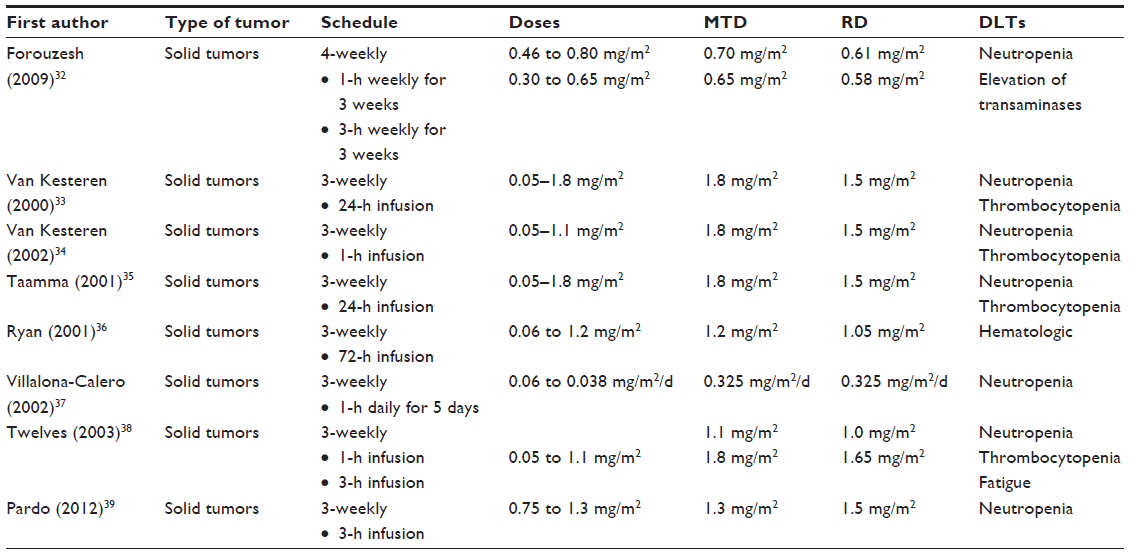 | Table 1 Phase I studies with trabectedin as single agent |
Neutropenia and thrombocytopenia were reported as the most frequent dose-limiting toxicities (DLT). Hepatotoxicity consisted mainly of elevation of transaminases; this toxicity was consistently reported to increase with trabectedin area under the curve (AUC), although it was always reversible (duration between 3–4 weeks), and not dose limiting.32 Moreover, hepatotoxicity was even lower when the dose was divided over 5 days compared to the single-dose schedule.37
Escalation of trabectedin was also investigated in patients with alteration of hepatic function stratified according to the basal levels of alkaline phosphatase (AP); MTD was defined only in the subgroup of cases with AP between normal and ≤1.5 mg/m2, and results equivalent to 1.3 mg/m2, while there were critical issues in full enrollment of patients with more elevated AP level. Interestingly enough, there was no difference in serum level of markers of liver function (bilirubin, transaminases, etc) according to different serum AP levels;39 this finding has been confirmed in a pharmacokinetic analysis in which no relation was evident between AP or transaminase levels with trabectedin pharmacokinetics.31 In conclusion, as far as the available data are concerned, trabectedin (at the dose of 1.3 mg/m2) can be utilized in patients with mild hepatic dysfunction, who nevertheless should be closely monitored.39
A summary of the Phase I studies with trabectedin in combination is provided in Table 2. The rationale for exploring the combination of trabectedin with platinum compounds relies on the available evidences about the molecular target of the two drugs. Platinum agents hit the major groove of DNA, thus inducing DNA damage which is repaired by the HRR system. On the other hand, trabectedin activity requires an efficient NER machinery.23,25 Moreover, earlier preclinical studies have demonstrated a synergistic effect of the combination.45 In 2009, Sessa et al46 published a Phase I study investigating cisplatin (40 mg/m2, Days 1 and 8, every 21 days) combined with escalating dose of trabectedin (from 0.3 to 0.7 mg/m2); 39 patients were treated in the dose-finding step, and ten patients were enrolled in the expansion part of the study. MTD of trabectedin was 0.7 mg/m2, with neutropenia representing the most common DLT, while the recommended doses (RD) were 0.5 mg/m2 and 0.6 mg/m2 in the previously treated and naïve patients, respectively. Other toxicities were represented by nausea/vomiting (67%), asthenia/fatigue (55%), and reversible transaminases elevation (51%). Confirmed partial responses were reported in four of 13 evaluable ovarian cancer patients, and in one patient with uterine leiomyosarcoma.46 Since the 3-weekly administration was considered to be responsible for a higher neutropenia and liver toxicity, a Phase I study utilizing the 3-weekly schedule was planned47; the regimen consisted of cisplatin at a fixed dose of 75 mg/m2, 1-hour intravenous infusion, followed by escalating doses of trabectedin in a 3-hour infusion, with both drugs administered on day 1. Two DLTs, grade 4 neutropenia longer than 7 days duration and grade 3 vomiting despite standard antiemetic therapy, occurred at the starting dose of trabectedin (0.75 mg/m2); therefore, the immediately lower dose (trabectedin 0.60 mg/m2) was evaluated in a total of eight patients. No DLTs occurred, and this was declared the recommended dose. Two out of five ovarian cancer patients experienced partial response to treatment (response rate, 17.0%; lasting 4.1 and 8.4 months, respectively), and it is worth noting that five patients had stable disease (including sarcoma, ovarian and breast cancer patients). These results, which are comparable to those obtained with the weekly schedule, and also with results from the Phase I study utilizing trabectedin combined with carboplatin, showed that the 3-weekly schedule is feasible, tolerable, and able to produce some antitumor activity.
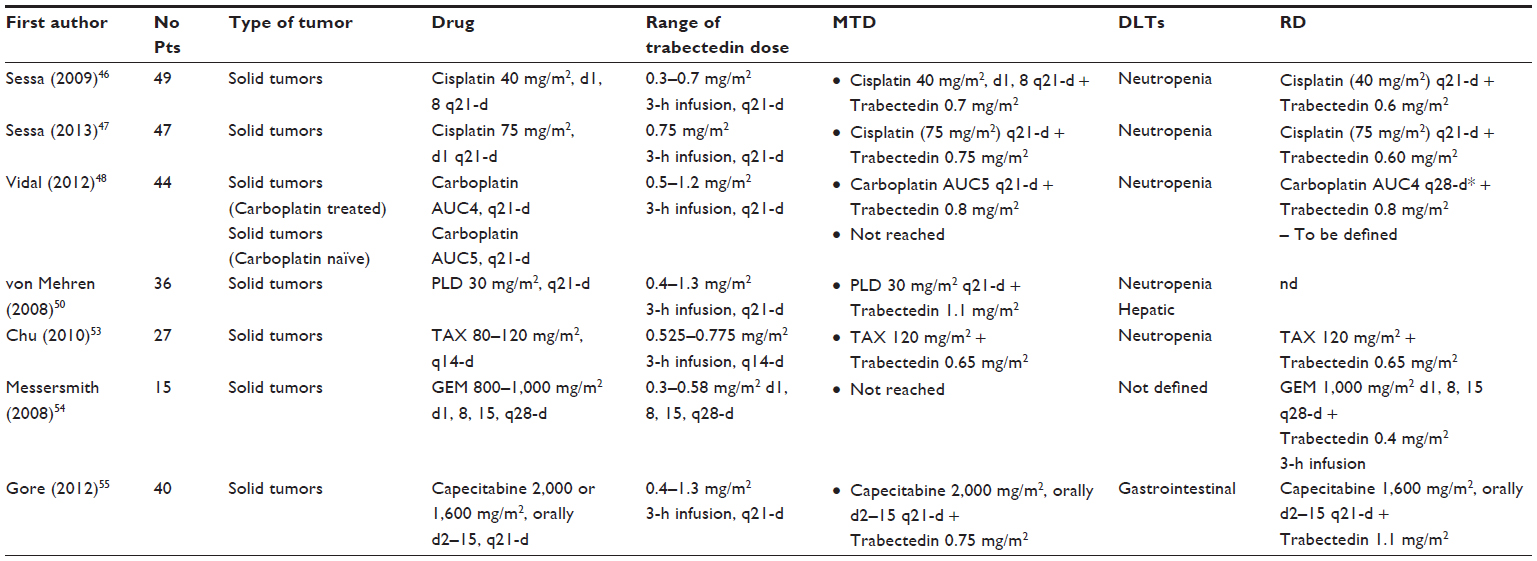 | Table 2 Phase I studies with trabectedin in combination |
The combination trabectedin-carboplatin was investigated in a Phase I study focused on the two different clinical settings:48 carboplatin-pretreated patients who received carboplatin AUC 4 (Group 1) and carboplatin-naïve patients who received carboplatin AUC 5 (Group 2), both as a 1-hour infusion followed by trabectedin at the dose range from 0.5–1.2 mg/m2 in the 3-weekly schedule. A total of 44 patients were enrolled and treated: in Group 1 (N=18), at the trabectedin dose of 1.0 mg/m2, only one out of ten patients had DLTs; however, given the high frequency of dose delays which resulted in 66.7% of rescheduling, the recommended dose was established to be 0.8 mg/m2 combined with carboplatin AUC 4. In Group 2, DLTs occurred at trabectedin 0.8 mg/m2 (3/8 patients), 1.0 mg/m2 (3/10 patients), and 1.2 mg/m2 (2/2 patients) with cumulative hematological toxicity associated with an important number of transfusions. In this group, neither the MTD nor the RD were established, thus requiring further investigation. Promising antitumor activity was found for this combination, especially in patients with advanced ovarian cancer and soft tissue sarcoma. In particular, in Group 1 patients, two partial responses and six stable disease were documented, while in Group 2 there were two partial responses and 14 stable disease.
One of the most promising trabectedin combinations is represented by the association with PLD: indeed, in vitro and in vivo studies had earlier suggested an additive or even a weak synergism between the two drugs according to type of experimental models.49 The combination of trabectedin plus PLD has been investigated in a Phase I study including 36 patients. Patients were administered PLD (30 mg/m2, 1-hour infusion), followed by one of the six trabectedin doses (0.4, 0.6, 0.75, 0.9, 1.1 and 1.3 mg/m2) infused over 3 hours and repeated every 21 days until evidence of complete response, disease progression, or unacceptable toxicity.50 The MTD of trabectedin was 1.1 mg/m2. Neither drug had its PK affected by concomitant administration compared with trabectedin and PLD each given as a single agent. The most common grade 3–4 toxicities were neutropenia (31%) and elevated transaminases (31%). Six patients responded (one complete response, five partial responses), with an objective response rate of 16.7%, and 14 patients (39.0%) have had stable disease for more than 4 months, including two out of four ovarian cancer patients enrolled.
As far as combination of paclitaxel and trabectedin is concerned, some earlier evidences from preclinical studies had already shown that the two drugs were able to exert a sequence-dependent synergistic cytotoxicity in sarcoma and breast cancer cells in vitro,51,52 and an additive effect in breast cancer xenografts.52 The Phase I study published by Chu et al53 investigated dose escalation of trabectedin (0.525–0.775 mg/m2, 3-hour infusion) plus paclitaxel (80–120 mg/m2), every 14 days. As expected, neutropenia was the main DLT. At the doses of paclitaxel 120 mg/m2, and trabectedin 0.65 mg/m2, there was only one DLT in eleven patients; however, given the long-lasting grade 4 neutropenia and the long recovery time, these doses were considered as the RD. As far as antitumor activity is concerned, response was seen in only one patient, but there were ten cases of stable disease which lasted >10 months in five patients.
Trabectedin and gemcitabine were administered on Day 1, 8, and 15 of a 28-day cycle at planned doses of 0.3–0.58 mg/m2 and 800–1,000 mg/m2 in the Phase I study by Messersmith et al54 There were no DLTs with trabectedin 0.4 mg/m2 and gemcitabine 1,000 mg/m2; however, the high frequency of dose modifications (reduction, delay) limited the determination of MTD for the weekly schedule, thus soliciting an alternative schedule of treatment. Finally, the Phase I study by Gore et al55 investigated trabectedin (0.4–1.3 mg/m2) and capecitabine (2,000 mg/m2/day orally every 3 weeks). Since gastrointestinal DLT toxicity were documented with 2,000 mg/m2/day capecitabine, the dose was reduced to 1,600 mg/m2/day, and escalation of trabectedin was started at 0.6 mg/m2. MTD resulted at 1,600 mg/m2/day plus trabectedin 1.1 mg/m2. No pharmacokinetic interaction was documented between the two drugs. One patient had partial response, while 18 patients had long-lasting stable disease.
Overall, the combination of trabectedin with the most commonly used agent in ovarian cancer was demonstrated to be feasible and endowed with some antitumor activity. None of the described combinations showed an influence of each of the two drugs as far as pharmacokinetic features are concerned.
Phase II studies with trabectedin as single agent or in combination
A summary of Phase II studies using trabectedin as a single agent in ovarian cancer56–58 is presented in Table 3. When considering the whole series in each study, the response rate ranges between 16.3% and 38.9%, although the relatively high patient heterogeneity likely represents a major limit for reliable comparisons. Indeed, the percentage of patients already administered up to two previous lines of chemotherapy ranged between 28.3% and 40.7%. More importantly, the rate of cases defined as bearing platinum-resistant versus platinum-sensitive disease largely differed across the studies; in particular, the fraction of platinum-resistant patients ranged between 3.8% up to 55.0%. Therefore, a more reliable analysis of results should focus on specific clinical settings. In the subgroup of platinum-resistant patients, the rate of response was, as expected, low (range: 6.3%–7.0%), while in patients with platinum-sensitive disease, trabectedin was able to induce objective response between 29.0% and 43.0%. A pooled analysis of the available Phase II studies59 reported an objective response rate of 26.0%, a median duration of response of 5.5 months, and a very encouraging rate of stable disease in almost 30% of cases. Interestingly enough, patients administered the 3-weekly schedule exhibited a higher response rate compared to cases administered the weekly regimen (36.0% versus 16.0%, P=0.0001). Finally, it has to be emphasized that trabectedin activity does not seem to be related to the amount of previous chemotherapy lines. In this context, a recently published large, retrospective study on 98 heavily treated recurrent ovarian cancer patients (median number of previous treatments: 4) reported an objective response rate of 27.5%, a rate which did not vary according to number of previously administered chemotherapy lines.60 In the pooled analysis of data relative to the 3-weekly regimens, median progression-free survival (PFS) was calculated to be 5.6 months with both the 24-hour and the 3-hour infusions; on the other hand, median PFS was only 2.8 months in patients treated with the weekly schedule.59 This difference was also documented for OS; indeed, median OS was longer for the 3-weekly 24-hour infusion (median OS: 20.4 months) and the 3-hour infusion (median OS: 17.1 months) compared to the weekly schedule (median OS: 13.7 months).59
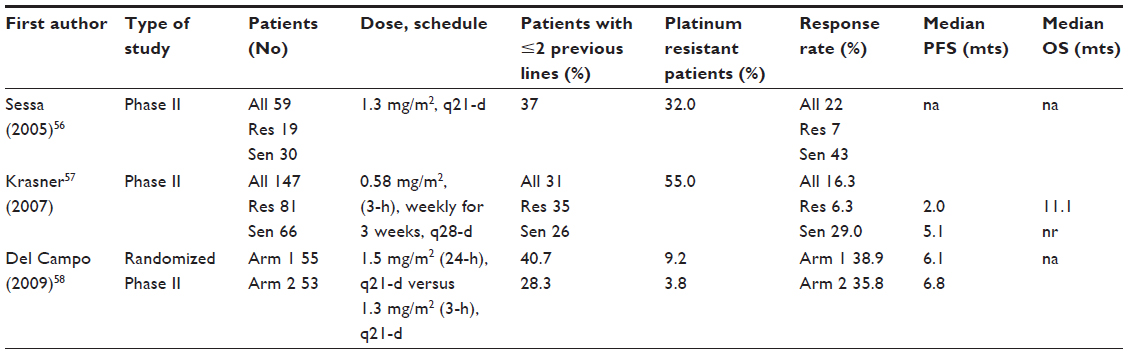 | Table 3 Phase II studies with trabectedin single agent or in combination |
As far as toxicity is concerned, reversible myelosuppression and transient elevation of transaminases were the most frequent hematological and nonhematological toxicities, respectively, regardless of the schedule of trabectedin administration (Table 4). In particular, grade 3–4 neutropenia was registered in 26% of patients from the pooled analysis of the three Phase II studies, while grade 3–4 elevation of AST and ALT was reported in 18% and 38% of cases, respectively.
 | Table 4 Toxicity from Phase II studies with trabectedin single agent or in combination |
Overall, all Phase II studies highlighted the promising activity of trabectedin single agent, especially in patients with platinum-sensitive disease, as well as a manageable and noncumulative toxicity profile of the drug.
Trabectedin: Phase III studies
Based on the strong rationale sustaining the association trabectedin/PLD and the favorable results from preclinical and Phase I studies, this combination was chosen for the direct comparison with PLD in the Phase III trial OVA-301 (NCT00113607). This study, planned in 2005, aimed at comparing trabectedin 1.1 mg/m2/PLD 30 mg/m2 every 21 days versus PLD 50 mg/m2 every 28 days in ovarian, peritoneal, and tubal cancer recurring/progressing after first-line chemotherapy,61 with the exclusion of refractory cases; patients were stratified according to the Eastern Cooperative Oncology Group Performance Status (0–1 versus 2) and platinum sensitivity (PFI <6 months versus PFI ≥6 months). Originally, the primary endpoint was OS, but it was later amended to PFS at the end of 2006. Secondary endpoints included response rates, duration of response, safety, and quality of life issues. Overall, 672 patients were enrolled (337 allocated to trabectedin/PLD versus 335 allocated to PLD). In the whole series, the response rate, as assessed by independent radiology review by RECIST (Response Evaluation Criteria in Solid Tumors), was significantly higher in trabectedin/PLD than PLD alone group (27.6% versus 18.8%, P=0.008). In platinum-resistant cases (n=242), no difference in response rate was observed in the combination versus PLD alone (13.4% versus 12.2%, respectively), while platinum-sensitive patients showed a higher response rate to trabectedin/PLD compared to PLD (35.3% versus 22.6%; P=0.0042).
Median PFS was 7.3 months for the combination arm and 5.8 months for the single agent arm, with a hazard ratio (HR) of 0.79 (95% confidence interval [CI]: 0.65–0.96; P=0.019). There was no difference in median PFS between the two arms in the platinum-resistant population; on the other hand, superiority of trabectedin/PLD versus PLD alone was shown in platinum-sensitive disease, since median PFS was 9.2 months versus 7.5 months, respectively, with a HR of 0.73 (95% CI: 0.56–0.95; P=0.017).
When analysis of PFS was performed in the subgroup of partially platinum-sensitive disease (N=214), the superiority of trabectedin/PLD versus the single agent was even more evident: median PFS was 7.4 versus 5.5 months, respectively; the HR was 0.65 (95% CI: 0.45–0.92; P=0.0152), which translated into a 35% reduction of progression of disease.62 Since then, however, it has become evident that an imbalance in the duration of PFI in favor of the PLD arm could have influenced outcome results. Mature data about OS have been published in 2012.63
After a median follow up of 47.4 months, 522 deaths were registered in the entire population: median OS was 22.2 months for trabectedin/PLD versus 18.9 months in PLD-treated patients, with a HR of 0.86 (95% CI: 0.72–1.02; P=0.0835).
However, subsequent analysis adjusting for PFI duration was performed and a statistically significant improvement of OS was shown favoring trabectedin/PLD with a HR of 0.82 (95% CI: 0.69–0.98; P=0.0285). Overall, in platinum-sensitive recurrent ovarian cancer patients there was no statistically significant difference according to treatment (median OS of 27 months for combination versus 24.2 months for single agent; HR: 0.83, 95% CI: 0.67–1.04, P=0.105). However, an additional unplanned exploratory analysis showed that patients with a PFI between 6–12 months showed the largest OS benefit from the combination, with a HR of 0.64 (95% CI: 0.47–0.86; P=0.0027).63
The selective superiority of trabectedin/PLD in the subset of partially platinum-sensitive patients could not be ascribed to the number of treatments administered after progression on the OVA-301 study: indeed, the percentage of patients receiving subsequent treatment after progression during the OVA-301 study was similar between the two arms (76% versus 77% for combination versus single agent arm); platinum-based regimens were less frequently administered to patients progressing after trabectedin/PLD than PLD (48% versus 55%). On the other hand, it was shown that median OS from the administration of subsequent platinum-based chemotherapy was longer in partially platinum-sensitive patients allocated to the trabectedin/PLD arm compared to those allocated to the PLD arm (median OS 13.3 months versus 9.8 months, respectively, HR: 0.63, P=0.0357), suggesting that the enhanced OS benefit with trabectedin/PLD compared to single agent PLD might be related to the prolongation of PFI.64 This hypothesis has fuelled the setup of Phase III randomized studies aiming at clarifying whether the artificial prolongation of PFI with a nonplatinum drug may improve the effectiveness of the overall treatment in patients with a PFI of 6–12 months after completion of primary chemotherapy. In particular, the MITO-8 study (NCT00657878) randomizes patients to the combination carboplatin/paclitaxel versus PLD or topotecan or gemcitabine) and implies a crossover for both arms at disease progression. On the other hand, the Inovatyon (International Ovarian Cancer Patients Trial With Yondelis) study (NCT01379989) investigates the superiority of trabectedin/PLD versus carboplatin/PLD in partially platinum-sensitive disease.
As far as toxicity issues are concerned, Grade 3/4 anemia, neutropenia, and thrombocytopenia were documented in 14%, 63%, and 18% of trabectedin/PLD cases, and were significantly more frequent compared to PLD alone. Among nonhematological toxicities, Grade 3/4 elevation of sGOT and sGPT was reported in 38% of cases, but it was described as of short duration, and of decreased magnitude with subsequent cycles. On the other hand, hand and foot syndrome was documented in only 4% of cases in the combination arm compared to 20% in the PLD alone arm. In spite of the increased hematological toxicity in the trabectedin/PLD group, there was no deterioration of QoL/patient-reported outcome, as well as in QLQ-C30, OV28, and EQ-5D questionnaires.65 Moreover, a very recent cost-effectiveness analysis based on the final survival data of the OVA-301 study confirmed a significant improvement of OS, and an increased cost-effectiveness ratio per quality-adjusted life-year compared to the original evaluation.66 Based on these results, trabectedin/PLD combination can be considered as the most effective nonplatinum-based combination in partially platinum-sensitive disease; this combination can be exploited not only to artificially prolong PFI with the aim to increase sensitivity to subsequent platinum treatment, but also to provide patients with an equivalent therapeutic alternative to platinum agents in case of residual neurotoxicity, history of platinum hypersensitivity, or repeated treatments with platinum regimens.
While awaiting for the identification of biomarkers predictive of trabectedin sensitivity,67 other specific ovarian cancer clinical settings more likely to benefit from trabectedin or trabectedin combinations are actively investigated; given the strong rationale sustaining the role of BRCA 1/2 mutation or BRCAness in conditioning responsiveness to trabectedin, the MITO-15 (Multicenter Italian Trials in Ovarian Cancer and Gynecology) study (NCT01772979) has been conducted to investigate the efficacy of single agent trabectedin in relapsed ovarian cancer with BRCA mutation or exhibiting the BRCAness phenotype. The trial has recently closed patient accrual, and analysis of data is ongoing. Finally, considering the recent introduction of bevacizumab in the first-line treatment, and the upcoming approval in the salvage setting, there is increasing interest in exploiting the combination of trabectedin with the antiangiogenetic drug. In this context, a Phase II study (NCT01735071) has been recently launched aimed at assessing the efficacy and safety of the combination trabectedin and bevacizumab with or without carboplatin in partially platinum-sensitive recurrent ovarian cancer patients.
Conclusion
Since its approval for treatment of partially platinum-sensitive recurrent disease, trabectedin has shown its potential for therapeutic exploitation in ovarian cancer; in particular, its peculiar mechanisms of action suggest its potential activity in specific subsets of ovarian cancer patients endowed with BRCA mutation or the so-called BRCAness phenotype (ie, serous, high-grade carcinomas; repeated response to platinum-based regimens); this is likely to enlarge in the future the clinical settings in which candidates can take advantage of even single agent trabectedin. Moreover, the acquisition of recent evidences supporting the efficacy of trabectedin in modulating tumor microenvironment opens new potential scenarios for combinations with agents directed against angiogenesis as well as molecules involved in macrophage activity.
Active search is ongoing in order to define biomarkers predictive of response to trabectedin treatment, and also to help clarify the biological mechanisms sustaining one of the special features of trabectedin activity, namely the achievement of long-lasting stable disease.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, MA J, Zou Z, Jemal A. Cancer statistics. CA Cancer J Clin. 2014;64(1):9–29. | |
Eisenkop SM, Spirtos NM, Friedman RL, Lin WC, Pisani AL, Perticucci S. Relative influences of tumor volume before surgery and the cytoreductive outcome on survival for patients with advanced ovarian cancer: a prospective study. Gynecol Oncol. 2003;90(2):390–396. | |
Harter P, Hilpert F, Mahner S, Heitz F, Pfisterer J, du Bois A. Systemic therapy in recurrent ovarian cancer: current treatment options and new drugs. Expert Rev Anticancer Ther. 2010;10(1):81–88. | |
Burger RA, Brady MF, Bookman MA, et al; Gynecologic Oncology Group. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365(26):2473–2483. | |
Perren TJ, Swart AM, Pfisterer J, et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365(26):2484–2496. | |
Pujade-Lauraine E. How to approach patients in relapse. Ann Oncol. 2012;23 Suppl 10:x128–x131. | |
Naumann RW, Coleman RL. Management strategies for recurrent platinum-resistant ovarian cancer. Drugs. 2011;71(11):1397–1412. | |
Pujade-Lauraine E, Hilpert F, Weber B, et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: The AURELIA open-label randomized phase III trial. J Clin Oncol. 2014;32(13):1302–1308. | |
Aghajanian C, Blank SV, Goff BA, et al. OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol. 2012;30(17):2039–2045. | |
Colombo N, Gore M. Treatment of recurrent ovarian cancer relapsing 6–12 months post platinum-based chemotherapy. Crit Rev Oncol Hematol. 2007;64(2):129–138. | |
Bookman MA. Extending the platinum-free interval in recurrent ovarian cancer: the role of topotecan in second-line chemotherapy. Oncologist. 1999;4(2):87–94. | |
Pignata S, Ferrandina G, Scarfone G, et al; SOCRATES and MITO investigators. Extending the platinum-free interval with a non-platinum therapy in platinum-sensitive recurrent ovarian cancer. Results from the SOCRATES Retrospective Study. Oncology. 2006;71(5–6):320–326. | |
Vincenzi B, Napolitano A, Frezza AM, Schiavon G, Santini D, Tonini G. Wide-spectrum characterization of trabectedin: biology, clinical activity and future perspectives. Pharmacogenomics. 2010;11(6):865–878. | |
Demetri GD, Chawla SP, von Mehren M, et al. Efficacy and safety of trabectedin in patients with advanced or metastatic liposarcoma or leiomyosarcoma after failure of prior anthracyclines and ifosfamide: results of a randomized phase II study of two different schedules. J Clin Oncol. 2009;27(25):4188–4196. | |
Monk BJ, Herzog TJ, Kaye SB, et al. Trabectedin plus pegylated liposomal doxorubicin (PLD) versus PLD in recurrent ovarian cancer: overall survival analysis. Eur J Cancer. 2012;48(15):2361–2368. | |
Monk BJ, Herzog TJ, Kaye SB, et al. Trabectedin plus pegylated liposomal Doxorubicin in recurrent ovarian cancer. J Clin Oncol. 2010;28(19):3107–3114. | |
European Medicines Agency (EMA). Assessment Report for Yondelis. International non-proprietary name/common name; Trabectedin procedure. No EMEA/H/C/000773/11/0008 [webpage on the Internet]. http://www.ema.europa.eu/humandocs/PDFs/EPAR/Yondelis/EMEA-H-773-II-08-AR.pdf. 2009. | |
Cuevas JC, Francesh A. Development of Yondelis (Trabectedin, ET-743). A semisynthetic process solves the supply problem. Nat Prod Rev. 2008;26(3):322–337. | |
Moore II BM, Seaman FC, Hurley LH. NMR-based model of an ecteinascidin 743-DNA adduct. J Am Chem Soc. 1997;119(23):5475–5476. | |
D’Incalci M, Galmarini CM. A review of trabectedin (ET-743): a unique mechanism of action. Mol Cancer Ther. 2010;9(8):2157–2163. | |
Takebayashi Y, Pourquier P, Zimonjic DB, et al. Antiproliferative activity of ecteinascidin 743 is dependent upon transcription-coupled nucleotide-excision repair. Nat Med. 2001;7(8):961–966. | |
Erba E, Bergamaschi D, Bassano L, et al. Ecteinascidin-743 (ET-743), a natural marine compound, with a unique mechanism of action. Eur J Cancer. 2001;37(1):97–105. | |
Tavecchio M, Simone M, Erba E, et al. Role of homologous recombination in trabectedin-induced DNA damage. Eur J Cancer. 2008;44(4):609–618. | |
Soares DG, Machado MS, Rocca CJ, et al. Trabectedin and its C subunit modified analogue PM01183 attenuate nucleotide excision repair and show activity toward platinum-resistant cells. Mol Cancer Ther. 2011;10(8):1481–1489. | |
Damia G, Silvestri S, Carrassa L, et al. Unique pattern of ET-743 activity in different cellular systems with defined deficiencies in DNA-repair pathways. Int J Cancer. 2001;92(4):583–588. | |
Schöffski P, Taron M, Jimeno J, et al. Predictive impact of DNA repair functionality on clinical outcome of advanced sarcoma patients treated with trabectedin: a retrospective multicentric study. Eur J Cancer. 2011;47(7):1006–1012. | |
Di Giandomenico S, Frapolli R, Bello E, et al. Mode of action of trabectedin in myxoid liposarcomas. Oncogene. 2013. | |
Allavena P, Signorelli M, Chieppa M, et al. Anti-inflammatory properties of the novel antitumor agent yondelis (trabectedin): inhibition of macrophage differentiation and cytokine production. Cancer Res. 2005;65(7):2964–2971. | |
Germano G, Frapolli R, Simone M, et al. Antitumor and anti-inflammatory effects of trabectedin on human myxoid liposarcoma cells. Cancer Res. 2010;70(6):2235–2244. | |
Germano G, Frapolli R, Belgiovine C, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23(2):249–262. | |
Perez-Ruixo JJ, Zannikos P, Hirankarn S, et al. Population pharmacokinetic meta-analysis of trabectedin (ET-743, Yondelis) in cancer patients. Clin Pharmacokinet. 2007;46(10):867–884. | |
Forouzesh B, Hidalgo M, Chu Q, et al. Phase I and pharmacokinetic study of trabectedin as a 1- or 3-hour infusion weekly in patients with advanced solid malignancies. Clin Cancer Res. 2009;15(10):3591–3599. | |
van Kesteren C, Cvitkovic E, Taamma A, et al. Pharmacokinetics and pharmacodynamics of the novel marine-derived anticancer agent ecteinascidin 743 in a phase I dose-finding study. Clin Cancer Res. 2000;6(12):4725–4732. | |
van Kesteren C, Twelves C, Bowman A, et al. Clinical pharmacology of the novel marine-derived anticancer agent Ecteinascidin 743 administered as a 1- and 3-h infusion in a phase I study. Anticancer Drugs. 2002;13(4):381–393. | |
Taamma A, Misset JL, Riofrio M, et al. Phase I and pharmacokinetic study of ecteinascidin-743, a new marine compound, administered as a 24-hour continuous infusion in patients with solid tumors. J Clin Oncol. 2001;19(5):1256–1265. | |
Ryan DP, Supko JG, Eder JP, et al. Phase I and pharmacokinetic study of ecteinascidin 743 administered as a 72-hour continuous intravenous infusion in patients with solid malignancies. Clin Cancer Res. 2001;7(2):231–242. | |
Villalona-Calero MA, Eckhardt SG, Weiss G, et al. A phase I and pharmacokinetic study of ecteinascidin-743 on a daily x 5 schedule in patients with solid malignancies. Clin Cancer Res. 2002;8(1):75–85. | |
Twelves C, Hoekman K, Bowman A, et al. Phase I and pharmacokinetic study of Yondelis (Ecteinascidin-743; ET-743) administered as an infusion over 1 h or 3 h every 21 days in patients with solid tumours. Eur J Cancer. 2003;39(13):1842–1851. | |
Pardo B, Salazar R, Ciruelos E, et al. Phase I and pharmacokinetic study of trabectedin 3-hour infusion every three weeks in patients with advanced cancer and alteration of hepatic function. Med Oncol. 2012;29(3):2240–2250. | |
Brandon EF, Meijerman I, Klijn JS, et al. In-vitro cytotoxicity of ET-743 (Trabectedin, Yondelis), a marine anti-cancer drug, in the Hep G2 cell line: influence of cytochrome P450 and phase II inhibition, and cytochrome P450 induction. Anticancer Drugs. 2005;16(9):935–943. | |
Levy RH, Thummel KE, Trager WF, Hansten PD, Eichelbaum M, editors. Metabolic Drug Interactions. Philadelphia: Lippincott Williams and Wilkins, 2000. | |
Reid JM, Kuffel MJ, Ruben SL, et al. Rat and human liver cytochrome P-450 isoform metabolism of ecteinascidin 743 does not predict gender-dependent toxicity in humans. Clin Cancer Res. 2002;8(9):2952–2962. | |
Beumer JH, Lopez-Lazaro L, Schellens JH, Beijnen JH, van Tellingen O. Evaluation of human plasma protein binding of trabectedin (Yondelis, ET-743). Curr Clin Pharmacol. 2009;4(1):38–42. | |
Yondelis, INN-trabectedin – Europa [webpage on the Internet]. http://www.ema.europa.eu/docs/it_IT/document./WC500045832.pdf. | |
D’Incalci M, Colombo T, Ubezio P, et al. The combination of yondelis and cisplatin is synergistic against human tumor xenografts. Eur J Cancer. 2003;39(13):1920–1926. | |
Sessa C, Cresta S, Noberasco C, et al. Phase I clinical and pharmacokinetic study of trabectedin and cisplatin in solid tumours. Eur J Cancer. 2009;45(12):2116–2122. | |
Sessa C, Del Conte G, Christinat A, et al. Phase I clinical and pharmacokinetic study of trabectedin and cisplatin given every three weeks in patients with advanced solid tumors. Invest New Drugs. 2013;31(5):1236–1243. | |
Vidal L, Magem M, Barlow C, et al. Phase I clinical and pharmacokinetic study of trabectedin and carboplatin in patients with advanced solid tumors. Invest New Drugs. 2012;30(2):616–628. | |
Meco D, Colombo T, Ubezio P, et al. Effective combination of ET-743 and doxorubicin in sarcoma: preclinical studies. Cancer Chemother Pharmacol. 2003;52(2):131–138. | |
von Mehren M, Schilder RJ, Cheng JD, et al. A phase I study of the safety and pharmacokinetics of trabectedin in combination with pegylated liposomal doxorubicin in patients with advanced malignancies. Ann Oncol. 2008;19(10):1802–1809. | |
Takahashi N, Li WW, Banerjee D, Scotto KW, Bertino JR. Sequence-dependent enhancement of cytotoxicity produced by ecteinascidin 743 (ET-743) with doxorubicin or paclitaxel in soft tissue sarcoma cells. Clin Cancer Res. 2001;7(10):3251–3257. | |
Takahashi N, Li W, Banerjee D, et al. Sequence-dependent synergistic cytotoxicity of ecteinascidin-743 and paclitaxel in human breast cancer cell lines in vitro and in vivo. Cancer Res. 2002;62(23):6909–6915. | |
Chu Q, Mita A, Forouzesh B, et al. Phase I and pharmacokinetic study of sequential paclitaxel and trabectedin every 2 weeks in patients with advanced solid tumors. Clin Cancer Res. 2010;16(9):2656–2665. | |
Messersmith WA, Jimeno A, Ettinger D, et al. Phase I trial of weekly trabectedin (ET-743) and gemcitabine in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2008;63(1):181–188. | |
Gore L, Rivera E, Basche M, et al. Phase I combination study of trabectedin and capecitabine in patients with advanced malignancies. Invest New Drugs. 2012;30(5):1942–1949. | |
Sessa C, De Braud F, Perotti A, et al. Trabectedin for women with ovarian carcinoma after treatment with platinum and taxanes fails. J Clin Oncol. 2005;23(9):1867–1874. | |
Krasner CN, McMeekin DS, Chan S, et al. A Phase II study of trabectedin single agent in patients with recurrent ovarian cancer previously treated with platinum-based regimens. Br J Cancer. 2007;97(12):1618–1624. | |
Del Campo JM, Roszak A, Bidzinski M, et al; Yondelis Ovarian Cancer Group. Phase II randomized study of trabectedin given as two different every 3 weeks dose schedules (1.5 mg/m2 24 h or 1.3 mg/m2 3 h) to patients with relapsed, platinum-sensitive, advanced ovarian cancer. Ann Oncol. 2009;20(11):1794–1802. | |
del Campo JM, Sessa C, Krasner CN, et al. Trabectedin as single agent in relapsed advanced ovarian cancer: results from a retrospective pooled analysis of three phase II trials. Med Oncol. 2013;30(1):435. | |
Ferrandina G, Salutari V, Vincenzi B, et al. Trabectedin as single agent in the salvage treatment of heavily treated ovarian cancer patients: a retrospective, multicenter study. Gynecol Oncol. 2013;130(3):505–510. | |
Monk BJ, Herzog TJ, Kaye SB, et al. Trabectedin plus pegylated liposomal Doxorubicin in recurrent ovarian cancer. J Clin Oncol. 2010;28(19):3107–3114. | |
Poveda A, Vergote I, Tjulandin S, et al. Trabectedin plus pegylated liposomal doxorubicin in relapsed ovarian cancer: outcomes in the partially platinum-sensitive (platinum-free interval 6–12 months) subpopulation of OVA-301 phase III randomized trial. Ann Oncol. 2011;22(1):39–48. | |
Monk BJ, Herzog TJ, Kaye SB, et al. Trabectedin plus pegylated liposomal doxorubicin (PLD) versus PLD in recurrent ovarian cancer: overall survival analysis. Eur J Cancer. 2012;48(15):2361–2368. | |
Kaye SB, Colombo N, Monk BJ, et al. Trabectedin plus pegylated liposomal doxorubicin in relapsed ovarian cancer delays third-line chemotherapy and prolongs the platinum-free interval. Ann Oncol. 2011;22(1):49–58. | |
Krasner CN, Poveda A, Herzog TJ, et al. Patient-reported outcomes in relapsed ovarian cancer: results from a randomized Phase III study of trabectedin with pegylated liposomal doxorubicin (PLD) versus PLD alone. Gynecol Oncol. 2012;127(1):161–167. | |
Fisher M, Gore M. Cost-effectiveness of trabectedin plus pegylated liposomal doxorubicin for the treatment of women with relapsed platinum-sensitive ovarian cancer in the UK: analysis based on the final survival data of the OVA-301 trial. Value Health. 2013;16(4):507–516. | |
Monk BJ, Kaye SB, Poveda A, et al. Nibrin is a marker of clinical outcome in patients with advanced serous ovarian cancer treated in the phase III OVA-301 trial. Gynecol Oncol. 2014;132(1):176–180. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.