")
Back to Archived Journals » Core Evidence » Volume 10
Clinical utility of tadalafil in the treatment of pulmonary arterial hypertension: an evidence-based review
Authors Henrie A, Nawarskas J, Anderson J
Received 2 April 2015
Accepted for publication 10 June 2015
Published 2 November 2015 Volume 2015:10 Pages 99—109
DOI https://doi.org/10.2147/CE.S58457
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Adam M Henrie, James J Nawarskas, Joe R Anderson
College of Pharmacy, University of New Mexico, Albuquerque, NM, USA
Abstract: Pulmonary arterial hypertension (PAH) is a chronic and disabling condition characterized by an elevated pulmonary vascular resistance and an elevated mean pulmonary arterial pressure. Despite recent improvements in treatment availability, PAH remains challenging to treat, burdensome for patients, and ultimately incurable. Tadalafil is a phosphodiesterase-5 inhibitor that is administered once daily by mouth for the treatment of PAH. Current treatment guidelines recommend tadalafil as an option for patients with World Health Organization functional class II or III PAH. In a placebo-controlled clinical trial, patients taking tadalafil demonstrated significantly improved exercise capacity as measured by the 6-minute walk distance. Patients also experienced decreased incidence of clinical worsening, increased quality of life, and improved cardiopulmonary hemodynamics. Uncontrolled studies and smaller trials have indicated a possible role for tadalafil as a suitable alternative to sildenafil and as a beneficial add-on option when used in combination with other treatments for PAH. Tadalafil is generally safe and well tolerated. Adverse events are typically mild-to-moderate in intensity, and discontinuation rates are usually low. The purpose of this review is to provide an evidence-based evaluation of the clinical utility of tadalafil in the treatment of PAH.
Keywords: tadalafil, phosphodiesterase-5 inhibitor, pulmonary arterial hypertension
Introduction
Pulmonary arterial hypertension (PAH) is a rare, however, debilitating chronic condition that remains progressive and incurable despite recent approval of several novel treatment options. In PAH, blood flow through the lungs is impaired, resulting in increased pulmonary vascular resistance (PVR) and an elevated mean pulmonary arterial pressure (mPAP). These factors often lead to pulmonary vasculature fibrosis and eventual right ventricular heart failure.1 Pulmonary hypertension is classified by the World Health Organization (WHO) into five groups based on a diversity of etiologies, including group 1: PAH, which is idiopathic, heritable, drug/toxin induced, or associated with connective tissue disease, portal hypertension, HIV infection, or congenital heart disease; group 2: pulmonary hypertension due to left heart disease; group 3: pulmonary hypertension due to lung disease; group 4: chronic thromboembolic pulmonary hypertension; and group 5: pulmonary hypertension with unclear multifactorial mechanisms.2
Patients with PAH commonly present with dyspnea on exertion; however, additional symptoms, including fatigue, weakness, angina, syncope, palpitations, and lower extremity edema, may also be present.3 The severity of PAH is based upon patient symptoms and activity level and is categorized by the WHO functional classification scheme which is as follows: WHO Class I patients have no limitation in physical activity and do not experience symptoms (dyspnea, fatigue, chest pain, or near syncope) with ordinary physical activity; WHO Class II patients have a slight limitation in physical activity experiencing symptoms with ordinary physical activity; WHO Class III patients have a marked limitation in physical activity experiencing symptoms with less than ordinary activity; and WHO Class IV patients are unable to carry out any physical activity without symptoms and experience symptoms at rest or with very slight activity.4 The 6-minute walk distance (6MWD) and the Borg dyspnea scale are often employed by clinicians and researchers to quantify a patient’s exercise capacity and breathing difficulty as well as to gage response to treatment. Cardiopulmonary hemodynamics, including PVR, mPAP, pulmonary arterial wedge pressure, and cardiac index (CI), are also obtained and assessed throughout the disease diagnostic and monitoring process.5
PAH is associated with poor survival, increased morbidity, and diminished quality of life (QoL).6,7 Patients often require a multidisciplinary approach to treatment in which needs relating to physical care as well as emotional and social care can be met.7,8 Currently, a number of guidelines exist to aid clinicians in treating patients with PAH.3,4,7,9 Although the available guidelines vary slightly in content, they generally agree in treatment approach. Goals of the treatment include improvement of symptoms, QoL, and survival.3 Patients who have less severe disease and who respond during acute vasoreactivity testing are initially started on a trial therapy with an oral calcium channel blocker (CCB). Patients who either fail to respond during acute vasoreactivity testing, fail to maintain a response while taking an oral CCB, or present with more severe diseases are considered for treatment with other approved options. Prostacyclin analogs (epoprostenol, iloprost, treprostinil), endothelin receptor antagonists (ERAs; ambrisentan, bosentan, macitentan), phosphodiesterase-5 (PDE-5) inhibitors (sildenafil, tadalafil), and a soluble guanylate cyclase (sGC) stimulator (riociguat) are all currently available for the treatment of PAH (Table 1). Oral agents, such as ERAs, PDE-5 inhibitors, treprostinil, and riociguat, are recommended for patients with WHO functional class II. Patients with WHO functional class III are treated with either an oral medication or a prostacyclin analog (inhaled or parenteral). Patients at higher risk, WHO functional class IV, should be treated with intravenous epoprostenol.9 Variations from this approach as well as treatment after inadequate clinical response to initial therapy can include strategies such as combination therapy and are preferentially implemented at expert PAH care centers according to the clinician’s judgment.4 Adjunctive therapies, including diuretics, anticoagulants, digoxin, or supplemental oxygen, may also be employed to better achieve treatment goals.
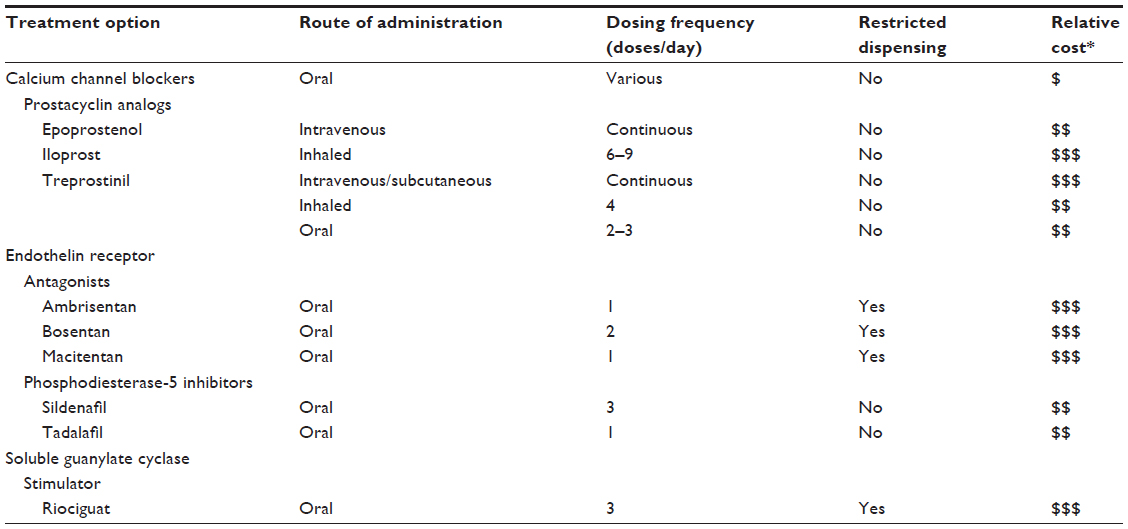 | Table 1 Treatment options for pulmonary arterial hypertension |
Although the causal mechanisms for the pathogenesis of PAH remain unclear, the progression of PAH is the result of pulmonary arterial endothelial dysfunction leading to an imbalance between key pulmonary vascular mediators, such as decreased nitric oxide (NO) and prostracylin and increased endothelin-1.11,12 This imbalance leads to increased thrombosis, vasoconstriction, and proliferation of smooth muscle and endothelial cells within the pulmonary vasculature.12 Current treatments are directed at correcting the imbalance in these mediators.
While the body of evidence supporting PAH treatment is continually growing, there remain several important limitations to current treatment recommendations.13 PAH has many etiologies, including some which remain grouped as idiopathic. Studies have used a broad definition of PAH in patient recruitment resulting in efficacy data from a heterogeneous selection of etiologies. As such, there are limitations in knowledge about specific treatment efficacy in each form of PAH.4 Furthermore, a few studies have evaluated the relative utility of available treatment options in head-to-head comparisons. This has resulted in a paucity of data supporting the use of a specific agent as first-line treatment.9,14 In addition to limitations in treatment recommendations, currently available treatment options can be at times challenging and burdensome to use. With the exception of CCBs, there are no means to effectively predict benefit from the available treatment options.4 Treatment options can be overly burdensome to patients as a result of high cost, frequent dosing, and inconvenient administration. Several agents are contraindicated in pregnancy and possess clinically important drug interactions.4,9 These limitations and others drive in part the need for novel treatment options. The purpose of this review is to provide an evidence-based evaluation of the clinical utility of tadalafil in the treatment of PAH.
Tadalafil
As mentioned previously, NO production and release is decreased as a result of endothelial dysfunction.15 NO causes vasodilation of the pulmonary vasculature by increasing the production of the second messenger cyclic guanosine monophosphate (cGMP) via activation of sGC (Figure 1).16 cGMP activates protein kinase G, which in turn activates sarcolemmal potassium channels. This results in intracellular hyperpolarization and inhibition of voltage-gated calcium channels ultimately leading to vasorelaxation.17 Thus, the decrease in NO availability seen in PAH results in decreased cGMP and increased pulmonary arterial vasoconstriction. PDE-5 is the predominant phosphodiesterase in the pulmonary vasculature and is responsible for the rapid degradation and inactivation of cGMP.18,19 Treatment with PDE-5 inhibitors (sildenafil or tadalafil) results in a competitive inhibition of cGMP degradation resulting in increased cGMP concentrations.20 The increased cGMP concentrations lead to relaxation of pulmonary vascular smooth muscle and vasodilation of the pulmonary vascular bed.16 Currently, sildenafil (Revatio®) and tadalafil (Adcirca®) are the only PDE-5 inhibitors approved by the United States Food and Drug Administration for the treatment of WHO group 1 PAH.21,22 Other PDE-5 inhibitors, such as vardenafil, have not been approved for the treatment of PAH, and the limited evidence available using these drugs in the treatment of PAH prohibit meaningful comparison to tadalafil. Several pharmacokinetic and pharmacodynamic differences exist between sildenafil and tadalafil (Table 2). One of the main pharmacokinetic differences is the long plasma half-life (t1/2) of 17.5 hours with tadalafil, which facilitates once-daily dosing as compared to the required three times daily dosing of sildenafil.22,23 When compared to sildenafil, the less frequent dosing of tadalafil is advantageous in terms of patient convenience and adherence.24 Tadalafil is primarily metabolized by the cytochrome P450 3A (CYP3A) enzyme subfamily to an inactive metabolite. As such, concomitant administration with medications that induce or inhibit CYP3A may result in significant drug–drug interactions.21,25 Bosentan, an ERA used in the treatment of PAH, induces CYP3A4 and has been demonstrated to decrease tadalafil serum concentrations and exposure.25 Although tadalafil is not primarily eliminated renally, starting doses of tadalafil require adjustment in patients with a creatinine clearance (CrCl) of 31–80 mL/min, and tadalafil should be avoided in patients with a CrCl <30 mL/min or in patients on hemodialysis.21
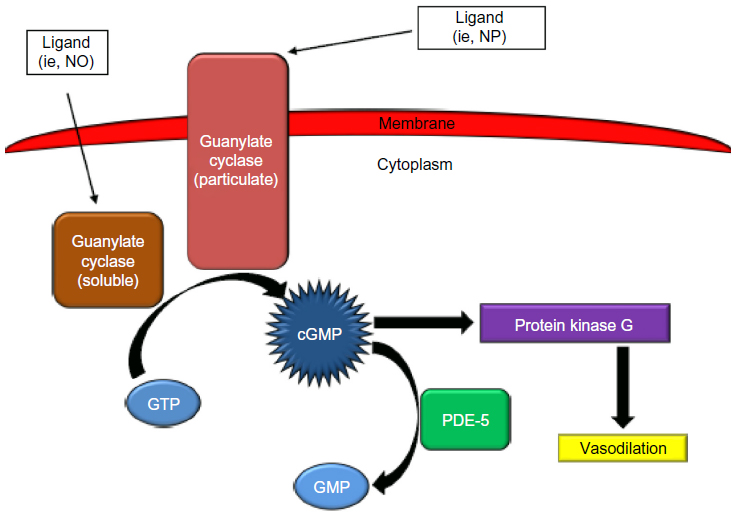 | Figure 1 The role of phosphodiesterase-5 (PDE-5) in the intracellular signaling pathway of the pulmonary vasculature. |
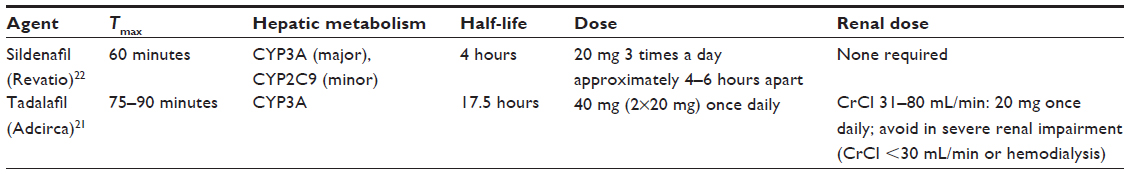 | Table 2 Phosphodiesterase-5 inhibitors used for the treatment of pulmonary hypertension |
The pharmacodynamic differences between sildenafil and tadalafil are based on each agent’s selectivity for various PDE isozymes.26 Tadalafil is >1,000-fold more selective for PDE-5 than for PDE-1–4 and PDE-7–10, 780-fold more selective for PDE-5 than for PDE-6, and only seven-fold more selective for PDE-5 than for PDE-11. In comparison, sildenafil is 41-fold more selective for PDE-5 than for PDE-1, seven-fold more selective for PDE-5 than for PDE-6, and 203-fold more selective for PDE-5 than for PDE-11. These differences in selectivity may result in the differences in adverse effects dependent on the isozyme location and activity. PDE-6 is expressed only in the retina and is important for visual transduction. Inhibition of PDE-6 can result in visual disturbances, which have been more frequently reported with sildenafil than with tadalafil. PDE-1 is expressed in the brain, myocardial cells, and vascular smooth muscle cells. Inhibition of PDE-1 may cause vasodilatation, flushing, and tachycardia. PDE-11 is expressed in the liver, kidney, skeletal muscle, prostate, and testes. Tadalafil is less selective for PDE-11 than sildenafil and is more likely to be associated with back pain and myalgias than with sildenafil.
Tadalafil, as well as other PDE-5 inhibitors, has several pharmacodynamic drug–drug interactions of concern. Tadalafil does not significantly lower systemic blood pressure; however, concomitant administration of tadalafil with medications that do lower systemic blood pressure can result in potentiation of hypotension. Coadministration of tadalafil with nitrates is contraindicated due to the risk of significant hypotension, and, because of the long t1/2 of tadalafil, nitrates should not be administered for 48 hours following tadalafil administration.21 Caution is warranted when tadalafil is used in combination with certain antihypertensives such as alpha-adrenergic blocking agents (eg, doxazosin).21 PDE-5 inhibitors, including tadalafil, are contraindicated with riociguat, a recently approved agent for the treatment of PAH.27 Riociguat is a sGC stimulator that results in increased production of cGMP and resultant arterial vasodilation (Figure 1). Because PDE-5 inhibitors decrease degradation of cGMP, there is a risk of profound hypotension if administered concomitantly with riociguat. Though not documented, it would be expected that coadministration of tadalafil with prostacyclin analogs would increase the risk of hypotension and should be used with caution.
As of 2015, a monthly supply of Adcirca 40 mg once daily is estimated to cost about US$2,700. While there is currently no economic evidence comparing tadalafil to other PAH treatment options, several analyses have demonstrated a favorable cost-effective profile for sildenafil when compared to other PAH treatment options.28,29 Tadalafil may also be a cost-effective option; however, further research is needed to provide insight into tadalafil’s comparative cost-effectiveness. PAH treatment guidelines recommend using tadalafil in patients who are WHO functional class II or III.3,4,7,9 In recent years, a number of studies have provided insight into the clinical efficacy and effectiveness of tadalafil in the treatment of PAH (Table 3).24,30–41
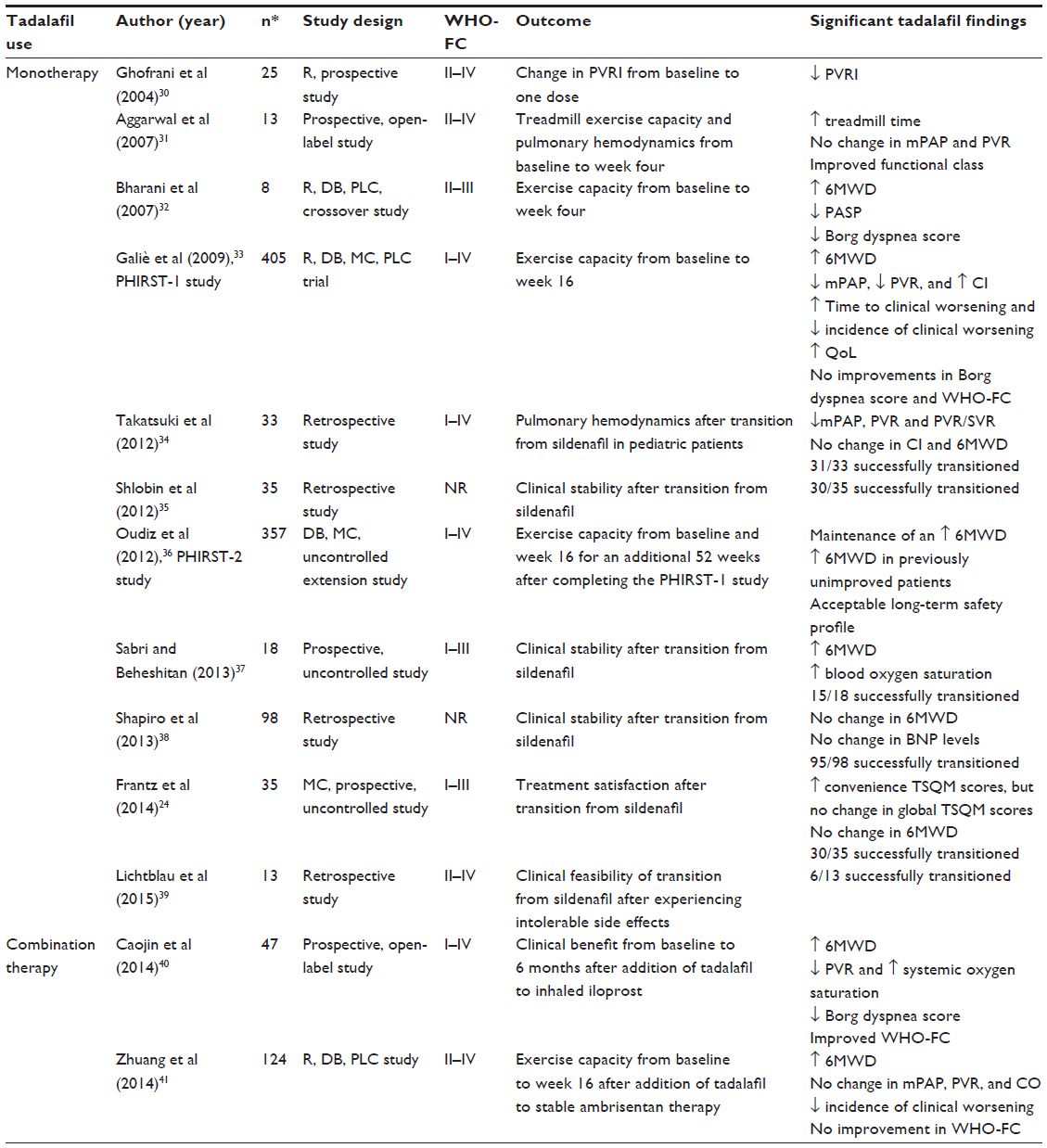 | Table 3 Chronological summary of studies demonstrating tadalafil clinical efficacy |
Clinical efficacy
Monotherapy
The earliest available literature providing evidence for the clinical utility of tadalafil in treating PAH came from case studies, as well as small prospective studies. From 2004 to 2008, a number of case studies were published that detailed the successful use of tadalafil in a total of 17 patients. From these various case reports, it is evident that tadalafil showed potential in providing PAH patients with improved exercise capacity.42–46 In 2004, a small prospective study gave insight into tadalafil’s potential to alter cardiopulmonary hemodynamics by demonstrating that one dose of tadalafil affected cardiopulmonary hemodynamics in a manner similar to that of sildenafil.30
In 2007, results from two small prospective studies from India were published.31,32 The first study examined the effects of adding tadalafil to the current regimens of 13 patients with PAH caused by congenital disease, chronic pulmonary thromboembolism, and primary pulmonary hypertension.31 Baseline treadmill exercise capacity following Bruce protocol as well as hemodynamics were compared to those measured after 4 weeks of daily tadalafil doses of 20 mg or 40 mg. Treadmill exercise time significantly increased from a mean of 350.54 seconds to 479.54 seconds (P<0.01). Improvements in functional class were seen in eleven of 13 patients. Patients also experienced a favorable although nonsignificant reduction in key measures of cardiopulmonary hemodynamics, including mPAP and mean PVR. The second study released that year showed benefit in eight patients with PAH related to congenital left to right shunt lesions during a randomized, double-blind, placebo-controlled crossover study.32 Patients received treatment with tadalafil 20 mg daily or placebo daily for 4 weeks. After completion of the first treatment arm of the study, patients underwent a washout period of 2 weeks, and then received the other treatment option for another 4 weeks. Outcome measures included change in 6MWD, pulmonary artery systolic pressure, Borg dyspnea score, and WHO functional class from baseline to week 4. There was a significant increase in 6MWD of 136.38 m on tadalafil compared to an increase of 46.5 m on placebo (P=0.001). Additionally, significant decreases in pulmonary artery systolic pressure and Borg dyspnea scores were seen in patients while taking tadalafil.
In 2009, results from Pulmonary Arterial Hypertension and Response to Tadalafil (PHIRST)-1 Phase III clinical trial were published.33 The PHIRST-1 was a randomized, double-blind, multicenter, placebo-controlled, dose-ranging trial in 405 PAH patients with disease that was idiopathic or heritable in nature or related to anorexigen use, connective tissue disease, HIV infection, or congenital shunts. Patients were randomized to take placebo, tadalafil 2.5 mg, tadalafil 10 mg, tadalafil 20 mg, or tadalafil 40 mg once daily for 16 weeks. Patients who were receiving treatment with prostacyclin therapy were excluded; however, patients on stable treatment with bosentan could participate in the study. The primary endpoint was the change from baseline to week 16 in 6MWD. Secondary endpoints included changes in WHO functional class, time from randomization to clinical worsening, Borg dyspnea score, QoL, cardiopulmonary hemodynamics, and safety. The 6MWD, Borg dyspnea score, and WHO functional class were measured every 4 weeks during the study. Tadalafil doses of 10 mg, 20 mg, and 40 mg significantly improved the mean placebo-corrected 6MWD by 20 m (P=0.047), 27 m (P=0.028), and 33 m (P<0.001), respectively, at week 16. No significant changes were seen in WHO functional class or Borg dyspnea scores. Time to clinical worsening was significantly increased (P=0.041), and incidence of clinical worsening significantly decreased (P=0.038) only in the tadalafil 40 mg group when compared to placebo. Significant improvements in QoL as measured using validated questionnaires were also seen in patients taking tadalafil 40 mg.47 Cardiopulmonary hemodynamics, including mPAP and PVR, were significantly decreased in the 93 patients for whom data were available while taking tadalafil doses of 20 mg and 40 mg. A significant improvement in CI was also seen in patients taking tadalafil 40 mg.
The patients who had completed PHIRST-1 and the patients who had discontinued PHIRST-1 while taking placebo were eligible for continued participation in a double-blind, 52-week, uncontrolled extension study known as the PHIRST-2 study.36 Durability of efficacy and long-term safety were evaluated in the 357 patients who participated. If the patients were stable on tadalafil 20 mg from the PHIRST-1 study, they continued this treatment for 52 weeks. All the other patients received tadalafil 40 mg daily for 52 weeks. Durability of efficacy was assessed by comparing 6MWD across the PHIRST-1 and PHIRST-2 studies. WHO functional class and factors associated with clinical worsening were also assessed in patients who had received treatment with tadalafil 20 mg or tadalafil 40 mg in PHIRST-1. Improvements in 6MWD that were observed at the end of the PHIRST-1 study were maintained at week 52. In patients who had completed the PHIRST-1 study with placebo, tadalafil 2.5 mg, or tadalafil 10 mg, 6MWD improved at the end of week 52; however, the 6MWD did not improve to a level similar to that seen at the conclusion of PHIRST-1 in patients taking tadalafil doses of 20 mg or 40 mg. Multivariate analysis identified race, PAH etiology, duration of bosentan use, age, baseline WHO functional class, and 6MWD at PHIRST-1 baseline as the risk factors for clinical worsening.
In 2014, two post hoc analyses of the PHIRST studies were published.48,49 The first analysis looked at safety and efficacy differences in patients <65 (n=177) vs ≥65 (n=66) years of age at the time of PHIRST-1 study entry and who received placebo, tadalafil 20 mg, or tadalafil 40 mg.48 At the completion of the 16-week PHIRST-1 study, patients <65 years old had significantly increased 6MWD, while patients ≥65 years old had a nonsignificant increase in 6MWD. By the end of the 52-week PHIRST-2 study, 6MWD was significantly increased in both the age groups. There was also an observed trend of decreased incidence of clinical worsening; however, at no point did this trend become significant in either age group alone. The second analysis examined factors for association in achieving a clinically relevant response during the PHIRST-1 study.49 A clinically relevant response was defined as obtaining a minimal important difference (MID) in either 6MWD (MID = an increase >33 m) or in questionnaire scores measuring QoL (MID = a >5-unit increase in summary score). Using an adjusted logistic regression model, the patient factors of younger age, male sex, and lower baseline 6MWD were identified as being associated with a greater likelihood of achieving a clinically relevant response. A PAH etiology of connective tissue disorder was associated with a lower likelihood of achieving a clinically relevant response.
In recent years, several uncontrolled prospective and retrospective studies evaluating the use of tadalafil after transitioning from sildenafil have been published.24,34,35,37–39 These studies reported data from patients taking a final dose of sildenafil in the evening, and then starting tadalafil the next day. A successful transition from sildenafil to tadalafil was obtained in a combined total of 207 out of 232 patients. Generally, these studies showed that there was no change in 6MWD after transitioning. One study prospectively evaluated patient satisfaction after transitioning as measured by the Treatment Satisfaction Questionnaire for Medication (TSQM). Scores relating to convenience after transitioning were increased; however, global TSQM scores did not change.24 Other studies demonstrated the clinical feasibility of transitioning to tadalafil in unique samples involving pediatric patients as well as in patients who had previously experienced intolerable side effects while taking sildenafil.34,39
In summary, the majority of evidence supporting the efficacy of tadalafil in PAH treatment comes from a large randomized clinical trial, the associated 52-week extension study which followed as well as from post hoc analyses of that clinical trial. In total, these studies provide strong evidence that tadalafil 40 mg significantly improves 6MWD for a sustained period of time. Additionally, tadalafil 40 mg improves cardiopulmonary hemodynamics, time to clinical worsening, and QoL.33,36,48,49 Several uncontrolled studies have provided some evidence supporting the effectiveness of switching from sildenafil to tadalafil.24,34,35,37–39
Combination therapy
Evidence for the use of tadalafil in combination with other PAH treatments was initially furnished by pharmacokinetic studies as well as case reports. In 2008, Wrishko et al showed that coadministration of tadalafil with bosentan, a P450 inducer, for 10 days in 14 healthy subjects decreased tadalafil exposure as measured by area under the curve (AUC) by 41.5% as well as decreased the maximum plasma concentration (Cmax) by 26.6%.25 Conversely, coadministration of tadalafil with ambrisentan in 23 healthy subjects did not result in decreased tadalafil plasma concentrations, including AUC or Cmax.50 Tadalafil pharmacokinetics were prospectively evaluated in 23 pediatric patients, 16 of whom were also taking bosentan. Along with age and estimated glomerular filtration rate, concomitant bosentan use was not found to have an effect on oral clearance (CL/F).51 From 2008 to 2011, four case studies detailed the successful and effective use of tadalafil in combination with a prostacyclin (n=4), sitaxentan (n=6), or a combination of bosentan and beraprost (n=1).52–55
A post hoc analysis of the PHIRST-1 study determined the effect of bosentan background therapy.56 Treatment-naïve patients who received placebo (n=37), tadalafil 20 mg (n=37), and tadalafil 40 mg (n=37) as well as patients with background bosentan exposure who received placebo (n=45), tadalafil 20 mg (n=45), and tadalafil 40 mg (n=42) were evaluated from baseline to week 16 for placebo-adjusted change in 6MWD, changes in functional class, incidence of clinical worsening, and cardiopulmonary hemodynamics. Only treatment-naïve patients receiving tadalafil 40 mg experienced a significant increase in placebo-adjusted 6MWD (P<0.01). Treatment-naïve patients receiving tadalafil 20 mg and bosentan background patients experienced an increase in placebo-adjusted 6MWD that failed to reach statistical significance. Background bosentan patients taking placebo experienced an increase in 6MWD of 19 m, while treatment-naïve patients taking placebo experienced a decrease in 6MWD of 3 m. The authors suggested that failure to show statistical significance in placebo-adjusted 6MWD in the background bosentan subgroup receiving tadalafil might be due, in part, to the increased 6MWD in background bosentan patients receiving placebo as well as the small sample sizes employed in the analyses. Another possible explanation for the decreased benefit in the background bosentan patients is reduced tadalafil serum concentrations as a result of CYP3A4 induction by bosentan. The incidence of clinical worsening was significantly lower in treatment-naïve patients receiving tadalafil 40 mg compared to that found in treatment-naïve patients receiving placebo. Changes in functional class and cardiopulmonary hemodynamics for all the subgroups also improved numerically but failed to reach statistical significance.
In 2014, results were published from two prospective studies from the People’s Republic of China examining the efficacy of tadalafil when used in various combinations with other PAH treatments.40,41 The first study was a prospective, uncontrolled, open-label evaluation of 47 patients with PAH secondary to congenital heart disease.40 Patients received a low dose of iloprost administered as 2.5 μg inhaled six times a day for 6 months and then were started on tadalafil 5 mg once daily in combination with iloprost for an additional 6 months. Outcome measures included change in 6MWD, Borg dyspnea score, oxygen saturation, WHO functional class, and cardiopulmonary from baseline to 6 months of monotherapy with iloprost. These same measures were assessed again after an additional 6 months in order to compare results of iloprost monotherapy with combination therapy. Iloprost monotherapy resulted in significant improvements in 6MWD, Borg dyspnea score, oxygen saturation, and WHO functional class. Following 6 months of combination therapy, there were additional significant improvements in these same measures when compared to values obtained at the end of 6 months of monotherapy. The 6MWD significantly increased from a mean of 457.22 m after monotherapy to a mean of 490.09 m (P<0.05) after combination therapy. Additionally, WHO functional class, Borg dyspnea scores, and measures of cardiopulmonary hemodynamics, including PVR, decreased significantly, while systemic oxygen saturation during exercise increased significantly.
The second study conducted exclusively in the People’s Republic of China was a randomized, double-blind, placebo-controlled study evaluating the benefits of adding tadalafil in 124 stable patients who had previously received ambrisentan 10 mg daily for 4 months.41 The study included patients with idiopathic or heritable PAH or with PAH related to anorexigen use, connective tissue disease, or repaired congenital heart disease. Patients were randomized to either placebo or tadalafil 40 mg daily for 16 weeks in addition to ambrisentan. Outcome measures included change in 6MWD, WHO functional class, incidence of clinical worsening, and cardiopulmonary hemodynamics assessed every 4 weeks from baseline to week 16. The 6MWD was found to be significantly increased from baseline at weeks 8, 12, and 16 in patients receiving tadalafil combination, whereas the 6MWD was not found to be significantly increased from baseline at any week in patients receiving placebo. When compared between groups at week 16, 6MWD was found to be significantly greater by 36.1 m (P=0.042) in patients receiving tadalafil combination compared to patients receiving placebo combination. By week 16, there were nine fewer incidences of clinical worsening in the tadalafil treatment group (P=0.046); however, there were no significant improvements in WHO functional class or cardiopulmonary hemodynamics.
In summary, subgroup analysis of the PHIRST-1 study indicates that there may be some additional benefit in using tadalafil in combination with bosentan; however, results from this analysis failed to be statistically significant and conclusions were based on small sample sizes.56 Some evidence is available supporting tadalafil use in combination with iloprost.40 Finally, as demonstrated by one randomized study, tadalafil may provide additional benefit when used in combination with ambrisentan in increasing 6MWD and decreasing incidence of clinical worsening.41 As opposed to bosentan, ambrisentan does not induce CYP3A and would not be expected to decrease tadalafil serum concentrations nor therapeutic efficacy. Strong evidence for tadalafil use in combination therapy is lacking, and larger controlled studies will need to be performed in order to better understand what role, if any, tadalafil has when used in combination with PAH therapies.
Safety and tolerability
Tadalafil is considered to be safe and typically well tolerated when taken for the treatment of PAH. The most common adverse event (AE) associated with tadalafil use is headache.21 Approximately 15%, 32%, and 42% of patients during the PHIRST-1 study experienced headache while taking placebo, tadalafil 20 mg, and tadalafil 40 mg, respectively. These values dropped to 14% and 16% during the 52-week PHIRST-2 extension study for tadalafil 20 mg and tadalafil 40 mg, respectively. Other AEs, including myalgias and flushing, were reported at a lesser frequency than headache and were typically mild-to-moderate in intensity. Discontinuation rates during the PHIRST-1 study were ~16% and were similar in all treatment groups. Discontinuation rates after 52 weeks of therapy remained similar to those seen during the PHIRST-1 study.33,36 Furthermore, discontinuation rates in elderly patients did not appear to differ from those observed in younger patients.48 When tadalafil was used as an alternative after transitioning from sildenafil, tolerability of tadalafil was reported in the majority of patients.24,34,35,37–39 Additionally, one study reported that about half of the patients who had experienced intolerable AEs while taking sildenafil were able to tolerate and successfully transition to tadalafil.39 Although data are limited, incidence of AEs appears to be similar in pediatric population.34
Studies evaluating tadalafil safety when used in combination with other PAH treatment options did not report a significantly increased incidence of AEs for tadalafil combination use with bosentan, ambrisentan, and inhaled iloprost. Discontinuation rates also remained similar to those rates observed in patients on tadalafil monotherapy.40,41,56 In addition to mild-to-moderate AEs, rare but serious AEs associated with tadalafil may occur, including vision loss, hearing loss, and priapism.21
Conclusion
Tadalafil is a convenient and generally well tolerated once-a-day PDE-5 inhibitor for treating patients with PAH. There is strong evidence that tadalafil at doses of 40 mg daily improves exercise capacity for sustained periods of time when used as monotherapy with WHO group 1 PAH patients with WHO functional class II or III symptoms. Tadalafil may also be beneficial at improving cardiopulmonary hemodynamics, QoL, and time to clinical worsening. Data from uncontrolled studies indicate that tadalafil may be a suitable alternative to sildenafil for patients desiring decreased pill burden. While the role for tadalafil in combination therapy appears promising, evidence-based recommendations for tadalafil use in combination therapy are currently difficult given the lack of evidence from large controlled trials.
Disclosure
The authors report no conflicts of interest in this work.
References
Chin KM, Rubin LJ. Pulmonary arterial hypertension. J Am Coll Cardiol. 2008;51(16):1527–1538. | |
Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25):D34–D41. | |
McLaughlin VV, Archer SL, Badesch DB, et al; American College of Cardiology Foundation Task Force on Expert Consensus Documents, American Heart Association, American College of Chest Physicians, American Thoracic Society, Inc., Pulmonary Hypertension Association. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009;53(17):1573–1619. | |
Taichman DB, Ornelas J, Chung L, et al. Pharmacologic therapy for pulmonary arterial hypertension in adults: CHEST guideline and expert panel report. Chest. 2014;146(2):449–475. | |
Badesch DB, Champion HC, Gomez-Sanchez MA. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(Suppl 1):S55–S66. | |
D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension: results from a national prospective registry. Ann Intern Med. 1991;115(5):343–349. | |
Galie N, Hoeper MM, Humbert M, et al; ESC Committee for Practice Guidelines (CPG). Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J. 2009;30(20):2493–2537. | |
Howard LS, Ferrari P, Mehta S. Physicians’ and patients’ expectations of therapies for pulmonary arterial hypertension: where do they meet? Eur Respir Rev. 2014;23(134):458–468. | |
Galiè N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62(Suppl D):D60–D72. | |
Lexicomp Online® [database on the Internet]. Lexi-Drugs®, Hudson, Ohio: Lexi-Comp, Inc.; April 1, 2015. Available from: http://online.lexi.com/crlsql/servlet/crlonline. Accessed October 9, 2015. | |
Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109:159–165. | |
Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–1665. | |
Hemnes AR. Pulmonary arterial hypertension treatment guidelines: new answers and even more questions. Chest. 2014;146(2):239–241. | |
Tonelli AR, Zein J, Ioannidis JP. Geometry of the randomized evidence for treatments of pulmonary hypertension. Cardiovasc Ther. 2013; 31(6):e138–e146. | |
Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333:214–221. | |
Moncada S, Higgs A. The l-arginine-nitric oxide pathway. N Engl J Med. 1993;329:2002–2012. | |
Archer SL, Huang JM, Hampl V, Nelson DP, Shultz PJ, Weir EK. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1994;91:7583–7587. | |
Beavo JA. Cyclic nucleotide phosphodiesterases: functional implications of multiple isoforms. Physiol Rev. 1995;75:725–748. | |
Corbin JD, Beasley A, Blount MA, Francis SH. High lung PDE5: a strong basis for treating pulmonary hypertension with PDE5 inhibitors. Biochem Biophys Res Commun. 2005;334:930–938. | |
Francis SH, Corbin JD. Molecular mechanisms and pharmacokinetics of phosphodiesterase-5 antagonists. Curr Urol Rep. 2003;4:457–465. | |
Adcirca® (tadalafil) tablets for oral administration [package insert]. Indianapolis, IN: Eli Lily and Company; 2014. | |
Revatio® (sildenafil) tablets, for oral use [package insert]. New York, NY: Pfizer Labs; 2012. | |
Forgue ST, Patterson BE, Bedding AW, et al. Tadalafil pharmacokinetics in healthy subjects. Br J Clin Pharmacol. 2006;61:280–288. | |
Frantz RP, Durst L, Burger CD, et al. Conversion from sildenafil to tadalafil: results from the sildenafil to tadalafil in pulmonary arterial hypertension (SITAR) study. J Cardiovasc Pharmacol Ther. 2014; 19(6):550–557. | |
Wrishko RE, Dingemanse J, Yu A, Darstein C, Phillips DL, Mitchell MI. Pharmacokinetic interaction between tadalafil and bosentan in healthy male subjects. J Clin Pharmacol. 2008;48(5):610–618. | |
Bischoff E. Potency, selectivity, and consequences of nonselectivity of PDE inhibition. Int J Impot Res. 2004;16(Suppl 1):S11–S14. | |
Adempas® (riociguat) tablets, for oral use [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc.; 2014. | |
Garin MC, Clark L, Chumney EC, Simpson KN, Highland KB. Cost-utility of treatments for pulmonary arterial hypertension: Markov state-transition decision analysis model. Clin Drug Investig. 2009;29(10):635–646. | |
Chen YF, Jowett S, Barton P, et al. Clinical and cost-effectiveness of epoprostenol, iloprost, bosentan, sitaxentan and sildenafil for pulmonary arterial hypertension within their licensed indications: a systematic review and economic evaluation. Health Technol Assess. 2009;13(49):1–320. | |
Ghofrani HA, Voswinckel R, Reichenberger F, et al. Differences in hemodynamic and oxygenation responses to three different phosphodiesterase-5 inhibitors in patients with pulmonary arterial hypertension. J Am Coll Cardiol. 2004;44(7):1488–1496. | |
Aggarwal P, Patial RK, Negi PC, Marwaha R. Oral tadalafil in pulmonary artery hypertension: a prospective study. Indian Heart J. 2007; 59(4):329–335. | |
Bharani A, Patel A, Saraf J, Jain A, Mehrotra S, Lunia B. Efficacy and safety of PDE-5 inhibitor tadalafil in pulmonary arterial hypertension. Indian Heart J. 2007;59(4):323–328. | |
Galiè N, Brundage BH, Ghofrani HA, et al; Pulmonary Arterial Hypertension and Response to Tadalafil (PHIRST) Study Group. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119(22):2894–2903. | |
Takatsuki S, Calderbank M, Ivy DD. Initial experience with tadalafil in pediatric pulmonary arterial hypertension. Pediatr Cardiol. 2012;33(5):683–688. | |
Shlobin OA, Brown AW, Weir N, Ahmad S, Lemma M, Nathan SD. Transition of PH patients from sildenafil to tadalafil: feasibility and practical considerations. Lung. 2012;190(5):573–578. | |
Oudiz RJ, Brundage BH, Galiè N, et al; PHIRST Study Group. Tadalafil for the treatment of pulmonary arterial hypertension a double-blind 52-week uncontrolled extension study. J Am Coll Cardiol. 2012;60(8):768–774. | |
Sabri MR, Beheshitan E. Comparison of the therapeutic and side effects of tadalafil and sildenafil in children and adolescents with pulmonary arterial hypertension. Pediatr Cardiol. 2014;35(4):699–704. | |
Shapiro S, Traiger G, Hill W, Zhang L, Doran AK. Safety, tolerability, and efficacy of overnight switching from sildenafil to tadalafil in patients with pulmonary arterial hypertension. Cardiovasc Ther. 2013;31(5):274–279. | |
Lichtblau M, Harzheim D, Ehlken N, et al. Safety and long-term efficacy of transition from sildenafil to tadalafil due to side effects in patients with pulmonary arterial hypertension. Lung. 2015;195(1):105–112. | |
Caojin Z, Yigao H, Tao H, et al. Effects of low doses of aerosolized iloprost combined with tadalafil in treatment of adult congenital heart disease with severe pulmonary arterial hypertension. Chin Med J (Engl). 2014;127(5):975–977. | |
Zhuang Y, Jiang B, Gao H, Zhao W. Randomized study of adding tadalafil to existing ambrisentan in pulmonary arterial hypertension. Hypertens Res. 2014;37(6):507–512. | |
Tay EL, Geok-Mui MK, Poh-Hoon MC, Yip J. Sustained benefit of tadalafil in patients with pulmonary arterial hypertension with prior response to sildenafil: a case series of 12 patients. Int J Cardiol. 2008;125(3):416–417. | |
Singh TS, Kumar D, Basu S, Panja M, Mitra B. Tadalafil in the management of severe pulmonary artery hypertension. Indian Heart J. 2006;58(1):52–53. | |
de Carvalho AC, Hovnanian AL, Fernandes CJ, Lapa M, Jardim C, Souza R. Tadalafil as treatment for idiopathic pulmonary arterial hypertension. Arq Bras Cardiol. 2006;87(5):e195–e197. | |
Affuso F, Palmieri EA, Di Conza P, Guardasole V, Fazio S. Tadalafil improves quality of life and exercise tolerance in idiopathic pulmonary arterial hypertension. Int J Cardiol. 2006;108(3):429–431. | |
Palmieri EA, Affuso F, Fazio S, Lembo D. Tadalafil in primary pulmonary arterial hypertension. Ann Intern Med. 2004;141(9):743–744. | |
Pepke-Zaba J, Beardsworth A, Chan M, Angalakuditi M. Tadalafil therapy and health-related quality of life in pulmonary arterial hypertension. Curr Med Res Opin. 2009;25(10):2479–2485. | |
Berman-Rosenzweig E, Arneson C, Klinger JR. Effects of dose and age on adverse events associated with tadalafil in the treatment of pulmonary arterial hypertension. Pulm Circ. 2014;4(1):45–52. | |
Mathai SC, Hassoun PM, Puhan MA, Zhou Y, Wise RA. Gender differences in response to tadalafil in pulmonary arterial hypertension. Chest. 2015;147(1):188–197. | |
Spence R, Mandagere A, Harrison B, Dufton C, Boinpally R. No clinically relevant pharmacokinetic and safety interactions of ambrisentan in combination with tadalafil in healthy volunteers. J Pharm Sci. 2009; 98(12):4962–4974. | |
Kohno H, Ichida F, Hirono K, et al. Plasma concentrations of tadalafil in children with pulmonary arterial hypertension. Ther Drug Monit. 2014;36(5):576–583. | |
Bendayan D, Shitrit D, Kramer MR. Combination therapy with prostacyclin and tadalafil for severe pulmonary arterial hypertension: a pilot study. Respirology. 2008;13(6):916–918. | |
Affuso F, Cirillo P, Ruvolo A, Carlomagno G, Fazio S. Long term combination treatment for severe idiopathic pulmonary arterial hypertension. World J Cardiol. 2010;2(3):68–70. | |
Faruqi S, Fathi H, Morice AH. Combination of sitaxentan and tadalafil for idiopathic pulmonary arterial hypertension following relapse on bosentan. Int J Cardiol. 2010;144(3):e43–e45. | |
Maki H, Yao A, Inaba T. Initial and programmed combination therapy with oral drugs for severe idiopathic pulmonary arterial hypertension. Int Heart J. 2011;52(5):323–326. | |
Barst RJ, Oudiz RJ, Beardsworth A, et al; Pulmonary Arterial Hypertension and Response to Tadalafil (PHIRST) Study Group. Tadalafil monotherapy and as add-on to background bosentan in patients with pulmonary arterial hypertension. J Heart Lung Transplant. 2011;30(6):632–643. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.