")
Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 11
Clinical Utility of Opicapone in the Management of Parkinson’s Disease: A Short Review on Emerging Data and Place in Therapy
Authors Azevedo Kauppila L, Pimenta Silva D, Ferreira JJ
Received 12 February 2021
Accepted for publication 21 April 2021
Published 11 May 2021 Volume 2021:11 Pages 29—40
DOI https://doi.org/10.2147/DNND.S256722
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Thomas Müller
Linda Azevedo Kauppila,1 Daniela Pimenta Silva,1 Joaquim J Ferreira2– 4
1Department of Neurosciences and Mental Health, Centro Hospitalar Universitário Lisboa Norte, Lisbon, Portugal; 2CNS - Campus Neurológico, Torres Vedras, Portugal; 3Instituto de Medicina Molecular João Lobo Antunes, Lisbon, Portugal; 4Laboratory of Clinical Pharmacology and Therapeutics, Faculdade de Medicina da Universidade de Lisboa, Lisbon, Portugal
Correspondence: Joaquim J Ferreira
Laboratório de Farmacologia Clínica e Terapêutica, Faculdade de Medicina de Lisboa, Av. Prof. Egas Moniz, Lisboa, 1649-028, Portugal
Email [email protected]
Abstract: Parkinson’s disease (PD) is a prevalent neurodegenerative disorder, and levodopa (L-dopa) remains the most efficacious drug treatment for PD and a gold-standard for symptom control. Nonetheless, a significant majority of PD patients develop motor fluctuations over their disease course, with a significant impact on quality-of-life, meaning control of such complications translates into a fundamental clinical need. Catechol-O-methyl transferase (COMT) inhibitors (COMT-i) are used as first-line adjuvant therapy to L-dopa for end-of-dose (EoD) motor fluctuations, since they increase L-dopa availability in the brain by inhibiting its peripheral metabolism. Opicapone (OPC), a once-daily, long-acting COMT-i, is the most recent and potent of its class, having been licensed in Europe in 2016 as an add-on to preparations of L-dopa/DOPA decarboxylase inhibitors in PD patients with EoD motor fluctuations. More recently, it has also received approval in the USA and Japan in 2020. Two high-quality positive efficacy studies (double-blind Phase III clinical trials) established OPC efficacy with significant reduction in OFF time (average 60 minutes vs placebo), without concomitant increase of distressing dyskinesias during ON time. These beneficial effects were sustained in open-label extension studies, without unexpected safety issues or adverse events, with dyskinesia having been the most frequent complaint. OPC also avoids liver toxicity and gastrointestinal issues compared with previous COMT-i. In this review, we aimed to cover OPC’s lifecycle (synthesis to commercialization), its clinical pharmacological data, safety, tolerability and pharmacovigilance evidence, and discuss its role in the management of motor fluctuations in PD as well as its emerging place in international recommendations.
Keywords: Parkinson’s disease, opicapone, motor fluctuations, COMT inhibitor, safety, tolerability
Introduction
Parkinson’s disease is the second most common neurodegenerative disease worldwide.1 Although no available therapies alter the underlying neurodegenerative process, symptomatic therapies can improve patient quality-of-life. Levodopa (L-Dopa) continues to be the most effective treatment for motor symptoms of PD.2 With sustained treatment with L-Dopa high doses and longer disease duration, motor complications emerge with a significant impact on patient’s quality-of-life.3–6
Catechol-O-methyl transferase (COMT) inhibitors are currently used as add-on therapy to L-Dopa for the treatment of end-of-dose motor fluctuations, as they inhibit peripheral L-Dopa metabolism and increase its delivery to the brain.7 A hydrophilic 1,2,4-oxadiazole analog with a pyridine N-oxide residue at position 3 was developed to provide high COMT inhibitory potency and to avoid cell toxicity risk,8 giving rise to the development of opicapone (OPC), a third generation COMT inhibitor.
Here we aim to overview OPC’s lifecycle, since its conception to commercialization, the evidence about its efficacy, tolerability and safety profile, and to discuss its role in the management of motor fluctuations in PD in current clinical practice.
Opicapone Lifecycle: From Conception to Commercialization
Opicapone (OPC) is a hydrophilic 1,2,4-oxadiazole analog with the chemical name 2,5-dichloro-3-(5-[3,4-dihydroxy-5-nitrophenyl]-1,2,4-oxadiazol-3-yl)-4,6-dimethylpyridine 1-oxide, also known as BIA 9–1067.8
Developmental Phase
OPC was developed to improve the limitations from other catechol-O-methyltransferase inhibitors (COMT-i), namely to reduce the risk of toxicity, improve peripheral tissue selectivity, and provide a more convenient dosage regimen.8
In vitro COMT inhibition by OPC was evaluated in rat liver and brain homogenates and compared to TLC. OPC achieved significant and maintained peripheral COMT inhibition up to 24 hours, with no measurable effect on central COMT.8 The concentration-dependent peripheral inhibitory potency was then evaluated in rats by giving increasing doses (0.03–3 mg/kg). OPC was found to be more potent than TLC, with an ED50 of 1.05±0.04 mg/kg.8 The pharmacodynamic interaction with L-dopa was also compared, and OPC conferred a more conservative but sustained increase in L-dopa levels,8 virtually over the entire 24 hour period, thanks to a slow complex dissociation rate and a high binding affinity (sub-picomolar Kd).9
Preclinical Studies
In vitro and ex vivo Studies
In liver homogenates from rats administered with OPC, Tolcapone (TLC) and Entacapone (ENT), OPC demonstrated a stronger and more sustained COMT inhibitory effect.10 One hour after administration, COMT inhibition was 99% with OPC vs 82% and 68% with TLC and ENT, respectively, at the same dose levels. Nine hours after administration, there was no COMT inhibition with ENT, TLC produced only a minimal inhibitory effect (16%), and OPC continued to inhibit COMT activity by 91%.10 Equally the evaluation on brain catecholamines showed a significant increase of L-Dopa and dopamine, as OPC behaved as a tight-binding inhibitor of human recombinant S-COMT with an inhibitory 10-fold lower than the one obtained for TLC.10
Another study explored the pharmacological properties of OPC using rat (brain, kidney, liver, and erythrocytes) and human (hepatocytes) tissues. There was a very long and strong peripheral COMT activity inhibition (>80%), both of the hepatic and kidney enzymes.11 Upon oral administration of L-Dopa/benserazide, OPC led to a prolonged and sustained increase in the peripheral and central bioavailability of L-Dopa, from 2 hours up to 24 hours, with concomitant reduction of 3-O-methyldopa (3-OMD) levels.11 The same study assessed the risk for cytotoxicity, OPC providing the lowest evidence for liver toxicity, and a larger safety margin when compared to TLC or ENT, as shown by the low potential in decreasing both the ATP content and mitochondrial membrane potential in human primary hepatocytes after a 24 hour incubation period.11
OPC was also studied in the Cynomolgus monkey with implanted microdialysis probes in the substantia nigra, dorsal striatum, and prefrontal cortex.12 There was a significant increase in brain and systemic bioavailability of levodopa and related metabolites: L-Dopa systemic exposure was increased by 2-fold, and the more pronounced effects in the brain were observed in the prefrontal cortex with a 2.3-fold increase in the Area Under the Curve (AUC). The marked inhibitory effect of OPC upon peripheral COMT was also evidenced by a 5-fold reduction in plasma 3-OMD levels, and a 76–84% reduction in erythrocyte COMT activity.12
The results here described were in line with the data obtained in the initial development phase of the compound.8 On the basis of these promising results in animals, human pharmacology studies were conducted to evaluate the tolerability, pharmacokinetics and pharmacodynamics of OPC in healthy subjects.
Phase I Clinical Trials
To date, as many as 33 phase I trials have been conducted on OPC use, with more than 1,000 subjects exposed.
In a human pharmacology study in young healthy male volunteers, OPC was well-tolerated and presented dose-proportional kinetics, following single oral doses (10–1,200 mg).13 Despite the low plasma exposure (terminal elimination half-life of 0.8–3.2 hours), the levels of erythrocyte COMT inhibition were sustained far beyond the observable point of plasma drug clearance (observed half-life in human erythrocytes was 61.6 hours), independent of dose administration.13 This is probably due to the long residence time of the reversible complex formed between COMT and OPC, as shown by the slow dissociation process (Koff = 1.9×10−6 s−1).9
Following an 8-day once-daily multiple-dose regimen up to 30 mg OPC, sulfation appears to be the main metabolic pathway for OPC in humans, and bile is likely the main route of excretion.14,15 The systemic exposure increased in an approximately dose-proportional manner, with an apparent terminal half-life of 1.0–1.4 hours, maximum S-COMT inhibition (Emax) between 69.9% and 98.0% following the last dose of OPC, and a half-life in excess of 100 hours, which was dose-independent.14 These results replicated the notion of a putative underlying rate constant comparable to the estimated dissociation rate constant of the COMT-OPC complex.9,13,14
Comparing the S-COMT inhibitory effect described in these studies, OPC was much stronger than what is reported for TLC and ENT in healthy subjects: Emax was 72% and 80% for TLC 100 mg and 200 mg,16 respectively; 65% for ENT 200 mg;17 and S-COMT activity returned to baseline approximately 18 hours after TLC16 and 8 hours after ENT.17 Remarkably, 24 hours after the last dose of OPC, S-COMT activity was still decreasing about 42.8%, 52.4%, 56.8%, and 64.9% in the 5 mg, 10 mg, 20 mg, and 30 mg OPC groups, respectively.14
Regarding the effect on the L-Dopa pharmacokinetics profile throughout a day, repeated doses of OPC or concomitant administration with ENT were compared in healthy subjects. OPC was found to significantly increase the L-Dopa minimum plasma concentration (Cmin) and its systemic extent of exposure (AUC0–24).18 The reduced clearance and prolonged half-life of L-Dopa, with any OPC dose, resulted in a dose-dependent increase of L-Dopa Cmin of at least 2.5-fold.18 In addition, OPC may enhance stable L-Dopa plasma concentrations as it increases Cmin with no impact on L-Dopa fluctuation (as assessed by Cmax-Cmin).18 When compared to ENT, OPC provided a superior response upon the bioavailability of L-Dopa associated to more pronounced, long-lasting, and sustained COMT inhibition.18
A food-effect study was conducted to investigate the effect of food on the oral bioavailability of OPC.13 The authors concluded that the rate and extent of systemic exposure to OPC significantly decreased following concomitant ingestion of a high-fat high-calorie meal. This was replicated in a further study in healthy subjects,19 however OPC COMT inhibition was not affected by concomitant administration of a moderate meal.19
The effect of moderate liver impairment on pharmacokinetics and pharmacodynamics of OPC was assessed in an open-label, parallel-group trial with individuals with moderate hepatic dysfunction (Child–Pugh category B, score to 7–9) and matched with healthy individuals.20 The bioavailability of a single dose of OPC 50 mg was significantly higher in patients with moderate chronic hepatic impairment.20 The authors hypothesized that a reduced first-pass effect would be the reason for the increased exposure to OPC (AUC and Cmax), however no dose adjustment was needed in mild-to-moderate hepatic impairment due to its systemic elimination before the subsequent dose administration.20 Notably, there were no liver enzyme increments in any individual, none of them showed evidence of hepatic disease worsening, and the other phase I trials showed no changes in liver function monitoring.21
The effect of OPC on cardiac repolarization was also evaluated in 64 healthy subjects.22 There was no clinically relevant effect of OPC 50 and 800 mg vs placebo on cardiac depolarization or repolarization, and therapeutic (50 mg) as well as supratherapeutic (800 mg) had no QT-prolonging effect,22 further supporting its cardiac safety.
Reported drug-related adverse events (AEs) were mild in severity and clinical safety tests did not raise any safety concerns.13,14,18,21 The most common AEs were somnolence, headache, and gastrointestinal complaints, including nausea, vomiting, and diarrhea.21
Overall, these studies clearly suggest that OPC has a favorable safety, pharmacokinetic and pharmacodynamic profile, adequate for a once-daily regimen, which in the treatment of PD patients represent an advantage over ENT and TLC, prompting further studies in PD patients.
Phase II Clinical Trials
Two phase II clinical trials explored the effect of single and repeat-dose OPC on L-Dopa pharmacokinetics in Parkinson’s disease (PD) patients with end-of-dose motor fluctuations.23,24
These two multicenter, double-blind randomized controlled trials were the first to study the effects of once daily doses of OPC on L-Dopa pharmacokinetics in PD patients.23,24 OPC was found to significantly increase the extent of L-Dopa systemic exposure (as assessed by AUC) under both 15 and 30 mg OPC doses.23 Furthermore, peak L-Dopa exposure (as assessed by Cmax) was significantly increased under 30 mg OPC.23 Maximum COMT inhibition (Emax) ranged from 52% (5 mg OPC) to 80% (30 mg OPC)23 and 100% (100 mg OPC).24 Maximum decrease in the plasma 3-OMD was observed following administration of 100 mg OPC.24
Although the study was not designed for motor outcomes, it found a dose-dependent and statistically significant decrease in absolute OFF time and increase in ON time without dyskinesias compared with placebo.23,24 OPC was well tolerated when administered with standard release 100/25 mg L-Dopa/carbidopa or L-Dopa/benserazide.23,24
Phase III Clinical Trials: BIPARK-I and BIPARK-II
The efficacy of oral once-daily OPC as an adjunctive therapy to L-Dopa in patients with PD and EoD motor fluctuations was evaluated in double-blind, multinational, Phase 3 trials (BIPARK I and BIPARK II; total treatment duration 14–15 weeks).25,26 BIPARK I also evaluated the noninferiority of OPC to ENT.21,25,27 In BIPARK I and BIPARK II, adjunctive OPC 50 mg/day provided better efficacy than add-on placebo in terms of improving motor fluctuations, with OPC significantly reducing time in the off state and increasing time in the on state.21,25–27
BIPARK I, which enrolled 600 PD patients with motor fluctuations, showed that OPC 50 mg was more efficacious than placebo in reducing OFF time and non-inferior to ENT.25 The mean reduction in the time spent in the OFF state in a day was 116.8 minutes for OPC 50 mg, 96.3 minutes for ENT, and 56.0 minutes for placebo.25 This corresponds to a reduction in time in the OFF state of 60.8 minutes daily versus placebo.25 Compared to ENT, OPC 50 mg showed a tendency for a greater magnitude of effect in OFF-time reduction and performed better for both CGI-C and the PGI-C, suggesting a greater clinical benefit.25 Treatment-emergent AE (TEAE) were reported in 62 (54%) of 115 in the OPC 50 mg group; the most common ones were dyskinesia (five patients in the placebo group, 10 in the ENT group, 17 in the OPC 5 mg group, nine in the OPC 25 mg group, 18 in the OPC 50 mg group), insomnia (in 1, 7, 2, 7, and 7 patients, respectively), and constipation (in 3, 5, 4, 0, and 7 patients, respectively).25 Serious AE were reported in six patients in the placebo group, eight in the ENT group, four each in the OPC 5 mg and 50 mg groups, and one in the OPC 25 mg group.25 In BIPARK II, a total of 427 patients were randomized to 25 or 50 mg of OPC or placebo.26 In this study, OPC 25 and 50 mg were also more efficacious than placebo in reducing OFF state time (1.7, 2.0, and 1.1 hours, respectively),21,26 with the adjusted treatment difference vs placebo being significant for the 50 mg OPC group (treatment effect, −54.3 minutes; p=0.008), but not for the 25 mg OPC group (treatment effect, −37.2 minutes; p=0.11).26 The most frequent AE in OPC (25 and 50 mg) vs placebo groups were dyskinesia (30, 36, and 11 patients, respectively), constipation (12, 10, and 2 patients), and dry mouth (13, 6, and 1 patient), as had already been identified in BIPARK.25,26
In summary, in two phase III clinical trials, OPC 50 mg demonstrated to be superior to placebo in OFF-time reduction without increasing ON time with troublesome dyskinesias in PD patients with EoD motor fluctuations. This benefit was maintained during 1-year follow-up with no major issues on its safety profile, even among older PD patients.25,26,28
No study has specifically assessed OPC effects on non-motor symptoms, but results from BIPARK I and BIPARK II studies showed a non-significant improvement in NMSS total score, which was more evident with OPC 50 mg, and numerical differences favoring OPC for both sleep and fatigue domains.25,26
BIPARK-I and BIPARK-II Extension Studies
In the 1-year, open-label (OL) extension studies of BIPARK I (n=495)29,30 and BIPARK II (n=286),26 OPC and L-Dopa dosages were adjusted based on clinical responses and/or associated AE. In patients receiving adjunctive OPC during the double-blind and extension phases (ie, OPC–OPC group), the beneficial effects of OPC on motor fluctuations that were observed in the double-blind phases were maintained during the 1-year extension phases of BIPARK I and BIPARK II.26,29,30 Patients who switched from adjunctive placebo or ENT to adjunctive OPC at the end of the double-blind phase experienced significant (p<0.05) improvements in motor fluctuations during the 1-year BIPARK I extension phase.29 Add-on OPC treatment was associated with maintained improvements from baseline in UPDRS activities of daily living (Part II) and motor (Part III) scores during the open-label extension of BIPARK I, with mean UPDRS II ON and OFF (OFF score; p=0.0043 vs prior placebo) and UPDRS III (p=0.0456 vs prior placebo) scores improving from baseline by −2.2, −4.4, and −7.4 points, respectively.27,30
Considering the BIPARK II OL phase, off-time reduction was maintained throughout this period, with −126.3 minutes at 1-year open-label end point; the adjusted mean change from beginning to finish of the OL phase in off-time was −18.31 (95% CI=−43.56–6.95) minutes.26 Mean (SD) total on-time also increased by 24.9 (156.4) minutes, without or with unbothersome dyskinesia.26 During the OL phase, mean (SD) on-time associated with distressing dyskinesia increased by only 6.0 (129.1) minutes.26 With the conclusion of the 1-year BIPARK II extension phase, no worsening of non-motor symptoms during adjunctive OPC treatment was identified, with a mean improvement in NMSS total score of −4.2.31
Data from the BIPARK I and II double-blind and OL studies were combined in a more recent study in 2019.32 Double-blind OPC treatment significantly reduced OFF time from a starting point of 6.1–6.6 OFF hours. The mean (95% CI) treatment effect vs placebo was −35.1 minutes (p=0.0106) and −58.1 minutes (p<0.0001) for the 25 and 50 mg doses, respectively.32 OFF time reductions were accompanied by noteworthy increases in ON time without distressing dyskinesia (p<0.05 and p<0.0001 for the 25 and 50 mg doses, correspondingly), and no significant differences were noted for ON time with bothersome dyskinesia.32 During the OL phase, patients previously treated with OPC 50 mg maintained a positive effect of this treatment in their patient diary registries, while patients treated with 25 mg further improved with OPC dose optimization, and switch from placebo to OPC resulted in important OFF time reductions and increased ON time.32 Therefore, over the course of 1 year of OL treatment, OPC steadily reduced OFF time and increased ON time without it translating into an increase of distressing dyskinesia occurrence.32
Safety and Tolerability Data from Phase III Clinical Trials
OPC is generally well tolerated, with no clinically relevant effects on hepatobiliary function and no serious cases of hepatotoxicity having been previously reported in clinical trials.27 In a pooled and integrated safety analysis of BIPARK I and II and its extension studies, the most common TEAEs in OPC recipients (25 or 50 mg/day) were dyskinesia, constipation, insomnia, and dry mouth, most of which were of mild-to-moderate intensity and had no dose relationship.27,32
In both BIPARK I and II studies, the most common TEAE was dyskinesia, but it showed a tendency to decrease with time after visit four in all OPC groups (5, 25, and 50 mg).21
In the OL 1-year extension phase of BIPARK II, the most common TEAEs were dyskinesia (21.5%), worsening PD (17.0%), falls (9.1%), CK increase (7.4%), insomnia (5.7%), and orthostatic hypotension (5.4%). Thirty-two (9.1%) patients withdrew the study due to AE. The most frequent TEAEs leading to study withdraw were dopaminergic effects: three patients (0.8%) for dyskinesia, three (0.8%) for hallucinations, one (0.3%) for orthostatic hypotension, and one (0.3%) for worsening of PD. Most of these were mild-to-moderate, with the incidence of severe TEAE having been 11.3% (n=40).33 A total of five deaths occurred, which were considered to be unrelated to the treatment drug, specified below.21,26,33
Less than 1% of OPC receivers developed impulse control disorder (ICD) AE, and OPC treatment did not appear to increase the risk of ICDs or suicidality with longer-term treatment.27 No unanticipated safety concerns were recognized during the 1-year extension studies.27 Continued use of OPC once-daily during 1-year (25 mg or 50 mg) was globally harmless and well tolerated, reinforcing its clinical utility in the management of PD motor fluctuations.34
In a subgroup analysis of pooled safety data in older PD patients (> 70 years old, 29% of the 766 BIPARK patients), OPC also seemed to be safe and well-tolerated, with most frequent TEAEs compared to younger patients having been hallucinations, visual hallucinations and weight decrease (adjusted to placebo).28 The OPC group had a higher discontinuation rate due to TEAEs and the most common were dyskinesia (2.6%) and vomiting (2.6%).28
BIPARK studies’ data were also pooled in order to better understand OPC’s role on hepatobiliary function.35 No significant hepatic blood work changes from baseline to endpoint were detected in any treatment group.35 The occurrence of possibly clinically meaningful values was low and similar for placebo and OPC groups,35 with hepatic-related AE having been lower in OPC groups (1.2%) vs placebo (3.1%).21 Notably, none of the hepatic events in the OPC groups led to the drug discontinuation.21
Another pooled analysis from BIPARK studies was conducted to assess potential OPC cardiac safety through ECG parameters. No significant differences among groups were determined from baseline concerning QTcF/QTcB, PR, and heart rate (HR). The placebo-adjusted changes in QT/QTc were <1 ms for both OPC doses, and all upper limits of the two-sided 90% CI were significantly below the threshold of 10 ms. ECG morphological abnormalities were observed in rare subjects and in comparable proportions across groups. Moreover, neither arrhythmias nor myocardial ischemia were verified. To conclude, OPC presented no clinically noteworthy effects on cardiac conduction and repolarization as assessed by changes in QT/QTc, maximum QRS duration, PR intervals, and HR.21
No deaths occurred during BIPARK I; one death from pneumonia was registered in the placebo group during the double-blind phase of BIPARK II.21 In the OL phase of BIPARK II, deaths were reported due to septic shock, traumatic brain injury, cerebral hemorrhage, small cell lung cancer, and an unknown cause, but none were deemed to be associated to treatment.21,26
Commercialization
On the basis of the clinical development program comprising 27 Phase I studies, two Phase II studies and two Phase III studies, on April 2016, the European Medicines Agency adopted a positive opinion recommending granting for marketing authorization of Ongentys®.36 In June 2016, the European Commission authorized OPC as an adjunct therapy to levodopa/DOPA decarboxylase inhibitors (DDCIs) in adult patients with PD and EoD motor fluctuations.
In Europe, OPC is currently marketed in Germany, the United Kingdom, Spain, Portugal, and Italy. On April 2020, OPC was approved by the US Food and Drug Administration.37
In Japan, OPC received manufacturing and marketing approval on June 2020.38 The formulation developed by ONO Pharmaceutical Co. Ltd. (Osaka, Japan) is a tablet formulation, as opposed to the capsule used in clinical trials BIPARK I and II and marketed in approved countries. A phase I open-label, randomized, 2-way, 2-period crossover clinical trial compared the pharmacokinetic characteristics of both formulations of OPC, after single oral administration to healthy Japanese volunteers.39 Compared with similar capsule doses (25 and 50mg), the proposed smaller tablet formulation was associated with higher Cmax and AUClast/0-∞ values, but similar t1/2 and tmax values.39 OPC tablets were further studied in Japanese PD patients with motor fluctuations in a randomized placebo-controlled study, the double-blind part of COMFORT-PD (COMt-inhibitor Findings from Opicapone Repeated Treatment for Parkinson’s Disease).40 The study design was similar to BIPARK I and II clinical trials. Recruited Japanese PD patients had lower OFF-time and used lower L-Dopa doses.40 The main conclusion of this study is that once-daily adjunct OPC tablets at doses of 25 and 50 mg significantly reduced OFF-time compared with placebo among Japanese L-Dopa treated patients with PD and motor fluctuations, with no dose dependency between OPC tablet dosages.40 Overall, the efficacy results in this study are broadly consistent with those seen in BIPARK I and II, however the least squares mean changes in OFF-time were numerically lower in Japanese patients than in the BIPARK I and II studies for OPC 25 mg, 50 mg, and placebo.26,40
Post Hoc Analysis from Clinical Trials
Recent Fluctuators
PD patients enrolled in BIPARK-I and II were divided in subgroups according to disease- and therapy-related characteristics,41 to evaluate the added benefit of OPC 50 mg across the motor fluctuations spectrum of PD. Five hundred and twenty-two patients were randomized to placebo (PLC) (n=257) and OPC 50 mg (n=265). OPC 50 mg was superior to PLC in all subgroups, except the one treated with 700 mg/day L-Dopa or more (p=0.064). OPC 50 mg was more efficacious in patients who have lower disease duration (less than 8 years of duration), lower Hoehn and Yahr (HY) (less than 2.5), lower duration of motor fluctuations (onset in 1 year or less), and lower L-Dopa intakes (less than 4) and daily amount (less than 700 mg).41 Furthermore, there was a trend towards a lower incidence of dopaminergic-related TEAEs in the same subgroups of patients.41
COMTi-naïve patients from BIPARK-I who were ‘recent fluctuators’ (ie, patients having an onset of motor fluctuations in 1 year or less of study baseline) were further included in a post-hoc analysis.42 The least-squares (LS) mean (standard error – SE) reduction from baseline in absolute OFF-time was 124 minutes (21.0) for the OPC group compared to 84.1 (21.4) and 56.7 (21.8) minutes for ENT and PLC groups, respectively.42 The LS mean (SE) increase in absolute ON-time from baseline was 131.2 minutes (21.1) for the OPC group, as opposed to both ENT and PLC groups [85.5 (21.4) and 32.7 (21.9) minutes, respectively].42 Overall OPC 50 mg demonstrated added benefit as a first adjunctive COMTi, in comparison with PLC and ENT, in “recent fluctuators”.
A more conservative analysis including 97 COMT-naïve “recent fluctuator” patients further demonstrated that OPC 50 mg numerically decreased the absolute OFF-time and significantly increased absolute ON-time (by approximately 1 hour), compared with ENT, when used as first adjunctive COMTi in patients recently diagnosed with motor fluctuations.43
Levodopa Monotherapy
A Full Analysis Set from BIPARK-I and II combining PLC and OPC groups demonstrated an approximately 2-fold greater improvement, compared to PLC, in the mean change from baseline in OFF-time when OPC 50 mg was added to any of the levodopa-containing treatment regimens, namely L-Dopa only, L-Dopa plus dopamine agonists (DA) and L-Dopa plus monoamine oxidase B inhibitor (MAO-Bi).44 Interestingly, OFF-time reduction was greater when OPC 50 mg was used as first add-on to L-Dopa.44
Further exploratory post-hoc analysis in patients treated with L-Dopa in monotherapy was performed.45 The LS mean reduction from baseline in absolute OFF-time was 109.2 minutes (95% CI, –147.9 to –70.4) for the OPC group compared to 40.3 minutes (95% CI, –80.7 to 0) for the PLC group.45 Similarly, the LS mean increase in absolute ON-time from baseline was 96.7 minutes (95% CI, 58.1 to 135.3) for the OPC group, while the PLC group experienced a gain of 16.9 minutes (95% CI, –23.3 to 57.0).45 Thus, OPC 50 mg was efficacious in decreasing the absolute OFF-time [absolute difference = 68.8 minutes (95% CI, –124.8 to –12.8)] and increasing the absolute ON-time [absolute difference = 79.8 minutes (95% CI, 24.3 to 135.4)].45 These results confirm that OPC 50 mg may be a viable option as a first-line adjunctive therapy in PD patients with motor fluctuations treated with L-Dopa in monotherapy.45
Opicapone in Clinical Practice
OPTIPARK Clinical Trial
OPTIPARK was a Phase IV, prospective, open-label, single-arm trial conducted in Germany and the UK under clinical practice conditions.46 It included a broad and heterogeneous population of fluctuating PD patients, mirroring a clinical setting.
Four hundred and ninety-five PD patients with motor fluctuations were treated with OPC 50 mg for 3 (Germany) or 6 (UK) months in addition to their current L-Dopa and other antiparkinsonian treatments.46 After 3 months of treatment with OPC 50 mg, 71.3% showed clinical improvement as judged by the investigators (CGI-C), and for the 95 UK patients who were assessed at 6 months, 85.3% were also judged as improved since commencing treatment.46 Similarly, 76.9% of patients self-rated (PGI-C) improvement after 3 months, as well as 79.8% of patients from the UK subgroup after 6 months of treatment with OPC.46 UPDRS motor and activities of daily living (ADL) scores had a clinically significant improvement which may indicate that treatment with OPC not only increases ON time, but also improve the quality of ON time.46 Treatment with OPC was also associated with a small but significant improvement in overall quality-of-life, as assessed by PDQ-8, and nonmotor symptoms.46 Importantly, a subgroup analysis for CGI-C at Month 3 confirmed improvements regardless of age or concomitant use of dopamine agonists with or without MAO-B inhibitors at baseline.46
OPC 50 mg was generally well-tolerated, with most events reported as mild or moderate in severity. While the most common reason for withdrawal from the study was AEs (17.0%), nausea was the most frequent, affecting only 2% of patients.46 About 45.1% treatment-emergent adverse events (TEAEs) were considered to be at least possibly related to OPC; dyskinesia (11.5%) and dry mouth (6.5%) were the most frequently reported.46 Although dyskinesia was reported as a TEAE in 11.5% of patients, only five patients (1%) discontinued from the study due to dyskinesia, most patients remained on the same total daily L-Dopa dose.46
In general, the results of this large open-label study in PD patients with motor fluctuations are the first to confirm the effectiveness, safety and tolerability of once-daily OPC 50 mg as used in routine clinical practice.
Opicapone in the Elderly
Data available on the use of OPC in the elderly is scarce. As mentioned above, there is a subgroup analysis of pooled safety data in older PD patients (>70 years old) from both BIPARK extension studies. This group of patients had more frequent TEAEs and a greater discontinuation rate due to TEAEs.28
Management of PD in geriatric patients is a great concern, due to numerous comorbidities and polypharmacy that may increase undesirable pharmacological interactions and adverse effects, hampering optimal treatment.47 To investigate if OPC is safe and effective in PD patients of 75 years old or more, in a real-world setting (OPC plus standard of care), a multinational, multicenter, prospective non-interventional study is being conducted (ClinicalTrials.gov Identifier: NCT03959540). Conclusions about its use in clinical practice in this population may be drawn afterwards.
Pharmacovigilance Data
Safety and tolerability data from BIPARK-I and BIPARK-II and its 1-year open label extension studies were mentioned above, and published on April 28, 2016 in the Public Assessment Report (PAR) by EMA (EMA/343011/2016).36
Treatment-emergent adverse events (TEAEs) were detailed in the open-label clinical setting OPTIPARK study, replicating some of the data from clinical trials. Overall, 371 (74.9%) patients experienced TEAEs, which were mostly assessed as mild or moderate, and as expected for a dopaminergic therapy in patients with PD. Two hundred and twenty-three (45.1%) were considered at least possibly related to treatment, the most frequent ones (>5%) being dyskinesia (11.5%), dry mouth (6.5%), and dizziness (4.8%).46 Other TEAEs possibly treatment-related were nausea (4.4%), constipation (4.0%), insomnia (2.4%), hallucination (2.2%), fall (2.0%), and diarrhea (0.6%).46 The frequency of serious treatment-related TEAEs was low; seven patients (1.4%) had more than one of the following events: anxiety, visual hallucination, psychotic disorder, dizziness, hypertension, hypotension, tachycardia and femoral neck fracture.46 When analyzed by age, 56.6% from the older group (age above 67.7 years old) experienced any TEAEs, having also the highest proportion of those who discontinued treatment due to a TEAE.
A post-hoc analysis from OPTIPARK demonstrated that the majority of at least possibly drug-related TEAEs were reported during the first week of OPC treatment, and from the third week onwards the incidence of these TEAEs was consistently low (<4%).48 Within the first week, dyskinesia was the most frequently reported (6.5%) but with very low impact on patient discontinuation (<0.5%).48
Post-marketing data on safety and tolerability is reported in the European public assessment report (EPAR) for Ongentys®, last updated on May 20, 2020.49 Dyskinesia is consistently the most frequent TEAE (17.7%).49 Moreover, “common” TEAE (with no specified frequency) are other nervous system disorders (dizziness, headache, somnolence), psychiatric disorders (abnormal dreams, visual hallucination, insomnia), orthostatic hypotension, gastrointestinal disorders (constipation, dry mouth, vomiting), muscle spasms, and increased blood creatine phosphokinase.49
Lastly, unpublished raw data of cumulative serious and nonserious adverse reactions of OPC are registered in EudraVigilance, the European database of suspected adverse drug reaction reports. As of November 7, 2020, the number of individual cases was 535. The most frequent cumulative events were the central nervous system disorders (n=211) followed by psychiatric disorders (n=170), gastrointestinal complaints (n=122), and general disorders and administration site conditions (n=119).50
Opicapone and International Recommendations
Since OPC is a fairly recent drug, its inclusion in international recommendations is still emerging.
The last European Academy of Neurology recommendations on the therapeutic management of PD were published in 2013 (at that time, an EFNS/MDS-ES review);51 therefore, OPC was not yet included in this update, since OPC was first commercialized in Europe in June 2016 by BIAL.15,21,27,52 UK’s National Institute for Health and Care Excellence (NICE) has an evidence summary for OPC available online,53 published in 2017, but this drug has not been specifically addressed in NICE guidelines “Parkinson's disease in adults: diagnosis and management“, which have been published in 2017 and updated in September 2020.54 COMT-i are mentioned, in particular ENT and TLC, with no information concerning OPC.54
The AAN April 2006 guideline, “Practice Parameter: Treatment of Parkinson disease with motor fluctuations and dyskinesia (an evidence-based review)”, was retired in February 2018, since it had not been updated or reaffirmed in 5 years or less after the previous publication or reaffirmation. Nevertheless, OPC was only FDA approved in April 2020.37
OPC was included in the most recent MDS update on treatments for the motor symptoms of PD.55 OPC was deemed efficacious and clinically useful for treating motor fluctuations in clinical practice, based on its evaluation in two high-quality positive efficacy studies (BIPARK I and II)25,26 and one high-quality pharmacokinetic study which included motor outcomes.23 There were no safety concerns, reckoning an acceptable risk and no need of specialized monitoring.55 To date, OPC has not yet been assessed in nonfluctuating PD patients.
Management Proposal for Motor Fluctuations
Opicapone’s Place in Therapy
Motor complications seem to affect about 40% of patients after 4–6 years of L-Dopa treatment.3 Occurrence of motor complications appears to be related to longer disease duration4 and to higher daily doses of L-Dopa treatment.4,5 Dyskinesia and motor fluctuations, two manifestations of motor complications associated to L-Dopa treatment, have a substantial impact on the quality-of-life of PD patients,6 being imperative to address them appropriately.
Motor fluctuations are characterized by periods of reduced benefit from the medication (“off” time), which may be simple predictable wearing-off and/or early-morning off periods, or more complex unpredictable on–off, dose-failure, “delayed-on”, beginning-of-dose worsening or end-of-dose rebound.56 Wearing-off and early-morning off periods are the most common motor fluctuations,57,58 characterized by the re-emergence of parkinsonian symptoms toward the end of the treatment interval between individual doses of L-Dopa and in the morning before the first L-Dopa dose, respectively.56 Wearing-off worsens with disease progression and will eventually affect up to 80% of patients with more than 10 years of disease duration.57 Strategies for reducing the time that medication is not optimally effective include L-Dopa dose and frequency adjustment, adding COMTi or MAO-Bi or adding another dopaminergic medication, in order to increase dopamine availability.51,59
On the basis of international recommendations and guidelines51,54,55 on the management of motor fluctuations, and evidence from OPC clinical trials and post-hoc analysis, we propose the flowchart in Figure 1 to approach motor fluctuations in clinical practice, including the role of OPC.
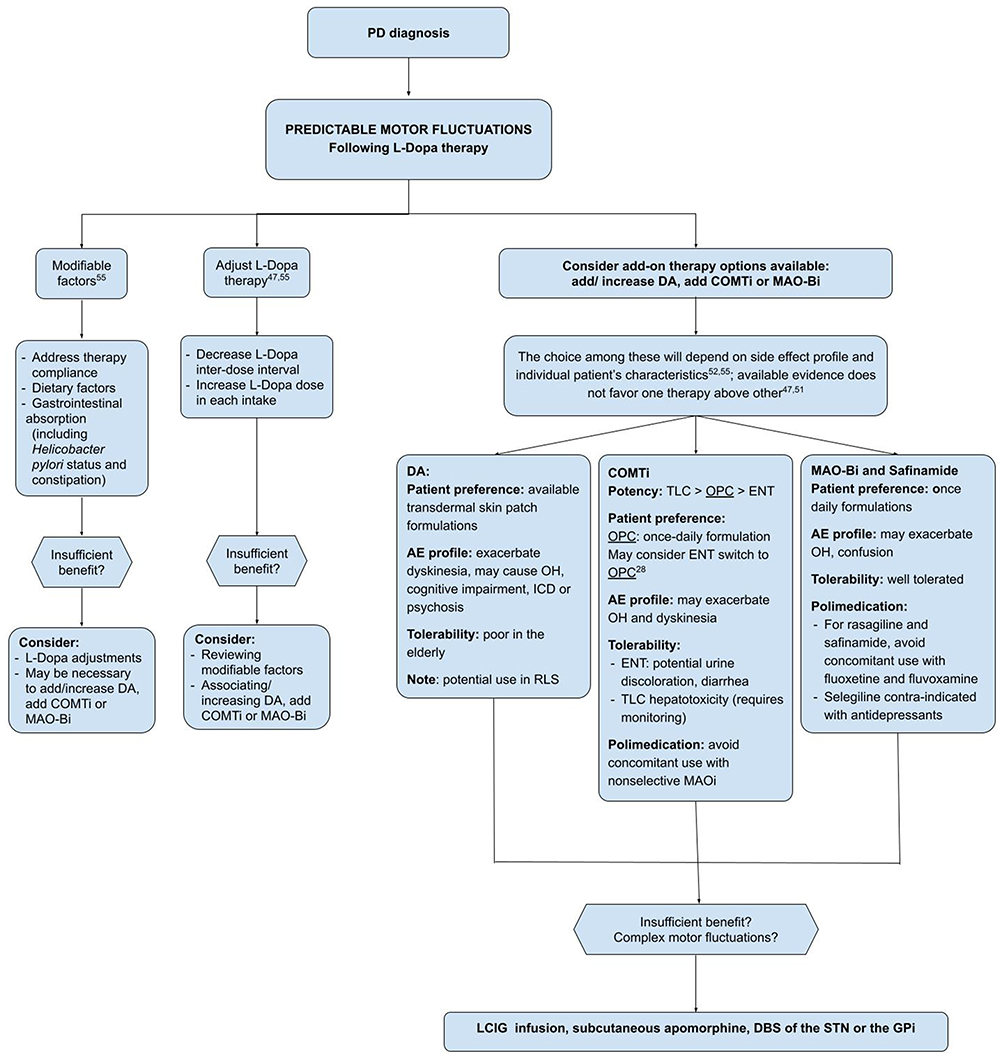 |
Figure 1 Management proposal for motor fluctuations, considering factors that influence add-on treatment choice. Abbreviations: DA, dopamine agonists; COMTi, catechol-O-methyltransferase inhibitor; MAOB-I, monoamine oxidase B inhibitor; OPC, opicapone; TLC, tolcapone; ENT, entacapone; ICD, impulse control disorder; RLS, restless leg syndrome; OH, orthostatic hypotension; LCIG, levodopa/carbidopa intestinal gel; DBS, deep brain stimulation. |
Conclusion
OPC is a purely peripheral COMT inhibitor with an unprecedented duration of action, with favorable pharmacodynamics with L-Dopa,8 resulting in stable and sustained plasma L-Dopa levels over prolonged periods, allowing a single daily dose regimen.
In clinical trials, OPC proved its efficacy in significantly reducing OFF time, by approximately 1 hour, without increasing troublesome dyskinesias during ON time, with sustained effects over 1 year. OPC is well-tolerated and adverse effects are those expected from dopaminergic stimulation.
Data from a “real world” clinical trial (OPTIPARK) confirmed its efficacy, safety and tolerability, making OPC a fairly robust therapy for the treatment of end-of-dose motor fluctuations in patients with PD. To date, pharmacovigilance data did not raise any unexpected safety concerns.
Abbreviations
3-OMD, 3-O-methyldopa; ADL, activities of daily living; AE, adverse event; AUC, area under the curve; CGI-C, clinician global impression of change; COMT, catechol-O-methyltransferase; COMT-I, catechol-O-methyltransferase inhibitor; DA, dopamine agonists; ECG, electrocardiogram; EMA, European Medicines Agency; ENT, entacapone; EoD, end of dose; HY stage, Hoehn and Yahr stage; L-Dopa, levodopa; Levodopa/DDCi, levodopa/DOPA decarboxylase inhibitors; MAO-Bi, monoamino oxidase B inhibitor; MDS, Movement Disorder Society; NICE, National Institute for Health and Care Excellence; OL, open-label; OPC, opicapone; PGI-C, Patients’ Global Impression of Change; PLC, placebo; TEAE, treatment-emergent adverse event; TLC, tolcapone.
Disclosure
The authors Daniela Pimenta Silva and Linda Azevedo Kauppila report no conflicts of interest in this work.
Joaquim J. Ferreira has held consultancy functions with GlaxoSmithKline, Novartis, TEVA, Lundbeck, Solvay, Abbott, AbbVie, BIAL, Merck-Serono, Merz, Ipsen, Biogen, NeuroDerm, Zambon, Sunovion, Affiris, ONO; has received lecture fees from Biogen, BIAL, Sunovion, ONO, Zambon, AbbVie; has received grants from GlaxoSmithKline, Grunenthal, MSD, Allergan, Novartis, Fundação MSD (Portugal), Medtronic and Teva; has been employed by Faculdade de Medicina de Lisboa and CNS – Campus Neuológico.
References
1. Dorsey ER, Constantinescu R, Thompson JP, et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68(5):384–386. doi:10.1212/01.wnl.0000247740.47667.03
2. LeWitt PA. Levodopa therapy for Parkinson’s disease: pharmacokinetics and pharmacodynamics. Mov Disord. 2015;30(1):64–72. doi:10.1002/mds.26082
3. Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord. 2001;16(3):448–458. doi:10.1002/mds.1090
4. Cilia R, Akpalu A, Sarfo FS, Cham M, Amboni M, Cereda E. The modern pre-levodopa era of Parkinson’s disease: insights into motor complications from sub-Saharan Africa. Brain. 2014;137:2731–2742. doi:10.1093/brain/awu195
5. Fahn S, Oakes D, Shoulson I, Parkinson Study Group. Levodopa and the progression of Parkinson’s disease. N Engl J Med. 2004;351(24):2498–2508. doi:10.1056/NEJMoa033447
6. Chapuis S, Ouchchane L, Metz O, Gerbaud L, Durif F. Impact of the motor complications of Parkinson’s disease on the quality of life. Mov Disord. 2005;20(2):224–230. doi:10.1002/mds.20279
7. Müller T. Catechol-O-methyltransferase inhibitors in Parkinson’s disease. Drugs. 2015;75(2):157–174. doi:10.1007/s40265-014-0343-0
8. Kiss LE, Ferreira HS, Torrão L, et al. Discovery of a long-acting, peripherally selective inhibitor of catechol-O-methyltransferase. J Med Chem. 2010;53(8):3396–3411. doi:10.1021/jm1001524
9. Palma PN, Bonifácio MJ, Loureiro AI, Soares-da-silva P. Computation of the binding affinities of catechol-O-methyltransferase inhibitors: multisubstate relative free energy calculations. J Comput Chem. 2012;33(9):970–986. doi:10.1002/jcc.22926
10. Bonifacio MJ, Torrão L, Loureiro AI, Wright LC, Soares-da-silva P. Opicapone: characterization of a novel peripheral long-acting catechol-O-methyl transferase inhibitor. Parkinsonism Relat Disord. 2012;18:S125. doi:10.1016/S1353-8020(11)70567-3
11. Bonifácio MJ, Torrão L, Loureiro AI, Palma PN, Wright LC, Soares-da-silva P. Pharmacological profile of opicapone, a third-generation nitrocatechol catechol-O-methyl transferase inhibitor, in the rat. Br J Pharmacol. 2015;172(7):1739–1752. doi:10.1111/bph.13020
12. Bonifácio MJ, Sutcliffe JS, Torrão L, Wright LC, Soares-da-silva P. Brain and peripheral pharmacokinetics of levodopa in the cynomolgus monkey following administration of opicapone, a third generation nitrocatechol COMT inhibitor. Neuropharmacology. 2014;77:334–341. doi:10.1016/j.neuropharm.2013.10.014
13. Almeida L, Rocha JF, Falcão A, et al. Pharmacokinetics, pharmacodynamics and tolerability of opicapone, a novel catechol-O-methyltransferase inhibitor, in healthy subjects: prediction of slow enzyme-inhibitor complex dissociation of a short-living and very long-acting inhibitor. Clin Pharmacokinet. 2013;52(2):139–151. doi:10.1007/s40262-012-0024-7
14. Rocha JF, Almeida L, Falcão A, et al. Opicapone: a short lived and very long acting novel catechol-O-methyltransferase inhibitor following multiple dose administration in healthy subjects. Br J Clin Pharmacol. 2013;76(5):763–775. doi:10.1111/bcp.12081
15. Fabbri M, Ferreira JJ, Lees A, et al. Opicapone for the treatment of Parkinson’s disease: a review of a new licensed medicine. Mov Disord. 2018;33(10):1528–1539. doi:10.1002/mds.27475
16. Dingemanse J, Jorga KM, Schmitt M, et al. Integrated pharmacokinetics and pharmacodynamics of the novel catechol-O-methyltransferase inhibitor tolcapone during first administration to humans*. Clin Pharmacol Ther. 1995;57(5):508–517. doi:10.1016/0009-9236(95)90035-7
17. Keränen T, Gordin A, Karlsson M, et al. Inhibition of soluble catechol-O-methyltransferase and single-dose pharmacokinetics after oral and intravenous administration of entacapone. Eur J Clin Pharmacol. 1994;46(2):151–157. doi:10.1007/BF00199880
18. Rocha J-F, Falcão A, Santos A, et al. Effect of opicapone and entacapone upon levodopa pharmacokinetics during three daily levodopa administrations. Eur J Clin Pharmacol. 2014;70(9):1059–1071. doi:10.1007/s00228-014-1701-2
19. Santos A, Falcão A, Ferreira J, et al. Effect of food on Opicapone pharmacokinetics and pharmacodynamics [abstract]. Mov Disord. 2017;32(suppl 2).
20. Rocha JF, Santos A, Falcão A, et al. Effect of moderate liver impairment on the pharmacokinetics of opicapone. Eur J Clin Pharmacol. 2014;70(3):279–286. doi:10.1007/s00228-013-1602-9
21. Castro Caldas A, Teodoro T, Ferreira JJ. The launch of opicapone for Parkinson’s disease: negatives versus positives. Expert Opin Drug Saf. 2018;17(3):331–337. doi:10.1080/14740338.2018.1433659
22. Pinto R, l’Hostis P, Patat A, et al. Evaluation of opicapone on cardiac repolarization in a thorough QT/QTc study. Clin Pharmacol Drug Dev. 2015;4(6):454–462. doi:10.1002/cpdd.188
23. Ferreira JJ, Rocha J-F, Falcão A, et al. Effect of opicapone on levodopa pharmacokinetics, catechol-O-methyltransferase activity and motor fluctuations in patients with Parkinson’s disease. Eur J Neurol. 2015;22(5):815–25, e56. doi:10.1111/ene.12666
24. Rocha J-F, Ferreira JJ, Falcão A, et al. Effect of 3 single-dose regimens of opicapone on levodopa pharmacokinetics, Catechol-O-methyltransferase activity and motor response in patients with parkinson disease. Clin Pharmacol Drug Dev. 2016;5(3):232–240. doi:10.1002/cpdd.217
25. Ferreira JJ, Lees A, Rocha J-F, Poewe W, Rascol O, Soares-da-silva P. Opicapone as an adjunct to levodopa in patients with Parkinson’s disease and end-of-dose motor fluctuations: a randomised, double-blind, controlled trial. Lancet Neurol. 2016;15(2):154–165. doi:10.1016/S1474-4422(15)00336-1
26. Lees AJ, Ferreira J, Rascol O, et al. Opicapone as adjunct to levodopa therapy in patients with parkinson disease and motor fluctuations: a randomized clinical trial. JAMA Neurol. 2017;74(2):197–206. doi:10.1001/jamaneurol.2016.4703
27. Scott LJ. Opicapone: a review in parkinson’s disease. Drugs. 2016;76(13):1293–1300. doi:10.1007/s40265-016-0623-y
28. Lees A, Ferreira J, Lopes N, et al. Efficacy and safety of opicapone in patients over 70 years with Parkinson’s disease and motor fluctuations [abstract]. Mov Disord. 2015;30(Suppl1: 258). doi:10.1002/mds.26295
29. Ferreira J, Lees A, Tolosa E, et al. Switching double-blind opicapone, entacapone or placebo to open-label opicapone: efficacy results of the 1-year extension of study BIPARK I [abstract]. Mov Disord. 2016;31(Suppl.2).
30. Ferreira J, Lees A, Rascol O, et al. Activities of daily living and motor scores of the UPDRS in fluctuating Parkinson’s disease patients treated with opicapone [abstract]. Mov Disord. 2016;31(Suppl.2).
31. Oliveira C, Lees A, Ferreira J, et al. Opicapone and non-motor symptoms in Parkinson’s disease: results from a double-blind, randomized, placebo-controlled study and open-label extension. [abstract]. Mov Disord. 2015;30(Suppl1: S173). doi:10.1002/mds.26296
32. Ferreira JJ, Lees A, Rocha J-F, Poewe W, Rascol O, Soares-da-silva P. Long-term efficacy of opicapone in fluctuating Parkinson’s disease patients: a pooled analysis of data from two phase 3 clinical trials and their open-label extensions. Eur J Neurol. 2019;26(7):953–960. doi:10.1111/ene.13914
33. Costa R, Oliveira C, Pinto R, et al. One-year open-label efficacy and safety of opicapone inParkinson’s disease BIPARK-II study [abstract]. Mov Disord. 2014;29(Suppl1: 630). doi:10.1002/mds.25914
34. Lees A, Ferreira JJ, Rocha J-F, et al. Safety profile of opicapone in the management of parkinson’s disease. J Parkinsons Dis. 2019;9(4):733–740. doi:10.3233/JPD-191593
35. Lopes N, Ferreira J, Lees A, et al. Hepatic safety of opicapone in Parkinson’s disease patients. Mov Disord. 2015;30(Suppl1: 263). doi:10.1002/mds.26295
36. European Medicines Agency. Ongentys: EPAR - Public assessment report; 2016. Available from: https://www.ema.europa.eu/en/documents/assessment-report/ongentys-epar-public-assessment-report_en.pdf.
37. BIAL. FDA approves Opicapone; 2020. Available from: https://www.bial.com/com/media/news/fda-approves-opicapone.
38. BIAL. Opicapone approved in Japan; 2020. Available from: https://www.bial.com/com/media/news/opicapone-approved-in-japan.
39. Nomoto M, Takeda A, Iwai K, Nishimura A, Hattori N. Pharmacokinetic comparison of capsule and tablet formulations of opicapone in healthy japanese subjects: Phase 1 Study. Clin Pharmacol Drug Dev. 2021;10(2):173–179. doi:10.1002/cpdd.802
40. Takeda A, Takahashi R, Tsuboi Y, et al. Randomized, controlled study of opicapone in japanese parkinson’s patients with motor fluctuations. Mov Disord. 2021;36(2):415–423. doi:10.1002/mds.28322
41. Ebersbach G, Ferreira J, Antonini A, et al. Opicapone’s added benefit as a first-line adjunctive therapy to levodopa and when used promptly in the motor fluctuations spectrum of Parkinson’s disease: a post-hoc analysis of BIPARK-I and II [abstract]. Mov Disord. 2020;35(suppl 1).
42. Lees A, Ferreira J, Poewe W, et al. Efficacy and safety/tolerability of opicapone in catechol-O-methyltransferase inhibitor-naïve Parkinson’s disease patients recently diagnosed with motor fluctuations [abstract]. Mov Disord. 2020;35(suppl 1).
43. Ferreira J, Lees A, Ebersbach G, et al. Efficacy of opicapone compared to entacapone in catechol-O-methyltransferase inhibitor-naïve Parkinson’s disease patients recently diagnosed with motor fluctuations: a post-hoc conservative analysis [abstract]. Mov Disord. 2020;35(suppl 1).
44. Antonini A, Ebersbach G, Rascol O, et al. Efficacy of opicapone in different levodopa-containing treatment regimens in Parkinson’s disease patients with motor fluctuations [abstract]. Mov Disord. 35(suppl 1).
45. Ferreira J, Poewe W, Antonini A, et al. Opicapone as first-line adjunctive levodopa treatment in parkinson’s disease patients with motor fluctuations: findings from BIPARK-I and II combined post-hoc analysis [abstract]. Mov Disord. 2020;35(suppl 1).
46. Reichmann H, Lees A, Rocha J-F, Magalhães D, Soares-da-silva P. Effectiveness and safety of opicapone in Parkinson’s disease patients with motor fluctuations: the OPTIPARK open-label study. Transl Neurodegener. 2020;9(1):9. doi:10.1186/s40035-020-00187-1
47. Klietz M, Greten S, Wegner F, Höglinger GU. Safety and tolerability of pharmacotherapies for parkinson’s disease in geriatric patients. Drugs Aging. 2019;36(6):511–530. doi:10.1007/s40266-019-00654-z
48. Lees A, Reichmann H, Rocha JF, Magalhães D, Soares-da-silva P. Onset of drug-related adverse events in parkinson’s disease patients with motor fluctuations treated with opicapone in clinical practice: OPTIPARK post-hoc analysis [abstract]. Mov Disord. 2020;35(Suppl1).
49. Ongentys® (opicapone) Summary of product characteristics: bial - Portela Ca, S.A; 2020. Available from: https://www.ema.europa.eu/en/documents/product-information/ongentys-epar-product-information_en.pdf.
50. European Medicines Agency. EudraVigilance; 2021. Available from: www.adrreports.eu. Accessed
51. Ferreira JJ, Katzenschlager R, Bloem BR, et al. Summary of the recommendations of the EFNS/MDS-ES review on therapeutic management of Parkinson’s disease. Eur J Neurol. 2013;20(1):5–15. doi:10.1111/j.1468-1331.2012.03866.x
52. Salamon A, Zádori D, Szpisjak L, Klivényi P, Vécsei L. Opicapone for the treatment of Parkinson’s disease: an update. Expert Opin Pharmacother. 2019;20(18):2201–2207. doi:10.1080/14656566.2019.1681971
53. Christopher Kobylecki HT. Parkinson’s disease with end-of-dose motor fluctuations: opicapone; 2017. Available from: nice.org.uk/guidance/es9.
54. National Institute for Health and Care Excellence. Parkinson’s disease in adults: diagnosis and management. (NICE Guideline No. 71). London: National Institute for Health and Care Excellence (NICE); 2017 Jul. Avaialble from: https://www.ncbi.nlm.nih.gov/books/NBK447153/.
55. Fox SH, Katzenschlager R, Lim S-Y, et al. International Parkinson and movement disorder society evidence-based medicine review: update on treatments for the motor symptoms of Parkinson’s disease. Mov Disord. 2018;33(8):1248–1266. doi:10.1002/mds.27372
56. Aquino CC, Fox SH. Clinical spectrum of levodopa-induced complications. Mov Disord. 2015;30(1):80–89. doi:10.1002/mds.26125
57. Stocchi F, Antonini A, Barone P, et al. Early DEtection of wEaring off in Parkinson disease: the DEEP study. Parkinsonism Relat Disord. 2014;20(2):204–211. doi:10.1016/j.parkreldis.2013.10.027
58. Rizos A, Martinez-Martin P, Odin P, et al. Characterizing motor and non-motor aspects of early-morning off periods in Parkinson’s disease: an international multicenter study. Parkinsonism Relat Disord. 2014;20(11):1231–1235. doi:10.1016/j.parkreldis.2014.09.013
59. Fackrell R, Carroll CB, Grosset DG, et al. Noninvasive options for ‘wearing-off’ in Parkinson’s disease: a clinical consensus from a panel of UK Parkinson’s disease specialists. Neurodegener Dis Manag. 2018;8(5):349–360. doi:10.2217/nmt-2018-0020
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.