")
Back to Journals » Patient Preference and Adherence » Volume 8
Clinical utility and patient perspectives on the use of extended half-life rFIXFc in the management of hemophilia B
Authors Miguelino M, Powell J
Received 10 May 2014
Accepted for publication 18 June 2014
Published 8 August 2014 Volume 2014:8 Pages 1073—1083
DOI https://doi.org/10.2147/PPA.S54951
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Maricel G Miguelino, Jerry S Powell
Division of Hematology and Oncology, University of California Davis Medical Center, Sacramento, CA, USA
Abstract: Hemophilia B is an X-linked genetic disease caused by mutation of the gene for coagulation protein factor IX (FIX), with an incidence of approximately once every 30,000 male births in all populations and ethnic groups. When severe, the disease leads to spontaneous life threatening bleeding episodes. When untreated, most patients die from bleeding complications before 25 years of age. Current therapy requires frequent intravenous infusions of therapeutic recombinant or plasma-derived protein concentrates containing FIX. Most patients administer the infusions at home every few days, and must limit their physical activities to avoid abnormal bleeding when the FIX activity levels are below normal. After completing the pivotal Phase III clinical trial, a new therapeutic FIX preparation that has been engineered for an extended half-life in circulation, received regulatory approval in March 2014 in Canada and the US. This new FIX represents a major therapeutic advance for patients with hemophilia B. The half-life is prolonged due to fusion of the native FIX molecule with the normal constant region of immunoglobulin G. This fusion molecule then follows the normal immunoglobulin recirculation pathways through endothelial cells, resulting in prolonged times in circulation. In the clinical trials, over 150 patients successfully used eftrenonacog alfa regularly for more than 1 year to prevent spontaneous bleeding, to successfully treat any bleeding episodes, and to provide effective coagulation for major surgery. All infusions were well tolerated and effective, with no inhibitors detected and no safety concerns. This promising therapy should allow patients to use fewer infusions to maintain appropriate FIX activity levels in all clinical settings.
Keywords: factor IX, hemophilia B, prophylaxis, genetic coagulation defects, extended half-life
Introduction
Hemophilia B is an X-linked genetic disorder in which there is a mutation in the gene encoding the coagulation protein factor IX (FIX), one of the critical serine proteases in the coagulation cascade. When severe, defined as FIX clotting activity <1%, patients will be at risk for spontaneous, life threatening bleeding episodes. Individuals with moderate hemophilia B (1%–5% clotting activity) or with mild hemophilia B (6%–49% activity clotting activity) will usually suffer abnormal bleeding only after minor trauma or surgery. The incidence of hemophilia B is approximately one in 30,000 male births, consistently seen in various geographic and ethnic populations studied.1
When untreated, patients with severe hemophilia B have a short life expectancy of approximately 25 years, but over the past several decades, the clinical management for hemophilia B has improved dramatically.2,3 Factor replacement therapy has reduced the morbidity and mortality of hemophilia B through reduction in the frequency of bleeding episodes and improvement in the quality of life.4,5 However, while individuals with hemophilia B have nearly normal life expectancy compared to individuals with normal coagulation, deaths still occur at higher rates due to bleeding episodes.6 Regular intravenous infusions of FIX concentrates 2–3 times each week, termed prophylactic therapy, reduce the development of hemophilic arthropathy and are now the standard of care for children and increasingly for adults as well.7,8 The development of recombinant factors has provided a safe and reproducible source of the factors, as well as increased the supply, but these therapies are expensive, as the annual costs have risen to ~$150,000 per patient in the US. Although prophylaxis is the recommended standard for treatment, these rigorous regimens are difficult and adherence remains a problem.9 Many patients find it difficult to spend 20 minutes every other morning to mix and infuse intravenous factor concentrates. In addition, convenient access to peripheral veins remains difficult, and many children require use of central venous access devices, with the concomitant risks of sepsis and thrombosis.10,11 Despite recent promising success in gene therapy for hemophilia B, a cure for hemophilia is not yet available.12 Thus, improved FIX products are needed.
Various approaches are in development to improve the treatment of hemophilia B.13–18 Eftrenonacog alfa is the first extended half-life FIX approved for treatment of hemophilia B, in March 2014 in Canada and the US. The protein is composed of a single molecule of recombinant FIX (rFIX) covalently fused to the dimeric Fc domain of immunoglobulin (Ig) G1 (rFIXFc).15,17 This approach combines two molecular structures, each with a long history of safety and efficacy in clinical use.19 rFIX currently is available from two different pharmaceutical manufacturers, with the first rFIX approved for clinical use nearly 20 years ago. The IgG constant region (Fc) has been molecularly engineered to create fusion proteins with prolonged circulating half-life for other products currently used clinically (eg, etanercept, romiplostim) and others in development.19,20 After nearly a decade of experience in clinical use, those protein fusions support the safety of the Fc fusion approach. In addition, there are animal models that suggest that the Fc fusion approach may be less immunogenic and perhaps even tolerogenic compared to the parent compound.21,22 In hemophilia, the clinical trials currently getting started should provide interesting and definitive human data for this question.
With Fc fusion proteins, the neonatal Fc receptor interacting with the endogenous IgG recycling pathway delays lysosomal degradation of IgG and the fusion proteins, recycling them back into circulation and thus prolonging the plasma half-life.23,24 The Fc domain has a half-life of 3 weeks. Although Fc fusions are typically expressed as homodimers formed through a disulfide bond, these dimeric structures were not effective for large clotting factors. Fusion of the monomeric form of the IgG1 Fc to human FIX, factor FVIIa (FVIIa), and B domain-deleted FVIII has demonstrated increases in plasma half-life.24 In addition, the larger sized molecules are expected to experience slower renal clearance.25 An additional advantage during manufacture is that the Fc domain can improve the solubility and stability of the partner molecule both in vitro and in vivo, and allows for easy cost-effective purification by protein-G/A affinity chromatography.17
Preclinical studies of Alprolix™ (rFIXFc)
Eftrenonacog alfa contains a single molecule of FIX attached through recombinant technology to the constant region (Fc) of IgG. There are no intervening linker sequences. A human cell line, HEK-293H cells, is used for expression, and co-transfected with the expression cassette for PC5,26 a processing enzyme, to ensure full cleavage of the FIX propeptide. Cell lines are grown in serum-free suspension media in the presence of vitamin K. Purification of rFIXFc monomer is by column chromatography with the use of a protein A capture step and two anion exchange steps, Fractogel® DEAE (EMD Millipore, Billerica, MA, USA) and Q Sepharose® (GE Healthcare Life Sciences, Little Chalfont, UK). The last ion exchange step involves pseudo-affinity elution17 from a Q Sepharose resin with low ionic strength CaCl2 to obtain rFIXFc with highest specific activity.
Biochemical characterization of rFIXFc
After expression of rFIXFc, proper propeptide processing was confirmed by Lys-C peptide mapping. The rFIXFc chain contains 641 amino acids, and the Fc chain contains 227 amino acids. The extensive posttranslational modification of FIX was also found for rFIXFc after expression in HEK-293H cells.27 In addition, rFIXFc was comparable with rFIX, for Ser-158 phosphorylation and Tyr-155 sulfation. N-linked glycosylation fingerprints indicated that the FIX-derived glycans are not fully sialylated, similar to rFIX. rFIXFc possessed a greater degree of β-hydroxylation of Asp-64 than did rFIX or plasma-derived FIX.
Animal pharmacokinetic studies of rFIXFc compared with rFIX
Coagulation activity and rFIXFc protein levels assayed in FIX-deficient mice demonstrated that rFIXFc had measurable activity for much longer than rFIX after single injection.17 Similar results showing prolonged half-life were seen in rats, normal mice, FIX deficient dogs and cynomolgus monkeys as well.17 In neonatal receptor null knockout mice, rFIXFc showed a half-life similar to the half-life of rFIX in these animals of 16.5 hours,17 demonstrating that the mechanism of half-life extension was through the neonatal receptor. The animal pharmacokinetic studies consistently demonstrated that the terminal half-life of rFIXFc was three-fold to four-fold longer than rFIX across species.
Phase I study in patients with hemophilia B
Shapiro et al reported the first-in-human Phase I/IIa clinical trial of rFIXFc.15 This open-label, dose-escalation trial (clinicaltrials.gov identifier NCT00716716) in previously treated adult subjects with hemophilia B examined the safety and pharmacokinetics of rFIXFc in patients with hemophilia B. Inclusion and exclusion criteria were typical for clinical trials in hemophilia B. Male subjects ≥18 years of age with severe (defined as ≤2 IU/dL FIX coagulant activity) hemophilia B and ≥150 prior documented exposure days to other FIX products were included. Patients with a history of inhibitors or allergic reactions associated with FIX or intravenous immunoglobulin, concurrent autoimmune disease, coagulation disorder other than hemophilia B, or were taking medications that could affect hemostasis were excluded.
Safety was evaluated by physical examination, vital signs, electrocardiogram, and observation for adverse events, neutralizing antibodies, or laboratory changes over time. FIX activity was determined by the one-stage activated partial thromboplastin time clotting assay with the use of Normal Reference Plasma as a calibrator. FIX inhibitor was measured by Nijmegen-modified Bethesda assay, and the anti-rFIXFc antibodies were evaluated by a specific bridging electrochemiluminescent immunoassay.
Subjects with a variety of hemophilia B genotypes, such as stop codon/nonsense and missense mutations, were included. Several subjects had near absence of FIX antigen that correlated with markedly reduced FIX activity, whereas others with missense genotypes had more antigen than activity, indicating a dysfunctional circulating protein. A total of 16 adverse events, such as dysgeusia and mild headache, were reported by seven subjects, distributed evenly across treatment groups, and most were mild or moderate. No clinically relevant changes occurred in laboratory values, QT interval, or vital signs. No allergic reactions, FIX inhibitors, or anti-rFIXFc antibodies were detected. There were no reports of thrombosis during the study.
Pharmacokinetics in the Phase I clinical trial
A dose-proportional increase in FIX activity was observed immediately after infusion of eftrenonacog alfa (ie, rFIXFc). FIX activity exhibited bi-exponential decay after infusion of rFIXFc and was characterized by a short distribution (α) phase followed by a log-linear elimination (β) phase. The mean elimination half-life was dose independent over the therapeutic dose range tested, ie, 53.5 hours, 57.6±8.27 hours (half-life ± standard deviation), and 56.5±14.1 hours at 25 IU/kg, 50 IU/kg, and 100 IU/kg, respectively. The mean elimination half-life reported for rFIX (BeneFIX® Wyeth BioPharma, Andover, MA USA) is 19.3±4.97 hours (range 11.1–36.4 hours).28 The average time to 3% above baseline was 3.81 days, 6.28±1.11 days, and 8.53±1.58 days after doses of 25 IU/kg, 50 IU/kg, and 100 IU/kg, respectively. At 168 hours (1 week) after dosing, the plasma FIX activity was sustained at an average of 1.11 IU/dL, 2.47±0.911 IU/dL, and 4.65±1.73 IU/dL above baseline for 25 IU/kg, 50 IU/kg, and 100 IU/kg, respectively. The parameters for mean residence time, clearance, and volume of distribution at steady state were all independent of dose. The mean residence time for all dose groups was 71.9±9.66 hours (range 53.2–85.9 hours), whereas the corresponding value reported for rFIX was 26.0±6.07 hours (range 15.8–46.1 hours). The mean clearance of rFIXFc was 3.18 mL/hour/kg, whereas the reported value for rFIX was 8.40±2.01 mL/hour/kg, and the mean volume of distribution at steady state of rFIXFc was 227±57.1 mL/kg (range 162–296 mL/kg). Furthermore, each 1 IU/kg infused rFIXFc raised plasma FIX activity by 0.930±0.179 IU/dL on average. The corresponding values reported for rFIX were 0.75±0.23 IU/dL per IU/kg (range 0.34–1.38 IU/dL per IU/kg).
Plasma rFIXFc antigen levels were measured by an rFIXFc-specific enzyme-linked immunosorbent assay, and the results were in parallel with the activity results. The plasma rFIXFc activity, as measured by the one-stage clotting assay, correlated well with rFIXFc antigen by enzyme-linked immunosorbent for all subjects as a group (R2=0.946, P<0.001). This Phase I clinical trial confirmed that rFIXFc might offer a viable therapeutic approach for hemophilia B.
Phase III clinical study in patients with severe hemophilia B
The Phase III nonrandomized, open-label, multicenter study was designed to compare the pharmacokinetics of rFIXFc with those of recombinant FIX and to assess the safety and efficacy of repeated administration of rFIXFc for the prevention and treatment of bleeding in adolescents and adults with severe hemophilia B (ClinicalTrials.gov number NCT01027364).29 The participants were representative of the general adult population with severe hemophilia B, all were ≥12 years of age and had severe hemophilia B (endogenous FIX level of ≤2 IU/dL or ≤2% of normal levels). Representative of different clinical regimens in use, the study included four treatment groups: Group One received weekly dose-adjusted prophylaxis (50 IU/kg of body weight of rFIXFc to start) with the dose adjusted as needed clinically, Group Two received interval-adjusted prophylaxis (100 IU/kg every 10 days to start) with the interval adjusted as needed, Group Three received treatment as needed (episodic or on-demand treatment) for bleeding episodes (20–100 IU/kg) with the dose adjusted according to bleeding severity, and Group Four received treatment as needed for surgical procedures. In a subgroup of Group One participants, comparative sequential pharmacokinetic assessments of rFIX (BeneFIX) and rFIXFc were performed following infusion of a dose of 50 IU/kg, and repeated at week 26. The primary efficacy endpoint was the annualized bleeding rate, and safety endpoints included the development of inhibitors and adverse events. There were 123 patients enrolled who were demographically diverse and with a genotype profile as expected for this population. There were 5,243 rFIXFc administrations during the study, and overall 96.6% of participants in the prophylaxis groups were adherent to their treatment regimen, most of whom self-administered rFIXFc at home.
The pharmacokinetics subgroup included 22 subjects. The terminal half-life of rFIXFc was significantly longer than that of rFIX (geometric mean 82.1 versus 33.8 hours; P<0.001). The incremental recovery levels for rFIXFc and rFIX were similar. The time to reach a FIX level of 1 IU/dL (1%) was 11.2 days with rFIXFc and 5.1 days with rFIX. The pharmacokinetic findings at week 26 were similar. In the group that received prophylaxis dosing based on individualized intervals to maintain trough FIX levels above 3% (Group Two), over half of the participants had dosing intervals of ≥14 days during the last 3 months on the study. In the weekly prophylaxis group, the median dose was 45 IU/kg. As expected, the reduction in the annualized bleeding rate with prophylaxis as compared with episodic treatment was consistent across all demographic and disease-based subgroups. Participants in Groups One and Two with the highest bleeding frequency before study entry had median annualized bleeding rates of 2.05 and 2.76, respectively, while enrolled in the study. Among the participants receiving prophylaxis, 23.0% in Group One and 42.3% in Group Two had no bleeding episodes during the study. There were no reports of vascular thrombotic events, serious hypersensitivity, or anaphylaxis, and there were no deaths during the study. No unique safety concerns were identified in participants undergoing major surgery. Thus, adverse events observed in this study were consistent with those expected in the population of persons with hemophilia.30
Surgery experience during the Phase III clinical study
In the Phase III study,29 rFIXFc also demonstrated safe and effective perioperative hemostasis during the perioperative period in subjects requiring major – mostly orthopedic – surgery. In 14 major surgeries performed in twelve participants, including knee replacements, the hemostatic response during the perioperative period was rated by investigators or surgeons as excellent (for 13 surgeries) or good (for one). Blood loss was consistent with similar surgeries in subjects without hemophilia. Dosing was determined locally by the investigator based on individual rFIXFc pharmacokinetic profiles, type of surgery, and clinical status, and followed using local laboratory assay results. The strong correlation (R2=0.9586, P<0.001) between observed and predicted FIX activity suggests surgery did not impact on rFIXFc pharmacokinetic properties. No unique safety concerns were identified during surgical rehabilitation. rFIXFc was not associated with inhibitor formation in any subject, all of whom had prior exposure to FIX products. In general, for previously treated patients after 150 exposure days, the risk of developing an inhibitor after switching to another FIX product is very low. Thus, it is not surprising that no inhibitors were detected for subjects switching to the fusion protein, composed of two natural proteins without any new or foreign epitopes. There was no evidence of allergic reactions or thrombogenicity as reported previously for rFIX.31
Implications from the clinical studies for treatment of hemophilia B
Previous studies have reported clear medical benefits with early individualized prophylactic therapy in hemophilia.5,32 Not all individuals with severe hemophilia B can adhere to the strict requirements of frequent infusions with previous short-acting therapies. To achieve wider use, prophylaxis therapy needs to be effective, convenient, simple, and safe. Introduction of rFIXFc replacement therapy with an extended half-life may represent an important step toward achieving these goals (Figure 1).33 Moreover, a FIX product with an extended half-life may have an additional advantage in the treatment of episodic bleeds through a potential reduction in the number of follow-up treatments needed to support complete healing.34 These advantages were recognized by the subjects who participated in the Phase III clinical study.29 When given the choice of continuing on with treatment with eftrenonacog alfa on an extension study after the Phase III clinical trial, or returning to treatment with their previous shorter half-life FIX product, none of the subjects chose to return to treatment with their previous FIX product, and all chose to continue with the extended half-life product.
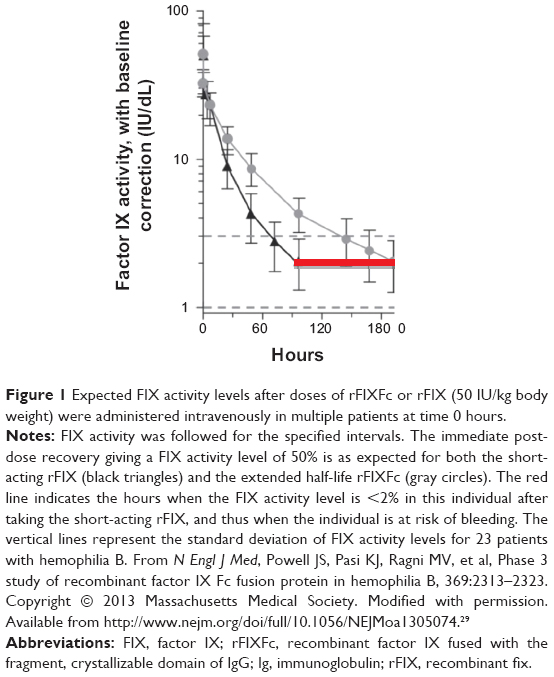 | Figure 1 Expected FIX activity levels after doses of rFIXFc or rFIX (50 IU/kg body weight) were administered intravenously in multiple patients at time 0 hours. |
As rFIXFc is the first extended half-life product, it is expected to replace current FIX products for children and adults with hemophilia B. It is important also to evaluate dosing strategies to understand how to efficiently initiate prophylaxis with rFIXFc. It is reassuring that none of the subjects experienced any clinical difficulties switching to the new product in the clinical trials. Since there is significant individual variation in risk of bleeding, selecting one dose and interval regimen is suboptimal as it may expose patients to avoidable bleeding events or overtreatment. The use of patient information (eg, previous therapy) to guide empiric selection of dosing regimen with a new FIX therapy may prove to be a more systematic and efficient approach.
The Phase III clinical study results provide insight into the pharmacodynamics of rFIXFc in patients with hemophilia B. The maximum concentration was reached immediately after infusion, suggesting a rapid onset of action similar to rFIX. This rapid onset was reflected clinically, as patients reported relief of pain from joint bleeding that was as rapid with rFIXFc as they experienced with rFIX. The in vivo recovery observed for rFIXFc may represent an improvement compared with that reported for rFIX, and may be attributable to the more human posttranslational modification(s) from the human cell line used to produce rFIXFc, or to the Fc moiety. Data from the pharmacokinetic analyses of the clinical trials may provide a means of optimizing individualized prophylactic treatment to achieve target trough levels and to reduce peak/trough variation.35–37 The results suggest that once weekly dosing of rFIXFc at 20 IU/kg, or every 10 days at 40 IU/kg, or every 2 weeks at 100 IU/kg is sufficient to maintain a mean trough of 1% above baseline in many adult patients with hemophilia B. In addition, one must consider the heterogeneity of reported clinical breakthrough bleeding events relative to trough level of plasma FIX activity,38 as well as the heterogeneity in pharmacokinetic parameters of individual subjects. Thus, prophylaxis dosing requires individual adjustment, taking into account the many other variables that may contribute to risk of bleeding, such as more intense activity levels. Indeed, with an extended half-life product there may be opportunities to infuse more often or with higher doses to achieve higher trough levels for times of increased activity.
An rFIX product with a longer half-life than currently available FIX products would be expected to require fewer injections to maintain target FIX levels, thus reducing the need for repeated venous access, potentially improving the acceptance of prophylactic regimens by patients with hemophilia B. Reduced frequency of dosing would be expected to reduce the number of pediatric patients in whom central venous access devices are required, thus reducing the significant medical complications that accompany use of central access devices; infections, sepsis, and thrombosis. These advantages should also increase adherence to prophylactic regimens in the pediatric population. Further, the increased half-life should decrease repeated dosing in the treatment of episodic bleeding or in surgical settings.
The initial pricing of eftrenonacog alfa has been announced, a “wholesale acquisition cost” of 2.85 USD per IU,39 and its use for routine prophylaxis appears to be about the same annual cost as for current rFIX products. Certainly, actual clinical use will determine the final pricing structure and impact how rapidly the hemophilia community adopts use of this new FIX product. In addition, experience in other clinical settings will be needed. Use in patients with mild and moderate hemophilia B has not been examined yet; but no safety issues would be expected. Hemophilia B patients who have a history of developing an inhibitor or an allergic reaction to any FIX product were excluded from the clinical trials, so use in this population has not been examined. Female carriers of hemophilia B with low FIX activity levels, including pregnant women, were also excluded from the clinical trials. Finally, there are no data on use in patients >65 years of age, and for use in previously untreated patients with severe hemophilia B. Studies with new factor products in previously untreated patients will be critical to assess potential immunogenicity of the new products. No special problems in any of these populations would be expected, but clinical experience has not been reported.
Within a couple of years, eftrenonacog alfa should become the cornerstone in the treatment of hemophilia B. The increased half-life should significantly improve the quality of life of patients with hemophilia B. Joint health will improve and higher trough levels of FIX as a result of the increased half-life should allow more normal activities in patients with hemophilia B. The clinical trials for gene therapy of FIX deficiency continue slowly to make progress, and it is expected that within a couple of years patients will have the options of using one of the new FIX products with increased half-life or participating in gene therapy trials. Additional clinical experience is needed to guide their choice.
Currently, close to 75% of the world’s population of hemophilia B patients lack access to any FIX product for routine treatment of bleeding episodes.40 A major advantage of the introduction of the new FIX products will be to encourage the manufacturers of current FIX products to create new markets for these products in the underserved populations.
Other new products in development for hemophilia B
Two other extended half-life FIX products have completed Phase III clinical trials and are expected to be submitted for regulatory approval in 2015. N9-GP is an rFIX molecule with a 40 kDa polyethylene glycol (PEG) molecule attached to the activation peptide of FIX.13 During activation, the PEG moiety is removed, leaving the wild-type activated FIX. The toxicology program did not identify any PEG-related safety issues. The Phase III trial demonstrated overall safety and efficacy. In the Phase I trial, there was one serious adverse event reported as probably being related to N9-GP, a hypersensitivity reaction (nausea, vomiting, paresthesias, facial swelling, and diaphoresis, but no changes in blood pressure or pulse) that occurred during administration of N9-GP in a 25-year-old male patient who had no history of inhibitors nor allergic reactions to his previous plasma-derived FIX product. After the event, the patient continued with his previous plasma-derived FIX product without any complications.13
Another approach to extend the half-life of FIX uses fusion of the coagulation protein to albumin, which has a half-life of approximately 20 days. The increase in half-life mediated by albumin fusion proteins also results from their interaction with the salvage neonatal Fc receptor present on many cell types, in addition to the slower renal clearance for larger-sized molecules.25 The albumin fusion approach uses recombinant technology to covalently attach the clotting factor to albumin through the FIX activation peptide. Thus, during the activation of the coagulation cascade at the site of clotting, the FIX activation peptide is cleaved, releasing the albumin part of the fusion so that only the native FIX remains at the site of clotting. Animal studies, toxicology, and the Phase I clinical trials have demonstrated safety and efficacy with extended half-life.18,41–43 The Phase III trial has finished enrollment.
FVIII extended half-life products in clinical trials
While the focus is on hemophilia B, it may be useful to review briefly new developments in hemophilia to provide context (Table 1). For extension of half-life for FVIII, efforts have pursued conjugating the factor protein at one or a few specific sites with PEG, a hydrophilic polymer, or use recombinant technology to fuse the factor protein with the Fc fragment of IgG. Fusion of FVIII to albumin has failed so far to preserve effective coagulation activity. Fusion of FVIII to Fc, similar to the efforts for FIX described above, was more successful.44 Some of these new approaches to new products for hemophilia may also have the additional advantage of inducing tolerance to FVIII.45 Although any of these approaches has substantially increased the plasma half-life of factors FIX and FVIIa, only limited half-life extension has been observed for FVIII. This is likely due to the different mechanisms of clearance between FIX/FVIIa and FVIII. Previous studies demonstrated that FIX clearance is likely mediated through interaction of the amino terminal Gla domain residues 3–11 with endothelial/collagen IV sites, although the specific mechanism remains unclear.46 In contrast, clearance of FVIII is very rapid in the absence of von Willebrand factor (vWF), but in the presence of vWF, FVIII is stabilized in the plasma with a half-life of approximately 12 hours. The clearance of FVIII from the plasma is likely dependent on the dominant role that vWF plays in regulating the clearance of FVIII, and thus conversely vWF interaction may limit the effectiveness of the different methods to extend the half-life of FVIII.
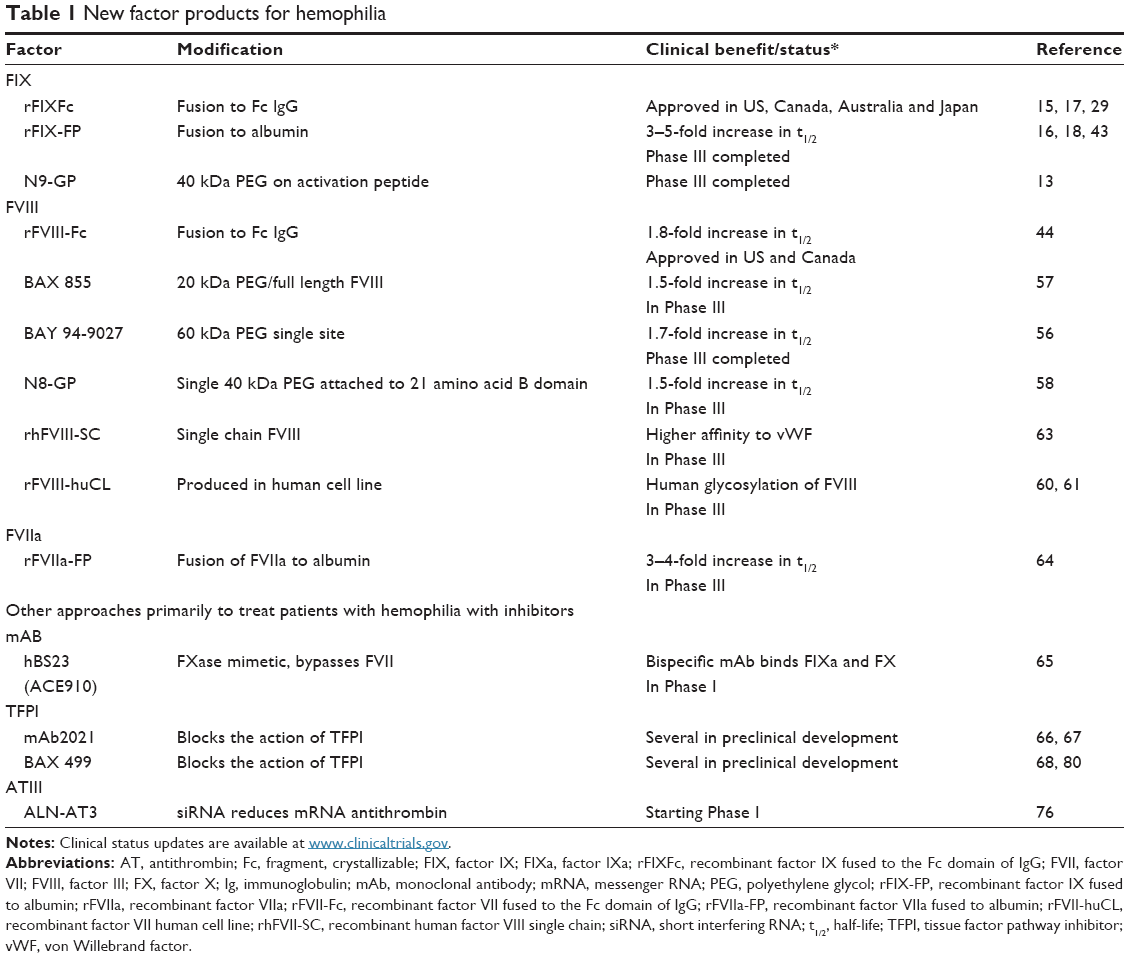 | Table 1 New factor products for hemophilia3 |
Approaches that use PEGylation
PEGylation involves the covalent attachment of PEG to a protein.47 The most common method of PEG addition is through covalent attachment to lysine residues or N-terminal amines, but this often reduces activity of the protein and the extent of PEG addition is variable, producing a heterogeneous product and complicating reliable synthesis for consistent effectiveness. A new approach involving targeted, site-specific PEGylation has significant advantages.48 One such approach is attachment of PEG–maleimide to cysteine residues. For FVIII, the PEGylation was highly specific and the site of PEGylation on the FVIII molecule was critical to PEGylation efficiency, preservation of the coagulation activity, and improvement in pharmacokinetic parameters.49–51
Nearly a dozen PEGylated protein therapeutics have been approved for use, including anti-TNFα (tumor necrosis factor-α) monoclonal antibody fragment antigen-binding fragment,52 VEGF (vascular endothelial growth factor) aptamer, Epoetin β, and IFNα-2α (interferon-α-2α).53 Although no long-term safety concerns due to the PEGylation have arisen with any of the approved therapeutics, appropriate clinical observations will be needed as the clinical trials progress for the coagulation proteins. There are several PEGylated coagulation factors that are in clinical development. In hemophilia animal models of bleeding, all of these newer PEGylated FVIII molecules have full coagulation activity, and in early clinical trial experience, there are no indications of adverse events, toxicity, and/or immunogenicity in previously treated patients with hemophilia. Extensive safety data for PEGylated proteins containing high molecular weight PEG do not indicate any safety concerns after chronic use in animal models or patients.54 However, there are differences in how the PEGylation is achieved, and subtle clinical effects may become apparent through additional clinical experience.
Small PEG molecules are more rapidly cleared than large ones. Larger PEG molecules do not penetrate into tissues as well as smaller ones. With >10 kDa PEG, there is increased pinocytotic uptake into macrophages and Kupffer cells; with >30 kDa PEG, renal clearance decreases; and with >50 kDa PEG, liver clearance increases.54,55 All of the PEGylated FVIII products in clinical trials currently use PEG adducts >15 kDa; specific toxicology experiments for each will need to be evaluated. For example, there was no toxicity associated with acute high-dose administration of BAY 94-9027, a site specific PEG-conjugated FVIII molecule. Most experts consider PEG to be inert in the human body.56
Recently, a summary was reported for toxicology and preclinical results for BAX 855, a full-length recombinant FVIII that is PEGylated by chemical means at specific lysine residues. Assessment of animal toxicity was based on mortality, clinical observations, clinical pathology, male fertility in rats, organ weights, and pathology evaluations. No PEG-related effects were observed.57 In a Phase I clinical trial, Novo Nordisk demonstrated a glycoPEGylated FVIII that increased plasma half-life by 1.6-fold. A single dose of up to 75 IU/kg N8-GP was well tolerated in patients with hemophilia A, with no safety concerns.58 Recently, the top level results for the Phase III clinical trial have been reported to the press59 – in 184 previously treated hemophilia A subjects, one neutralizing inhibitor developed. Further details will be needed to assess the significance of this finding.
Two other new FVIII products in clinical trial need to be included in this discussion. One is a recombinant FVIII manufactured in a human cell line, and currently in Phase III in adults and children and in a clinical trial for previously untreated patients with hemophilia A.60,61 The potential advantages are improved half-life and possibly a lower rate of inhibitor development in previously untreated patients due to human glycosylation patterns. Another new FVIII product was modified by recombinant technology so that the FVIII is a single-chain molecule. It has a higher affinity to vWF, which may translate into a longer half-life, and it is currently in a Phase III clinical trial.16,42,62,63
FVIIa products in clinical trials
FVIIa has been used commonly for nearly 20 years for the treatment of patients with inhibitory antibodies, and now there are other FVIIa products in development. Using the same fusion technology described above for FIX, recombinant FVIIa fusion protein is a promising extended half-life product that is in Phase III clinical trial development.64
Other novel approaches for hemophilia therapies
There are also several novel approaches being pursued in preclinical work or early Phase I trials. These new efforts to ameliorate the coagulation defect of hemophilia are intriguing, but much remains to be determined as they enter clinical development.
Inhibition of tissue factor pathway inhibitor
One novel approach in development involves manipulation of the antithrombotic pathways that control coagulation. The tissue factor/FVIIa/factor Xa complex forms small amounts of thrombin to initiate coagulation. Tissue factor pathway inhibitor (TFPI) inhibits this complex through its two Kunitz domains: Kunitz domain 1 interacts with FVIIa and Kunitz domain 2 interacts with factor Xa. A monoclonal antibody to the second Kunitz domain neutralizes the inhibitory effect of TFPI on extrinsic pathway activation. Subcutaneous administration of this antibody (monoclonal antibody 2021) 24 hours before injury prevented bleeding in hemophilic animals.65–67
Recently, several therapeutic agents that inhibit TFPI have been described as potential hemophilia treatments. One is a nucleic acid aptamer that binds tightly and specifically to TFPI and inhibits its function in vitro and in vivo.68 A non-anticoagulant sulfated polysaccharide BAX 513 demonstrated efficacy in hemophilia dogs.69 TFPI antagonist peptides have been designed and have demonstrated hemostatic efficacy in vitro and in vivo in mouse models.68,70–72 Each type of inhibitor of TFPI provides unique advantages and interactions with clotting factors. There is some concern that inhibition of TFPI by certain inhibitor types may worsen bleeding tendencies. Further studies in animals and humans will be needed to sort out the therapeutic opportunities of these potentially exciting approaches.
Targeting antithrombin
Another novel approach is represented by the development of ALN-AT3, a synthetic, N-acetylgalactosamine-conjugated RNA interference therapeutic designed to suppress liver production of antithrombin messenger RNA after subcutaneous injection. Reducing antithrombin levels has the potential to reduce the stoichiometric inhibition of thrombin and thus improve hemostasis for patients with hemophilia.73 Subcutaneous administration of ALN-AT3 resulted in dose-dependent and reversible reduction of circulating antithrombin, with a single-dose median effective dose of ~1 mg/kg in multiple species.74,75 Reduction in antithrombin was associated with significant increases in thrombin generation and enhanced hemostasis in hemophilia A and B mouse models. In a microvessel laser injury in hemophilia A and B mice, functional improvements in hemostasis were observed after treatment with ALN-AT3. Stable hemostatic plug formation was observed at all sites of injury in treated animals. Extensive toxicology studies of ALN-AT3 have been conducted in several species, and as expected, the toxicity observed in wild-type animals was due to the increased procoagulant pharmacology of ALN-AT3. Greater than 90% reduction in antithrombin in wild-type animals led to thrombotic events. In contrast, reduction by ALN-AT3 to 5% antithrombin levels was well tolerated in hemophilia A and B mice, with no evidence of thrombosis.76 Further studies are required in models that have functional FVIII and/or FIX to demonstrate long-term safety, and a Phase I clinical trial is planned.
Future concerns of novel molecules
A major concern in hemophilia A is the development of neutralizing antibodies (inhibitors) that prevent further use of therapeutic FVIII1. Current options for treatment of these patients with inhibitors are very expensive. Therefore, there are significant potential implications as the clinical trials with the new therapies evolve. It may develop that the extended half-life products will have a lower inhibitor rate, or perhaps a higher rate of developing inhibitors. These issues will only be resolved by planned clinical trials in previously untreated patients.
Recombinant FVIIa has not produced neutralizing antibodies in hemophilia patients, probably because they have native FVIIa and do not recognize the infused FVIIa as a foreign protein.77 Several pharmaceutical companies have development programs to generate molecularly modified FVIIa proteins with either longer half-life or improved efficacy. Two programs (Novo Nordisk and Bayer)78,79 with the most clinical trial data to date were based on molecular constructs that introduced conservative amino acid substitutions into native FVIIa. After no problems were seen in extensive animal testing and Phase I clinical trials, both programs were terminated in Phase III trials, due to the detection of neutralizing inhibitors. These results will need further investigation, but serve as a potential warning for programs based on molecular alterations of clotting proteins.
Conclusion
The past quarter-century has witnessed a tremendous expansion in our understanding of mechanisms that regulate hemostasis in vivo, and the development of extended half-life factor products with promise to improve treatment of hemophilia. Eftrenonacog alfa is the first fruit from these advances. Its marked increase in half-life for FIX is expected to allow less frequent infusions of factor, allow higher factor trough levels so there may be increased potential for preventing spontaneous bleeding, and perhaps most importantly, allow individuals with hemophilia B to have a more normal quality of life. In the near future, there will be other factor products with extended half-life for both hemophilia A and B, and there may be alternative approaches for preventing bleeding in hemophilia that may offer even greater advantages. Future concerns include: (1) how the costs of these new factor products will influence their clinical use; (2) if there will be any issues with inhibitor formation with the new factor products in previously untreated patients; and (3) if there will be any reason for concern in using these new factor products in an increasingly elderly hemophilia population. As over the past 25 years, progress in hemophilia will depend on enthusiastic participation by physicians and patients in appropriate clinical trials to obtain clear answers to guide future hemophilia treatment.
Disclosure
JSP receives clinical trial support from Bayer, Octapharma, NovoNordisk, Baxter, CSL Behring, and Biogen Idec. MGM declares no conflicts of interest in this work.
References
Powell JS, Rodgers GM. Inherited coagulation disorders. In: Greer JP, Arber DA, Glader B, et al, editors. Wintrobe’s Clinical Hematology. Philadelphia, PA: Wolters Kluwer; 2014:1143–1185. | ||
Giangrande P. Haemophilia B: Christmas disease. Expert Opin Pharmacother. 2005;6(9):1517–1524. | ||
Mannucci PM, Mendolicchio L, Gringeri A. Use of prophylaxis to prevent complications of hemophilia. Adv Exp Med Biol. 2001;489:59–64. | ||
Nilsson IM, Berntorp E, Lofqvist T, Pettersson H. Twenty-five years’ experience of prophylactic treatment in severe haemophilia A and B. J Intern Med. 1992;232(1):25–32. | ||
Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535–544. | ||
Darby SC, Kan SW, Spooner RJ, et al. Mortality rates, life expectancy, and causes of death in people with hemophilia A or B in the United Kingdom who were not infected with HIV. Blood. 2007;110(3):815–825. | ||
White GC 2nd, Courter S, Bray GL, Lee M, Gomperts ED. A multicenter study of recombinant factor VIII (Recombinate) in previously treated patients with hemophilia A. Thromb Haemost. 1997;77(4):660–667. | ||
Feldman BM, Pai M, Rivard GE, et al. Tailored prophylaxis in severe hemophilia A: interim results from the first 5 years of the Canadian Hemophilia Primary Prophylaxis Study. J Thromb Haemost. 2006;4(6):1228–1236. | ||
Hacker MR, Geraghty S, Manco-Johnson M. Barriers to compliance with prophylaxis therapy in haemophilia. Haemophilia. 2001;7(4):392–396. | ||
Manco-Johnson MJ, Riske B, Kasper CK. Advances in care of children with hemophilia. Semin Thromb Hemost. 2003;29(6):585–594. | ||
Gater A, Thomson TA, Strandberg-Larsen M. Haemophilia B: impact on patients and economic burden of disease. Thromb Haemost. 2011; 106(3):398–404. | ||
Nathwani AC, Tuddenham EG, Rangarajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365(25):2357–2365. | ||
Negrier C, Knobe K, Tiede A, Giangrande P, Moss J. Enhanced pharmacokinetic properties of a glycoPEGylated recombinant factor IX: a first human dose trial in patients with hemophilia B. Blood. 2011;118(10):2695–2701. | ||
Martinowitz U, Shapiro A, Quon DV, et al. Pharmacokinetic properties of IB1001, an investigational recombinant factor IX, in patients with haemophilia B: repeat pharmacokinetic evaluation and sialylation analysis. Haemophilia. 2012;18(6):881–887. | ||
Shapiro AD, Ragni MV, Valentino LA, et al. Recombinant factor IX-Fc fusion protein (rFIXFc) demonstrates safety and prolonged activity in a Phase I/IIa study in hemophilia B patients. Blood. 2012;119(3):666–672. | ||
Metzner HJ, Pipe SW, Weimer T, Schulte S. Extending the pharmacokinetic half-life of coagulation factors by fusion to recombinant albumin. Thromb Haemost. 2013;110(5):931–939. | ||
Peters RT, Low SC, Kamphaus GD, et al. Prolonged activity of factor IX as a monomeric Fc fusion protein. Blood. 2010;115(10):2057–2064. | ||
Santagostino E, Negrier C, Klamroth R, et al. Safety and pharmacokinetics of a novel recombinant fusion protein linking coagulation factor IX with albumin (rIX-FP) in hemophilia B patients. Blood. 2012;120(12):2405–2411. | ||
Schmidt SR. Fusion-proteins as biopharmaceuticals – applications and challenges. Curr Opin Drug Discov Devel. 2009;12(2):284–295. | ||
Huang C. Receptor-Fc fusion therapeutics, traps, and MIMETIBODY technology. Curr Opin Biotechnol. 2009;20(6):692–699. | ||
Borel Y, Fritsche R, Borel H, Dahlgren U, Dalhman-Hoglund A, Hanson LA. Parenteral and oral administration of tolerogens: protein–IgG conjugates. Ann N Y Acad Sci. 1996;778:80–87. | ||
Borel H, Borel Y. A novel technique to link either proteins or peptides to gammaglobulin to construct tolerogens. J Immunol Methods. 1990;126(2):159–168. | ||
Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7(9):715–725. | ||
Rath T, Baker K, Dumont JA, et al. Fc-fusion proteins and FcRn: structural insights for longer-lasting and more effective therapeutics. Crit Rev Biotechnol. Epub 2013 Oct 24. | ||
Kontermann RE. Strategies for extended serum half-life of protein therapeutics. Curr Opin Biotechnol. 2011;22(6):868–876. | ||
Lusson J, Vieau D, Hamelin J, Day R, Chretien M, Seidah NG. cDNA structure of the mouse and rat subtilisin/kexin-like PC5: a candidate proprotein convertase expressed in endocrine and nonendocrine cells. Proc Natl Acad Sci U S A. 1993;90(14):6691–6695. | ||
Bond M, Jankowski M, Patel H, et al. Biochemical characterization of recombinant factor IX. Semin Hematol. 1998;35(2 Suppl 2):11–17. | ||
Lissitchkov T, Matysiak M, Zavilska K, et al. Head-to-head comparison of the pharmacokinetic profiles of a high-purity factor IX concentrate (AlphaNine®) and a recombinant factor IX (BeneFIX®) in patients with severe haemophilia B. Haemophilia. 2013;19(5):674–678. | ||
Powell JS, Pasi KJ, Ragni MV, et al. Phase 3 study of recombinant factor IX Fc fusion protein in hemophilia B. N Engl J Med. 2013;369(24):2313–2323. | ||
Lambert T, Recht M, Valentino LA, et al. Reformulated BeneFix: efficacy and safety in previously treated patients with moderately severe to severe haemophilia B. Haemophilia. 2007;13(3):233–243. | ||
Roth DA, Kessler CM, Pasi KJ, Rup B, Courter SG, Tubridy KL. Human recombinant factor IX: safety and efficacy studies in hemophilia B patients previously treated with plasma-derived factor IX concentrates. Blood. 2001;98(13):3600–3606. | ||
van den Berg HM, Fischer K, van der Bom JG. Comparing outcomes of different treatment regimens for severe haemophilia. Haemophilia. 2003;9(Suppl 1):27–31. | ||
Lillicrap D. Improvements in factor concentrates. Curr Opin Hematol. 2010;17(5):393–397. | ||
Hoffman M, Whinna HC, Monroe DM. Circulating tissue factor accumulates in thrombi, but not in hemostatic plugs. J Thromb Haemost. 2006;4(9):2092–2093. | ||
Carlsson M, Bjorkman S, Berntorp E. Multidose pharmacokinetics of factor IX: implications for dosing in prophylaxis. Haemophilia. 1998;4(2):83–88. | ||
Kisker CT, Eisberg A, Schwartz B. Prophylaxis in factor IX deficiency product and patient variation. Haemophilia. 2003;9(3):279–284. | ||
Bjorkman S. Prophylactic dosing of factor VIII and factor IX from a clinical pharmacokinetic perspective. Haemophilia. 2003;9(Suppl 1):101–108. | ||
Ahnstrom J, Berntorp E, Lindvall K, Bjorkman S. A 6-year follow-up of dosing, coagulation factor levels and bleedings in relation to joint status in the prophylactic treatment of haemophilia. Haemophilia. 2004;10(6):689–697. | ||
Weisman R. Biogen Idec prices its newly approved hemophilia drug. The Boston Globe. 2014 April 19. | ||
Skinner MW, Myles E. World Federation of Hemophilia: 50 years of advancing treatment for all. Haemophilia. 2013;19(4):475–480. | ||
Weimer T, Wormsbacher W, Kronthaler U, Lang W, Liebing U, Schulte S. Prolonged in-vivo half-life of factor VIIa by fusion to albumin. Thromb Haemost. 2008;99(4):659–667. | ||
Schulte S. Innovative coagulation factors: albumin fusion technology and recombinant single-chain factor VIII. Thromb Res. 2013;131(Suppl 2):S2–S6. | ||
Santagostino E. PROLONG-9FP clinical development program – Phase I results of recombinant fusion protein linking coagulation factor IX with recombinant albumin (rIX-FP). Thromb Res. 2013;131(Suppl 2):S7–S10. | ||
Mahlangu J, Powell JS, Ragni MV, et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood. 2013;123(3):317–325. | ||
Scott DW, Pratt KP, Miao CH. Progress toward inducing immunologic tolerance to factor VIII. Blood. 2013;121(22):4449–4456. | ||
Gui T, Lin HF, Jin DY, et al. Circulating and binding characteristics of wild-type factor IX and certain Gla domain mutants in vivo. Blood. 2002;100(1):153–158. | ||
Abuchowski A, van Es T, Palczuk NC, Davis FF. Alteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. J Biol Chem. 1977;252(11):3578–3581. | ||
Caliceti P, Veronese FM. Pharmacokinetic and biodistribution properties of poly(ethylene glycol)–protein conjugates. Adv Drug Deliv Rev. 2003;55(10):1261–1277. | ||
Shen BW, Speigel PC, Chang CH, et al. The tertiary structure and domain organization of coagulation factor VIII. Blood. 2008;111(3):1240–1247. | ||
Ngo JC, Huang M, Roth DA, Furie BC, Furie B. Crystal structure of human factor VIII: implications for the formation of the factor IXa–factor VIIIa complex. Structure. 2008;16(4):597–606. | ||
Mei B, Pan C, Jiang H, et al. Rational design of a fully active, long-acting PEGylated factor VIII for hemophilia A treatment. Blood. 2010;116(2):270–279. | ||
Horton S, Walsh C, Emery P. Certolizumab pegol for the treatment of rheumatoid arthritis. Expert Opin Biol Ther. 2012;12(2):235–249. | ||
Alconcel SNS, Baas AS, Maynard HD. FDA-approved poly(ethylene glycol)–protein conjugate drugs. Polym Chem. 2011;2(7):1442–1448. | ||
Ivens IA, Baumann A, McDonald TA, Humphries TJ, Michaels LA, Mathew P. PEGylated therapeutic proteins for haemophilia treatment: a review for haemophilia caregivers. Haemophilia. 2013;19(1):11–20. | ||
Tang L, Leong L, Sim D, et al. von Willebrand factor contributes to longer half-life of PEGylated factor VIII in vivo. Haemophilia. 2013;19(4):539–545. | ||
Coyle TE, Reding MT, Lin JC, Michaels LA, Shah A, Powell J. Phase I study of BAY 94-9027, a PEGylated B-domain-deleted recombinant factor VIII with an extended half-life, in subjects with hemophilia A. J Thromb Haemostat. 2014;12(4):488–496. | ||
Turecek PL, Bossard MJ, Graninger M, et al. BAX 855, a PEGylated rFVIII product with prolonged half-life. Development, functional, and structural characterisation. Hamostaseologie. 2012;32(Suppl 1): S29–S38. | ||
Tiede A, Brand B, Fischer R, et al. Enhancing the pharmacokinetic properties of recombinant factor VIII: first-in-human trial of glycoPEGylated recombinant factor VIII in patients with hemophilia A. J Thromb Haemost. 2013;11(4):670–678. | ||
Novo Nordisk A/S. Novo Nordisk reports positive results from first phase 3 trial with N8-GP, a long-acting factor VIII for treatment of haemophilia A [company announcement]. Bagsværd: Novo Nordisk A/S; 2014 [March 19]. Available from: http://www.novonordisk.com/include/asp/exe_news_attachment.asp?sAttachmentGUID=31c49463-5e29-4262-9e4d-f8c47e96a57b. Accessed July 18, 2014. | ||
Sandberg H, Kannicht C, Stenlund P, et al. Functional characteristics of the novel, human-derived recombinant FVIII protein product, human-cl rhFVIII. Thromb Res. 2012;130(5):808–817. | ||
Kannicht C, Ramstrom M, Kohla G, et al. Characterisation of the post-translational modifications of a novel, human cell line-derived recombinant human factor VIII. Thromb Res. 2013;131(1):78–88. | ||
Schulte S. Challenges for new haemophilia products from a manufacturer’s perspective. Thromb Res. Epub 2013 Dec 21. | ||
Zollner SB, Raquet E, Muller-Cohrs J, et al. Preclinical efficacy and safety of rVIII-SingleChain (CSL627), a novel recombinant single-chain factor VIII. Thromb Res. 2013;132(2):280–287. | ||
Zollner S, Schuermann D, Raquet E, et al. Pharmacological characteristics of a novel, recombinant fusion protein linking coagulation factor VIIa with albumin (rVIIa-FP). J Thromb Haemost. 2014;12(2): 220–228. | ||
Muto A, Yoshihashi K, Takeda M, et al. Anti-factor IXa/X bispecific antibody (ACE910): hemostatic potency against ongoing bleeds in a hemophilia A model and the possibility of routine supplementation. J Thromb Haemostat. 2014;12(2):206–213. | ||
Hilden I, Lauritzen B, Sorenson BB, et al. Hemostatic effect of a monoclonal antibody mAb 2021 blocking the interaction between FXa and TFPI in a rabbit hemophilia model. Blood. 2012;119(24):5871–5878. | ||
Agerso H, Overgaard RV, Peterson MB, et al. Pharmacokinetics of an anti-TFPI monoclonal antibody (concizumab) blocking the TFPI interaction with the active site of FXa in cynomolgus monkeys after iv and sc administration. Eur J Pharm Sci. 2014;56:65–69. | ||
Gorczyca ME, Nair SC, Jilma B, et al. Inhibition of tissue factor pathway inhibitor by the aptamer BAX499 improves clotting of hemophilic blood and plasma. J Thromb Haemost. 2012;10(8):1581–1590. | ||
Prasad S, Lillicrap D, Labelle A, et al. Efficacy and safety of a new-class hemostatic drug candidate, AV513, in dogs with hemophilia A. Blood. 2008;111(2):672–679. | ||
Knappe S, Gorczyca ME, Jilma B, et al. Plasmatic tissue factor pathway inhibitor is a major determinant of clotting in factor VIII inhibited plasma or blood. Thromb Haemost. 2013;109(3):450–457. | ||
Peraramelli S, Thomassen S, Heinzmann A, et al. Direct inhibition of factor VIIa by TFPI and TFPI constructs. J Thromb Haemost. 2013; 11(4):704–714. | ||
Dockal M, Hartmann R, Fries M, et al. Small peptides blocking inhibition of factor Xa and tissue factor-factor VIIa by tissue factor pathway inhibitor (TFPI). J Biol Chem. 2014;289(3):1732–1741. | ||
Bolliger D, Szlam F, Suzuki N, Matsushita T, Tanaka KA. Heterozygous antithrombin deficiency improves in vivo haemostasis in factor VIII-deficient mice. Thromb Haemost. 2010;103(6):1233–1238. | ||
Peyvandi F, Garagiola I, Seregni S. Future of coagulation factor replacement therapy. J Thromb Haemost. 2013;11(Suppl 1):84–98. | ||
Sehgal A, Vaishnaw A, Fitzgerald K. Liver as a target for oligonucleotide therapeutics. J Hepatol. 2013;59(6):1354–1359. | ||
Akinc A, Sehgal A, Hettinger J, et al. An RNAi Therapeutic Targeting Antithrombin Increases Thrombin Generation in Nonhuman Primates. Available from: http://www.alnylam.com/web/wp-content/uploads/2012/12/ALNY-ASH-AT3posterXpanel-Dec2012-r.pdf. Accessed 18 July, 2014. | ||
Konkle BA, Ebbesen LS, Erhardtsen E, et al. Randomized, prospective clinical trial of recombinant factor VIIa for secondary prophylaxis in hemophilia patients with inhibitors. J Thromb Haemost. 2007;5(9):1904–1913. | ||
Lentz SR, Ehrenforth S, Karim FA, et al. Recombinant factor VIIa analogue in management of hemophilia with inhibitors: results from a multicenter, randomized, controlled trial of vatreptacog alfa. J Thromb Haemost. Epub 2014 Jun 16. | ||
Mahlangu JN, Koh PL, Ng HJ, Lissitchkov T, Hardtke M, Schroeder J. The TRUST Trial: Anti-Drug Antibody Formation In a Patient With Hemophilia With Inhibitors After Receiving The Activated Factor VII Product Bay 86-6150. Blood. 2013;122(21):573. | ||
Parunov LA, Soshitova NP, Fadeeva OA, et al. Drug-drug interaction of the anti-TFPI aptamer BAX499 and factor VIII: Studies of spatial dynamics of fibrin clot formation in hemophilia A. Thrombo Res. 2014;133(1):112–119. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.