")
Back to Journals » OncoTargets and Therapy » Volume 11
Clinical use of FLT3 inhibitors in acute myeloid leukemia
Authors Sutamtewagul G , Vigil CE
Received 25 April 2018
Accepted for publication 15 June 2018
Published 16 October 2018 Volume 2018:11 Pages 7041—7052
DOI https://doi.org/10.2147/OTT.S171640
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Arseniy Yuzhalin
Grerk Sutamtewagu, Carlos E Vigil
Division of Hematology, Oncology and Blood and Marrow Transplantation, University of Iowa Hospitals and Clinics, Iowa City, IA, USA
Abstract: Acute myeloid leukemia (AML) is a highly heterogeneous disease. Mutation with internal tandem duplication of fms-like tyrosine kinase-3 (FLT3-ITD) is one of the two most common driver mutations and the presence of FLT3-ITD delivers poor prognosis. A number of ongoing clinical efforts are focused on FLT3 inhibitor use to improve the outcomes of this otherwise difficult leukemia. Midostaurin has been shown to improve outcomes in FLT3-mutated AML in the frontline setting. Several FLT3 inhibitors, especially second-generation agents, have shown clinically meaningful activity in relapsed or refractory AML and in patients not amenable to intensive therapy. In this article, we briefly review the biology of FLT3 in the physiological state and its role in leukemogenesis. We present a detailed review of current clinical evidence of FLT3 inhibitors and their use in the induction, treatment of relapsed or refractory disease, and maintenance setting.
Keywords: fms-like tyrosine kinase 3, FLT3 inhibitor, FLT3-ITD mutation, leukemia, myeloid, acute, protein kinase inhibitors
Introduction
Acute myeloid leukemia (AML) is a highly heterogeneous disease defined mainly by cytogenetic or mutational characteristics.1 Mutation with internal tandem duplication of fms-like tyrosine kinase-3 (FLT3-ITD) is one of the two most common driver mutations, along with NPM mutation, identified in 22% of a large study cohort of AML.2 FLT3-ITD is one of the earliest molecular markers described in AML, first reported in 19963 and later associated with marked leukocytosis, higher blast percentage, increased risk of relapse, and poor overall survival (OS).4
A high total mutant level adversely impacts the rate of relapse and OS, especially when the allelic ratio is >50%.5–7 The 2017 European Leukemia Net risk stratification classified FLT3-ITD into low (<50%) and high (≥50%) allelic burden by DNA fragment analysis; therefore, AML with mutated NPM1 and FLT3-ITDlow was reclassified into a favorable risk category.8 Hematopoietic stem cell transplant (HSCT) is not recommended in first remission in these favorable risk patients since no OS benefit is found.9 However, this allelic burden cut-point is being challenged as there are some data indicating that even low allelic burden by this definition is not necessarily associated with better prognosis.10
Patients with wild-type NPM1 and FLT3-ITDhigh have a poor prognosis, with 5-year relapse rate as high as 79% and 5-year OS rate of only 15%.5 With allogeneic HSCT, relapse is still high, with a 2-year cumulative incidence of relapse of 44%–46%.9 These data indicate the unmet clinical need and warrant experimental therapeutic approaches.
FLT3 is a targetable tyrosine kinase. Several clinical trials have focused on various FLT3 inhibitors for each setting of AML treatment – frontline therapy, treatment of relapsed refractory disease, and as maintenance therapy after allogeneic HSCT. Moreover, many second-generation FLT3 inhibitor trials are ongoing, as reviewed later in the text.
Function of FLT3 in normal hematopoiesis
FLT3 belongs to the class III receptor tyrosine kinase (RTK) family that also includes c-KIT, colony-stimulating factor one receptor (formerly FMS), and platelet-derived growth factor receptors α and β (PDGFRA and PDGFRB). Class III RTKs have five immunoglobulin G-like motifs within their extracellular domain. The outer three act as a ligand-binding domain and the remaining two tyrosine kinase domains (TKDs) are located in the C-terminal, intracytoplasmic side, and are usually in an auto-inhibited state when they are not bound to their ligands. Upon ligand binding, class III RTKs oligomerize (homodimerize in the case of FLT3), causing transphosphorylation of tyrosine residues within the TKDs, leading to their activation and signal transduction through PI3K and MAPK pathways.11 FLT3 function is involved in normal hematopoiesis and is normally expressed in early hematopoietic progenitor cells including uncommitted precursors, early progenitors with lymphoid and myeloid potential, common myeloid progenitors, granulocyte-macrophage progenitors (GMPs), and common lymphoid progenitors. FLT3 is not expressed in the megakaryocyte-erythrocyte progenitor. However, knockout mice with Flt3−/− have shown no significant abnormality in hematopoiesis aside from a reduction in B-cell progenitors, indicating that presence of FLT3 signaling is not essential for hematopoiesis or granulopoiesis.12 FLT3 signaling is important in the growth of pre-GMPs but with significant heterogeneity among stem cells.13 FLT3 signaling also plays a crucial role in B-lymphopoiesis.14
FLT3 mutation in myeloid neoplasms
FLT3 has been found to be overexpressed in many AMLs, myelodysplastic syndrome, B-lineage acute lymphoblastic leukemias, and, in a smaller proportion, other leukemias.15 Aberrancy in the FLT3 gene is one of the most common abnormalities and is among the first genetic abnormalities identified in AML. There are two major types of FLT3 mutations: internal tandem duplication (ITD) and point mutations in the TKD.
FLT3-ITD is more common than the TKD mutation. It can involve highly variable numbers of in-frame base pair duplications, from three to more than 400 in the juxta-membrane domain. Elongation of this juxta-membrane domain allows homodimerization of RTKs without ligand binding, which results in constitutive activation. In addition to the PI3K and MAPK pathways, suppression of antiapoptotic FOXO31 can confer survival advantage, and activation of STAT5 may lead to genomic instability through generation of reactive oxygen species and subsequent double-strand DNA breakage, both of which may be associated with poor outcomes.4,14 STAT5 activation is an example of a different qualitative action of the mutated FLT3 that would not usually occur with binding of ligand to wild-type FLT3.16 The role of FLT3-ITD in leukemogenesis has been clarified in several cell lines and in vivo studies. A knock-in approach in which human FLT3-ITD was inserted into the normal Flt3 locus in mice showed that mice with Flt3wt/ITD developed splenomegaly, leukocytosis, and expansion of myeloid cells, all of which are characteristics of myeloproliferative disease, indicating that FLT3-ITD can enhance myeloid expansion but not at a level adequate to cause AML.17
FLT3-TKD mutation is less common, found in approximately 5%–10% of all AML cases. Several FLT3-TKD point mutations can cause ligand-independent RTK activation. The most common mutation occurs in the TKD2 at the D835 gatekeeper position.18 These point mutations in the intracellular TKD may signal through different transduction pathways from FLT3-ITD that could explain its smaller effect on prognosis. FLT3-TKD mutation seems to have prognostic significance only through interaction with other genes. For example, presence of both KMT2A (formerly MLL) partial tandem duplication and FLT3-TKD is associated with poorer prognosis, while co-occurrence of NPM1 and FLT3-TKD is associated with improved OS.2
Wild-type FLT3 overexpression is also found in up to 70%–100% of cases of AML. Clinical data on prognosis of FLT3 overexpression are scarce, though association with poor prognosis has been reported.19 Overexpression of FLT3 may partly explain the beneficial effect of FLT3 inhibition in wild-type FLT3 AML.
While FLT3 mutations, especially FLT3-ITD, are common in acute promyelocytic leukemia, adverse effects on clinical outcomes are not consistent among published studies.20
Therapeutic agents targeting FLT3: pharmacology and development
Due to the high frequency and adverse significance of FLT3, several FLT3 tyrosine kinase inhibitors (FLT3 inhibitors) have been developed. These agents act via competitive inhibition with adenosine triphosphate on the TKD, resulting in decreased autophosphorylation and its successive activation.
First-generation FLT3 inhibitors are relatively nonspecific for FLT3 and usually inhibit other class III RTKs such as KIT and PDGFR.21 Off-target inhibition may be associated with increased toxicity and modest clinical benefit. First-generation FLT3 inhibitors include tandutinib (MLN518, CT53518), sunitinib, sorafenib, midostaurin (PKC412), and lestaurtinib (CEP701).
Second-generation FLT3 inhibitors, including quizartinib (AC220), crenolanib (CP-868–596), ponatinib, pacritinib (SB1518), and gilteritinib (ASP2215), are more potent and selective. These drugs are currently undergoing testing in various clinical settings (Table 1).
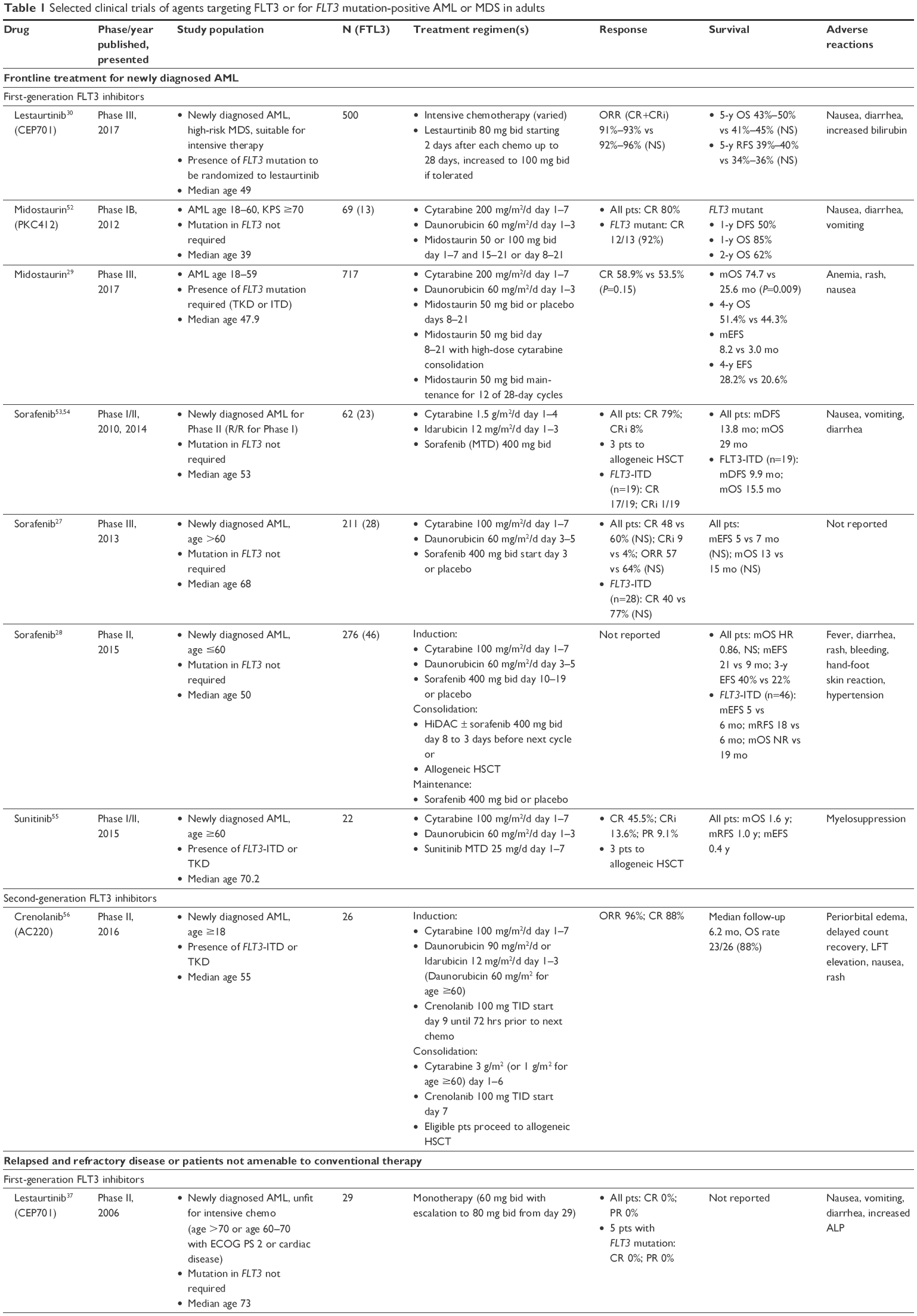 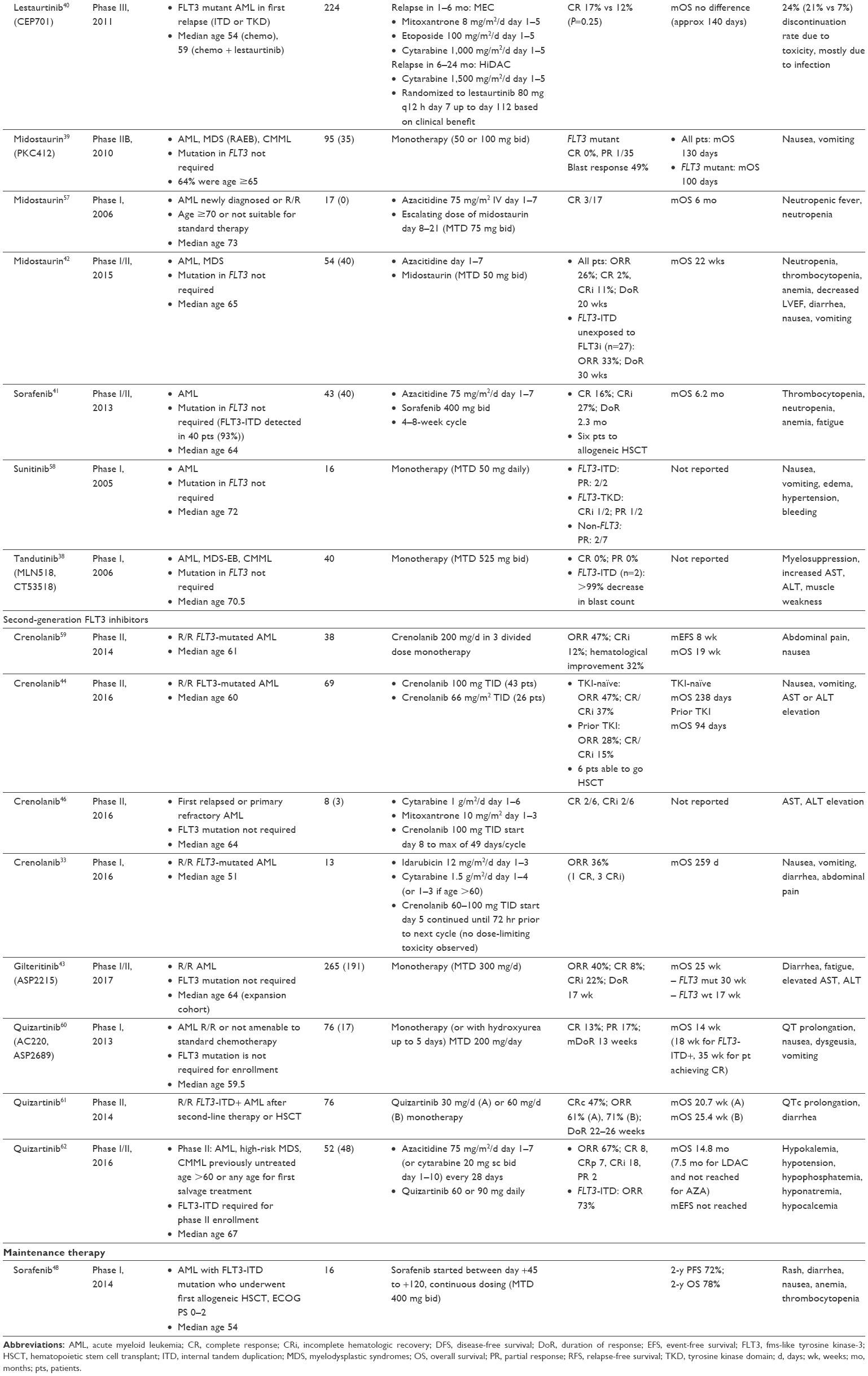 | Table 1 Selected clinical trials of agents targeting FLT3 or for FLT3 mutation-positive AML or MDS in adults |
There are several FLT3 inhibitors for AML in current Phase I or Phase I/II development including E6201 (dual MEK/FLT3 inhibitor, NCT05418000), TAK-659 (dual SYK/FLT3 inhibitor, NCT02323113), SKLB1028 (multikinase inhibitor, NCT02859948), and CT053PTSA (multikinase inhibitor, NCT03125876). Most other non-TKI agents targeting FLT3 are in preclinical development, including FLYSYN (chimeric and Fc-optimized IgG1 antibody to FLT3, NCT02789254), miR-150 nanoparticles,19 FLT3-redirected chimeric antigen receptor T-cell immunotherapy,22 and arsenic trioxide.23
FLT3 inhibitor in combination with intensive induction chemotherapy in newly diagnosed AML
AML with FLT3 mutation, especially FLT3-ITD, has a relatively high relapse rate. Although one could hypothesize that targeting FLT3 in the upfront treatment of FLT3-mutated AML may result in better outcome, there are still several problems. First, certain first-generation FLT3 inhibitors may have inadequate inhibitory properties that may halt the clinical efficacy shown in lestaurtinib. Second, there are multiple subclones present even at the time of diagnosis of AML,24 and FLT3 mutation is known to be a later event and present in only some subclones. In vitro data have shown that in newly diagnosed FLT3-mutated AML, the AML cells did not seem to be highly “addicted” to FLT3 signaling as in the relapsed setting.25 Monotherapy with selective FLT3 inhibitor in this setting is very unlikely to yield a complete remission; thus, combination with standard induction chemotherapy along with the use of multi-targeted TKIs is likely needed. However, complete in vivo inhibition of FLT3 by the first-generation TKI is difficult due to excess toxicity from this off-target inhibition. It has been proposed that the use of first-generation TKI during induction followed by more specific second-generation maintenance could be the optimal approach.26
First-generation FLT3 inhibitors
Sorafenib is a multikinase inhibitor against intracellular Raf kinases and cell surface kinase receptors (VEGFR-1, VEGFR-2, VEGFR-3, PDGFR-β, cKIT, FLT3, and RET). These cell surface kinase receptors are present in AML and bone marrow stromal cells, supporting AML cell proliferation and survival. Serve et al reported a Phase III study of sorafenib sequentially administered after intensive chemotherapy in elderly patients.27 Twenty-eight of 211 patients had FLT3-ITD-positive AML. There was no difference in response rate, event-free survival (EFS) or OS in either the overall or the FLT3-mutated populations. A similar treatment schedule was also tested in a younger population in a randomized Phase II study.28 This showed statistically significant improvement in 3-year EFS in the sorafenib group vs placebo (40% and 22%, respectively; P=0.013). However, the sorafenib treatment also demonstrated an increase in toxicity, especially diarrhea and skin rash. There was no difference in OS. In a small subgroup with FLT3-ITD (n=43), sorafenib seemed to result in better, though not statistically significant, relapse-free survival (RFS) and OS.
Midostaurin is also a multikinase inhibitor, originally developed to target protein kinase C. It was also found to inhibit wild-type and mutant (both ITD and TKD) FLT3, PDGFR-α and β, VEGFR-2, and KIT (wild-type and D816V mutant). The RATIFY study (CALGB 10603), a Phase III, randomized, controlled trial, has shown that midostaurin, added to intensive induction and consolidation therapy followed by 1-year maintenance, leads to a significant improvement of OS (median OS 74.7 vs 25.6 months, P=0.009) and EFS, even though complete response (CR) rates were not different between midostaurin and placebo arms.29 The OS benefit does not seem to vary by the type of FLT3 mutation (ITD-high, ITD-low, or TKD) and is maintained in both censored and uncensored analyses at the time of transplant. Midostaurin was also well tolerated, with the most common adverse events being nausea and skin rash. Anemia was also found to be more common in the midostaurin group. In clinical practice, most patients also complain of its distinctive unpleasant odor. The results of the RATIFY study led to the approval of midostaurin by the US FDA in April 2017 for use in newly diagnosed FLT3 mutation-positive AML. Currently, this is the only approved FLT3 inhibitor.
Lestaurtinib is another first-generation FLT3 inhibitor with broad-spectrum activity, including tropomyosin receptor kinase A, JAK2, and other kinases similar to midostaurin. However, one Phase III, randomized study of lestaurtinib administered sequentially to intensive induction chemotherapy demonstrated no OS or RFS benefit.30 A correlative study as a part of the trial showed OS and RFS benefit in patients who achieved at least 85% FLT3 inhibition. The difference in outcomes between midostaurin and lestaurtinib indicates the importance of the level of FLT3 inhibition and possibly maintenance use after induction chemotherapy.
Second-generation FLT3 inhibitors
Crenolanib is a selective FLT3 inhibitor and is active against both ITD and TKD mutants.31 It also inhibits PDGFR but not c-KIT. This dual FLT3 inhibition feature is important since it has been observed that relapse after initial response to FLT3 inhibitor could emerge from the acquired TKD mutation, especially at the D835 and F691 gatekeeper positions.32 Preliminary results of an ongoing Phase II study of crenolanib sequentially added to an intensive induction regimen (NCT02283177) have shown high efficacy in newly diagnosed FLT3-mutated AML, demonstrating an overall response rate (ORR) of 96% (CR 88%) and an OS rate of 88%, with a median follow-up of 6.2 months.33 Moreover, this combination has also resulted in minimal residual disease-negative status (measured by flow cytometry) in 80% of patients.34 A Phase III study of crenolanib vs midostaurin in this setting has been planned (NCT03258931). One of the drawbacks of crenolanib is its short half-life (6–8 hours) without any accumulation after chronic dosing; thus, a thrice-daily dosing regimen is required.
Gilteritinib is a pan-FLT3 and AXL inhibitor. A Phase I clinical trial is ongoing to evaluate the combination of gilteritinib and cytarabine/idarubicin induction followed by consolidation and gilteritinib maintenance (NCT02236013). Preliminary results show a CR rate of 82.6% in FLT3-mutated patients.35 It is being tested for maintenance after first CR and in combination with azacitidine for patients not amenable to intensive treatment.
Quizartinib is a selective inhibitor of FLT3-ITD but is not active against the TKD variant, as demonstrated in patients who experienced relapse after CR from quizartinib acquired the TKD mutation (either D385 or F691).36 A Phase III QuANTUM-First trial is ongoing to evaluate its efficacy in newly diagnosed patients (NCT02668653).
FLT3 inhibitor in relapsed/refractory AML or patients not amenable to conventional therapy
As already noted, relapse of FLT3-mutated subclones is addicted to FLT3 signaling.25 It is more reasonable to use a potent selective FLT3 inhibitor, especially the agents that possess an activity against both FLT3-ITD and TKD variants, such as gilteritinib and crenolanib. Overall response rates of first-generation FLT3 inhibitors to date have ranged from only 0%–3% as monotherapy (lestaurtinib,37 tandutinib,38 and midostaurin39) to 20%–40% when combined with azacitidine or other chemotherapy.40–42 Although the response rate of second-generation FLT3 inhibitor appears to be 40%–50% as monotherapy, duration of response (DoR) is relatively short (around 20 weeks); thus, benefit without allogeneic HSCT may not be clinically meaningful. Monotherapy with second-generation FLT3 inhibitors usually induces complete response with incomplete hematologic recovery (CRi) for reasons not yet elucidated.43 Some monotherapy clinical trials include newly diagnosed patients who are not fit for intensive therapy.
First-generation FLT3 inhibitors
Midostaurin monotherapy has yielded a disappointing response rate in relapsed/refractory patients. A phase IIB study demonstrated a 71% “blast response” rate (defined by >50% reduction in peripheral blood or bone marrow blast count) in FLT3-mutated AML; however, no patient experienced CR and only a few experienced partial response (PR).39 An attempt to improve response in combination with azacitidine has demonstrated an increase in response rate (33%) with DoR of 30 weeks in a Phase I/II study.42
Sorafenib in combination with azacitidine was tested in a Phase I/II study, with a 16% CR rate and 27% CRi.41 In this study, six patients were able to undergo allogeneic HSCT. This combination is quite well tolerated despite several patients experiencing grade 3–4 rash. One of the benefits of sorafenib is that it is readily available for off-label use.
Second-generation FLT3 inhibitors
Crenolanib monotherapy is currently in ongoing trials in multiply relapsed or refractory AML (NCT01657682, NCT01522469).44 To date, TKI-naïve patients have had a better response (ORR 50%: 39% CRi and 11% PR) compared to patients who previously received TKIs (ORR 31%, CRi 17%). Median OS has been highest in the TKI-naïve, FLT3-ITD group (238 days) and worst in dual FLT3-ITD and TKD patients who previously received TKI (63 days). Elevations in aspartate aminotransferase and alanine aminotransferase have been common, but of low grade, and the rate of drug discontinuation due to adverse events is low. Crenolanib in combination with idarubicin and high-dose cytarabine as induction followed by allogeneic HSCT or consolidation and crenolanib maintenance so far has shown one CR and three CRi in eleven evaluable patients (NCT02400281).45 Another combination Phase I/II study with high-dose cytarabine and mitoxantrone (HAM) showed two CR and two CRi from eight evaluable patients and was well tolerated (NCT02626338).46 A randomized Phase III study with this combination is ongoing (NCT02298166).
Gilteritinib monotherapy was studied in a Phase I/II trial for relapsed or refractory AML.43 Most of the patients in this trial (191 of 256) harbored a FLT3 mutation. ORR was 49% in patients who had FLT3 mutation with 9% CR and 27% CRi. DoR was 20 weeks and median OS was 30 weeks. Only 19% of the patients underwent HSCT. Patients who received prior TKI had lower ORR (37%) and CR/CRi (26%). DoR was also shorter (14 weeks). Gilteritinib seems to be well tolerated. For less amenable patients, combination of gilteritinib and azacitidine is being tested in a Phase II/III study (NCT02752035).
Quizartinib monotherapy was shown to be effective in relapsed or refractory AML with FLT3-ITD mutation in the final results of a Phase II study, showing an ORR of 61% with a composite CR rate of 47%.47 A Phase III QuANTUM-R study is currently evaluating the efficacy of quizartinib monotherapy in this setting compared to standard salvage chemotherapy (NCT02039726). Combination therapy trial with omacetaxine has also been planned (NCT03135054).
FLT3 inhibitor as maintenance therapy after induction/consolidation or allogeneic HSCT
Maintenance therapy is usually given to prevent relapse or prolong RFS. Ideal maintenance agent not only has to have efficacy but also has to be minimally toxic and relatively safe for long-term use. Currently, there are no Phase III data available to guide its use after allogeneic HSCT and only Phase I data were published for sorafenib. Several FLT3 inhibitor protocols have integrated maintenance therapy after achieving first complete remission.
First-generation FLT3 inhibitors
In a Phase I study, sorafenib was used as maintenance therapy after HSCT in FLT3-ITD AML.48 Maximal tolerated dose was 400 mg twice a day. One-year progression-free survival was 85%. Five out of 22 patients in this study discontinued treatment due to toxicity. A presented abstract from a similar study also showed that sorafenib was well-tolerated and seemed to be effective in the peri-transplant setting.49 Sorafenib is also currently undergoing investigation in patients with FLT3-ITD mutated AML in complete or partial remission after induction therapy (NCT01578109).
Midostaurin maintenance for one year after combination therapy with induction and consolidation was included in the treatment protocol of the RATIFY trial.29 This was thought to contribute to the efficacy and high transplant rate. Nevertheless, unplanned post hoc analysis of patients who received maintenance therapy within the RATIFY trial showed no difference in disease-free survival or OS in patients receiving midostaurin maintenance vs placebo.50 Maintenance therapy seems to be well tolerated with a low rate of discontinuation due to adverse events (8% for midostaurin vs 6% for placebo). Of note, maintenance midostaurin is not included in the FDA-approved indication and usage guidelines. An extension clinical trial is planned to explore the benefit of midostaurin maintenance in elderly patients after allogeneic HSCT (NCT02723435).
Second-generation FLT3 inhibitors
At the time of this writing, there are several planned or ongoing clinical trials using second-generation FLT3 inhibitor as maintenance therapy after first CR including gilteritinib (NCT02997202) and quizartinib (NCT02668653), or after allogeneic HSCT including crenolanib (NCT02400255) and gilteritinib (NCT02997202).
Resistance to FLT3 inhibition
Primary resistance to TKI treatment is not well elucidated. Secondary resistance is almost universally seen after prolonged use in various types of malignancies, usually due to emerging mutation in or near the kinase domain of the RTK. Some TKI-resistant mutations have a predominant pattern, eg, ABL1 T315I in TKI-treated chronic myeloid leukemia or EGFR T790M in EGFR TKI-treated non-small-cell lung cancer. As previously noted, FLT3 mutation is not the early event in the evolution of AML and thus not required for AML growth and proliferation; thus, loss of FLT3 mutation is common and may result in loss of response to FLT3 inhibitor (FLT3 independence). Several secondary RTK-resistant mutations have been described after quizartinib use and could be heterogeneous even within a single patient.36 Most of the mutations after quizartinib use happen at D835 residue. However, a subanalysis of the RATIFY study suggested that a FLT3-independent mechanism, such as loss of FLT3-ITD, is the main finding at relapse after midostaurin use.51
Conclusion
Several clinical efforts in FLT3 inhibition are currently underway, either as combination or monotherapy for controlling AML. The incorporation of midostaurin into the induction regimen, showing early significant improvement in EFS and OS, appears promising. Early phase studies especially on more selective FLT3 inhibitors as single agent or in combination with hypomethylating agents have shown potential benefits in patients who are not candidates for intensive therapy as well as those with relapsed disease.
The role of maintenance FLT3 inhibition is being explored in several ongoing studies. Confirmatory clinical trial results are eagerly awaited and may have the potential to change clinical practice in FLT3-mutated leukemia.
Disclosure
The authors report no conflicts of interest in this work.
References
Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. | ||
Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016;374(23):2209–2221. | ||
Nakao M, Yokota S, Iwai T, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10(12):1911–1918. | ||
Small D. FLT3 mutations: biology and treatment. Hematology Am Soc Hematol Educ Program. 2006:178–184. | ||
Gale RE, Green C, Allen C, et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood. 2008;111(5):2776–2784. | ||
Whitman SP, Archer KJ, Feng L, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B study. Cancer Res. 2001;61(19):7233–7239. | ||
Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99(12):4326–4335. | ||
Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447. | ||
Ho AD, Schetelig J, Bochtler T, et al. Allogeneic Stem Cell Transplantation Improves Survival in Patients with Acute Myeloid Leukemia Characterized by a High Allelic Ratio of Mutant FLT3-ITD. Biol Blood Marrow Transplant. 2016;22(3):462–469. | ||
Harada Y, Nagata Y, Kihara R, et al. Prognostic analysis according to the 2017 ELN risk stratification by genetics in adult acute myeloid leukemia patients treated in the Japan Adult Leukemia Study Group (JALSG) AML201 study. Leuk Res. 2018;66:20–27. | ||
Berenstein R. Class III Receptor Tyrosine Kinases in Acute Leukemia – Biological Functions and Modern Laboratory Analysis. Biomark Insights. 2015;10(Suppl 3):1–14. | ||
Mackarehtschian K, Hardin JD, Moore KA, Boast S, Goff SP, Lemischka IR. Targeted disruption of the flk2/flt3 gene leads to deficiencies in primitive hematopoietic progenitors. Immunity. 1995;3(1):147–161. | ||
Böiers C, Buza-Vidas N, Jensen CT, et al. Expression and role of FLT3 in regulation of the earliest stage of normal granulocyte-monocyte progenitor development. Blood. 2010;115(24):5061–5068. | ||
Tsapogas P, Mooney CJ, Brown G, Rolink A. The Cytokine Flt3-Ligand in Normal and Malignant Hematopoiesis. Int J Mol Sci. 2017;18(6):E1115. | ||
Carow CE, Levenstein M, Kaufmann SH, et al. Expression of the hematopoietic growth factor receptor FLT3 (STK-1/Flk2) in human leukemias. Blood. 1996;87(3):1089–1096. | ||
Mizuki M, Schwable J, Steur C, et al. Suppression of myeloid transcription factors and induction of STAT response genes by AML-specific Flt3 mutations. Blood. 2003;101(8):3164–3173. | ||
Li L, Piloto O, Nguyen HB, et al. Knock-in of an internal tandem duplication mutation into murine FLT3 confers myeloproliferative disease in a mouse model. Blood. 2008;111(7):3849–3858. | ||
Hospital MA, Green AS, Maciel TT, et al. FLT3 inhibitors: clinical potential in acute myeloid leukemia. Onco Targets Ther. 2017;10:607–615. | ||
Jiang X, Bugno J, Hu C, et al. Targeted Treatment of FLT3-Overexpressing Acute Myeloid Leukemia with MiR-150 Nanoparticles Guided By Conjugated FLT3 Ligand Peptides. Blood. 2015;126:3784. | ||
Molica M, Breccia M. FLT3-ITD in acute promyelocytic leukemia: clinical distinct profile but still controversial prognosis. Leuk Res. 2015;39(4):397–399. | ||
Lancet JE. FLT3 inhibitors for acute myeloid leukemia. Clin Adv Hematol Oncol. 2015;13(9):573–575. | ||
Chien CD, Sauter CT, Ishii K, et al. Preclinical Development of FLT3-Redirected Chimeric Antigen Receptor T Cell Immunotherapy for Acute Myeloid Leukemia. Blood. 2016;128:1072. | ||
Nagai K, Hou L, Li L, et al. Arsenic Trioxide Synergizes with FLT3 Tyrosine Kinase Inhibitors to Kill FLT3-ITD+ Leukemic Cells through Depressed Production and Degradation of FLT3 Protein. Blood. 2016;128:1568. | ||
Grimwade D, Ivey A, Huntly BJ. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. 2016;127(1):29–41. | ||
Pratz KW, Sato T, Murphy KM, Stine A, Rajkhowa T, Levis M. FLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood. 2010;115(7):1425–1432. | ||
Levis M. How and when to use FLT3 inhibitors. Clin Lymphoma Myeloma Leuk. 2017;17:S18–S21. | ||
Serve H, Krug U, Wagner R, et al. Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: results from a randomized, placebo-controlled trial. J Clin Oncol. 2013;31(25):3110–3118. | ||
Röllig C, Serve H, Hüttmann A, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015;16(16):1691–1699. | ||
Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med. 2017;377(5):454–464. | ||
Knapper S, Russell N, Gilkes A, et al. A randomized assessment of adding the kinase inhibitor lestaurtinib to first-line chemotherapy for FLT3-mutated AML. Blood. 2017;129(9):1143–1154. | ||
Smith CC, Lasater EA, Lin KC, et al. Crenolanib is a selective type I pan-FLT3 inhibitor. Proc Natl Acad Sci U S A. 2014;111(14):5319–5324. | ||
Zimmerman EI, Turner DC, Buaboonnam J, et al. Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood. 2013;122(22):3607–3615. | ||
Ohanian M, Kantarjian HM, Borthakur G, et al. Efficacy of a Type I FLT3 Inhibitor, Crenolanib, with Idarubicin and High-Dose Ara-C in Multiply Relapsed/Refractory FLT3+AML. Blood. 2016;128:2744. | ||
Stone RM, Collins R, Tallman MS, et al. Effect of cytarabine/anthracycline/crenolanib induction on minimal residual disease (MRD) in newly diagnosed FLT3 mutant AML. Journal of Clinical Oncology. 2017;35:7016. | ||
Pratz K, Cherry M, Altman JK, et al. Preliminary Results from a Phase 1 Study of Gilteritinib in Combination with Induction and Consolidation Chemotherapy in Subjects with Newly Diagnosed Acute Myeloid Leukemia (AML). Blood. 2017;130:722. | ||
Smith CC, Paguirigan A, Jeschke GR, et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood. 2017;130(1):48–58. | ||
Knapper S, Burnett AK, Littlewood T, et al. A phase 2 trial of the FLT3 inhibitor lestaurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy. Blood. 2006;108(10):3262–3270. | ||
Deangelo DJ, Stone RM, Heaney ML, et al. Phase 1 clinical results with tandutinib (MLN518), a novel FLT3 antagonist, in patients with acute myelogenous leukemia or high-risk myelodysplastic syndrome: safety, pharmacokinetics, and pharmacodynamics. Blood. 2006;108(12):3674–3681. | ||
Fischer T, Stone RM, Deangelo DJ, et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28(28):4339–4345. | ||
Levis M, Ravandi F, Wang ES, et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood. 2011;117(12):3294–3301. | ||
Ravandi F, Alattar ML, Grunwald MR, et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood. 2013;121(23):4655–4662. | ||
Strati P, Kantarjian H, Ravandi F, et al. Phase I/II trial of the combination of midostaurin (PKC412) and 5-azacytidine for patients with acute myeloid leukemia and myelodysplastic syndrome. Am J Hematol. 2015;90(4):276–281. | ||
Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol. 2017;18:1061–1075. | ||
Cortes JE, Kantarjian HM, Kadia TM, et al. Crenolanib besylate, a type I pan-FLT3 inhibitor, to demonstrate clinical activity in multiply relapsed FLT3-ITD and D835 AML. Journal of Clinical Oncology. 2016;34:7008. | ||
Ohanian M, Kantarjian HM, Borthakur G, et al. Efficacy of a Type I FLT3 Inhibitor, Crenolanib, with Idarubicin and High-Dose Ara-C in Multiply Relapsed/Refractory FLT3+ AML. Blood. 2016;128:2744. | ||
Iyer SP, Jethava Y, Karanes C, Eckardt JR, Collins R. Safety Study of Salvage Chemotherapy High-Dose Ara-C/Mitoxantrone (HAM) and Type I FLT3-TKI Crenolanib in First Relapsed/Primary Refractory AML. Blood. 2016;128:3983. | ||
Schiller GJ, Tallman MS, Goldberg SL, et al. Final results of a randomized phase 2 study showing the clinical benefit of quizartinib (AC220) in patients with FLT3-ITD positive relapsed or refractory acute myeloid leukemia. Journal of Clinical Oncology. 2014;32:7100. | ||
Chen YB, Li S, Lane AA, et al. Phase I trial of maintenance sorafenib after allogeneic hematopoietic stem cell transplantation for fms-like tyrosine kinase 3 internal tandem duplication acute myeloid leukemia. Biol Blood Marrow Transplant. 2014;20(12):2042–2048. | ||
Pratz KW, Gojo I, Karp JE, et al. Prospective Study of Peri-Transplant Use of Sorafenib As Remission Maintenance for FLT3-ITD Patients Undergoing Allogeneic Transplantation. Blood. 2015;126:3164. | ||
Larson RA, Mandrekar SJ, Sanford BL, et al. An Analysis of Maintenance Therapy and Post-Midostaurin Outcomes in the International Prospective Randomized, Placebo-Controlled, Double-Blind Trial (CALGB 10603/RATIFY [Alliance]) for Newly Diagnosed Acute Myeloid Leukemia (AML) Patients with FLT3 Mutations. Blood. 2017;130:145. | ||
Schmalbrock LK, Cocciardi S, Dolnik A, et al. Clonal Evolution of FLT3-ITD Positive AML in Patients Treated with Midostaurin in Combination with Chemotherapy within the Ratify (CALGB 10603) and AMLSG 16-10 Trials. Blood. 2017;130:182. | ||
Stone RM, Fischer T, Paquette R, et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia. 2012;26(9):2061–2068. | ||
Ravandi F, Arana Yi C, Cortes JE, et al. Final report of phase II study of sorafenib, cytarabine and idarubicin for initial therapy in younger patients with acute myeloid leukemia. Leukemia. 2014;28(7):1543–1545. | ||
Ravandi F, Cortes JE, Jones D, et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol. 2010;28(11):1856–1862. | ||
Fiedler W, Kayser S, Kebenko M, et al. A phase I/II study of sunitinib and intensive chemotherapy in patients over 60 years of age with acute myeloid leukaemia and activating FLT3 mutations. Br J Haematol. 2015;169(5):694–700. | ||
Wang ES, Stone RM, Tallman MS, Walter RB, Eckardt JR, Crenolanib CR. A Type I FLT3 TKI, Can be Safely Combined with Cytarabine and Anthracycline Induction Chemotherapy and Results in High Response Rates in Patients with Newly Diagnosed FLT3 Mutant Acute Myeloid Leukemia (AML). Blood. 2016;128:1071. | ||
Cooper BW, Kindwall-Keller TL, Craig MD, et al. A phase I study of midostaurin and azacitidine in relapsed and elderly AML patients. Clin Lymphoma Myeloma Leuk. 2015;15(7):428–432. | ||
Fiedler W, Serve H, Döhner H, et al. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood. 2005;105(3):986–993. | ||
Randhawa JK, Kantarjian HM, Borthakur G, et al. Results of a Phase II Study of Crenolanib in Relapsed/Refractory Acute Myeloid Leukemia Patients (Pts) with Activating FLT3 Mutations. Blood. 2014;124:389. | ||
Cortes JE, Kantarjian H, Foran JM, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol. 2013;31(29):3681–3687. | ||
Schiller GJ, Tallman MS, Goldberg SL, et al. Final results of a randomized phase 2 study showing the clinical benefit of quizartinib (AC220) in patients with FLT3-ITD positive relapsed or refractory acute myeloid leukemia. Journal of Clinical Oncology. 2014;32:7100. | ||
Abdelall W, Kantarjian HM, Borthakur G, et al. The Combination of Quizartinib with Azacitidine or Low Dose Cytarabine Is Highly Active in Patients (Pts) with FLT3-ITD Mutated Myeloid Leukemias: Interim Report of a Phase I/II Trial. Blood. 2016;128:1642. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.