")
Back to Journals » OncoTargets and Therapy » Volume 10
Clinical targeting recombinant immunotoxins for cancer therapy
Authors Li M, Liu ZS , Liu XL, Hui Q, Lu SY, Qu LL, Li YS, Zhou Y, Ren HL , Hu P
Received 13 February 2017
Accepted for publication 10 May 2017
Published 20 July 2017 Volume 2017:10 Pages 3645—3665
DOI https://doi.org/10.2147/OTT.S134584
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Meng Li,1,* Zeng-Shan Liu,1,* Xi-Lin Liu,1,* Qi Hui,2,* Shi-Ying Lu,1 Lin-Lin Qu,1 Yan-Song Li,1 Yu Zhou,1 Hong-Lin Ren,1 Pan Hu1
1Key Laboratory of Zoonosis Research, Ministry of Education, Institute of Zoonosis, College of Veterinary Medicine, China-Japan Union Hospital, The First Hospital, Jilin University, Changchun, 2School of Pharmacy, Wenzhou Medical University, Wenzhou, People’s Republic of China
*These authors contributed equally to this work
Abstract: Recombinant immunotoxins (RITs) are proteins that contain a toxin fused to an antibody or small molecules and are constructed by the genetic engineering technique. RITs can bind to and be internalized by cells and kill cancerous or non-cancerous cells by inhibiting protein synthesis. A wide variety of RITs have been tested against different cancers in cell culture, xenograft models, and human patients during the past several decades. RITs have shown activity in therapy of several kinds of cancers, but different levels of side effects, mainly related to vascular leak syndrome, were also observed in the treated patients. High immunogenicity of RITs limited their long-term or repeat applications in clinical cases. Recent advances in the design of immunotoxins, such as humanization of antibody fragment, PEGylation, and modification of human B- and T-cell epitopes, are overcoming the above mentioned problems, which predict the use of these immunotoxins as a potential therapeutic method to treat cancer patients.
Keywords: targeted therapy, hematologic malignancies, solid tumors, vascular leak syndrome, immunogenicity
Introduction
Recombinant immunotoxins (RITs) are chimeric proteins for cancer therapy that contain a toxin fused to a targeting moiety. After the initial success of antibody therapy for cancer, monoclonal antibodies (Mabs) were used to link with the toxin molecules, which have higher specificity in targeting and higher potency in killing cancer cells. Thirty-five years ago, RITs were created by chemically conjugating a whole protein toxin to an Mab or a protein toxin devoid of its natural binding domain.1,2 Other immune proteins such as cytokines and growth factors have also been conjugated and genetically fused to toxins.3 The traditional immunotoxins coupled the toxin and the targeting moiety by chemical method, but the cumbersome steps and the high cost encouraged the development of novel RITs. Nowadays, these RITs are generally synthesized by recombinant DNA techniques through constructing conditional expression plasmid and expressing interest protein in Escherichia coli rapidly and efficiently. RITs combine the cell-killing power of toxin and the specificity of antibody therapies. The careful design of both target moiety and toxin is the key of a successful therapy, because each type of cancer cell expresses different kinds of surface antigens. Compared with other commonly used immunotherapies, RITs’ cytocidal action is not dependent on antibody or complement-dependent cytotoxicity, but it is dependent on the specificity of the targeting moiety and the high activity of the toxin moiety, which will induce development of resistance. RITs directly target the surface of cancer cells, and 1 single internalized toxin molecule could kill the cell. Based on this, patients would not develop myelosuppression dose-limiting toxicity (DLT). Some RITs have been approved by the US Food and Drug Administration (FDA) for the treatment of cancer, such as denileukin diftitox (ONTAK®). This review considers RITs with different target moieties and toxins directed to cancer cells and focuses on those that have been tested clinically in the recent decades.
Mechanism of action of bacterial toxins
The target moiety facilitates the transfer and internalization of RITs into cancer cells. For this purpose, various bacterial toxins have been used in RITs targeting cancer cells, and among these toxins, Pseudomonas exotoxin (PE) and Diphtheria toxin (DT) are the most commonly used bacterial toxins. Nearly all toxins kill cancer cells by enzymatically inhibiting protein synthesis. Similarly, both PE and DT also inhibit adenosine diphosphate (ADP)-ribosylate elongation factor 2 (EF-2) in the cytosol,4 which is a critical component of the protein synthesis machinery. Each toxin catalyzes the ADP-ribosylate of his-699 of EF-2, which is posttranslationally modified to a diphthamide residue.5 Despite their similar action, PE and DT differ greatly in amino acid sequence, and PE’s binding domain is located near to its amino terminal, but DT is located on the opposite terminal.
PE is a single-chain protein, consisting of 613 amino acids in length, which is further composed of 3 functional domains.6,7 Domain Ia (amino acids 1–252) is the binding domain, domain II (amino acids 253–364) mediates translocating the toxin to the cytosol, and domain III (amino acids 400–613) contains the ADP-ribosylating enzyme that inactivates EF-2 in the cytosol (Figure 1). The function of domain Ib (amino acids 365–399) is unclear. PE kills the cells following these steps (Figure 2): 1) Lys613 is removed by a carboxypeptidase in the plasma.8 2) Domain Ia binds to the α2 macroglobulin receptor on animal cells and internalizes via endosomes to the Golgi.9 3) The protease furin cleaves domain II between amino acids 279 and 280.10 4) The disulfide bond that joins the 2 fragments generated by proteolysis is reduced.11 5) Amino acids 609–612 (arginine–alutamic acid–aspartic acid–leucine, REDL) bind to an intracellular sorting receptor that transports the carboxy terminal fragment from the Golgi to the endoplasmic reticulum (ER).12,13 6) Amino acids 280–313 mediate translocation of the toxin to the cytosol.14,15 7) The ADP-ribosylating enzyme in amino acids 400–602 inactivates EF-2.4 8) Though inhibition of protein synthesis is sufficient to induce cell death eventually, cell death from toxins is facilitated by apoptosis.16,17
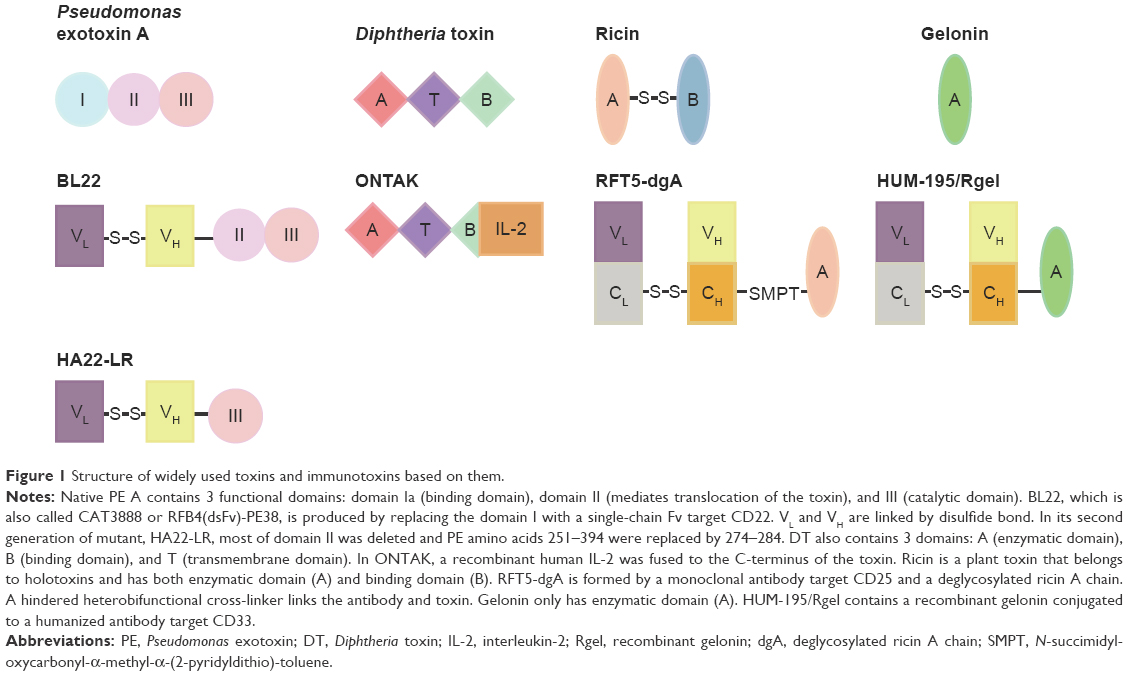 | Figure 1 Structure of widely used toxins and immunotoxins based on them. |
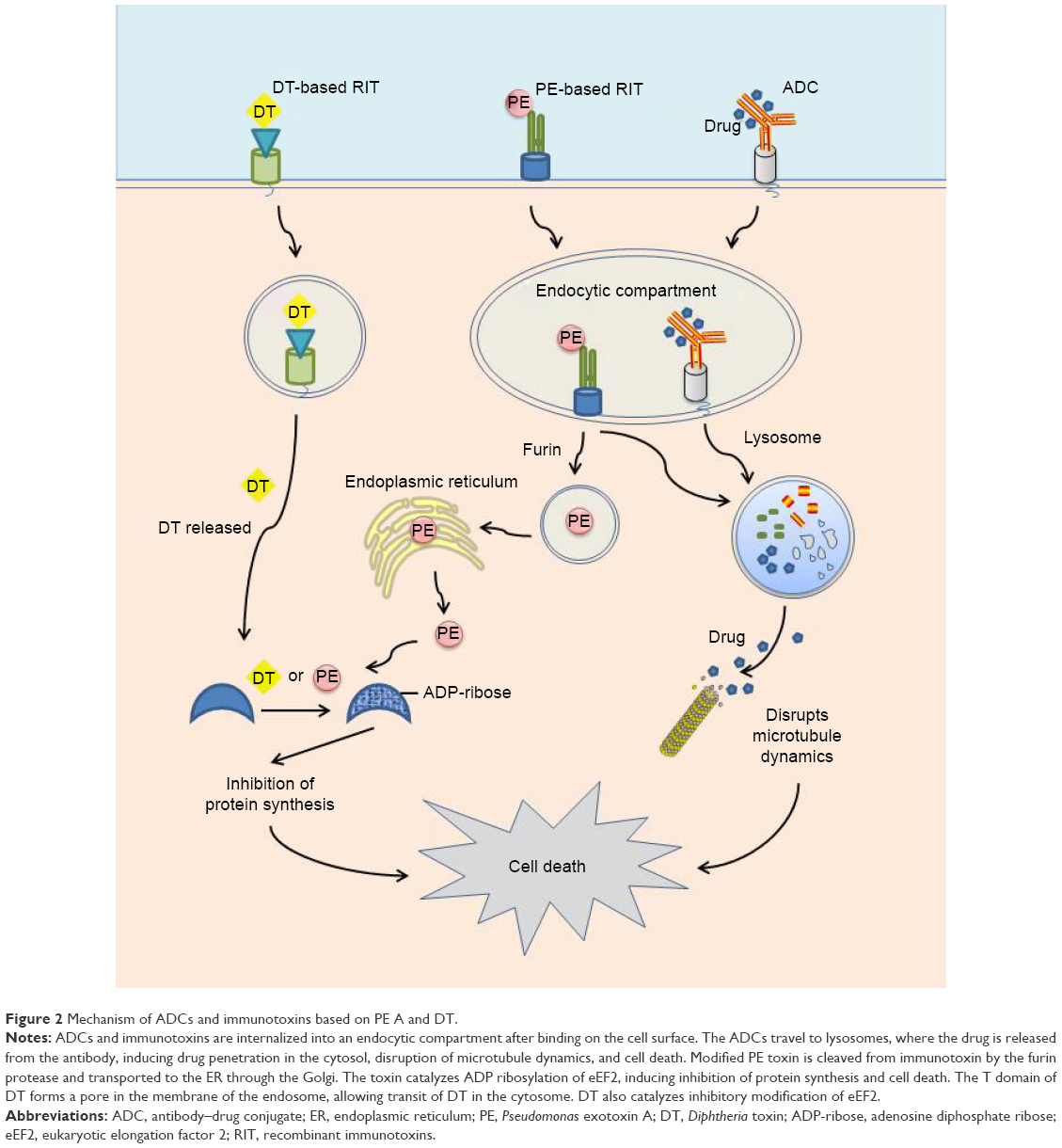 | Figure 2 Mechanism of ADCs and immunotoxins based on PE A and DT. |
Full-length 535-amino acid DT is a single-chain protein containing an enzymatic A domain (amino acids 1–193) and a binding B domain (amino acids 482–535).18 A translocation or transmembrane (T) domain is located in the center of the molecule (Figure 1).19 DT undergoes the following steps to kill cells (Figure 2): 1) DT is proteolytically cleaved outside the cell between Arg193 and Ser194,20 which is within a disulfide loop formed by Cys186 and Cys201. 2) DT binds on the cell surface via carboxyl residues 482–535 to a complex of heparin-binding epidermal growth factor (EGF)-like growth factor precursor and CD9.18 3) DT internalizes in an endosome and unfolds at low pH,21 and the disulfide bond between amino acids 186 and 201 is reduced. 4) The TH8 (amino acids 326–347) and TH9 (amino acids 358–376) domains form a hairpin that inserts in the membrane of the endosome and forms a channel through which the enzymatic fragment translocates to the cytosol.22 5) In the cytosol, nicotinamide adenine dinucleotide (NAD) binds to the active-site cleft of DT (amino acids 34–52), and the ADP ribose of NAD is transferred to EF-2.23,24 6) Similar to PE, DT also induces cell death by apoptosis.17
It has been shown that one or only a few of those toxin molecules delivered to the cytosol are sufficient to kill a target cell.25 However, the inhibition of protein synthesis by toxins was thought to be lethal recently.26,27 Because some toxin-treated cancer cells appear to survive from toxin treatment which suggests the existence of resistance to RIT-mediated apoptosis. Therefore, assays that focus more precisely on the mechanisms of cell death have been developed by these toxins later. The activities of bacterial toxin and RITs have been connected to apoptosis in some cell systems, but the mechanisms have not been extensively stated.17,28,29 Keppler et al indicated that a PE-based RIT, B3(Fv)-PE38, kills MCF-7 breast cancer cell line by 2 mechanisms: one is due to the inhibition of protein synthesis caused by inactivation of EF-2 and the other requires caspase activation.30 There was also evidence that PE toxin-induced apoptosis of human mast cells involves downregulation of anti-apoptotic proteins and activation of caspase-8 and −3 pathways.31 When treated with BL22, B cells of chronic lymphocytic leukemia (CLL) underwent programmed cell death that was characterized by caspase-9 and caspase-3 activation, poly(adenosine diphosphate[ADP]-ribose)polymerase (PARP) cleavage, DNA fragmentation, and membrane flipping.32 From the above, cell death mediated by bacterial toxins, such as PE and DT, is often facilitated by caspase-dependent programmed cell death, in addition to the inhibition of protein synthesis.
Mechanism of action of plant toxins
Plant toxins are classified into 2 classes, holotoxins and hemitoxins. Holotoxins, or class I ribosome-inactivating proteins, include ricin, abrin, modeccin, and mistletoe lectin. Hemitoxins, which are also referred to as class II ribosome-inactivating proteins, include gelonin, saporin, bouganin, and bryodin.33 Plant toxins such as ricin and gelonin also arrest protein synthesis but by inactivating the ribosome instead of EF-2.34,35 As shown in Figure 1, holotoxins such as ricin contain both binding and catalytic domains, whereas hemitoxins contain only catalytic domains. Only the catalytic domains of both holotoxins and hemotoxins translocate to the cytosol, and hence the binding domains of holotoxins would be removed by reduction of the disulfide bond prior to translocation. Plant toxins have been reported to prevent the association of EF-1 and EF-2 with the ribosomal subunit by removing the base of A4324 in 28s ribosomal RNA (rRNA).36 Ricin also removes the neighboring base G4323. These toxin-mediated processes stimulate the apoptotic pathway, as well as the bacterial toxins, leading to cell death in the end. How plant toxins translocate to the cytosol from the cell surface is still unknown. The intracellular transport of ricin is dependent on sorting receptors that cycle between the ER and the Golgi.37
RITs targeting hematologic malignancies
Generally, receptors of cytokines and growth factors are overexpressed in all types of cancer cells. In hematologic malignancies, several receptors are overexpressed compared with normal blood cells and have been targeted successfully in several studies (Table 1). Here, we discuss the recently published RITs targeting hematologic malignancies in clinical application with different surface markers.
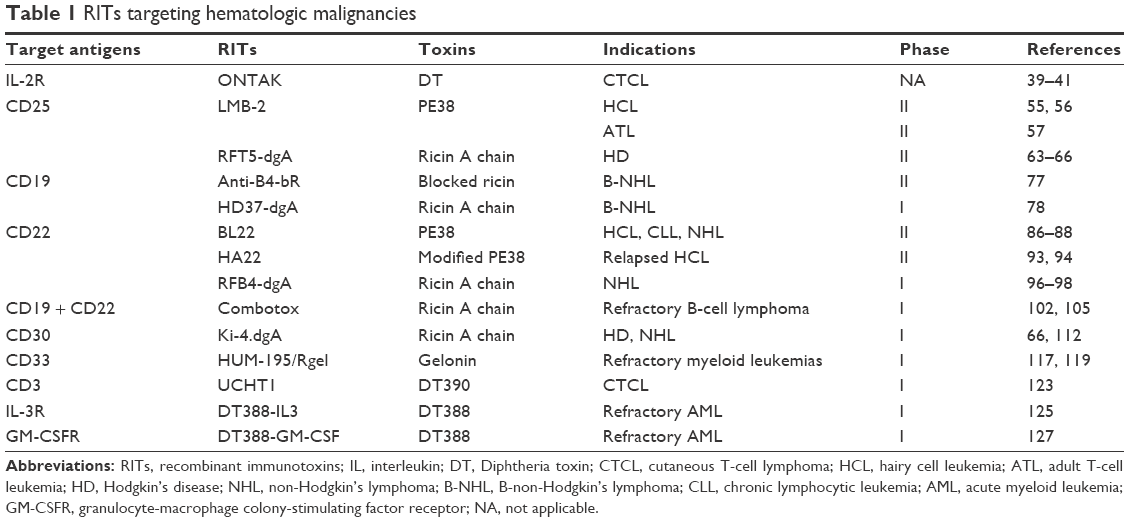 | Table 1 RITs targeting hematologic malignancies |
Interleukin-2 receptor (IL-2R)
Interleukin-2 (IL-2), one of the first lymphokines to be identified, plays a central role in the clonal expansion of activated T cells by interacting with its specific cell surface receptor (IL-2R). IL-2R, which binds the IL-2 with high affinity, is composed of 3 subunits (alpha [CD25], beta [CD122], and gamma [CD132]). IL-2R is overexpressed in hematologic malignancies such as adult T-cell leukemia (ATL), cutaneous T-cell lymphoma (CTCL), Hodgkin’s disease (HD), and other B- and T-cell leukemias and lymphomas. But only a small percentage of normal T cells are IL-2R positive. Thus, IL-2R has been broadly used to target leukemias and lymphomas.
Denileukin diftitox (ONTAK, DAB389IL-2)
Denileukin diftitox was granted initial approval in 2001 for the treatment of CTCL. Targeting domain of this RIT does not contain an antibody but a recombinant human IL-2 fused to the C-terminus of the DT toxin. The ligand targets those cells that express the IL-2R, which is transiently expressed on activated T cells but constitutively present in a number of hematologic malignancies. It does not cause generalized DT-related toxicity because the binding parts of the DT are replaced by the IL-2 segment targeting only those cells expressing IL-2R.38
In a single-arm Phase III trial, patients with IL-2R-positive CTCL received denileukin diftitox (9 or 18 μg/kg/d) for 5 consecutive days every 3 weeks. This study demonstrated 20% partial remission (PR), 10% complete regressions (CRs), and 30% overall response rate (ORR) in 71 patients. The duration of response ranged from 2.7 to >46.1 months with a median of 6.9 months. The side effects included flu-like symptoms, acute infusion-related events, and a vascular leak syndrome (VLS). Through measuring the neutralizing antibody of denileukin diftitox, there was no difference of tolerability at the 2 dose levels.39
A later Phase III trial confirmed and improved the response rate of denileukin diftitox with placebo.40 A total number of 144 patients with CTCL were assigned to 9 μg/kg/d denileukin diftitox, 18 μg/kg/d denileukin diftitox, or placebo infusion. The agents were administered for 5 consecutive days every 3 weeks. Compared with 15.9% ORR for placebo-treated patients (2% CR and 13.6% PR), 44% of patients (n=100) treated with denileukin diftitox achieved response (10% CR and 34% PR). Higher ORR was observed in the 18 μg/kg/d group than 9 μg/kg/d group (49.1% vs 37.8%, respectively). In addition, progression-free survival was significantly longer for patients treated with denileukin diftitox. This study demonstrated that denileukin diftitox had a significant and durable effect with an acceptable safety profile in patients with CTCL. Based on this study, Prince et al41 examined the efficacy and safety of denileukin diftitox in 36 patients with IL-2R low expression skin biopsies. The result demonstrated that the safety profile of denileukin diftitox in IL-2R low expression disease was similar to that in IL-2R-positive disease, which suggests that IL-2R low expression would not preclude clinical response to denileukin diftitox in patients with CTCL.
Denileukin diftitox has also been reported to reduce the percentage of regulatory T cells (Tregs) in the peripheral blood of patients with renal carcinoma, ovarian cancer, and melanoma.42–46 It is used infrequently because of poor tolerability and some kinds of side effects, but previously reported adverse effects can be managed effectively by supportive measures without dose reduction.47 Hence, denileukin diftitox can be used for the treatment of CTCL currently.
IL-2Rα (CD25)
The human IL-2Rα, also described as the Tac antigen or CD25, is a 55-kDa membrane glycoprotein (p55). The deduced amino acid sequence of IL-2Rα predicts a mature protein of 251 amino acids with a signal peptide of 21 amino acids in length. The amino-terminal 219 residues constitute the extracellular region. The next 19 residues constitute the membrane spanning region, and the carboxy-terminal 13 residues, the cytoplasmic region. Mutational analysis showed that the N-terminal 83 residues of the IL-2Rα, especially residues 1–6 and 35–43, were essential for its binding function.48
LMB-2 [anti-Tac(Fv)-PE38]
LMB-2, also named anti-Tac(Fv)-PE38, consists of a modified PE toxin and an antibody fragment. The antibody fragment, which contains the VH of anti-Tac fused to the VL via a 15-amino acid linker, selectively binds the α-subunits of IL-2R.49 LMB-2 has been shown to be cytotoxic to CD25+ malignant cells that were either established cell lines or directly obtained from patients with hematologic malignancies.50–53 LMB-2 produced CR in murine xenografts in vivo.49 Blood levels of LMB-2 causing tumor regression in mouse are well tolerated by monkeys.54
The Phase I trial of LMB-2 in patients with hematologic malignancies began in 1996.55 In this test, 4 patients with CD25+ hairy cell leukemia (HCL) received LMB-2 at 3 dose levels (30, 40, and 63 μg/kg every other day [QOD] ×3). All patients reacted to LMB-2 after a single cycle; 1 patient who received 2 cycles had a CR, marked by regression of HCL cells from blood and marrow, and did not relapse after 11 months. Three additional patients had 98%–99.8% reductions in malignant circulating cells. This study represents evidence that LMB-2 may be an effective new therapy for patients with CD25+ HCL.
To evaluate the pharmacokinetics, toxicity, immunogenicity, and antitumor activity of LMB-2, more patients with hematologic malignancies received dose levels that ranged from 2 to 63 μg/kg for a total of 59 cycles.56 Seven PRs were observed in patients with CTCL, HCL, HD, ATL, and CLL. One HCL patient achieved a durable CR for >20 months. All 4 patients with HCL responded to LMB-2. At the maximum tolerated dose (MTD; 40 μg/kg QOD ×3), toxicity was transient and included transaminase elevation and fever. Only 6 of 35 patients developed neutralizing antibodies after the first cycle. This study demonstrated that LMB-2 has clinical antitumor activity in CD25+ hematologic malignancies, especially HCL.
Although LMB-2 showed antitumor activity in Phase I trial, its application was limited by immunogenicity and rapid tumor growth between cycles. Currently, LMB-2 is being investigated in combination with fludarabine and cyclophosphamide for patients with ATL.57 In the previous report, treatment with fludarabine was associated with lower immunogenicity to murine antibodies, and the fludarabine–cyclophosphamide combination was associated with reductions in normal T and B cells. Patients received fludarabine (25 mg/m2) and cyclophosphamide (250 mg/m2) for 3 consecutive days before cycles began. Two weeks later, patients were treated cyclically every 3 weeks. Fludarabine and cyclophosphamide were administered on days 1, 2, and 3, followed by 30–40 μg/kg LMB-2 on days 3, 5, and 7. An ORR of 50% with 2 CRs and 2 PRs was achieved in 8 evaluable patients, 1 of 2 CRs recurred after 6 months only in a sanctuary site. The toxicity of LMB-2 was not increased by fludarabine and cyclophosphamide, while doses of fludarabine and cyclophosphamide used were also without DLT. With fludarabine and cyclophosphamide, normal T and B cells were reduced to 70% and 96% on average, respectively, which allowed more cycles to result in long-term remission. However, additional patients will be needed to determine if chemotherapy can delay immunogenicity of LMB-2 significantly.
RFT5-dgA
RFT5-dgA is an anti-CD25-ricin A chain RIT that is formed by a murine anti-CD25 monoclonal antibody (immunoglobulin G [IgG]) and a deglycosylated ricin A chain (dgA). RFT5-dgA was prepared by using the hindered heterobifunctional cross-linker to dgA.58 Bell et al evaluated the effect of depleting activated T cells with RFT5-dgA through an in vitro model of acute HIV infection, and the results showed that RFT5-dgA inhibited viral production by activated CD25+ HIV-infected cells and suppressed the breed of infection to uninfected T cells.59 The antitumor activity of this RIT was evaluated in severe combined immunodeficiency (SCID) mice with disseminated human Hodgkin’s lymphoma (HL)60 and in nude mice with subcutaneous solid HL.61 In these mice, 40 μg of RFT5-dgA decreased the diameter of >60% of solid Hodgkin and Reed–Sternberg (H-RS) tumors and inhibited the growth of HL in the majority of the treated mice. And RFT5-dgA was at least 7 times more effective than other ricin A chain-based RITs.62
RFT5-dgA was tested in a Phase I trial that involved 15 patients with refractory HL.63 In this dose escalation trial, all patients received RFT5-dgA intravenously on days 1, 3, 5, and 7 for doses per cycle of 5, 10, 15, or 20 mg/m2. After the treatment of 1–4 cycles, 7 of 15 patients made anti-ricin antibodies and 6 of 15 patients developed anti-mouse antibodies. Only 2 PRs were achieved, and 9 of 15 patients got progressive disease. Side effects were mainly related to VLS, which was characterized by decreases in serum albumin, hypotension, edema, myalgia, and tachycardia. In conclusion, RFT5-dgA showed encouraging efficacy at a dose level, and multiple cycles of treatment could be given without cumulative toxicities. In an extension of this Phase I trial, 5 additional patients were treated at the MTD (15 mg/m2).64 Tumor evaluations were performed after the end of completion of treatment. Overall, 2 patients at the MTD achieved PRs lasting 2 and 21 months, respectively. Of 20 patients, 10 showed progressive disease. Half of the patients developed anti-ricin antibodies or anti-mouse antibodies.
According to the preceding studies, more patients were enrolled in the subsequent trial at the MTD.65,66 A total of 27 patients were treated at this level, and all patients had signs of progressive disease before treatment with RFT5-dgA. Of 17 evaluable patients, 2 patients achieved PRs and 1 minor response (MR) and 5 stable diseases. Eleven of 16 patients receiving 2 or more cycles produced anti-ricin antibodies or anti-mouse antibodies, and the side effect in this study remained moderate and related to VLS.
CD19
CD19 is the hallmark differentiation antigen of the B lineage and has been proposed to serve as an important co-receptor molecule in conjunction with CD21 and CD81 for modifying signals generated through the B-cell antigen receptor complex.67,68 CD19 is a lineage-specific glycoprotein expressed on follicular dendritic cells and B cells. It is present on B cells from the earliest recognizable B-lineage cells during development to B-cell blasts but is lost on mature plasma cells. CD19 has been used to diagnose cancers that raised from cells – notably B-cell lymphomas.69 Treatments targeting CD19 have begun to enter trials from 2012.70,71 Most experimental anti-CD19 drugs work by exploiting the presence of CD19 to treat B-cell cancers specifically. But another study indicated that CD19 plays an active role in driving the growth of these cancers, which suggests that CD19 and its downstream signaling may be a better therapeutic target.72
Anti-B4-bR
Anti-B4-bR is an anti-CD19-blocked ricin (bR) RIT that contained an anti-B4 Mab chemically linked to intact ricin (A + B chain).73 The reactivity of anti-B4-bR with tissues lacking the B4 epitope was diminished by chemically blocking the natural binding site of the B chain.74 But the B chain of bR RITs can facilitate the efficient internalization of the A chain because both the ricin A and B chains have carbohydrate residues that are recognized by liver cells.75,76 Anti-B4-bR has been shown to be highly cytotoxic for B4-positive cells, and the cytotoxicity is restricted to these cells.73
Anti-B4-bR induced clinical responses when administered either as continuous infusions77 or as a daily bolus.73 Thirty-four patients with relapsed or refractory B-cell neoplasms (twenty-six NHL, four CLL, four ALL) received 7-day continuous infusion of anti-B4-bR at dose levels ranging from 10 to 70 mg/kg/d. Potentially, therapeutic serum levels of anti-B4-bR could be maintained for 4 days in patients treated at the MTD. And there were 2 CRs, 3 PRs, and 11 transient responses (TRs).77 Another Phase I trial that enrolled 25 patients with refractory B-cell malignancies was conducted to detail the toxicity and clinical response data when anti-B4-bR was administered as daily bolus infusions for 5 consecutive days.73 One CR, 2 PRs, and 8 mixed responses/TRs were observed after the treatment of anti-B4-bR. Human anti-mouse antibody and anti-ricin antibody were developed in 9 patients. These studies indicated that anti-B4-bR can be administered safely both by continuous infusion and as a daily bolus infusion. In a Phase I trial using anti-B4-bR immunotoxin, 11 of 12 patients with B-cell non-Hodgkin’s lymphoma (B-NHL) remained in CR to the treatment.
HD37-dgA
HD37-dgA was constructed by linking the murine HD37 Mab via a sterically hindered disulfide cross-linker, N-succinimidyl-oxycarbonyl-α-methyl-α-(2-pyridyldithio)-toluene (SMPT), to dgA. Stone et al78 used 2 regimens, intermittent bolus infusion and continuous infusion, for the administration of HD37-dgA to patients with NHL in 2 concomitant Phase I trials. In the intermittent bolus regimen, 2, 4, 8, 16, and 24 mg/m2 of HD37-dgA were divided into 4 equal doses administered every other day. Of 23 evaluable patients, 1 patient achieved CR that persisted >40 months and 1 patient achieved PR (overall 9%). In the continuous infusion regimen, HD37-dgA was administered continuously at 2 dose levels (9.6 and 19.2 mg/m2) for 8 days. One of 9 (11%) evaluable patients developed PR on the continuous infusion regimen. The MTD of each regimen was 16 and 19.2 mg/m2/8 d, respectively. The DLT mainly consisted of VLS, aphasia, and acrocyanosis on the 2 regimens. Almost 25% of patients on the bolus infusion regimen and 30% on the continuous infusion regimen developed antibody against mouse Ig and/or ricin A chain antibody. This test showed that HD37-dgA can be administered safely and can be used in the later clinical trial. In the following Phase II trial, HD37-dgA was used as adjuvant treatment for patients with relapsed B-NHL combined with anti-B4-bR; 26 of 49 patients remained in CR.
CD22
CD22 is a molecule belonging to the sialic acid-recognizing immunoglobulin lectin (SIGLEC) superfamily of lectins.79 It is a B-lineage-restricted surface molecule that modulates B-cell receptor signaling and mediates cellular adhesion. Mature B cells express CD22 on their surface, while it is found to a lesser extent on some immature B cells. It prevents the overactivation of the immune system and the development of autoimmune diseases.80 It is also expressed on B cells in most B-cell leukemias and lymphomas; therefore, it is thought to be a potential targeting therapy for B-cell leukemias and lymphomas.
BL22 [RFB4(dsFv)–PE38/CAT3888]
BL22, which is also named RFB4(dsFv)–PE38 or CAT3888, was the first RIT designed to kill CD22 overexpressed cells. It contained a single-chain Fv of the anti-CD22 BFR4 antibody81 fused to a 38 kDa portion of PE toxin. In preclinical studies, BL22 was cytotoxic to a wide variety of CD22+ cell lines82,83 and fresh malignant cells from patients.84 And it induced CRs in murine xenograft models at doses tolerated by cynomolgus monkeys.85
To assess the clinical activity of BL22, a dose escalation trial was approved.86 Of the 31 patients with B-cell cancers, 16 had HCL and were resistant to cladribine. Between 0.2 and 4.0 mg BL22 diluted in 50 mL of 0.2% albumin in 0.9% sodium chloride was administered intravenously every other day for a total of 3 doses. Of the 16 patients, 11 had CRs and 2 had PRs, while 6 of them had a CR after only one cycle of BL22. Surprisingly, only 4 of the 16 patients generated neutralizing antibodies against the BL22 after cycles 4, 1, 2, and 4, respectively.
In the following study, 46 patients with B-cell hematologic malignancies were enrolled, of whom 11 patients had CLL, 4 patients had NHL, and 31 patients had HCL.87 Of the 31 HCL patients, 16 had a high response rate to BL22 in an interim report.86 BL22 was administered QOD ×3 doses per cycle, dose levels were 3–50 μg/kg. There were no drug-related deaths, and the MTD was determined to be 40 μg/kg. Of the 31 patients with HCL, 19 (61%) CRs and 6 (19%) PRs were achieved, and the ORR was 81%. Only one CLL patient had a PR. The most common DLT was hemolytic uremic syndrome (HUS). HUS was observed in 4 HCL patients and 1 NHL patient during cycles 2/3 and cycle 1, respectively. In these Phase I trials, 65% of CRs were achieved after only one cycle of BL22, so retreatment may not be essential for all patients.
In the Phase II trial, BL22 was limited to 1 cycle and only retreated those patients who did not achieve the cytopenia level for CR. Thirty-six patients suffering from refractory/relapsed HCL were enrolled in this phase II trial.88 A dose rate of 40 μg/kg BL22 was given QOD ×3 on cycle 1. Patients without hematologic remission (HR) were retreated at 30 μg/kg QOD ×3 every 4 weeks for at least 8 weeks after cycle 1, while 50% of the patients (n=18) had responses after just 1 cycle (CR, 25%; PR, 25%), and 56% were retreated 2–13 cycles selectively. After retreatment, 17 CRs (47%), 5 HRs (14%), and 4 PRs (11%) for an ORR of 72% were observed. Compared with patients with spleens either absent or >200 mm (14 of 36), patients with lower baseline spleen height than 200 mm had higher CR (64% vs 21%) and ORR (95% vs 36%). In addition, the median time to relapse of cytopenias has not been reached after nearly 7 years.89
To evaluate the activity of BL22 in hematologic malignancies of children, Wayne et al90 conducted a pre-clinical and Phase I clinical study (Clinical Trials.gov number: NCT00077493). The results showed that BL22 was cytotoxic (median IC50 =9.8 ng/mL) to CD22+ fresh bone marrow or blasts from children with acute lymphoblastic leukemia (ALL) and also prolonged leukemia-free survival of xenografts. Although no obvious responses were achieved in adults, however, transient clinical activity was seen in most of the subjects.
BL22 has significant activity in HCL with a safety profile, but its activity in CLL, ALL, and NHL was limited.86,87 All cases in the Phase II trial generated neutralizing antibodies that neutralized the toxin portion of BL22, and it cannot achieve equal effect in children as well as in adults. So it is necessary to construct an improved RIT.
Moxetumomab pasudotox (CAT-8015/HA22)
BL22 achieved CR rates of 47%–61% in patients with HCL in Phase I and II trials.86–88 But it was less effective in patients with CLL87 who had lower CD22 expression. Consequently, hot-spot mutagenesis was used to increase the affinity of BL22, and the resulting protein was called moxetumomab pasudotox, CAT-8015, or HA22. Moxetumomab pasudotox contains Thr-His-Trp instead of Ser-Ser-Tyr at positions 100, 100A, and 100B in the antigen-binding site of VH. It was determined to have higher cytotoxicity and a 14-fold improved binding affinity, as a result of lower off-rate.91 Moxetumomab pasudotox has antitumor activity in animal xenograft models and a safety profile in cynomolgus monkeys in preclinical studies.92
To determine its safety and efficacy in the treatment of HCL, 28 patients were enrolled in a Phase I trial.93 Moxetumomab pasudotox was given at 5–50 μg/kg QOD ×3 for 1–16 cycles (median, 4 cycles), including 3 patients each at 5, 10, 20, and 30 μg/kg, 4 patients at 40 μg/kg, and 12 patients at 50 μg/kg. After all 114 cycles of moxetumomab pasudotox were administered, major responses were observed at all dose levels. The ORR ranges from 67% to 100% at each dose level without apparent correlation with dose. The ORR of 86% was achieved in 28 patients, whereas only one patient (5%) made neutralizing antibodies after cycle 1. Neutralizing antibodies were detected in 5 of 20, 1 of 13, and 1 of 9 patients after cycles 2, 3, and 4, respectively, but not in patients receiving 5–16 cycles. Thus, it permitted us to retreat most of the HCL patients to increase the chance and degree of response. Remarkable difference had been noted between moxetumomab pasudotox and BL22 in the results of clinical trials. In this HCL clinical trial, the highest dose for treatment was 50 μg/kg, a level higher than the MTD for BL22, and DLT was not observed. The ORR of moxetumomab pasudotox, 86%, was higher than the 72% ORR of the Phase II trial of BL22,88 and response rates at all dose levels were high.
Arons et al analyzed the plasma levels of 49 patients who achieved moxetumomab pasudotox to determine the relationship between response and high CD22 density on HCL cells.94 Those analyzed included the original 28 enrolled prior,93 an additional 20 at the highest dose level (50 μg/kg), and 1 additional patient enrolled prior. Moxetumomab pasudotox was administered to 49 patients at different dose levels every other day for 3 doses. There were 28 (57%) CRs and 88% ORR of 49 patients, while CRs at the highest dose level and lower doses were 64% and 44%, respectively. The difference in micro-residual disease (MRD)-free CR rate was significant (39% vs 6%, P=0.02). Correlations between dose level and both peak level and area under the curve were varied in each dose level. At the highest dose level, CR was more likely with lower volume of distribution and clearance. The pharmacokinetics analysis showed that moxetumomab pasudotox can achieve durable CR in relapsed and refractory HCL patients without MRD. This study also indicates that lower HCL tumor burden minimizes the CD22 “sink” effect allowing higher plasma levels and suggests that patients with lower tumor burden may improve the chance of the durable CR. Furthermore, no DLT or HUS cases were observed in any of the cycles administered to 49 patients.95 Moxetumomab pasudotox is the only known agent that can eliminate MRD in HCL in a high percentage of patients without causing myelosuppressive toxicities.
RFB4-dgA
CD22 molecules expressing the RFB4 epitope are present in 60%–70% of NHL cells. RFB4-dgA was prepared with the anti-CD22 Mab, RFB4, coupled to chemically dgA via the heterobifunctional cross-linker (SMPT). The antibodies used to construct RITs targeting RFB4 contain a mouse IgG1-k and a Fab′ of RFB4. RITs prepared by these antibodies have had notable potential for patients with NHL. Phase I studies of IgG-RFB4-dgA and Fab′-RFB4-dgA have been completed using an intermittent bolus regimen.96,97 Patients with relapsing NHL were treated with Fab′-RFB4-dgA, via 4-h bolus infusion regimen every second day, 5 of 14 (36%) evaluable patients achieved PRs.96 IgG-RFB4-dgA was tested in a similar NHL patient group treated by the same regimen; 1 CR and 5 PRs (ORR, 25%) were observed in 24 patients,97 and a continuous infusion Phase I study of IgG-RFB4-dgA has also been completed.98 In this subsequent Phase I trial, IgG-RFB4-dgA was administered over 8 days with comparable clinical responses (4 of 18 PRs, 22%).98 In all these trials, 20%–40% of the patients achieved responses and the DLT mainly consisted of VLS.99
Combotox (RFB4-dgA + HD37-dgA)
Combotox is a 1:1 mixture of HD37-dgA and RFB4-dgA, which are RITs that target the CD19 and CD22 antigens, respectively. A previous Phase I trial showed that RFB4-dgA induced 24% PRs and 1 long-lasting CR by continuous infusion regimen.98 A previous Phase I trial using continuous infusion of HD37-dgA also showed evidence of antitumor activity,78 concordant with the less potent action of it in vitro,100 and in SCID mice with Daudi lymphoma.101 Preclinical data showed that combotox is effective in killing cells in the Daudi disseminated lymphoma in SCID mice.101
To determine the MTD, clinical pharmacology, toxicity, and clinical responses of combotox, a Phase I trial was conducted involving 22 patients with refractory B-cell lymphoma.102 All patients expressed CD19 and CD22 on at least 30% of their tumor cells. Patients received a continuous infusion of combotox at 3 dose levels ranging from 10 to 30 mg/m2/192 h. After the treatment, only patients with circulating tumor cells (CTCs) in peripheral blood tolerated all doses without major toxicity, and prior therapies of these patients appeared to have little impact on toxicity. Analysis of the results indicated the fact that both lots of HD37-dgA tested in the trial had a tendency to aggregate after thawing, and the multimerization of HD37-dgA may have contributed to patient toxicity. Toxicities induced by combotox in this trial, including VLS and HUS, were similar to other types of RITs reported in previous trials. However, combotox appeared to be safe in patients with even minimal numbers of CTCs.102
Combotox, the mixture of HD37-dgA and RFB4-dgA, can bind to and kill human precursor-B-ALL blasts in vitro, and the combination was more effective than either RIT alone.103 Encouraging clinical data were observed in pediatric patients with pre-B-ALL.104 Hence, combotox is a new candidate for the treatment of patients with relapsed B-ALL. In the subsequent Phase I trial, combotox was administered to adult patients with refractory or relapsed B-ALL. Seventeen patients received combotox at 5 different dose levels (3, 5, 6, 7, and 8 mg/m2) by intravenous infusion. A cycle of treatment of combotox included 3 doses administered every other day. All patients experienced decreased peripheral blasts following the treatment of combotox, and 1 PR was observed.105 The DLT still related to VLS. Thus, combotox can be safely administered to adult patients with refractory B-ALL.
CD30
CD30 shows sequence homology to members of the tumor necrosis factor (TNF) receptor superfamily and is expressed only in activated but not resting T and B cells. It was initially described as a surface marker on neoplastic cells of HD.106 Screening for CD30 expression in normal and neoplastic cells led to recognition of CD30 as an activation-induced antigen mostly expressed on lymphoid cells and to the identification of a new category of NHL: CD30/Ki-1-positive anaplastic large-cell lymphoma (ALCL) by using monoclonal antibodies.107–109 The CD30/CD30 ligand system triggers cytolytic cell death in malignant lymphoma cell line and induces proliferation and cytokine production in T cells or neutrophils.110
Ki-4.dgA
Ki-4.dgA was constructed by linking the anti-CD30 Mab (ki-4) via a sterically hindered linker to dgA. Normal human organs revealed no major cross-reactivity of the anti-CD30 Mabs. Ki-4.dgA was 5 times more potent in vitro than other anti-CD30 dgA-RITs tested previously and showed high efficacy in the treatment of human HL in SCID mice.111 Therefore, ki-4.dgA was selected for a Phase I trial in patients with refractory CD30+ HL and NHL.
In the first Phase I trial that aimed to determine the MTD, DLT, antitumor activity, and pharmacokinetics of ki-4.dgA, 17 patients with relapsed CD30+ lymphoma received escalating doses (5, 7.5, or 10 mg/m2/cycle) QOD ×4 of the RIT for 1–3 cycles.66,112 One PR and 1 MR were achieved in 15 evaluable patients. Side effects and DLT were also associated with VLS, which is similar to RIT RFT5-dgA.65,66 Seven of 17 (40%) patients developed anti-ricin antibodies, and only one patient made anti-mouse antibodies.
RITs targeting other molecules
CD33 and HUM-195/Rgel
CD33 is a surface protein that is found on hematopoietic colony-forming cells but not in their more primitive precursors.113,114 CD33 has structural and binding characteristics that identify it as a member of the sialoadhesin family.115 Studies using samples from patients indicate that in 90% of patients with AML, leukemic cells express CD33.116 HUM-195/Rgel was prepared by the humanized anti-CD33 Mab, M195, conjugated to gelonin.117 HUM-195/Rgel was used in CD33+ cell lines and xenografts in nude mice.118 In a Phase I trial of refractory myeloid leukemias, HUM-195/Rgel induced a 38%–50% decrease in peripheral blood blasts or bone marrow blasts in 7 of 28 patients,117,119 and there was no CR or PR.
CD3 and UCHT1 (A-dmDT390-bisFv)
The CD3 antigen was first identified as a glycoprotein that is present on the surface of all human T lymphocytes.120 As more diverse labeling and immunoprecipitation methods were used, the structure of the CD3 antigen appeared to be more complex. The CD3 was expressed as a complex composed of 4 glycoproteins (CD3-γ, δ, ε, and ζ).121,122 CD3 is overexpressed in T-cell malignancies and is a potential target for the treatment of these diseases. UCHT1 (A-dmDT390-bisFv) is a kind of RIT composed of 2 single-chain Fv fragments of an anti-CD3ε Mab fused to DT390. UCHT1 was administered to 5 patients with CTCL by intravenous infusions.123 Two of 5 patients had PRs lasting 1 and >6 months. Anti-DT antibodies developed in all patients after 2 weeks, and the side effects of UCHT1 were fever, nausea, chills, hypoalbuminemia, and transaminasemia.
IL-3R and DT388-IL3
IL-3 is a cytokine that supports the proliferation and differentiation of myeloid progenitors, but it is absent from mature myeloid cells.124 It is overexpressed in myeloid leukemic progenitors and used as a therapeutic target. DT388-IL3, an RIT fused with a Met-His linker, showed antitumor activities in an SCID model of acute myeloid leukemia (AML).99 A Phase I trial of DT388-IL3 was constructed in patients with chemorefractory AML and myelodysplasia (MDS).125 One CR and 1 PR were observed out of 40 evaluable patients with AML and 1 PR out of 5 patients with MDS.
GM-CSFR and DT388-GM-CSF
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a cytokine responsible for the growth and differentiation of granulocytes and macrophages. GM-CSF receptor (GM-CSFR) is overexpressed in leukemic cells and targeted in leukemia.99 The RITs GM-CSF-PE38KDEL and DT388-GM-CSF exhibited specific cytotoxicity in leukemic cell lines and patients, and DT388-GM-CSF was more cytotoxic than GM-CSF-PE38KDEL.126 Thirty-one patients with refractory AML were treated with DT388-GM-CSF in a Phase I trial, all of those patients were resistant to chemotherapy.127 One CR and 2 PRs were observed among these patients, and the major DLT was cytokine release syndrome. Neutralizing antibodies against DT were observed in 28 of 31 patients.
RITs targeting solid tumors
RITs have produced many durable CRs in refractory HCL, where patients rarely develop neutralizing antidrug antibodies (ADAs) to the toxin component of the RIT. Targeting solid tumors with RITs is much more difficult than targeting hematologic malignancies. Not only because the cellular junctions between cells are tighter and the tumor was more tightly packed, but also the patients are less immunosuppressed and more likely to develop neutralizing antibodies to the toxin. Later, we discuss the recently published RITs targeting solid tumors (Table 2).
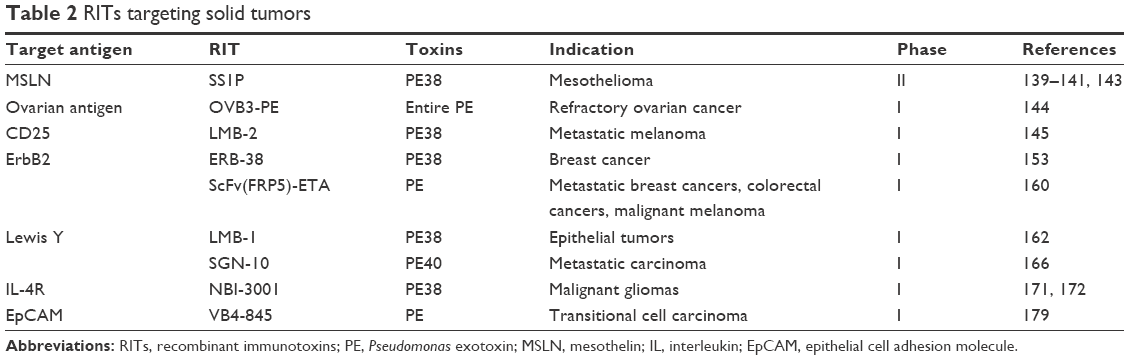 | Table 2 RITs targeting solid tumors |
Mesothelin (MSLN)
MSLN is a cell-surface glycoprotein that is normally expressed only in mesothelial cells, and it is also overexpressed in many solid tumors including mesothelioma,128 pancreatic adenocarcinoma,129,130 cholangiocarcinoma,131 nonmucinous ovarian cancer,132 triple-negative-type breast cancer,133 gastric cancer,134,135 cervical cancer,136 and lung adenocarcinoma.137 Otherwise, MSLN is not critical because MSLN knockout mice grow normally and have no discernible phenotype.138 All the above make MSLN one of the few targeting antigens with sufficient differential expression to allow safe treatment of solid tumor.
SS1P [SS1(dsFv)-PE38]
Phage display technology was used to generate new Fvs binding to MSLN. SS1P is an RIT obtained that underwent affinity improvement, which consists of the PE38 fragment fused to a murine anti-MSLN variable antibody fragment. SS1P was developed for systemic therapy of patients with MSLN-positive solid tumors. One Phase I, dose escalation study of single-agent SS1P was performed in 34 patients with MSLN-expressing advanced mesothelioma (n=20), ovarian (n=12), and pancreatic cancers (n=2).139 Dose level escalated from 8 to 60 μg/kg, and there were 3 patients enrolled at each dose level. The initial cohort of 17 patients received SS1P QOD ×6 doses, and the MTD was 18 μg/kg. DLT included urticaria (1 patient) and grade 3 VLS (2 patients). To allow further dose escalation, 17 patients were treated QOD ×3 doses and MTD was 45 μg/kg. But SS1P was well tolerated at the highest dose level with pleuritis as the DLT. Following this study, another Phase I trial was performed in patients with chemoresistant solid tumor expression MSLN.140 SSIP was administered by continuous infusion for 10 days and repeated cycles at 4-week intervals until the observation of neutralizing antibodies or progressive disease. Twenty-four patients received 4, 8, 12, 18, and 25 μg/kg/d ×10. One of 6 patients, who received the maximum dose level of SS1P, had DLT because of the reversible VLS. Neutralizing antibodies were observed in 18 (75%) of 24 patients, and 5 (21%) patients received a second cycle. Only one patient had a PR. These 2 Phase I trials showed similar efficacy and toxicity of SS1P by different administration schedule. The majority of patients developed ADAs after their first cycle. The main DLT included on-target pleura. These studies demonstrated the need for an effective means to suppress the host immune reaction to SS1P.
So SS1P was tested in patients with newly diagnosed malignant mesothelioma in combination with standard cisplatin and pemetrexed.141 Pemetrexed (500 mg/m2 on day 1) and cisplatin (75 mg/m2 on day 1) were administered every 3 weeks for up to 6 cycles with escalating doses of SS1P. SS1P was administered i.v. on days 1, 3, and 5 during only first 2 cycles at 4 dose levels from 25 to 55 μg/kg. In this study, the toxicity of the combination was similar to that observed with the single agent. Grade 3 toxicities associated with SS1P included fatigue, hypoalbuminemia, back pain, and hypotension. The grade 3 fatigue was dose limiting in only 1 patient at 55 μg/kg. Of 20 evaluable patients, 12 (60%) had a PR. Of 13 patients who received the MTD (45 μg/kg), 10 (77%) had a PR. This compares favorably to the response rate (41.3%) reported for pemetrexed and cisplatin alone.142 Overall, SS1P given with pemetrexed and cisplatin is safe and exhibits significant antitumor activity in patients with pleural mesothelioma, but the hematologic suppression caused by the chemotherapy failed to delay development of neutralizing ADAs.
In a pilot study, SS1P was tested in combination with the pentostatin plus cyclophosphamide immunosuppressive regimen.143 After depleting T and B cells through immunosuppressive regimen, antibody formation was largely delayed, thereby allowing multiple cycles of therapy with SS1P that was previously limited to 1 therapeutic cycle. The immune modulation regimen allowed 2 patients to receive 4 or 6 cycles of SS1P. In this study, of 10 patients with advanced, refractory mesothelioma, 3 patients experienced durable response that persisted for >18 months and 2 patients responded to chemotherapy after immunotoxin therapy. As a result, it is essential to reduce the immunogenicity of SS1P to develop better therapeutic effect.
Ovarian antigen and OVB3-PE
OVB3-PE is the first PE-based RIT that was tested in a clinical trial. It consisted of a murine antibody that targets an unknown antigen on ovarian cancer cells fused to the entire PE toxin,144 but it failed to be used in more patients after its first clinical trial. OVB3-PE was administered to 23 patients with refractory ovarian cancer intraperitoneally,144 but it showed a high level of non-specific toxicity, including central nervous system (CNS) toxicity, transient elevation of liver enzymes, and gastrointestinal (GI) toxicity. Moreover, 100% of the patients made antibodies against the PE toxin 14 days after the first therapy. Human anti-mouse antibodies were also detected in 75% of patients 28 days after therapy.
CD25 and LMB-2
LMB-2 is a CD25-directed PE-based RIT that was usually used to treat CD25+ hematologic malignancies. Considering CD25+ CD4+ Treg cells regulate peripheral self-tolerance and possess the ability to suppress antitumor responses,145 LMB-2 was administered to 8 patients with metastatic melanoma followed by Melanoma Antigen Recognized By T-cells 1 and gp100-specific peptide vaccination.145 LMB-2 administration resulted in a reduction in Treg numbers and did not augment the immune response to cancer vaccination. This study indicated that LMB-2 can selectively mediate a transient partial reduction in circulating and tumor-infiltrating Treg cells in vivo.
ErbB2
The ErbB receptors constitute a group of related transmembrane proteins that belong to the subclass I of the receptor tyrosine kinase superfamily. Four members of this family have been identified: ErbB/epidermal growth factor receptor (EGFR), ErbB2 (HER2/Neu), ErbB3 (HER3), and ErbB4 (HER4).146,147 Overexpression of ErbB glycoprotein and its tyrosine kinase activity induces loss of growth control and plays an important role in the development of several human cancers.148–150 ErbB2 has been reported to be minimally expressed in normal tissues.151 Thus, ErbB2 is an attractive target for immunotherapy.
ERB-38
ERB-38 is an RIT composed of the Fv portion of Mab e23 that reacts with ErbB2, fused to PE38, a truncated form of PE.152 The IC50 of ERB-38 is 0.2–4 ng/mL on the various ErbB2-positive tumor cell lines. ERB-38 is capable of causing CR in nude mice bearing epidermoid carcinoma and breast cancer. Then ERB-38 was administered in a Phase I trial in patients with advanced carcinoma.152 In this trial, 5 breast cancer patients and 1 esophageal cancer patient were treated with 3 doses of ERB-38. But hepatotoxicity was observed in all patients. Immunohistochemistry showed the presence of ErbB2 on hepatocytes.
ScFv(FRP5)-ETA
ScFv(FRP5)-ETA is a recombinant single-chain antibody toxin that contained a scFv portion of murine Mab FRP5, which recognizes the extracellular domain of ErbB2, linked to a truncated ETA.153 ScFv(FRP5)-ETA displayed potent antitumoral activity against a wide range of tumor cells including breast and ovarian carcinomas,153–155 prostate carcinomas,156 and squamous cell carcinomas.157,158 ScFv(FRP5)-ETA effectively inhibited growth of established murine tumor xenografts.153,155,157,158 ScFv(FRP5)-ETA was applied first in 11 patients with metastatic breast and colorectal cancers and with malignant melanoma from 4 clinical centers.159 ScFv(FRP5)-ETA was administered by intratumoral injection into cutaneous lesions for 7–10 days. Of 10 evaluated patients, treatment-induced shrinkage of tumors was observed in 6 patients and 4 CRs and 2 PRs among these 6 patients. Systemic liver toxicity was observed only in 1 patient treated at the highest daily dose levels.
Lewis Y
Lewis Y (LeY) is overexpressed as a surface membrane component of many solid tumors, and it is also expressed on gastrointestinal epithelium and in the pancreas.160
LMB-1
LMB-1 was the first RIT reported to have anti-tumor activity targeting an epithelial tumor.161 It is composed of an Mab that recognizes LeY, B3, chemically linked to PE38. The clinical test of LMB-1 was conducted in 38 patients with LeY-positive solid tumors who had failed conventional therapy. Objective antitumor activity was observed in only 5 patients, and 1 CR was observed in a patient with metastatic breast cancer. A tumor reduction >75% and resolution of clinical symptoms lasting for >6 months were observed in a colon cancer patient. The major toxicity of LMB-1 was VLS, but 33/38 of patients made antibodies against LMB-1 3 weeks after the first cycle of treatment.161
SGN-10 (BR96 sFv-PE40)
SGN-10 is an RIT consisting of sFv regions of the murine Mab, BR96, fused to a binding-defective portion of PE toxin (PE40).162 BR96 binds to the LeY carbohydrate antigen that is overexpressed on the surface of many human solid tumors.160 SGN-10 showed significant antitumor activity in murine xenografts of human breast and lung tumors and made CR in these tumor xenografts.163,164 On the basis of these favorable data, SGN-10 was developed for clinical trial in 46 patients with LeY-positive advanced carcinomas.165 In this test, cohorts of 3 patients were treated at each dose level on days 1, 4, 8, and 11, followed by a 14-day break. A treatment cycle was 28 days, and patient received 2 or more cycles until there was disease progression or unacceptable toxicity. No CR or PR was observed after 8-week treatment, although 31% patients had stable disease. In this study, DLT was due to GI toxicity rather than to VLS. The immunogenicity of the toxin portion limits the ability of SGN-10 by day 11 of therapy.165
IL-4R and NBI-3001 [IL-4(38-37)-PE38KDEL]
IL-4R is overexpressed on many different tumor surfaces. Human malignant glioma cell lines have been shown to express IL-4R,166,167 and primary cell lines from glioma also express IL-4R,168 but not normal brain cells. To treat these tumors that overexpress IL-4R on their surface, NBI-3001 was composed of circularly permuted IL-4 fused to a truncated portion of PE. NBI-3001 was highly cytotoxic to glioblastoma cell lines169 and made CRs in murine xenografts of human glioma in all the animals.170 NBI-3001 was first used in 9 patients with malignant high-grade gliomas.171 Six of 9 patients showed glioma necrosis, and one of them also showed extensive necrosis of tumor leading to CR. Additional patients were treated to determine the appropriate dose level for patients with malignant glioma.172 Thirty-one patients with astrocytoma were enrolled in this following trial, and 25 of them were diagnosed with glioblastoma multiforme (GBM), while the other 6 patients were diagnosed with anaplastic astrocytoma. The results showed decreased signal intensity in the tumor consistent with tumor necrosis after the treatment in most patients.
EpCAM and VB4-845
Epithelial cell adhesion molecule (EpCAM) is overexpressed in many carcinomas relative to normal tissue, as in the case of transitional cell carcinoma (TCC).173,174 TCC refers to those bladder tumors derived from urothelial tissue. And increase of EpCAM expression was regarded as these cancers progress from lower to higher grades.173,175,176 Thus, EpCAM is a potent clinically relevant antigen for targeted treatment of bladder cancer. VB4-845 is an RIT that targets EpCAM+ cancer cells. It contains an anti-EpCAM humanized scFv fused to a truncated form of PE that lacks the cell-binding domain.177 VB4-845 was tested in a clinical Phase I trial to determine its safety, tolerability, immunogenicity, and efficacy.178 Sixty bacillus Calmette-Guerin (BCG)-intolerant patients with TCC or in situ carcinoma were enrolled in this test. CRs were observed in 39% of patients after the treatment. Although the majority of patients developed antibodies against the toxin portion of VB4-845, VB4-845 therapy was safe and without the most adverse events.
Discussion
The introduction of the “magic bullet” concept by Paul Ehlrich led to the search for agents that can selectively target cancer cells. After the initial success of antibody therapy for cancer, Mabs reacting with cancer cells became widely available. The first RITs that consist of a protein toxin fused to a Mab targeting moiety were constructed in the early 1980s. From then on, toxins from a variety of plants and bacteria were investigated, as well as the continuous optimization of the targeting moiety. To make the RIT more suitable for clinical development, portions of the toxin that were not essential for processing or cytotoxic activity were deleted from the sequence, and point mutations were created in the native toxins to improve activity, limit immunogenicity, or reduce off-target toxicity. Nowadays, the generation of an RIT involves the genetic fusion of a modified form of the toxin and a cell-selective ligand. The ligand can be a recombinant antibody or an antibody fragment, carbohydrate antigen, growth factor, or tumor-associated antigen. To be superior to conventional treatments, the ligand must be directed toward antigens that are exclusively or at least preferentially expressed on tumor cells compared to normal tissues. Meanwhile, one or only a few of toxin molecules delivered to the cytosol is sufficient to kill a target cell. So RITs perfectly combine the high specificity of targeting ligand and the excellent cytotoxic activity of toxins.
As one of the most commonly used antibody drugs, antibody–drug conjugates (ADCs) also have the higher specificity and lower toxicity compared with standard chemotherapy. ADCs consist of an Mab chemically attached to a highly toxic chemotherapy agent for use in traditional systemic therapy for cancers.179 The antibody portion localizes the drug to the tumor but limits its deposition elsewhere, increases antitumor activity, and decreases systemic toxicity of the drug.180,181 Although ADCs have so many advantages, RITs have several favorable properties not shared by ADCs. First, the RITs induced the cell death by disrupting the process of protein synthesis (Figure 2). And RITs could bring cell death by activation of caspases, which means that RITs can also be used to treat apoptosis-resisted cancers. Second, the mechanism action of novel RIT allows easy combination with standard agents.179 Third, unlike chemotherapy agent used in ADCs, RITs can effectively kill nondividing cells, and RITs have little cross-resistance with other chemotherapy agents. And last, ADCs can cause off-target toxicity due to inappropriate payload, but RITs do not have this problem. In addition, RITs and ADCs share the same principles of selecting targets for therapy. But the requirement of differential expression is more stringent for RITs, so many targets suitable for ADCs are not suitable for RITs.
Because of higher specificity of RITs, selecting differential expressed surface markers is more essential than the category of toxin. Otherwise, RITs will induce some side effects and toxicities. Variety of toxicities has been observed with RITs that have limited the long-term treatment and efficacy in clinical practice. The most common toxicity for these agents is VLS characterized by weight gain, generalized edema, hypoalbuminemia, and orthostatic hypotension. RITs-mediated damage to endothelial cells appears to be responsible for VLS, because the cytotoxic protein must traverse endothelial cells to exit the blood vessels. Studies have shown that PE binds directly to endothelial cells, while truncated PE requires a ligand that reacts with the endothelium and a mutant form of PE showed less VLS.182,183 In addition, mutant toxins that lack enzymatic activity do not cause VLS, suggesting that VLS is an off-target effect of RITs.183 Ricin toxin contains short amino acid motifs that bind endothelial cells.184 Modification or deletion of these motif sequences reduced toxin-induced VLS.185,186 Most recently, Weldon et al deleted nearly entire PE domain II to prevent VLS, while preserving 2 of 3 putative endothelial binding motifs.187 Clinical factors may also induce the VLS. Clinical factors, such as administration of RITs, vary among patients that may affect the severity of VLS. Ricin-based RITs have reported more severe VLS than PE-based RITs. A retrospective study of HL patients showed that patients with a history of prior radiation therapy will have more frequent and more severe VLS.188 In addition to mutations of short amino acid motifs of toxin part, patients receiving premedication with dexamethasone have also been shown to have less severe VLS.189
As a novel class of immune therapeutics, RITs have been used in the treatment of many kinds of tumors. The clinical responses present a promising approach to fighting cancers. RITs produced limited responses in relapsed and refractory hematologic malignancies mainly because of their side effects and high immunogenicity. Nevertheless, their half-lives were too limited for diffusion to occur in solid tumors. Immunogenicity of protein drugs and antibody drugs suddenly attracts broad attention around the world. For regulatory agencies, immunogenicity assessments are required for the licensure of all biologics to ensure safety and efficacy of the proteins. The causes mainly include 2 aspects: 1) immunogenicity will affect safety and efficacy of the drugs, even life-threatening interaction with endogenous protein and 2) lacking of efficient predictive tools means ADAs can be detected only in late Phase III trials after significant expenses have accrued. Based on a wide range of clinical trials, the incidence of immunogenicity after 1 cycle of RIT ranges from 50% to 100% for solid tumors and from 0% to 40% for hematologic tumors.99 Patients with solid tumors are more likely to develop neutralizing antibodies to the toxin because of their less immunosuppression. The presence of neutralizing antibodies lowers the level of active RITs and their efficacy. There is a multitude of factors responsible for ADA formation against RITs. Studies on immunogenicity to RITs indicate that ADA had a strong correlation with a decrease in drug serum concentrations and resultant reduced efficacy.190 ADAs impact the PK of RITs in diverse ways. They can enhance clearance besides sustaining the circulation of RITs. ADA–RIT complexes circulating in the bloodstream trigger regular endogenous elimination processes that are mediated by the reticuloendothelial system, predominantly phagocytic cells in the liver and spleen. The complexes are internalized and undergo subsequent lysosomal degradation. At the same time, ADA–RIT complexes are often clearly slower than the free activated RIT, and compensatory upregulation of shed target may result in concentration increases in total, while “free” concentrations are actually decreasing.191 Moreover, the mechanism of action of an ADA-induced impact on the PK of RITs still needs further and extensive investigation.
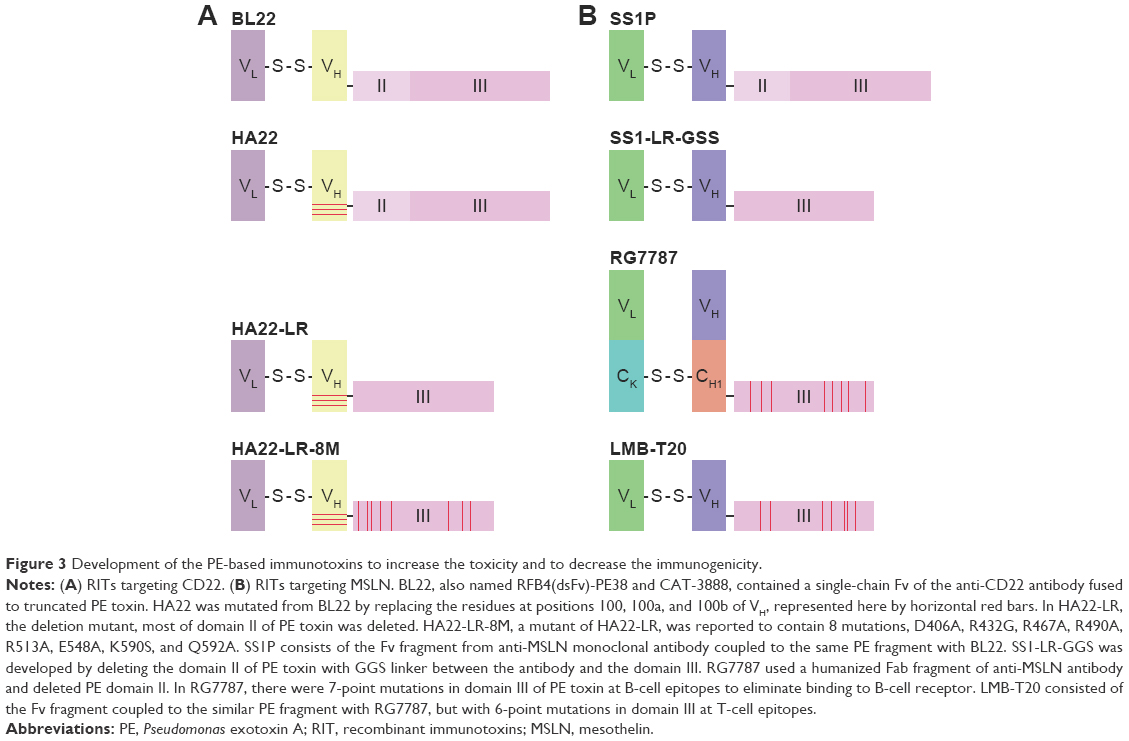
At present, several approaches have been used to prevent the development of neutralizing antibodies of RITs in patients. 1) To diminish the immunogenicity of RITs, the new generation of RITs consists of a humanized Fab or Fv fragment of antibody.192,193 It was reported that humanized RIT lost some of its epitopes.194 But the majority of the antibodies that have been found were against the toxin portion of RITs.195 Genetic engineered single-chain variable fragments (scFVs) also are fused to toxins instead of full-size antibodies.196 Compared with full-size Mab therapeutics, low immunogenicity scFV toxin therapeutics has several pros. Antibody fragments, such as scFVs, penetrate tissues and tumors more rapidly and deeply than full-size Mab. In addition, the scFVs have been suggested to permit binding to cryptic epitopes not accessible to full-size Mabs.197 2) The most useful method for some biologic agents, such as interferon198 and L-asparaginase,199 is PEGylation. Covalent attachment of polyethylene glycol (PEG) to RIT has been found to be useful in “masking” the immunogenic epitopes in the protein.200 PEGylation also prolongs the circulation of RIT by reducing renal clearance.201 3) Domain II of PE appeared to be the most immunogenic portion of the PE molecule.144 Mazor et al found that domain II of PE was sensitive to protease digestion and that almost all of domain II except the furin cleavage site (amino acids 274–284) could be removed without loss of activity.200 4) Liu et al202 identified the human B-cell epitopes of PE toxin by M13 phage display. Then they constructed a variant RIT with point mutations of the residues that make up the B-cell epitopes. The variant RIT, which with a deletion of domain II and 7-point mutations that modified human B-cell epitopes, had significantly reduced reactivity with human antisera and retained cytotoxic and antitumor activity.202 Liu et al used this approach to develop a new RIT, RG7787.202 The cytotoxic activity of RG7787 was significantly improved, but the immunogenicity results are not clear yet.193,203 5) Mazor et al204 used the similar approach to develop 2 variant RITs that have their T-cell epitopes removed or suppressed.205 The immunogenicity results suggested that removal of T-cell epitopes is more effective than the removal of B-cell epitopes.200 All these approaches were widely used in the production of new RITs, especially the deletion of domain II of PE toxin and modification of human B- or T-cell epitopes (Figure 3). The smaller molecular weight, the lower immunogenicity of RITs. So we constructed an RIT with small molecules that consisted of a PE38 toxin and 17 amino acids of amidated gastrin. This RIT, named rG17PE38KDEL, targets the overexpression of cholecystok II receptor (CCK2R) on gastric cancer cells, and it has lower immunogenicity in xenograft model, which means it can be used in further development and application.
 | Figure 3 Development of the PE-based immunotoxins to increase the toxicity and to decrease the immunogenicity. |
Recent advances in the design and administration of RITs are overcoming partial challenges. Serial modifications have been used to reduce nonspecific toxicities, to increase stability and to improve targeted cellular killing. To overcome another major challenge, immunogenicity, several approaches were developed in the past years. Onda et al206 developed the less immunogenic PE by engineering variant of the toxin. Based on this strategy, a bispecific ligand-directed toxin EGF4KDEL-7mut was developed, in which human EGF and IL-4 are linked to low immunogenic variant of PE38. It showed the dual benefit by increasing targeting specificity and reducing immunogenicity. Fused micromolecule or ligand also allowed long-term treatment of tumors. An RIT aiming at the treatment of gastric cancer, rG17PE38, was developed by our laboratory, in which amidated gastrin 17 (rG17) was linked to truncated modificatory PE38. It can suppress the growth of tumor and prolong the survival time in murine xenograft models, and the tumor-bearing mouse did not develop the neutralizing antibody against rG17PE38 after continuous infusion.
Several RITs have shown remarkable success against hematologic malignancies. It had been reported that RITs targeting MSLN produced major tumor regression in some patients with advanced mesothelioma.179 Although there is a further step to mitigate nonspecific toxicities and to enhance the activity of the toxin, we still can anticipate exciting successes in the future application of RITs based on appropriate combinations of cancers and selective target.
Conclusion
The success rate of immunotoxin therapy in clinical trials of leukemia has attracted more effects toward the newer and enhanced immunotoxins, but the immunotoxins targeting solid tumors did not prove effective as expected. Toxicity and immunogenicity remain major concerns, but recently they have been overcome partly by different strategies. As potential antineoplastic agents, immunotoxins are receiving more attention once again and they could be ideal molecules for combination therapy.
Acknowledgments
We thank Song Zhang, Jiang Chang, Chang Li, Ke Zhao, Yu-Ting Guan, Si-Yu Chen, Wen-Qiang He, and Yuan-Yuan Zhang for assistance with data collection and discussion. Furthermore, we express our gratitude to Dr Waqas Ahmad (Section of Epidemiology & Public Health, College of Veterinary and Animal Science, Jhang, Pakistan) for revising the paper for English language. This work was supported by the Key Project in Jilin Province (No 20120966), Specialized Research Fund for Doctoral Program of Colleges and Universities (No 20120061110078), the Fundamental Research Funds for the Platform Base Construction Project in Jilin University (No 2014ZKF01), National Natural Science Foundation Young Investigator Grant Program (No 81401953), the Youth Research Fund of Science and Technology Development Plan of Jilin Province (No 20160520162JH), and the Class General Financial and the special Financial Grant from the China Postdoctoral Science Foundation (No 2014M561303 and No 2015T80312).
Disclosure
The authors report no conflicts of interest in this work.
References
Moolten FL, Cooperband SR. Selective destruction of target cells by diphtheria toxin conjugated to antibody directed against antigens on the cells. Science. 1970;169(3940):68–70. | ||
Krolick KA, Villemez C, Isakson P, Uhr JW, Vitetta ES. Selective killing of normal or neoplastic B cells by antibodies coupled to the A chain of ricin. Proc Natl Acad Sci U S A. 1980;77(9):5419. | ||
Cawley DB, Herschman HR, Gilliland DG, Collier RJ. Epidermal growth factor-toxin A chain conjugates: EGF-ricin A is a potent toxin while EGF-diphtheria fragment A is nontoxic. Cell. 1980;22(2):563–570. | ||
Carroll SF, Collier RJ. Active site of Pseudomonas aeruginosa exotoxin A. Glutamic acid 553 is photolabeled by NAD and shows functional homology with glutamic acid 148 of diphtheria toxin. J Biol Chem. 1987;262(18):8707–8711. | ||
Phan LD, Perentesis JP, Bodley JW. Saccharomyces cerevisiae elongation factor 2. Mutagenesis of the histidine precursor of diphthamide yields a functional protein that is resistant to diphtheria toxin. J Biol Chem. 1993;268(12):8665–8668. | ||
Allured VS, Collier RJ, Carroll SF, McKay DB. Structure of exotoxin A of Pseudomonas aeruginosa at 3.0-Angstrom resolution. Proc Natl Acad Sci U S A. 1986;83(5):1320–1324. | ||
Hwang J, Fitzgerald DJ, Adhya S, Pastan I. Functional domains of Pseudomonas exotoxin identified by deletion analysis of the gene expressed in E. coli. Cell. 1987;48(1):129–136. | ||
Hessler JL, Kreitman RJ. An early step in Pseudomonas exotoxin action is removal of the terminal lysine residue, which allows binding to the KDEL receptor. Biochemistry (Mosc). 1997;36(47):14577–14582. | ||
Kounnas MZ, Morris RE, Thompson MR, FitzGerald DJ, Strickland DK, Saelinger CB. The alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein binds and internalizes Pseudomonas exotoxin A. J Biol Chem. 1992;267(18):12420–12423. | ||
Chiron MF, Fryling CM, FitzGerald DJ. Cleavage of Pseudomonas exotoxin and diphtheria toxin by a furin-like enzyme prepared from beef liver. J Biol Chem. 1994;269(27):18167–18176. | ||
McKee ML, FitzGerald DJ. Reduction of furin-nicked Pseudomonas exotoxin A: an unfolding story. Biochemistry (Mosc). 1999;38(50):16507–16513. | ||
Kreitman RJ, Pastan I. Importance of the glutamate residue of KDEL in increasing the cytotoxicity of Pseudomonas exotoxin derivatives and for increased binding to the KDEL receptor. Biochem J. 1995;307(1):29–37. | ||
Chaudhary VK, Jinno Y, FitzGerald D, Pastan I. Pseudomonas exotoxin contains a specific sequence at the carboxyl terminus that is required for cytotoxicity. Proc Natl Acad Sci U S A. 1990;87(1):308–312. | ||
Theuer C, Kasturi S, Pastan I. Domain II of Pseudomonas exotoxin A arrests the transfer of translocating nascent chains into mammalian microsomes. Biochemistry (Mosc). 1994;33(19):5894–5900. | ||
Theuer CP, Buchner J, FitzGerald D, Pastan I. The N-terminal region of the 37-kDa translocated fragment of Pseudomonas exotoxin A aborts translocation by promoting its own export after microsomal membrane insertion. Proc Natl Acad Sci U S A. 1993;90(16):7774–7778. | ||
Keppler-Hafkemeyer A, Kreitman RJ, Pastan I. Apoptosis induced by immunotoxins used in the treatment of hematologic malignancies. Int J Cancer. 2000;87(1):86–94. | ||
Brinkmann U, Brinkmann E, Gallo M, Pastan I. Cloning and characterization of a cellular apoptosis susceptibility gene, the human homologue to the yeast chromosome segregation gene CSE1. Proc Natl Acad Sci U S A. 1995;92(22):10427–10431. | ||
Rolf JM, Gaudin HM, Eidels L. Localization of the diphtheria toxin receptor-binding domain to the carboxyl-terminal Mr approximately 6000 region of the toxin. J Biol Chem. 1990;265(13):7331–7337. | ||
Choe S, Bennett MJ, Fujii G, et al. The crystal structure of diphtheria toxin. Nature. 1992;357(6375):216–222. | ||
Williams DP, Wen Z, Watson RS, Boyd J, Strom TB, Murphy JR. Cellular processing of the interleukin-2 fusion toxin DAB486-IL-2 and efficient delivery of diphtheria fragment A to the cytosol of target cells requires Arg194. J Biol Chem. 1990;265(33):20673–20677. | ||
D’Silva PR, Lala AK. Unfolding of diphtheria toxin identification of hydrophobic sites exposed on lowering of pH by photolabeling. J Biol Chem. 1998;273(26):16216–16222. | ||
Kaul P, Silverman J, Shen WH, et al. Roles of Glu 349 and Asp 352 in membrane insertion and translocation by diphtheria toxin. Protein Sci. 1996;5(4):687–692. | ||
Wilson BA, Blanke SR, Reich KA, Collier RJ. Active-site mutations of diphtheria toxin. Tryptophan 50 is a major determinant of NAD affinity. J Biol Chem. 1994;269(37):23296–23301. | ||
Bennett MJ, Eisenberg D. Refined structure of monomelic diphtheria toxin at 2.3 A resolution. Protein Sci. 1994;3(9):1464–1475. | ||
Yamaizumi M, Mekada E, Uchida T, Okada Y. One molecule of diphtheria toxin fragment a introduced into a cell can kill the cell. Cell. 1978;15(1):245–250. | ||
Mattoo AR, Fitzgerald DJ. Combination treatments with ABT-263 and an immunotoxin produce synergistic killing of ABT-263-resistant small cell lung cancer cell lines. Int J Cancer. 2013;132(4):978–987. | ||
Traini R, Ben-Josef G, Pastrana DV, et al. ABT-737 overcomes resistance to immunotoxin-mediated apoptosis and enhances the delivery of Pseudomonas exotoxin-based proteins to the cell cytosol. Mol Cancer Ther. 2010;9(7):2007–2015. | ||
Morimoto H, Bonavida B. Diphtheria toxin- and Pseudomonas A toxin-mediated apoptosis. ADP ribosylation of elongation factor-2 is required for DNA fragmentation and cell lysis and synergy with tumor necrosis factor-alpha. J Immunol. 1992;149(6):2089–2094. | ||
Kochi SK, Collier RJ. DNA fragmentation and cytolysis in U937 cells treated with diphtheria toxin or other inhibitors of protein synthesis. Exp Cell Res. 1993;208(1):296–302. | ||
Keppler-Hafkemeyer A, Brinkmann U, Pastan I. Role of caspases in immunotoxin-induced apoptosis of cancer cells. Biochemistry. 1998;37(48):16934–16942. | ||
Jenkins CE, Swiatoniowski A, Issekutz AC, Lin T-J. Pseudomonas aeruginosa exotoxin A induces human mast cell apoptosis by a caspase-8 and −3-dependent mechanism. J Biol Chem. 2004;279(35):37201–37207. | ||
Decker T, Oelsner M, Kreitman RJ, et al. Induction of caspase-dependent programmed cell death in B-cell chronic lymphocytic leukemia by anti-CD22 immunotoxins. Blood. 2004;103(7):2718–2726. | ||
Bolognesi A, Polito L, Tazzari PL, et al. In vitro anti-tumour activity of anti-CD80 and anti-CD86 immunotoxins containing type 1 ribosome-inactivating proteins. Br J Haematol. 2000;110(2):351–361. | ||
Walsh MJ, Dodd JE, Hautbergue GM. Ribosome-inactivating proteins. Virulence. 2013;4(8):774–784. | ||
Concanavalin A. Gelonin, a new inhibitor of protein synthesis, nontoxic to intact cells. J Biol Chem. 1980;255(14):6947–6953. | ||
Endo Y, Mitsui K, Motizuki M, Tsurugi K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 S ribosomal RNA caused by the toxins. J Biol Chem. 1987;262(12):5908–5912. | ||
Wesche J, Rapak A, Olsnes S. Dependence of ricin toxicity on translocation of the toxin A-chain from the endoplasmic reticulum to the cytosol. J Biol Chem. 1999;274(48):34443–34449. | ||
Foss FM. DAB 389 IL-2 (ONTAK): a novel fusion toxin therapy for lymphoma. Clin Lymphoma. 2000;1(2):110–116. | ||
Olsen E, Duvic M, Frankel A, et al. Pivotal phase III trial of two dose levels of denileukin diftitox for the treatment of cutaneous T-cell lymphoma. J Clin Oncol. 2001;19(2):376–388. | ||
Prince HM, Duvic M, Martin A, et al. Phase III placebo-controlled trial of denileukin diftitox for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2010;28(11):1870–1877. | ||
Prince HM, Martin AG, Olsen EA, Fivenson DP, Duvic M. Denileukin diftitox for the treatment of CD25 low-expression mycosis fungoides and Sézary syndrome. Leuk Lymphoma. 2013;54(1):69–75. | ||
Dannull J, Su Z, Rizzieri D, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest. 2005;115(12):3623–3633. | ||
Barnett B, Kryczek I, Cheng P, Zou W, Curiel TJ. Regulatory T cells in ovarian cancer: biology and therapeutic potential. Am J Reprod Immunol. 2005;54(6):369–377. | ||
Mahnke K, Schönfeld K, Fondel S, et al. Depletion of CD4+ CD25+ human regulatory T cells in vivo: kinetics of Treg depletion and alterations in immune functions in vivo and in vitro. Int J Cancer. 2007;120(12):2723–2733. | ||
Morse MA, Hobeika AC, Osada T, et al. Depletion of human regulatory T cells specifically enhances antigen-specific immune responses to cancer vaccines. Blood. 2008;112(3):610–618. | ||
Rasku MA, Clem AL, Telang S, et al. Transient T cell depletion causes regression of melanoma metastases. J Transl Med. 2008;6(1):1. | ||
Sue McCann MSN, Akilov OE, Geskin L. Adverse effects of denileukin diftitox and their management in patients with cutaneous T-cell lymphoma. Clin J Oncol Nurs. 2012;16(5):E164. | ||
Robb RJ, Rusk CM, Neeper MP. Structure-function relationships for the interleukin 2 receptor: location of ligand and antibody binding sites on the Tac receptor chain by mutational analysis. Proc Natl Acad Sci U S A. 1988;85(15):5654–5658. | ||
Kreitman RJ, Bailon P, Chaudihary VK, FitzGerald DJP, Pastan I. Recombinant immunotoxins containing anti-Tac(Fv) and derivatives of Pseudomonas exotoxin produce complete regression in mice of an interleukin-2 receptor-expressing human carcinoma. Blood. 1994;83(2):426–434. | ||
Krietman RJ, Chaudhary VK, Waldmann T, Willingham MC, FitzGerald DJ, Pastan I. The recombinant immunotoxin anti-Tac (Fv)-Pseudomonas exotoxin 40 is cytotoxic toward peripheral blood malignant cells from patients with adult T-cell leukemia. Proc Natl Acad Sci U S A. 1990;87(21):8291–8295. | ||
Kreitman RJ, Chaudhary VK, Waldmann TA, et al. Cytotoxic activities of recombinant immunotoxins composed of Pseudomonas toxin or diphtheria toxin toward lymphocytes from patients with adult T-cell leukemia. Leukemia. 1993;7(4):553–562. | ||
Saito T, Kreitman RJ, Hanada S, et al. Cytotoxicity of recombinant Fab and Fv immunotoxins on adult T-cell leukemia lymph node and blood cells in the presence of soluble interleukin-2 receptor. Cancer Res. 1994;54(4):1059–1064. | ||
Kreitman RJ, Batra JK, Seetharam S, Chaudhary VK, FitzGerald DJ, Pastan I. Single-chain immunotoxin fusions between anti-Tac and Pseudomonas exotoxin: relative importance of the two toxin disulfide bonds. Bioconjug Chem. 1993;4(2):112–120. | ||
Kreitman RJ, Pastan I. Targeting Pseudomonas exotoxin to hematologic malignancies. Semin Cancer Biol. 1995;6(5):297–306. Elsevier. | ||
Kreitman RJ, Wilson WH, Robbins D, et al. Responses in refractory hairy cell leukemia to a recombinant immunotoxin. Blood. 1999;94(10):3340–3348. | ||
Kreitman RJ, Wilson WH, White JD, et al. Phase I trial of recombinant immunotoxin anti-Tac (Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J Clin Oncol. 2000;18(8):1622–1636. | ||
Kreitman RJ, Singh R, Stetler-Stevenson M, Waldmann TA, Pastan I. Regression of adult T-cell leukemia with anti-CD25 recombinant immunotoxin LMB-2 preceded by chemotherapy. Blood. 2011;118(21):2575. | ||
Thorpe PE, Wallace PM, Knowles PP, et al. New coupling agents for the synthesis of immunotoxins containing a hindered disulfide bond with improved stability in vivo. Cancer Res. 1987;47(22):5924–5931. | ||
Bell KD, Ramilo O, Vitetta ES. Combined use of an immunotoxin and cyclosporine to prevent both activated and quiescent peripheral blood T cells from producing type 1 human immunodeficiency virus. Proc Natl Acad Sci U S A. 1993;90(4):1411–1415. | ||
Winkler U, Gottstein C, Schon G, et al. Successful treatment of disseminated human Hodgkin’s disease in SCID mice with deglycosylated ricin A-chain immunotoxins. Blood. 1994;83(2):466–475. | ||
Engert A, Martin G, Amlot P, Wijdenes J, Diehl V, Thorpe P. Immunotoxins constructed with anti-CD25 monoclonal antibodies and deglycosylated ricin a-chain have potent anti-tumour effects against human Hodgkin cells in vitro and solid Hodgkin tumours in mice. Int J Cancer. 1991;49(3):450–456. | ||
Engert A, Gottstein C, Winkler U, et al. Experimental treatment of human Hodgkin’s disease with ricin A-chain immunotoxins. Leuk Lymphoma. 1994;13(5–6):441–448. | ||
Engert A, Diehl V, Schnell R, et al. A phase-I study of an anti-CD25 ricin A-chain immunotoxin (RFT5-SMPT-dgA) in patients with refractory Hodgkin’s lymphoma. Blood. 1997;89(2):403–410. | ||
Schnell R, Vitetta E, Schindler J, et al. Clinical trials with an anti-CD25 ricin A-chain experimental and immunotoxin (RFT5-SMPT-dgA) in Hodgkin’s lymphoma. Leuk Lymphoma. 1998;30(5–6):525–538. | ||
Schnell R, Vitetta E, Schindler J, et al. Treatment of refractory Hodgkin’s lymphoma patients with an anti-CD25 ricin A-chain immunotoxin. Leukemia. 2000;14(1):129–135. | ||
Schnell R, Borchmann P, Staak JO, et al. Clinical evaluation of ricin A-chain immunotoxins in patients with Hodgkin’s lymphoma. Ann Oncol. 2003;14(5):729–736. | ||
Weiss A, Littman DR. Signal transduction by lymphocyte antigen receptors. Cell. 1994;76(2):263–274. | ||
Cambier JC, Pleiman CM, Clark MR. Signal transduction by the B cell antigen receptor and its coreceptors. Annu Rev Immunol. 1994;12(1):457–486. | ||
Scheuermann RH, Racila E. CD19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leuk Lymphoma. 1995;18(5–6):385–397. | ||
June CH. CAR T cells for leukemia and more. Cancer Res. 2012;72(8 suppl):L1–L3. | ||
Coghlan A. Novel gene therapy cures leukaemia in eight days. New Sci. 2013;217(2910):10. | ||
Chung EY, Psathas JN, Yu D, Li Y, Weiss MJ, Thomas-Tikhonenko A. CD19 is a major B cell receptor–independent activator of MYC-driven B-lymphomagenesis. J Clin Invest. 2012;122(6):2257–2266. | ||
Grossbard ML, Freedman AS, Ritz J, et al. Serotherapy of B-cell neoplasms with anti-B4-blocked ricin: a phase I trial of daily bolus infusion. Blood. 1992;79(3):576–585. | ||
Goldmacher VS, Scott CF, Lambert JM, et al. Cytotoxicity of gelonin and its conjugates with antibodies is determined by the extent of their endocytosis. J Cell Physiol. 1989;141(1):222–234. | ||
Blakey DC, Skilleter DN, Price RJ, Thorpe PE. Uptake of native and deglycosylated ricin A-chain immunotoxins by mouse liver parenchymal and non-parenchymal cells in vitro and in vivo. Biochim Biophys Acta. 1988;968(2):172–178. | ||
Blakey DC, Watson GJ, Knowles PP, Thorpe PE. Effect of chemical deglycosylation of ricin A chain on the in vivo fate and cytotoxic activity of an immunotoxin composed of ricin A chain and anti-Thy 1.1 antibody. Cancer Res. 1987;47(4):947–952. | ||
Grossbard ML, Lambert JM, Goldmacher VS, et al. Anti-B4-blocked ricin: a phase I trial of 7-day continuous infusion in patients with B-cell neoplasms. J Clin Oncol. 1993;11(4):726–737. | ||
Stone MJ, Sausville EA, Fay JW, et al. A phase I study of bolus versus continuous infusion of the anti-CD19 immunotoxin, IgG-HD37-dgA, in patients with B-cell lymphoma. Blood. 1996;88(4):1188–1197. | ||
Crocker PR, Clark EA, Filbin M, et al. Siglecs: a family of sialic-acid binding lectins. Glycobiology. 1998;8(2):v–vi. | ||
Hatta Y, Tsuchiya N, Matsushita M, Shiota M, Hagiwara K, Tokunaga K. Identification of the gene variations in human CD22. Immunogenetics. 1999;49(4):280–286. | ||
Li J-L, Shen G-L, Ghetie M-A, et al. The epitope specificity and tissue reactivity of four murine monoclonal anti-CD22 antibodies. Cell Immunol. 1989;118(1):85–99. | ||
Mansfield E, Chiron MF, Amlot P, Pastan I, FitzGerald DJ. Recombinant RFB4 single-chain immunotoxin that is cytotoxic towards CD22-positive cells. Biochem Soc Trans. 1997;25(2):709. | ||
Mansfield E, Amlot P, Pastan I, FitzGerald DJ. Recombinant RFB4 immunotoxins exhibit potent cytotoxic activity for CD22-bearing cells and tumors. Blood. 1997;90(5):2020–2026. | ||
Kreitman RJ, Margulies I, Stetler-Stevenson M, Wang Q-C, FitzGerald DJ, Pastan I. Cytotoxic activity of disulfide-stabilized recombinant immunotoxin RFB4 (dsFv)-PE38 (BL22) toward fresh malignant cells from patients with B-cell leukemias. Clin Cancer Res. 2000;6(4):1476–1487. | ||
Kreitman RJ, Wang Q-C, FitzGerald DJ, Pastan I. Complete regression of human B-cell lymphoma xenografts in mice treated with recombinant anti-CD22 immunotoxin RFB4 (dsFv)-PE38 at doses tolerated by cynomolgus monkeys. Int J Cancer. 1999;81(1):148–155. | ||
Kreitman RJ, Wilson WH, Bergeron K, et al. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N Engl J Med. 2001;345(4):241–247. | ||
Kreitman RJ, Squires DR, Stetler-Stevenson M, et al. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J Clin Oncol. 2005;23(27):6719–6729. | ||
Kreitman RJ, Stetler-Stevenson M, Margulies I, et al. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J Clin Oncol. 2009;27(18):2983–2990. | ||
Kreitman RJ, Pastan I. Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin Cancer Res. 2011;17(20):6398–6405. | ||
Wayne AS, Kreitman RJ, Findley HW, et al. Anti-CD22 immunotoxin RFB4(dsFv)-PE38 (BL22) for CD22-positive hematologic malignancies of childhood: preclinical studies and phase I clinical trial. Clin Cancer Res. 2010;16(6):1894–1903. | ||
Salvatore G, Beers R, Margulies I, Kreitman RJ, Pastan I. Improved cytotoxic activity toward cell lines and fresh leukemia cells of a mutant anti-CD22 immunotoxin obtained by antibody phage display. Clin Cancer Res. 2002;8(4):995–1002. | ||
Alderson RF, Kreitman RJ, Chen T, et al. CAT-8015: a second-generation Pseudomonas exotoxin A–based immunotherapy targeting CD22-expressing hematologic malignancies. Clin Cancer Res. 2009;15(3):832–839. | ||
Kreitman RJ, Tallman MS, Robak T, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol. 2012;30(15):1822–1828. | ||
Arons E, Stetler-Stevenson M, Wilson WH, FitzGerald DJP, Pastan I. Pharmacokinetic analysis of response in hairy cell leukemia treated by anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Blood. 2013;122(21):2871. | ||
Kreitman RJ, Arons E, Tallman MS, et al. High response rate of Moxetumomab pasudotox in relapsed/refractory hairy cell leukemia includes eradication of minimal residual disease: potential importance for outcome. Blood. 2015;126(23):4161. | ||
Vitetta ES, Stone M, Amlot P, et al. Phase I immunotoxin trial in patients with B-cell lymphoma. Cancer Res. 1991;51(15):4052–4058. | ||
Amlot PL, Stone MJ, Cunningham D, et al. A phase I study of an anti-CD22-deglycosylated ricin A chain immunotoxin in the treatment of B-cell lymphomas resistant to conventional therapy. Blood. 1993;82(9):2624–2633. | ||
Sausville EA, Headlee D, Stetler-Stevenson M, et al. Continuous infusion of the anti-CD22 immunotoxin IgG-RFB4-SMPT-dgA in patients with B-cell lymphoma: a phase I study. Blood. 1995;85(12):3457–3465. | ||
Kreitman RJ. Immunotoxins for targeted cancer therapy. AAPS J. 2006;8(3):E532–E551. | ||
Pezzutto A, Dörken B, Rabinovitch PS, Ledbetter JA, Moldenhauer G, Clark EA. CD19 monoclonal antibody HD37 inhibits anti-immunoglobulin-induced B cell activation and proliferation. J Immunol. 1987;138(9):2793–2799. | ||
Ghetie MA, Picker LJ, Richardson JA, Tucker K, Uhr JW, Vitetta ES. Anti-CD19 inhibits the growth of human B-cell tumor lines in vitro and of Daudi cells in SCID mice by inducing cell cycle arrest. Blood. 1994;83(5):1329–1336. | ||
Messmann RA, Vitetta ES, Headlee D, et al. A phase I study of combination therapy with immunotoxins IgG-HD37-deglycosylated ricin A chain (dgA) and IgG-RFB4-dgA (Combotox) in patients with refractory CD19 (+), CD22 (+) B cell lymphoma. Clin Cancer Res. 2000;6(4):1302–1313. | ||
Herrera L, Farah RA, Pellegrini VA, et al. Immunotoxins against CD19 and CD22 are effective in killing precursor-B acute lymphoblastic leukemia cells in vitro. Leukemia. 2000;14(5):853–858. | ||
Herrera L, Bostrom B, Gore L, et al. A phase 1 study of combotox in pediatric patients with refractory B-lineage acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2009;31(12):936–941. | ||
Schindler J, Gajavelli S, Ravandi F, et al. A phase I study of a combination of anti-CD19 and anti-CD22 immunotoxins (Combotox) in adult patients with refractory B-lineage acute lymphoblastic leukaemia: combotox in Adult ALL. Br J Haematol. 2011;154(4): 471–476. | ||
Schwab U, Stein H, Gerdes J, et al. Production of a monoclonal antibody specific for Hodgkin and Sternberg-Reed cells of Hodgkin’s disease and a subset of normal lymphoid cells. Nature. 1982;299:65–67. | ||
Andreesen R, Osterholz J, Lohr GW, Bross KJ. A Hodgkin cell-specific antigen is expressed on a subset of auto-and alloactivated T (helper) lymphoblasts. Blood. 1984;63(6):1299–1302. | ||
Falini B, Pileri S, Pizzolo G, et al. CD30 (Ki-1) molecule: a new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood. 1995;85:1. | ||
Schwarting R, Gerdes J, Durkop H, Falini B, Pileri S, Stein H. BER-H2: a new anti-Ki-1 (CD30) monoclonal antibody directed at a formol-resistant epitope. Blood. 1989;74(5):1678–1689. | ||
Wiley SR, Goodwin RG, Smith CA. Reverse signaling via CD30 ligand. J Immunol. 1996;157(8):3635–3639. | ||
Schneix R, Linnartz C, Katouzi AA, et al. Development of new ricin A-chain immunotoxins with potent anti-tumor effects against human Hodgkin cells in vitro and disseminated Hodgkin tumors in SCID mice using high-affinity monoclonal antibodies directed against the CD30 antigen. Int J Cancer. 1995;63(2):238–244. | ||
Schnell R, Staak O, Borchmann P, et al. A phase I study with an anti-CD30 ricin A-chain immunotoxin (Ki-4. dgA) in patients with refractory CD30+ Hodgkin’s and non-Hodgkin’s lymphoma. Clin Cancer Res. 2002;8(6):1779–1786. | ||
Andrews RG, Takahashi M, Segal GM, Powell JS, Bernstein ID, Singer JW. The L4F3 antigen is expressed by unipotent and multipotent colony-forming cells but not by their precursors. Blood. 1986;68(5):1030–1035. | ||
Andrews RG, Torok-Storb B, Bernstein ID. Myeloid-associated differentiation antigens on stem cells and their progeny identified by monoclonal antibodies. Blood. 1983;62(1):124–132. | ||
Freeman SD, Kelm S, Barber EK, Crocker PR. Characterization of CD33 as a new member of the sialoadhesin family of cellular interaction molecules. Blood. 1995;85(8):2005–2012. | ||
Bernstein ID, Singer JW, Andrews RG, et al. Treatment of acute myeloid leukemia cells in vitro with a monoclonal antibody recognizing a myeloid differentiation antigen allows normal progenitor cells to be expressed. J Clin Invest. 1987;79(4):1153–1159. | ||
Dean A, Talpaz M, Kantarjian H, et al. Phase I clinical trial of the anti-CD33 immunotoxin HuM195/rgel in patients (pts) with advanced myeloid malignancies. ASCO Meet Abstr. 2010;28(15_suppl):6549. | ||
Choudhary S, Mathew M, Verma RS. Therapeutic potential of anticancer immunotoxins. Drug Discov Today. 2011;16(11–12):495–503. | ||
Borthakur G, Rosenblum MG, Talpaz M, et al. Phase 1 study of an anti-CD33 immunotoxin, humanized monoclonal antibody M195 conjugated to recombinant gelonin (HUM-195/rGEL), in patients with advanced myeloid malignancies. Haematologica. 2013;98(2):217–221. | ||
van Agthoven A, Terhorst C, Reinherz E, Schlossman S. Characterization of T cell surface glycoproteins T1 and T3 present on all human peripheral T lymphocytes and functionally mature thymocytes. Eur J Immunol. 1981;11(1):18–21. | ||
Borst J, Prendiville MA, Terhorst C. Complexity of the human T lymphocyte-specific cell surface antigen T3. J Immunol. 1982;128(4):1560–1565. | ||
Borst J, Alexander S, Elder J, Terhorst C. The T3 complex on human T lymphocytes involves four structurally distinct glycoproteins. J Biol Chem. 1983;258(8):5135–5141. | ||
Frankel A, Zuckero S, Mankin A, et al. Anti-CD3 recombinant diphtheria immunotoxin therapy of cutaneous T cell lymphoma. Curr Drug Targets. 2009;10(2):104–109. | ||
Madhumathi J, Devilakshmi S, Sridevi S, Verma RS. Immunotoxin therapy for hematologic malignancies: where are we heading? Drug Discov Today. 2016;21(2):325–332. | ||
Frankel A, Liu J-S, Rizzieri D, Hogge D. Phase I clinical study of diphtheria toxin-interleukin 3 fusion protein in patients with acute myeloid leukemia and myelodysplasia. Leuk Lymphoma. 2008;49(3):543–553. | ||
Kreitman RJ, Pastan I. Recombinant toxins containing human granulocyte-macrophage colony-stimulating factor and either Pseudomonas exotoxin or diphtheria toxin kill gastrointestinal cancer and leukemia cells. Blood. 1997;90(1):252–259. | ||
Frankel AE, Powell BL, Hall PD, Case LD, Kreitman RJ. Phase I trial of a novel diphtheria toxin/granulocyte macrophage colony-stimulating factor fusion protein (DT388GMCSF) for refractory or relapsed acute myeloid leukemia. Clin Cancer Res. 2002;8(5):1004–1013. | ||
Chang K, Pai LH, Pass H, et al. Monoclonal antibody K1 reacts with epithelial mesothelioma but not with lung adenocarcinoma. Am J Surg Pathol. 1992;16(3):259–268. | ||
Hassan R, Laszik ZG, Lerner M, Raffeld M, Postier R, Brackett D. Mesothelin is overexpressed in pancreaticobiliary adenocarcinomas but not in normal pancreas and chronic pancreatitis. Am J Clin Pathol. 2005;124(6):838–845. | ||
Argani P, Iacobuzio-Donahue C, Ryu B, et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas identification of a new pancreatic cancer marker by Serial Analysis of Gene Expression (SAGE). Clin Cancer Res. 2001;7(12):3862–3868. | ||
Ordóñez NG. Application of mesothelin immunostaining in tumor diagnosis. Am J Surg Pathol. 2003;27(11):1418–1428. | ||
Hassan R, Kreitman RJ, Pastan I, Willingham MC. Localization of mesothelin in epithelial ovarian cancer. Appl Immunohistochem Mol Morphol. 2005;13(3):243–247. | ||
Tchou J, Wang L-C, Selven B, et al. Mesothelin, a novel immunotherapy target for triple negative breast cancer. Breast Cancer Res Treat. 2012;133(2):799–804. | ||
Baba K, Ishigami S, Arigami T, et al. Mesothelin expression correlates with prolonged patient survival in gastric cancer. J Surg Oncol. 2012;105(2):195–199. | ||
Einama T, Homma S, Kamachi H, et al. Luminal membrane expression of mesothelin is a prominent poor prognostic factor for gastric cancer. Br J Cancer. 2012;107(1):137–142. | ||
Chang K, Pastan I, Willngham MC. Frequent expression of the tumor antigen cak1 in squamous-cell carcinomas. Int J Cancer. 1992;51(4):548–554. | ||
Ho M, Bera TK, Willingham MC, et al. Mesothelin expression in human lung cancer. Clin Cancer Res. 2007;13(5):1571–1575. | ||
Bera TK, Pastan I. Mesothelin is not required for normal mouse development or reproduction. Mol Cell Biol. 2000;20(8):2902–2906. | ||
Hassan R, Bullock S, Premkumar A, et al. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res. 2007;13(17):5144–5149. | ||
Kreitman RJ, Hassan R, FitzGerald DJ, Pastan I. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin Cancer Res. 2009;15(16):5274–5279. | ||
Hassan R, Sharon E, Thomas A, et al. Phase 1 study of the antimesothelin immunotoxin SS1P in combination with pemetrexed and cisplatin for front-line therapy of pleural mesothelioma and correlation of tumor response with serum mesothelin, megakaryocyte potentiating factor, and cancer antigen 125. Cancer. 2014;120(21):3311–3319. | ||
Vogelzang NJ, Rusthoven JJ, Symanowski J, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol. 2003;21(14):2636–2644. | ||
Hassan R, Miller AC, Sharon E, et al. Major cancer regressions in mesothelioma after treatment with an anti-mesothelin immunotoxin and immune suppression. Sci Transl Med. 2013;5(208):208ra147. | ||
Pai LH, Bookman MA, Ozols RF, et al. Clinical evaluation of intraperitoneal Pseudomonas exotoxin immunoconjugate OVB3-PE in patients with ovarian cancer. J Clin Oncol. 1991;9(12):2095–2103. | ||
Powell DJ, Felipe-Silva A, Merino MJ, et al. Administration of a CD25-directed immunotoxin, LMB-2, to patients with metastatic melanoma induces a selective partial reduction in regulatory T cells in vivo. J Immunol. 2007;179(7):4919–4928. | ||
Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19(13):3159–3167. | ||
Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–137. | ||
Slamon D, Godolphin W, Jones L, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244(4905):707–712. | ||
Schneider PM, Hung M-C, Chiocca SM, et al. Differential expression of the c-erbB-2 gene in human small cell and non-small cell lung cancer. Cancer Res. 1989;49(18):4968–4971. | ||
Park J-B, Rhim JS, Park S-C, Kimm S-W, Kraus MH. Amplification, overexpression, and rearrangement of the erbB-2 protooncogene in primary human stomach carcinomas. Cancer Res. 1989;49(23):6605–6609. | ||
Press MF, Cordon-Cardo C, Slamon DJ. Expression of the HER-2/neu proto-oncogene in normal human adult and fetal tissues. Oncogene. 1990;5(7):953–962. | ||
Pai-Scherf LH, Villa J, Pearson D, et al. Hepatotoxicity in cancer patients receiving erb-38, a recombinant immunotoxin that targets the erbB2 receptor. Clin Cancer Res. 1999;5(9):2311–2315. | ||
Wels W, Harwerth I-M, Mueller M, Groner B, Hynes NE. Selective inhibition of tumor cell growth by a recombinant single-chain antibody-toxin specific for the erbB-2 receptor. Cancer Res. 1992;52(22):6310–6317. | ||
Spyridonidis A, Schmidt M, Bernhardt W, et al. Purging of mammary carcinoma cells during ex vivo culture of CD34+ hematopoietic progenitor cells with recombinant immunotoxins. Blood. 1998;91(5):1820–1827. | ||
Schmidt M, McWatters A, White RA, et al. Synergistic Interaction between an anti-p185HER-2 Pseudomonas exotoxin fusion protein [scFv(FRP5)–ETA] and ionizing radiation for inhibiting growth of ovarian cancer cells that overexpress HER-2. Gynecol Oncol. 2001;80(2):145–155. | ||
Wang L, Liu B, Schmidt M, Lu Y, Wels W, Fan Z. Antitumor effect of an HER2-specific antibody–toxin fusion protein on human prostate cancer cells. Prostate. 2001;47(1):21–28. | ||
Wels W, Beerli R, Hellmann P, et al. EGF receptor and p185erbB-2-specific single-chain antibody toxins differ in their cell-killing activity on tumor cells expressing both receptor proteins. Int J Cancer. 1995;60(1):137–144. | ||
Azemar M, Schmidt M, Arlt F, et al. Recombinant antibody toxins specific for ErbB2 and EGF receptor inhibit the in vitro growth of human head and neck cancer cells and cause rapid tumor regression in vivo. Int J Cancer. 2000;86(2):269–275. | ||
Azemar M, Djahansouzi S, Jäger E, et al. Regression of cutaneous tumor lesions in patients intratumorally injected with a recombinant single-chain antibody-toxin targeted to ErbB2/HER2. Breast Cancer Res Treat. 2003;82(3):155–164. | ||
Hellström I, Garrigues HJ, Garrigues U, Hellström KE. Highly tumor-reactive, internalizing, mouse monoclonal antibodies to Ley-related cell surface antigens. Cancer Res. 1990;50(7):2183–2190. | ||
Pai LH, Wittes R, Setser A, Willingham MC, Pastan I. Treatment of advanced solid tumors with immunotoxin LMB–1: an antibody linked to Pseudomonas exotoxin. Nat Med. 1996;2(3):350–353. | ||
Friedman PN, McAndrew SJ, Gawlak SL, et al. BR96 sFv-PE40, a potent single-chain immunotoxin that selectively kills carcinoma cells. Cancer Res. 1993;53(2):334–339. | ||
Friedman PN, Chace DF, Trail PA, Siegall CB. Antitumor activity of the single-chain immunotoxin BR96 sFv-PE40 against established breast and lung tumor xenografts. J Immunol. 1993;150(7):3054–3061. | ||
Siegall CB. Targeted therapy of carcinomas using BR96 sFv-PE40, a single-chain immunotoxin that binds to the Leyantigen. Semin Cancer Biol. 1995;6(5):289–295. | ||
Posey JA, Khazaeli MB, Bookman MA, et al. A phase I trial of the single-chain immunotoxin SGN-10 (BR96 sFv-PE40) in patients with advanced solid tumors. Clin Cancer Res. 2002;8(10):3092–3099. | ||
Puri RK, Leland P, Kreitman RJ, Pastan I. Human neurological cancer cells express interleukin-4 (IL-4) receptors which are targets for the toxic effects of IL4-Pseudomonas exotoxin chimeric protein. Int J Cancer. 1994;58(4):574–581. | ||
Liu H, Prayson RA, Estes ML, et al. In vivo eepression of the interleukin 4 receptor alpha by astrocytes in epilepsy cerebral cortex. Cytokine. 2000;12(11):1656–1661. | ||
Joshi BH, Plautz GE, Puri RK. Interleukin-13 receptor α chain: a novel tumor-associated transmembrane protein in primary explants of human malignant gliomas. Cancer Res. 2000;60(5):1168–1172. | ||
Puri RK, Hoon DS, Leland P, et al. Preclinical development of a recombinant toxin containing circularly permuted interleukin 4 and truncated Pseudomonas exotoxin for therapy of malignant astrocytoma. Cancer Res. 1996;56(24):5631–5637. | ||
Husain SR, Behari N, Kreitman RJ, Pastan I, Puri RK. Complete regression of established human glioblastoma tumor xenograft by interleukin-4 toxin therapy. Cancer Res. 1998;58(16):3649–3653. | ||
Rand RW, Kreitman RJ, Patronas N, Varricchio F, Pastan I, Puri RK. Intratumoral administration of recombinant circularly permuted interleukin-4-Pseudomonas exotoxin in patients with high-grade glioma. Clin Cancer Res. 2000;6(6):2157–2165. | ||
Weber F, Asher A, Bucholz R, et al. Safety, tolerability, and tumor response of IL4-Pseudomonas exotoxin (NBI-3001) in patients with recurrent malignant glioma. J Neurooncol. 2003;64(1–2):125–137. | ||
Balzar M, Winter MJ, de Boer CJ, Litvinov SV. The biology of the 17–1A antigen (Ep-CAM). J Mol Med. 1999;77(10):699–712. | ||
Litvinov SV, Balzar M, Winter MJ, et al. Epithelial cell adhesion molecule (Ep-CAM) modulates cell–cell interactions mediated by classic cadherins. J Cell Biol. 1997;139(5):1337–1348. | ||
Zorzos J, Zizi A, Bakiras A, et al. Expression of a cell surface antigen recognized by the monoclonal antibody AUA1 in bladder carcinoma: an immunohistochemical study. Eur Urol. 1994;28(3):251–254. | ||
Brunner A, Prelog M, Verdorfer I, Tzankov A, Mikuz G, Ensinger C. EpCAM is predominantly expressed in high grade and advanced stage urothelial carcinoma of the bladder. J Clin Pathol. 2008;61(3):307–310. | ||
Paolo CD, Willuda J, Kubetzko S, et al. A recombinant immunotoxin derived from a humanized epithelial cell adhesion molecule-specific single-chain antibody fragment has potent and selective antitumor activity. Clin Cancer Res. 2003;9(7):2837–2848. | ||
Kowalski M, Entwistle J, Cizeau J, et al. A phase I study of an intravesically administered immunotoxin targeting EpCAM for the treatment of nonmuscle-invasive bladder cancer in BCG-refractory and BCG-intolerant patients. Drug Des Devel Ther. 2010;4:313–320. | ||
Alewine C, Hassan R, Pastan I. Advances in anticancer immunotoxin therapy. Oncologist. 2015;20(2):176–185. | ||
Younes A, Bartlett NL, Leonard JP, et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med. 2010;363(19):1812–1821. | ||
Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367(19):1783–1791. | ||
Kreitman RJ, Hansen HJ, Jones AL, FitzGerald DJP, Goldenberg DM, Pastan I. Pseudomonas exotoxin-based immunotoxins containing the antibody LL2 or LL2-Fab′ induce regression of subcutaneous human B-cell lymphoma in mice. Cancer Res. 1993;53(4):819–825. | ||
Kuan CT, Pai LH, Pastan I. Immunotoxins containing Pseudomonas exotoxin that target LeY damage human endothelial cells in an antibody-specific mode: relevance to vascular leak syndrome. Clin Cancer Res. 1995;1(12):1589–1594. | ||
Baluna R, Rizo J, Gordon BE, Ghetie V, Vitetta ES. Evidence for a structural motif in toxins and interleukin-2 that may be responsible for binding to endothelial cells and initiating vascular leak syndrome. Proc Natl Acad Sci U S A. 1999;96(7):3957–3962. | ||
Smallshaw JE, Ghetie V, Rizo J, et al. Genetic engineering of an immunotoxin to eliminate pulmonary vascular leak in mice. Nat Biotechnol. 2003;21(4):387–391. | ||
Wang H, Song S, Kou G, et al. Treatment of hepatocellular carcinoma in a mouse xenograft model with an immunotoxin which is engineered to eliminate vascular leak syndrome. Cancer Immunol Immunother. 2007;56(11):1775–1783. | ||
Weldon JE, Xiang L, Zhang J, et al. A recombinant immunotoxin against the tumor-associated antigen mesothelin reengineered for high activity, low off-target toxicity, and reduced antigenicity. Mol Cancer Ther. 2013;12(1):48–57. | ||
Schindler J, Sausville E, Messmann R, Vitetta ES. The toxicity of deglycosylated ricin A chain-containing immunotoxins in patients with non-Hodgkin’s lymphoma is exacerbated by prior radiotherapy: a retrospective analysis of patients in five clinical trials. Clin Cancer Res. 2001;7:255–258. | ||
Siegall CB, Liggitt D, Chace D, Tepper MA, Fell PH. Prevention of immunotoxin-mediated vascular leak syndrome in rats with retention of antitumor activity. Proc Natl Acad Sci U S A. 1994;91(20):9514–9518. | ||
Büttel IC, Chamberlain P, Chowers Y, et al. Taking immunogenicity assessment of therapeutic proteins to the next level. Biologicals. 2011;39(2):100–109. | ||
Lee JW, Kelley M, King LE, et al. Bioanalytical approaches to quantify “Total” and “Free” therapeutic antibodies and their targets: technical challenges and PK/PD applications over the course of drug development. AAPS J. 2011;13(1):99–110. | ||
Bera TK, Onda M, Kreitman RJ, Pastan I. An improved recombinant Fab-immunotoxin targeting CD22 expressing malignancies. Leuk Res. 2014;38(10):1224–1229. | ||
Alewine C, Xiang L, Yamori T, Niederfellner G, Bosslet K, Pastan I. Efficacy of RG7787, a next-generation mesothelin-targeted immunotoxin, against triple-negative breast and gastric cancers. Mol Cancer Ther. 2014;13(11):2653–2661. | ||
Benhar I, Padlan EA, Jung S-H, Lee B, Pastan I. Rapid humanization of the Fv of monoclonal antibody B3 by using framework exchange of the recombinant immunotoxin B3 (Fv)-PE38. Proc Natl Acad Sci U S A. 1994;91(25):12051–12055. | ||
Nagata S, Pastan I. Removal of B cell epitopes as a practical approach for reducing the immunogenicity of foreign protein-based therapeutics. Adv Drug Deliv Rev. 2009;61(11):977–985. | ||
Bird RE, Hardman KD, Jacobson JW, et al. Single-chain antigen-binding proteins. Science. 1988;242(4877):423–427. | ||
Ward ES, Güssow D, Griffiths AD, Jones PT, Winter G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature. 1989;341(6242):544–546. | ||
Reddy KR. Development and pharmacokinetics and pharmacodynamics of pegylated interferon alfa-2a (40 kD). Semin Liver Dis. New York, NY: Thieme Medical Publishers, Inc; 2004;24:33–38. | ||
Graham ML. Pegaspargase: a review of clinical studies. Adv Drug Deliv Rev. 2003;55(10):1293–1302. | ||
Mazor R, Onda M, Pastan I. Immunogenicity of therapeutic recombinant immunotoxins. Immunol Rev. 2016;270(1):152–164. | ||
Inada Y, Furukawa M, Sasaki H, et al. Biomedical and biotechnological applications of PEG-and PM-modified proteins. Trends Biotechnol. 1995;13(3):86–91. | ||
Liu W, Onda M, Lee B, et al. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc Natl Acad Sci U S A. 2012;109(29):11782–11787. | ||
Hollevoet K, Mason-Osann E, Liu X, Imhof-Jung S, Niederfellner G, Pastan I. In vitro and in vivo activity of the low-immunogenic antimesothelin immunotoxin RG7787 in pancreatic cancer. Mol Cancer Ther. 2014;13(8):2040–2049. | ||
Mazor R, Eberle JA, Hu X, et al. Recombinant immunotoxin for cancer treatment with low immunogenicity by identification and silencing of human T-cell epitopes. Proc Natl Acad Sci U S A. 2014;111(23):8571–8576. | ||
Mazor R, Zhang J, Xiang L, et al. Recombinant immunotoxin with T-cell epitope mutations that greatly reduce immunogenicity for treatment of mesothelin-expressing tumors. Mol Cancer Ther. 2015;14(12):2789–2796. | ||
Onda M, Beers R, Xiang L, Nagata S, Wang QC, Pastan I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc Natl Acad Sci U S A. 2008; 105(32):11311–11316. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.