")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Clinical significance of DAPK promoter hypermethylation in lung cancer: a meta-analysis
Authors Li Y, Zhu M, Zhang X, Cheng D, Ma X
Received 23 November 2014
Accepted for publication 6 January 2015
Published 24 March 2015 Volume 2015:9 Pages 1785—1796
DOI https://doi.org/10.2147/DDDT.S78012
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Shu-Feng Zhou
Ying Li, Min Zhu, Xiaoju Zhang, Dongjun Cheng, Xitao Ma
Department of Respiratory and Critical Care Medicine, Henan Provincial People’s Hospital, Zhengzhou, People’s Republic of China
Abstract: Death-associated protein kinase 1 (DAPK) is an important serine/threonine kinase involved in various cellular processes, including apoptosis, autophagy, and inflammation. DAPK expression and activity are deregulated in a variety of diseases including cancer. Methylation of the DAPK gene is common in many types of cancer and can lead to loss of DAPK expression. However, the association between DAPK promoter hypermethylation and the clinicopathological significance of lung cancer remains unclear. In this study, we searched the MEDLINE, PubMed, Web of Science, and Scopus databases, systematically investigated the studies of DAPK promoter hypermethylation in lung cancer and quantified the association between DAPK promoter hypermethylation and its clinicopathological significance by meta-analysis. We observed that the frequency of DAPK methylation was significantly higher in lung cancer than in non-malignant lung tissues (odds ratio 6.02, 95% confidence interval 3.17–11.42, P<0.00001). The pooled results also showed the presence of a prognostic impact of DAPK gene methylation in lung cancer patients (odds ratio 3.63, 95% confidence interval 1.09–12.06, P=0.04). In addition, we summarized these findings and discuss tumor suppressor function, clinicopathological significance, and potential drug targeting of DAPK in lung cancer.
Keywords: lung, adenocarcinoma, squamous cell carcinoma, non-small cell lung cancer, death-associated protein kinase gene, DAPK, methylation, meta-analysis
Introduction
Lung cancer remains the leading cause of cancer-related death among men and women worldwide.1 It is estimated that 226,160 people in the USA have been diagnosed with lung cancer, and deaths for the year 2012 amounted to 160,340 people.2 In spite of advances in diagnosis and treatment, the prognosis is still poor. The unsatisfactory outcome of lung cancer may be attributed to late diagnosis of the disease and inefficient therapy for advanced tumors. With the discovery of screening markers specific for lung cancer, it may be possible to achieve earlier diagnosis and develop more informed choices for treatment.
The two major forms of lung cancer are non-small cell lung cancer (NSCLC), which accounts for about 85% of lung cancer, and small cell lung cancer, which accounts for about 15%. NSCLC can be subdivided into adenocarcinoma, squamous cell carcinoma, and large-cell carcinoma.3 Like genetic mutations in cancer, epigenetic modification is frequently observed in human cancer. The aberrant mechanisms include increased methylation of DNA, deacetylation of core histone proteins, and RNA interference.4 Hypermethylation in the promoter areas of tumor suppressor genes has been well established as a mechanism of transcriptional silencing in tumors, whereas tumors are also characterized by genome-wide hypomethylation.5 Death-associated protein kinase (DAPK) is a serine/threonine kinase involved in a variety of cellular processes, including apoptosis initiated by interferon (IFN)-γ, tumor necrosis factor (TNF)-α and Fas ligand,6,7 autophagy, and inflammation.8 DAPK function is deregulated in a variety of diseases including cancer, neuronal death, and stroke.8 As with many other tumor suppressor genes, the expression and function of DAPK is disrupted in various cancers. However, unlike genes such as p53,9 DAPK is commonly inactivated in cancer as a result of hypermethylation at the promoter of the DAPK gene rather than mutation;4,10 less frequently, loss of DAPK expression can also be due to homozygous deletion.11,12
Although methylation of the DAPK gene is found in various tumors, there is a big difference in frequency of methylation. It is logical to assume dysfunction of DAPK may be more crucial for the tumorigenesis of lung cancer because of the high frequency of methylation (on average 40.5%13–24) and prognostic effects.18 The aim of this study was to review the available publications to summarize the data by meta-analysis and characterize the clinical significance of DAPK gene promoter methylation in tumorigenesis of the lung.
Methods
Search strategy
MEDLINE, PubMed, Web of Science, Scopus, and Embase were searched in October 2014 using the search terms: “death-associated protein kinase”, “DAPK”, “methylation”, “lung cancer”, “adenocarcinoma”, and “squamous carcinoma”. Research papers identified through the search approach as described above were screened by titles first, then by the abstracts of the publications. After exclusion of non-relevant publications and identification of duplicates from the different databases, the remaining papers were evaluated in the full-text version for inclusion and exclusion criteria and for relevant articles in the reference lists. All clinical studies except case reports were chosen, for instance, randomized controlled trials, cohort studies, case-control studies, and case series. The language of publication was restricted to English and Chinese. All searched data were retrieved. Authors’ bibliographies and references of selected studies were also searched for other relevant studies. The most complete study was chosen to avoid duplication if the same patient populations were reported in multiple publications.
Selection criteria
We collected all eligible articles about the relationship between DAPK methylation and/or expression and the clinicopathological features and clinical outcomes in lung cancer patients in this meta-analysis. Studies meeting the following inclusion criteria were included: DAPK methylation and/or expression evaluated in lung tissues and other resources such as sputum, bronchial brush washing, and blood; research that revealed the relationship between DAPK methylation and/or expression and the clinicopathological parameters and prognosis of lung cancer; DAPK methylation and/or expression examined by methylation-specific polymerase chain reaction; articles published as full papers in English and Chinese; articles which provided sufficient information to estimate the hazard ratio for overall survival and 95% confidence interval (CI) and probabilities for overall survival where applicable. Exclusion criteria included the following: letters, reviews, case reports, conference abstracts, editorials, expert opinion, and non-English, non-Chinese language papers; articles having no information on overall survival or those for which the hazard ratio for overall survival could not be calculated from the given information; and all publications regarding in vitro/ex vivo studies, cell lines, and human xenografts.
Data extraction
The data extraction followed the procedure as described by the published literature.25 Two investigators independently extracted data from eligible studies. Disagreements were resolved by discussion and consensus. Two investigators reviewed all of the articles that met the inclusion and exclusion criteria. The following information was recorded for each study: the first author name, year of publication, sample source, number of cases, clinicopathological parameters, stage, DAPK methylation and/or expression, and patient survival. The study characteristics and clinical responses were summarized and turned into table format. Heterogeneity of investigation was evaluated to determine whether the data from the various studies could be analyzed in a meta-analysis.
Statistical analysis
The analysis was conducted using Stata version 12.0 (Stata Corp, College Station, TX, USA) and Review Manager version 5.2 (Cochrane Collaboration, Oxford, UK). Comparisons of dichotomous measures were done by pooled estimates of odds ratios (ORs) and their 95% CIs. A P-value <0.05 was considered to be statistically significant. Heterogeneity was examined by a Chi-square test with significance set at P<0.10; the total variation among the studies was estimated by I2. We used I2 statistic to assess heterogeneity. The I2 value is an estimate of the amount of variance due to between-study heterogeneity rather than chance (Cochran statistics). Substantial heterogeneity exists when I2 exceeds 50%. If there was heterogeneity among studies, we used a random effect model to pool the ORs; otherwise, a fixed effect model was selected.
The database search generated 47 articles from MEDLINE, PubMed, Web of Science, Scopus, and Embase. After initial screening of all titles, abstracts, and eligibility, 18 full-text studies were selected for more detailed assessment. The search of the article references did not produce additional publications. Eventually, 18 publications met the inclusion criteria for qualitative study and meta-analysis. The article search and study selection is shown in Figure 1.
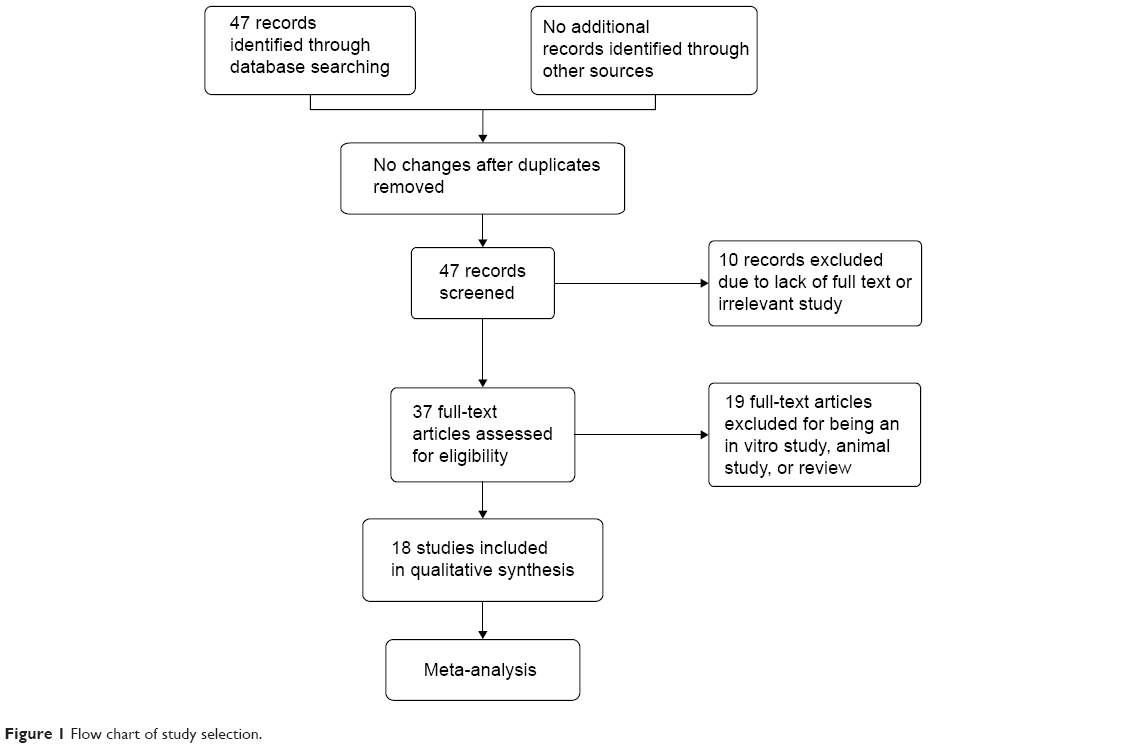 | Figure 1 Flow chart of study selection. |
Results
Identification of relevant studies
Forty-seven publications were identified by the search method as described above. Twenty-eight of those were excluded for being laboratory studies, non-original articles (review), lacking of matched controls, or studies irrelevant to the current analysis. Eventually, there were 18 studies included in final meta-analysis (Figure 1).
Study characteristics
Eighteen studies published from 2000 to 2014 were eligible for meta-analysis.13–23,26–32 One paper31 was written in Chinese with an abstract in English available on the PubMed website. A total of 1,270 lung cancer patients were enrolled, and 694 non-malignant lung tissues as controls were included, from the People’s Republic of China, Japan, Korea, Poland, Germany, and the USA. Their basic characteristics are summarized in Table 1. As described above, the database search generated 47 articles from MEDLINE, PubMed, Web of Science, Scopus, and Embase. The other 29 publications were excluded due to lack of full text, for being vitro/ex vivo studies, using cell lines or human xenografts, or being irrelevant studies.6,8,25,32–57
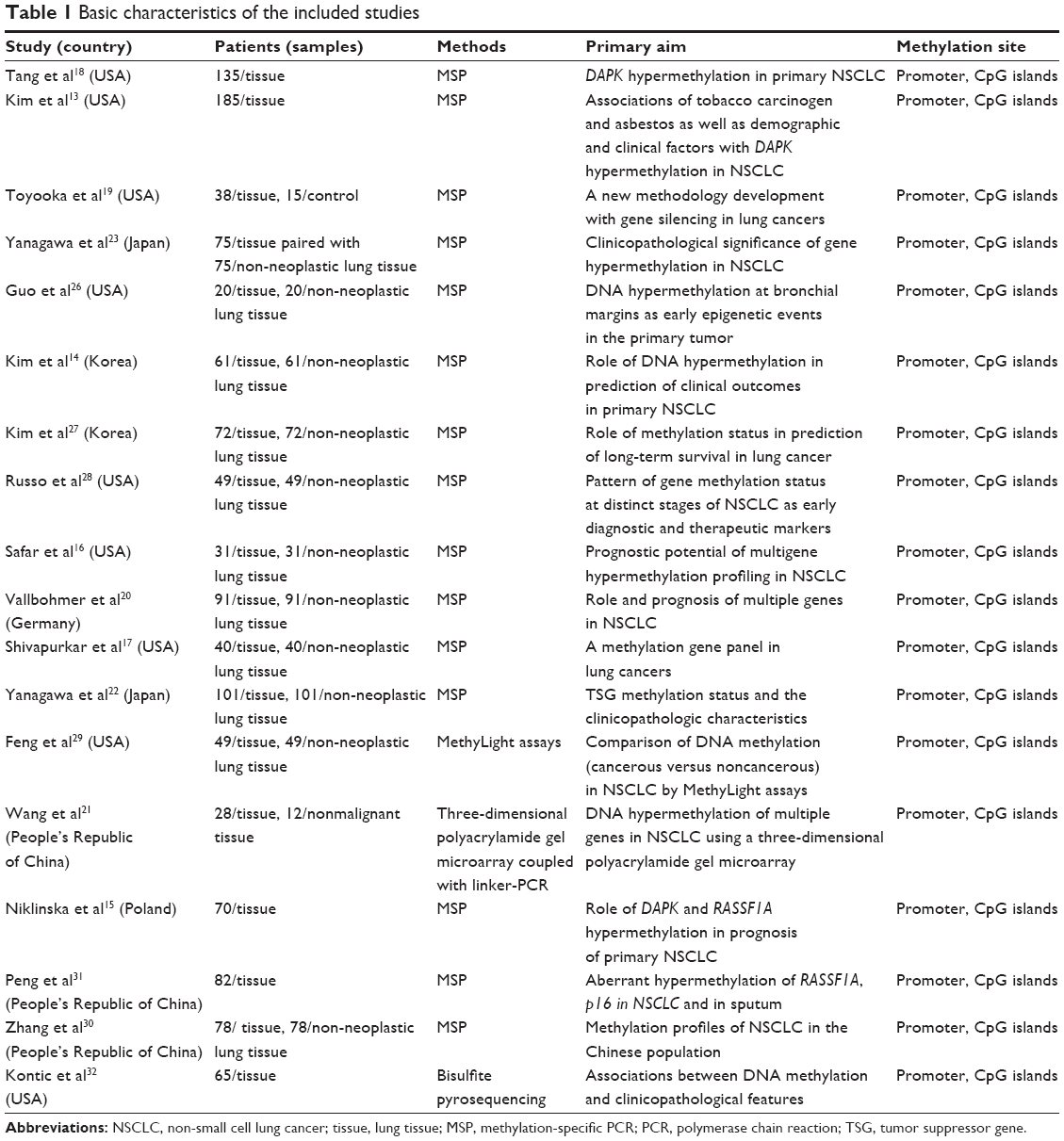 | Table 1 Basic characteristics of the included studies |
DAPK methylation and clinicopathological features
Inactivation of DAPK through methylation in lung cancer
In Figure 2, the first column has the study name; the second column is the proportion of DAPK methylation in lung cancers, and the third column is the proportion of DAPK methylation in normal controls. The weight in the fourth column is proportional to the inverse of the variance of the study (high variance, associated with a small study, meaning less weight is given to that study, and vice versa). The OR is shown numerically in the fifth column. In this case, the CI of the summary OR does not include 1.0 (it is 3.17–11.42), suggesting that the association is statistically significant. In the light of the “OR diagram” in the final column, the vertical line indicates an OR of 1.0. If DAPK methylation occurred more frequently in cancer tissue than in normal tissue, that would make the OR less than 1.0. The horizontal dots and bars represent the relative risk and 95% CI for each study. In summary, we observed that the frequency of DAPK methylation was significantly higher in lung cancer than in non-malignant lung tissues used as controls. The pooled OR from 13 studies including 733 lung cancer patients and 694 non-malignant lung tissues as controls shown in Figure 2 (OR 6.02, 95% CI 3.17–11.42, P<0.00001) indicates that inactivation of DAPK through methylation plays an important role in the pathogenesis of lung cancer. The overall methylation frequency of DAPK in lung cancer tissues was higher than that in normal controls, suggesting a potential role of DAPK methylation analysis in diagnosing lung cancer.
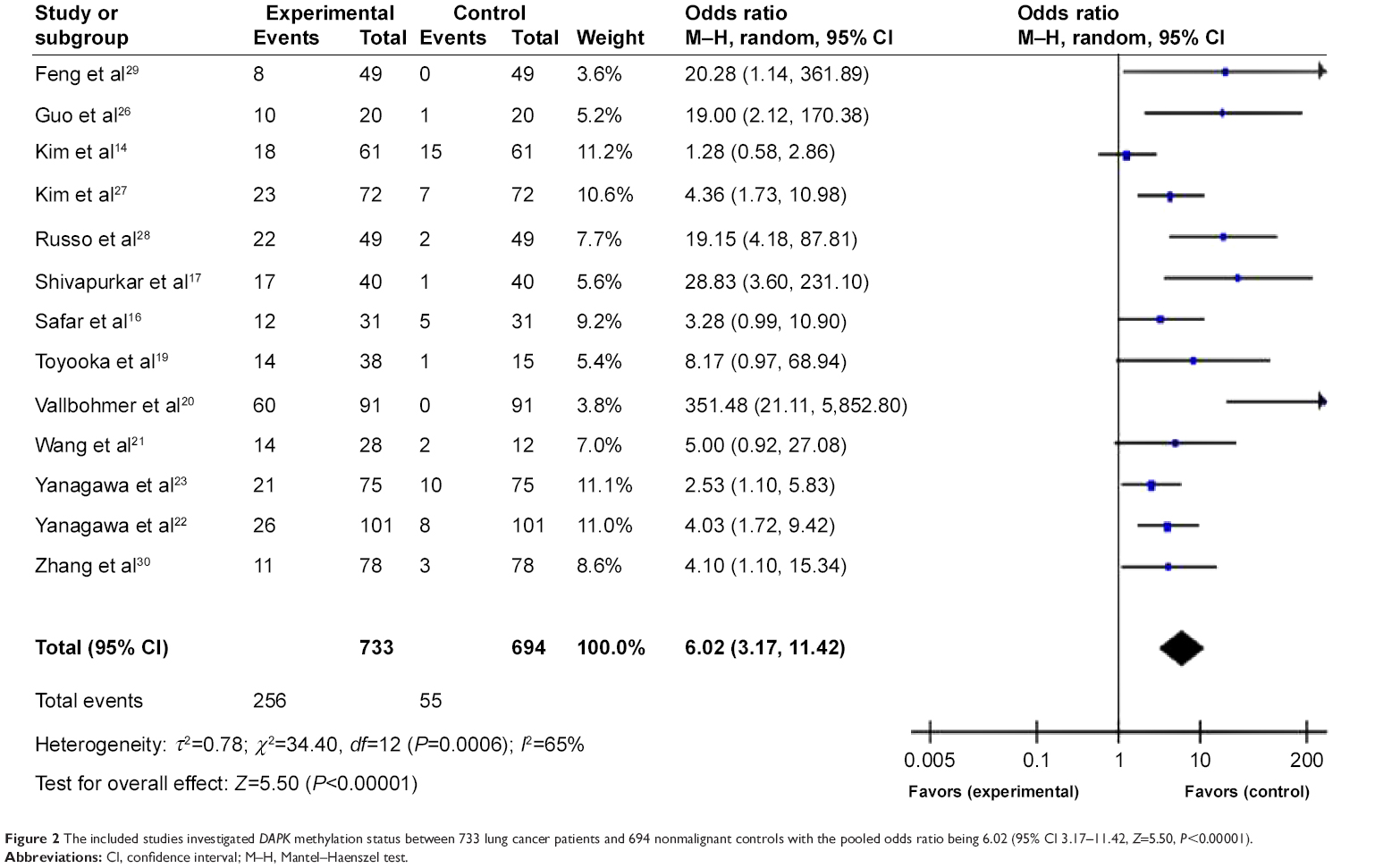 | Figure 2 The included studies investigated DAPK methylation status between 733 lung cancer patients and 694 nonmalignant controls with the pooled odds ratio being 6.02 (95% CI 3.17–11.42, Z=5.50, P<0.00001). |
Methylation of DAPK, histologic types, and disease stage
We further determined the possible associations between DAPK hypermethylation and clinicopathological features. The two major forms of lung cancer are NSCLC, which accounts for about 85% of lung cancer, and small cell lung cancer, which accounts for about 15%. NSCLC can be subdivided into adenocarcinoma, squamous cell carcinoma, and large-cell carcinoma.3 To determine whether methylation of DAPK is associated with histological subtypes, we analyzed these relationships in adenocarcinoma and squamous cell carcinoma separately. When the data were stratified by histology, adenocarcinomas showed the same frequent methylation of DAPK in tumors with squamous carcinoma histology as shown in Figure 3 (OR 0.91, 95% CI 0.64–1.29) from ten studies including 401 lung cancer patients and 263 non-malignant lung tissues. The OR is shown numerically in the fifth column, and the CI of the summary of OR does include 1.0 (it is 0.64–1.29), suggesting that the association is not statistically significant. The meta-analysis was performed to determine whether methylation of the promoter region of the DAPK gene is independently associated with tumor stage. When the data were stratified by disease stage using the tumor node metastasis staging system (TNM) (TNM I + II versus TNM III + IV), methylation of DAPK did not increase with increasing stage as shown in Figure 4 (OR 0.95, 95% CI 0.53–1.73). The OR is shown numerically in the fifth column, and the CI of the summary of OR does include 1.0 (it is 0.53–1.73), suggesting that the association is not statistically significant.
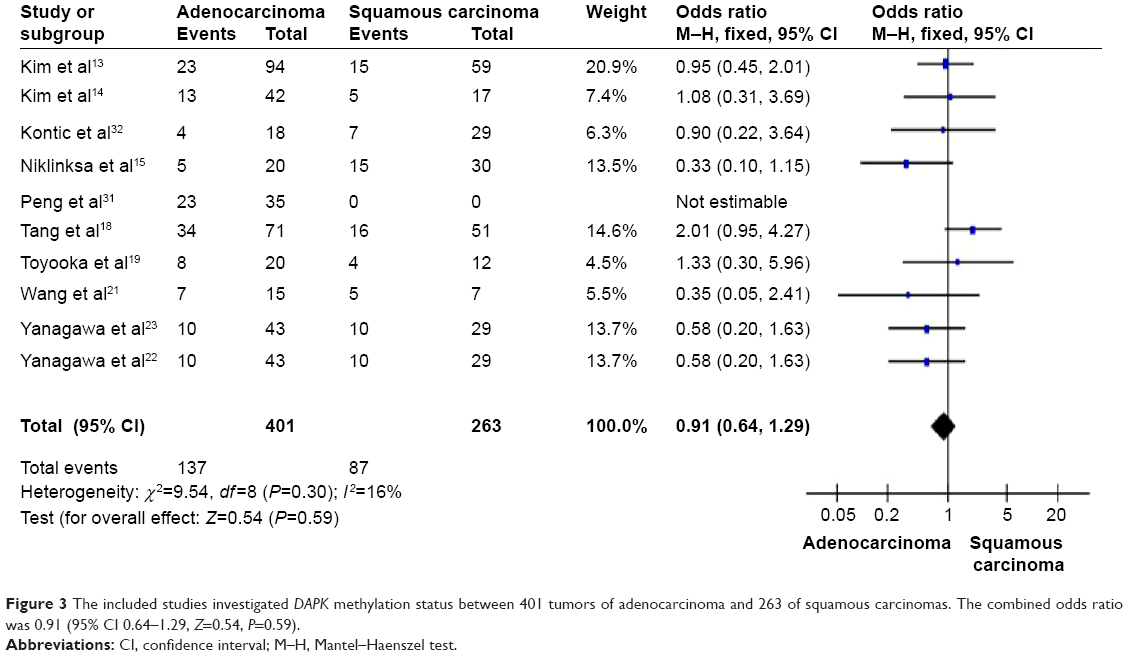 | Figure 3 The included studies investigated DAPK methylation status between 401 tumors of adenocarcinoma and 263 of squamous carcinomas. The combined odds ratio was 0.91 (95% CI 0.64–1.29, Z=0.54, P=0.59). |
 | Figure 4 Pooled results of methylation analysis of DAPK gene in different disease stages (TNM I + II versus TNM III + IV) in lung cancer. The pooled odds ratio is 0.95, 95% CI 0.53–1.73, Z=0.16, P=0.88). |
Prognostic value of DAPK gene methylation in lung cancer
We analyzed the relationship between methylation of the DAPK gene and patient survival. Three included studies14,15,18 estimated the relationship between overall survival in lung cancers and DAPK methylation. The pooled results (Figure 5) showed the presence of a prognostic impact of DAPK gene methylation in lung cancer patients (OR 3.63, 95% CI 1.09–12.06, Z=2.10, P=0.04). The hazard ratio is shown numerically in the fifth column, and the CI of the summary of hazard ratios does not include 1.0 (it is 1.09–12.06), suggesting that the association is statistically significant.
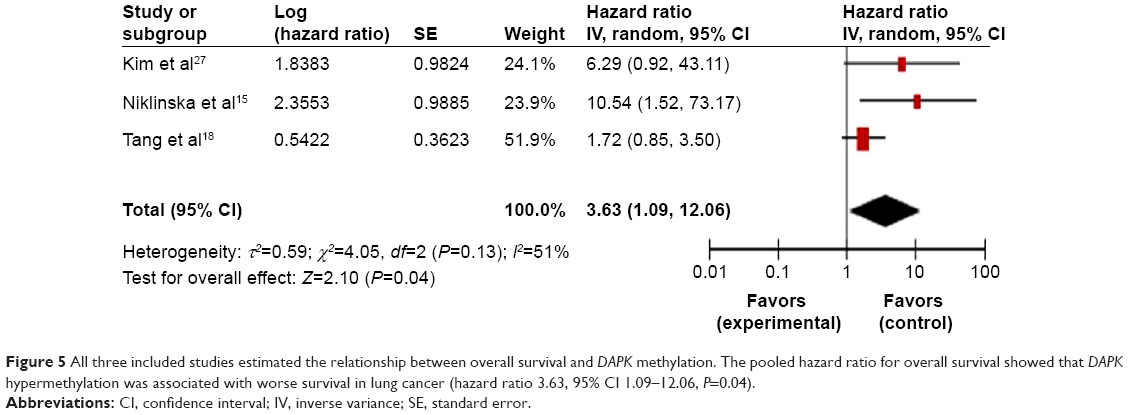 | Figure 5 All three included studies estimated the relationship between overall survival and DAPK methylation. The pooled hazard ratio for overall survival showed that DAPK hypermethylation was associated with worse survival in lung cancer (hazard ratio 3.63, 95% CI 1.09–12.06, P=0.04). |
Sensitivity analyses and publication bias
A sensitivity analysis, in which one study was removed at a time, was conducted to assess the stability of the results. The pooled ORs were not significantly changed, indicating the stability of our analyses. The funnel plots were largely symmetrical, suggesting there was no publication bias in the meta-analysis of DAPK gene methylation/expression and clinicopathological features (Figure 6).
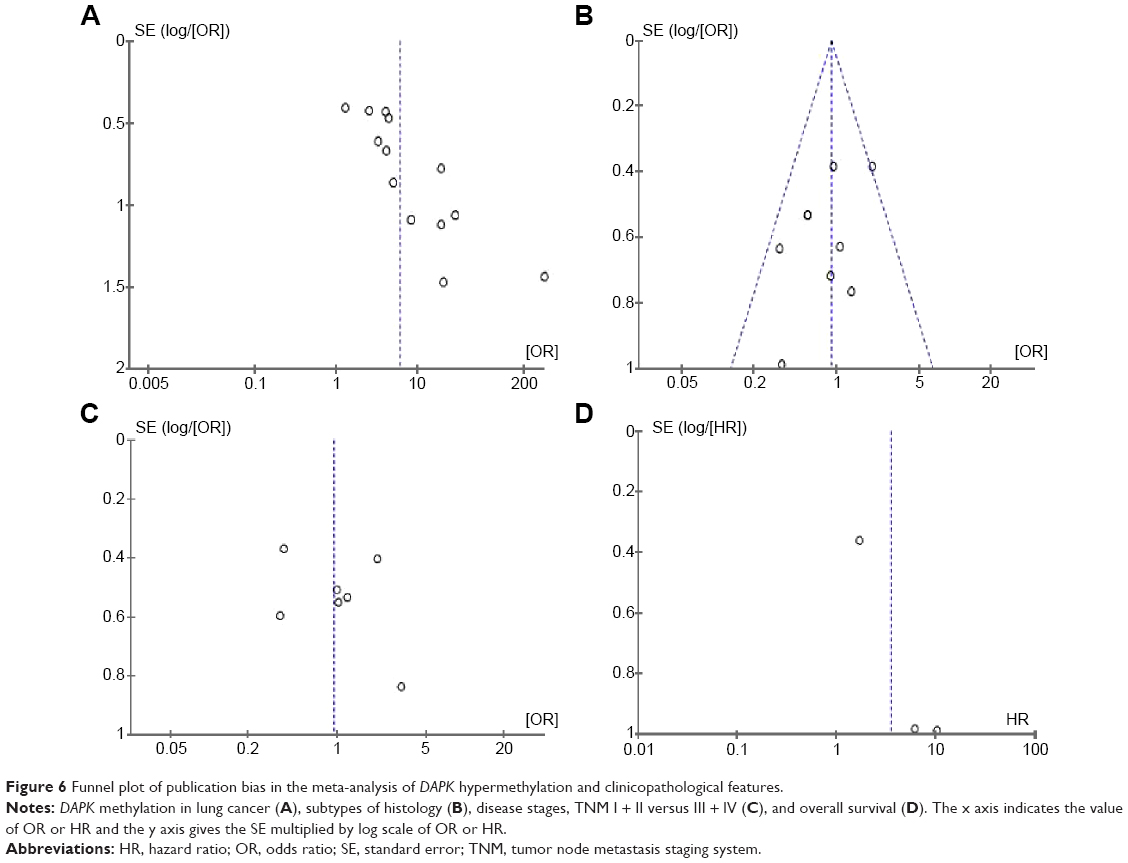 | Figure 6 Funnel plot of publication bias in the meta-analysis of DAPK hypermethylation and clinicopathological features. |
Discussion
DAPK is a Ca2+/calmodulin-regulated, 160 kDa serine/threonine, microfilament-bound kinase known to be activated by IFN-γ and TNF-α or Fas ligand-induced, and subsequently results in apoptosis.58–60 Aggressiveness of cancers has been associated with loss of DAPK expression61 mediated by methylation of the promoter region of the DAPK gene.10,62–64 From the present meta-analysis, we concluded that: DAPK gene inactivation through methylation plays an important role in the pathogenesis of lung cancer, and could be one of the determinants for its malignancy, as supported by higher DAPK methylation frequency in malignant lesions than in normal controls; overall survival tends to be shorter in lung cancer patients with epigenetic abnormalities of DAPK than in those with normal expression of DAPK gene; and no correlation exists between histological subtypes/disease stages (TNM I + II versus TNM III + IV) and hypermethylation status of the DAPK gene.
A meta-analysis is a more rigorous and methodologically complicated review than an overview or systematic review. There are two major reasons to do a meta-analysis; one is to quantitatively combine the results of previous studies to reach a summary estimate, and the other is to help guide further research. Based on the meta-analysis results, we suggest that DAPK hypermethylation should be regarded an early diagnostic marker for lung cancer and also a predictor for the prognosis of lung cancer. DAPK inhibitors, which are still in the early stages of evaluation, really need to be explored and developed.
Wild-type DAPK functions as an apoptosis inducer in a variety of cancers. However, some newly discovered splice variants of DAPK in cancer can antagonize the action of wild-type DAPK,65 which is similar to the situation occurring in p53 and p73 gene in cancer.66,67 In addition, in certain specific tissues or genetic environments, DAPK may be a survival promoter and promote tumorigenicity.68–71
Recent studies have revealed the mechanisms of DAPK-mediated cell death induced by TNF-α and IFN-γ,72 suggesting that DAPK can mediate the proapoptotic activity of TNF-α and IFN-γ via nuclear factor kappa B (NF-κB) signaling pathways. In the presence of DAPK, apoptosis induced by TNF-α or IFN-γ was additively increased, while the NF-κB activity induced by TNF-α or IFN-γ was inhibited. The activity of NF-κB was dependent on the level of DAPK, indicating the requirement of DAPK for activation of NF-κB.72
The mechanism for the role of DAPK has been largely attributed to promoter hypermethylation, which leads to gene silencing. However, recent studies indicate that DAPK expression can be detected in some types of cancer, but its function is still repressed, suggesting that regulation of DAPK function occurs in different ways. Homozygous deletions, allelic deletion, and point mutations have been found to attenuate DAPK expression.4 A positive correlation was also found between hyperphosphorylation of DAPK and activated Src kinase in colon cancer cell lines and primary tissue,73 suggesting a mechanism for inactivation of DAPK at the post-translational level in cancer cells.
The two major forms of lung cancer are NSCLC, which accounts for about 85% of lung cancers, and small cell lung cancer, which accounts for about 15%. NSCLC can be subdivided into adenocarcinoma, squamous cell carcinoma, and large-cell carcinoma.3 Our results are consistent with a previous publication reporting that the prevalence of DAPK gene hypermethylation was indistinguishable among the major histological subtypes of lung cancer.74
In addition to primary tumors, DAPK gene hypermethylation status has been detected in other sample sources. However, we were unable to undertake a meta-analysis due to the limited number of studies performed, so we summarize their data as follows. Two studies compared hypermethylation of DAPK in tumors, bronchial margins, and bronchoalveolar fluid.26,75,76 They found that there was significant concordance of the DAPK gene with regard to epigenetic changes between tumors, bronchial margin, and bronchial washings. Their data suggest that the frequencies of methylation of the DAPK gene found in bronchoalveolar fluid and in primary tumors were overall comparable. Their results can be interpreted as the bronchoalveolar fluid not reflecting the presence of tumor cells per se but as the bronchoalveolar fluid detecting a field effect present in the bronchial mucosa in close proximity to the tumor.
Peng et al31 determined the aberrant methylation status of DAPK along with RASSF1A and p16 gene promoter region in induced sputum from lung cancer patients, and found that joint detection for promoter hypermethylation of DAPK gene along with RASSF1A and p16 gene in induced sputum may be used as a simple and effective index of the diagnosis and prognosis of lung cancers, and can improve the positive rate. On the contrary, when DAPK, RASSF1A and CDH13 were observed to be significantly hypermethylated in lung tumors compared with matched normal tissue, however, these changes could not be detected in patients’ blood samples by Kontic et al32 suggesting these three genes are not suitable markers for the early detection of NSCLC, although they seem to play a role in the pathogenesis of NSCLC.
Given the central role of smoking in the development of lung cancer and the fact that DNA methylation is an early event in tumorigenesis,29 some biomarkers such as CCND2 and APC were investigated and demonstrated to be frequently hypermethylated in both cancerous and non-cancerous lung tissues from smokers with NSCLC, indicating that hypermethylation of these genes may be associated with chronic smoking status.29 However, no correlation was found between DAPK methylation and the smoking habits (smoking history) of individuals with NSCLC.76
DNA methylation and histone acetylation are the two most studied epigenetic changes in cancer. DAPK I inhibitors are still in the early stages of evaluation. DNA methyltransferase inhibitors, ie, azacitidine (5-azacytidine, 5AC), 5-aza-2′-deoxycytidine (decitabine) and pyrimidin-2-one β-ribofuranoside (zebularine) have been studied in the preclinical setting, and even in clinical trials.77,78 Under equivalent and clinically relevant treatment schemes, the three DNA methyltransferase inhibitors left a distinct gene expression profile in cells, indicating that these drugs are not identical in function. Differences in structure and cellular pharmacology between the three DNA methyltransferase inhibitors may account for the diversity observed. 5AC and decitabine have a nitrogen at position 5 of the pyrimidine ring instead of a carbon, but zebularine does not. 5AC is a ribonucleoside that is incorporated mainly into RNA, whereas decitabine and zebularine are incorporated only into DNA.78 Application of decitabine is limited by its high toxicity (myelosuppression and neurological toxicity) and instability in physiological solutions,79,80 while zebularine is less toxic and more stable, and can be taken orally.5,81 A Phase I study in advanced solid malignancies has found that a combination of erlotinib and 5AC was well tolerated, with promising clinical activity in lung, head and neck, and ovarian cancer. The recommended dose for Phase II study is erlotinib 150 mg daily and 5AC 75 mg/m2 daily on days 1–4 and 15–18 of a 28-day cycle.82 Another Phase I study involving lung, esophageal, or pleural cancer revealed that prolonged decitabine infusions can regulate gene expression in these primary thoracic malignancies.83 With respect to decitabine, Phase I, II, and III studies have been conducted. Chu et al84 observed that decitabine and valproic acid are an effective combination in reactivating silent genes by hypermethylation, as demonstrated by re-expression of fetal hemoglobin in patients with advanced stage IV NSCLC in a Phase I study; however, the continuous study was hampered by remarkably high neurological toxicity at a relatively low dosage. In another Phase I–II clinical trial, decitabine was reported to exert a remarkable chemotherapeutic action for NSCLC, but this action is delayed and prolonged, with the major toxicity being myelosuppression.85 In hematological malignancy, a randomized Phase II study demonstrated that addition of valproic acid to decitabine was not associated with an improved outcome in patients with myelodysplastic syndrome or elderly patients with acute myeloid leukemia.86 A post hoc analysis of a randomized Phase III study87 evaluated the impact of decitabine on transfusion dependence and survival in 485 elderly patients with newly diagnosed acute myeloid leukemia. More red blood cell and platelet transfusion-dependent patients at baseline became transfusion-independent with decitabine than did corresponding controls. In addition, patients who achieved transfusion independence with decitabine had increased treatment continuation, even in the absence of complete remission. Zebularine is now being tested in preclinical animal tumor or human cancer cell lines,88 but there is no phase study available yet.
DAPK was first identified in 1995,58 and DAPK gene methylation has been discovered in more than 30 types of cancers although the methylation rates vary.8 Re-expression of DAPK by demethylation in tumors may possibly bring clinical benefits as a potential drug target.8 Taken together, DAPK is an interesting therapeutic target in multiple human diseases including cancer. The development of specific compounds for activation of DAPK will be a promising strategy for targeting DAPK in the clinic.
Disclosure
The authors report no conflicts of interest in this work.
References
American Cancer Society, Cancer facts and figures 2013. Available from: http://www.cancer.org/research/cancerfactsfigures/cancerfactsfigures/cancer-facts-figures-2013. Accessed January 15, 2014. | ||
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. | ||
Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–1380. | ||
Michie AM, McCaig AM, Nakagawa R, Vukovic M. Death-associated protein kinase (DAPK) and signal transduction: regulation in cancer. FEBS J. 2010;277:74–80. | ||
Kim JS, Han J, Shim YM, Park J, Kim DH. Aberrant methylation of H-cadherin (CDH13) promoter is associated with tumor progression in primary nonsmall cell lung carcinoma. Cancer. 2005;104:1825–1833. | ||
Cohen O, Kimchi A. DAP-kinase: from functional gene cloning to establishment of its role in apoptosis and cancer. Cell Death Differ. 2001;8:6–15. | ||
Tang X, Wu W, Sun SY, Wistuba II, Hong WK, Mao L. Hypermethylation of the death-associated protein kinase promoter attenuates the sensitivity to TRAIL-induced apoptosis in human non-small cell lung cancer cells. Mol Cancer Res. 2004;2:685–691. | ||
Huang Y, Chen L, Guo L, Hupp TR, Lin Y. Evaluating DAPK as a therapeutic target. Apoptosis. 2014;19:371–386. | ||
Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. | ||
Katzenellenbogen RA, Baylin SB, Herman JG. Hypermethylation of the DAP-kinase CpG island is a common alteration in B-cell malignancies. Blood. 1999;93:4347–4353. | ||
Kawaguchi K, Oda Y, Saito T, et al. Death-associated protein kinase (DAP kinase) alteration in soft tissue leiomyosarcoma: promoter methylation or homozygous deletion is associated with a loss of DAP kinase expression. Hum Pathol. 2004;35:1266–1271. | ||
Simpson DJ, Clayton RN, Farrell WE. Preferential loss of death associated protein kinase expression in invasive pituitary tumours is associated with either CpG island methylation or homozygous deletion. Oncogene. 2002;21:1217–1224. | ||
Kim DH, Nelson HH, Wiencke JK, et al. Promoter methylation of DAP-kinase: association with advanced stage in non-small cell lung cancer. Oncogene. 2001;20:1765–1770. | ||
Kim YT, Lee SH, Sung SW, Kim JH. Can aberrant promoter hypermethylation of CpG islands predict the clinical outcome of non-small cell lung cancer after curative resection? Ann Thorac Surg. 2005;79:1180–1188. | ||
Niklinska W, Naumnik W, Sulewska A, Kozlowski M, Pankiewicz W, Milewski R. Prognostic significance of DAPK and RASSF1A promoter hypermethylation in non-small cell lung cancer (NSCLC). Folia Histochem Cytobiol. 2009;47:275–280. | ||
Safar AM, Spencer H 3rd, Su X, et al. Methylation profiling of archived non-small cell lung cancer: a promising prognostic system. Clin Cancer Res. 2005;11:4400–4405. | ||
Shivapurkar N, Stastny V, Suzuki M, et al. Application of a methylation gene panel by quantitative PCR for lung cancers. Cancer Lett. 2007; 247:56–71. | ||
Tang X, Khuri FR, Lee JJ, et al. Hypermethylation of the death-associated protein (DAP) kinase promoter and aggressiveness in stage I non-small-cell lung cancer. J Natl Cancer Inst. 2000;92:1511–1516. | ||
Toyooka S, Toyooka KO, Miyajima K, et al. Epigenetic down-regulation of death-associated protein kinase in lung cancers. Clin Cancer Res. 2003;9:3034–3041. | ||
Vallbohmer D, Brabender J, Yang D, et al. DNA methyltransferases messenger RNA expression and aberrant methylation of CpG islands in non-small-cell lung cancer: association and prognostic value. Clin Lung Cancer. 2006;8:39–44. | ||
Wang Y, Zhang D, Zheng W, Luo J, Bai Y, Lu Z. Multiple gene methylation of nonsmall cell lung cancers evaluated with 3-dimensional microarray. Cancer. 2008;112:1325–1336. | ||
Yanagawa N, Tamura G, Oizumi H, et al. Promoter hypermethylation of RASSF1A and RUNX3 genes as an independent prognostic prediction marker in surgically resected non-small cell lung cancers. Lung Cancer. 2007;58:131–138. | ||
Yanagawa N, Tamura G, Oizumi H, Takahashi N, Shimazaki Y, Motoyama T. Promoter hypermethylation of tumor suppressor and tumor-related genes in non-small cell lung cancers. Cancer Sci. 2003;94: 589–592. | ||
Zochbauer-Muller S, Fong KM, Virmani AK, Geradts J, Gazdar AF, Minna JD. Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer Res. 2001;61:249–255. | ||
Li J, Bi L, Lin Y, Lu Z, Hou G. Clinicopathological significance and potential drug target of p15INK4B in multiple myeloma. Drug Des Devel Ther. 2014;8:2129–2136. | ||
Guo M, House MG, Hooker C, et al. Promoter hypermethylation of resected bronchial margins: a field defect of changes? Clin Cancer Res. 2004;10:5131–5136. | ||
Kim YT, Park SJ, Lee SH, et al. Prognostic implication of aberrant promoter hypermethylation of CpG islands in adenocarcinoma of the lung. J Thorac Cardiovasc Surg. 2005;130:1378. | ||
Russo AL, Thiagalingam A, Pan H, et al. Differential DNA hypermethylation of critical genes mediates the stage-specific tobacco smoke-induced neoplastic progression of lung cancer. Clin Cancer Res. 2005;11: 2466–2470. | ||
Feng Q, Hawes SE, Stern JE, et al. DNA methylation in tumor and matched normal tissues from non-small cell lung cancer patients. Cancer Epidemiol Biomarkers Prev. 2008;17:645–654. | ||
Zhang Y, Wang R, Song H, et al. Methylation of multiple genes as a candidate biomarker in non-small cell lung cancer. Cancer Lett. 2011; 303:21–28. | ||
Peng Z, Shan C, Wang H. [Value of promoter methylation of RASSF1A, p16, and DAPK genes in induced sputum in diagnosing lung cancers]. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2010;35:247–253. Chinese. | ||
Kontic M, Stojsic J, Jovanovic D, et al. Aberrant promoter methylation of CDH13 and MGMT genes is associated with clinicopathologic characteristics of primary non-small-cell lung carcinoma. Clin Lung Cancer. 2012;13:297–303. | ||
Bradly DP, Gattuso P, Pool M, et al. CDKN2A (p16) promoter hypermethylation influences the outcome in young lung cancer patients. Diagn Mol Pathol. 2012;21:207–213. | ||
de Fraipont F, Levallet G, Creveuil C, et al. An apoptosis methylation prognostic signature for early lung cancer in the IFCT-0002 trial. Clin Cancer Res. 2012;18:2976–2986. | ||
Eisenberg-Lerner A, Kimchi A. DAPK silencing by DNA methylation conveys resistance to anti EGFR drugs in lung cancer cells. Cell Cycle. 2012;11:2051. | ||
Fujii M, Fujimoto N, Hiraki A, et al. Aberrant DNA methylation profile in pleural fluid for differential diagnosis of malignant pleural mesothelioma. Cancer Sci. 2012;103:510–514. | ||
Liu WB, Ao L, Zhou ZY, et al. CpG island hypermethylation of multiple tumor suppressor genes associated with loss of their protein expression during rat lung carcinogenesis induced by 3-methylcholanthrene and diethylnitrosamine. Biochem Biophys Res Commun. 2010;402:507–514. | ||
Millares L, Rosell A, Seto L, Sanz J, Andreo F, Monso E. Variability in the measurement of the methylation status of lung cancer-related genes in bronchial secretions. Oncol Rep. 2014;32:1435–1440. | ||
Ogawa T, Liggett TE, Melnikov AA, et al. Methylation of death-associated protein kinase is associated with cetuximab and erlotinib resistance. Cell Cycle. 2012;11:1656–1663. | ||
Scesnaite A, Jarmalaite S, Mutanen P, et al. Similar DNA methylation pattern in lung tumours from smokers and never-smokers with second-hand tobacco smoke exposure. Mutagenesis. 2012;27:423–429. | ||
Word B, Lyn-Cook LE Jr, Mwamba B, Wang H, Lyn-Cook B, Hammons G. Cigarette smoke condensate induces differential expression and promoter methylation profiles of critical genes involved in lung cancer in NL-20 lung cells in vitro: short-term and chronic exposure. Int J Toxicol. 2013;32:23–31. | ||
Buckingham L, Penfield Faber L, Kim A, et al. PTEN, RASSF1 and DAPK site-specific hypermethylation and outcome in surgically treated stage I and II nonsmall cell lung cancer patients. Int J Cancer. 2010;126:1630–1639. | ||
Han W, Wang T, Reilly AA, Keller SM, Spivack SD. Gene promoter methylation assayed in exhaled breath, with differences in smokers and lung cancer patients. Respir Res. 2009;10:86. | ||
Hirose Y, Kondo K, Takizawa H, et al. Aberrant methylation of tumour-related genes in thymic epithelial tumours. Lung Cancer. 2009;64: 155–159. | ||
Hoffmann AC, Kaifi JT, Vallbohmer D, et al. Lack of prognostic significance of serum DNA methylation of DAPK, MGMT, and GSTPI in patients with non-small cell lung cancer. J Surg Oncol. 2009;100:414–417. | ||
Jin Y, Xu H, Zhang C, et al. Combined effects of cigarette smoking, gene polymorphisms and methylations of tumor suppressor genes on non small cell lung cancer: a hospital-based case-control study in China. BMC Cancer. 2010;10:422. | ||
Katayama H, Hiraki A, Fujiwara K, et al. Aberrant promoter methylation profile in pleural fluid DNA and clinicopathological factors in patients with non-small cell lung cancer. Asian Pac J Cancer Prev. 2007;8:221–224. | ||
Licchesi JD, Westra WH, Hooker CM, Herman JG. Promoter hypermethylation of hallmark cancer genes in atypical adenomatous hyperplasia of the lung. Clin Cancer Res. 2008;14:2570–2578. | ||
Lin Q, Geng J, Ma K, et al. RASSF1A, APC, ESR1, ABCB1 and HOXC9, but not p16INK4A, DAPK1, PTEN and MT1G genes were frequently methylated in the stage I non-small cell lung cancer in China. J Cancer Res Clin Oncol. 2009;135:1675–1684. | ||
Liu Y, Lan Q, Shen M, et al. Aberrant gene promoter methylation in sputum from individuals exposed to smoky coal emissions. Anticancer Res. 2008;28:2061–2066. | ||
Pulling LC, Grimes MJ, Damiani LA, et al. Dual promoter regulation of death-associated protein kinase gene leads to differentially silenced transcripts by methylation in cancer. Carcinogenesis. 2009;30: 2023–2030. | ||
Zerilli F, Bonanno C, Shehi E, Amicarelli G, Adlerstein D, Makrigiorgos GM. Methylation-specific loop-mediated isothermal amplification for detecting hypermethylated DNA in simplex and multiplex formats. Clin Chem. 2010;56:1287–1296. | ||
Belinsky SA, Grimes MJ, Casas E, et al. Predicting gene promoter methylation in non-small-cell lung cancer by evaluating sputum and serum. Br J Cancer. 2007;96:1278–1283. | ||
Chai CY, Huang YC, Hung WC, Kang WY, Chen WT. Arsenic salts induced autophagic cell death and hypermethylation of DAPK promoter in SV-40 immortalized human uroepithelial cells. Toxicol Lett. 2007;173:48–56. | ||
Fischer JR, Ohnmacht U, Rieger N, et al. Prognostic significance of RASSF1A promoter methylation on survival of non-small cell lung cancer patients treated with gemcitabine. Lung Cancer. 2007;56:115–123. | ||
Liu Y, Gao W, Siegfried JM, Weissfeld JL, Luketich JD, Keohavong P. Promoter methylation of RASSF1A and DAPK and mutations of K-ras, p53, and EGFR in lung tumors from smokers and never-smokers. BMC Cancer. 2007;7:74. | ||
Tang M, Torres-Lanzas J, Lopez-Rios F, Esteller M, Sanchez-Cespedes M. Wnt signaling promoter hypermethylation distinguishes lung primary adenocarcinomas from colorectal metastasis to the lung. Int J Cancer. 2006;119:2603–2606. | ||
Deiss LP, Feinstein E, Berissi H, Cohen O, Kimchi A. Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev. 1995; 9:15–30. | ||
Cohen O, Feinstein E, Kimchi A. DAP-kinase is a Ca2+/calmodulin-dependent, cytoskeletal-associated protein kinase, with cell death-inducing functions that depend on its catalytic activity. EMBO J. 1997; 16:998–1008. | ||
Cohen O, Inbal B, Kissil JL, et al. DAP-kinase participates in TNF-alpha- and Fas-induced apoptosis and its function requires the death domain. J Cell Biol. 1999;146:141–148. | ||
Inbal B, Cohen O, Polak-Charcon S, et al. DAP kinase links the control of apoptosis to metastasis. Nature. 1997;390:180–184. | ||
Shiramizu B, Mick P. Epigenetic changes in the DAP-kinase CpG island in pediatric lymphoma. Med Pediatr Oncol. 2003;41:527–531. | ||
Ueki T, Toyota M, Sohn T, et al. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res. 2000;60:1835–1839. | ||
Sanchez-Cespedes M, Esteller M, Wu L, et al. Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer Res. 2000;60:892–895. | ||
Lin Y, Stevens C, Harrison B, Pathuri S, Amin E, Hupp TR. The alternative splice variant of DAPK-1, s-DAPK-1, induces proteasome-independent DAPK-1 destabilization. Mol Cell Biochem. 2009;328:101–107. | ||
Khoury MP, Bourdon JC. p53 isoforms: an intracellular microprocessor? Genes Cancer. 2011;2:453–465. | ||
Surget S, Khoury MP, Bourdon JC. Uncovering the role of p53 splice variants in human malignancy: a clinical perspective. Onco Targets Ther. 2013;7:57–68. | ||
Stevens C, Lin Y, Harrison B, et al. Peptide combinatorial libraries identify TSC2 as a death-associated protein kinase (DAPK) death domain-binding protein and reveal a stimulatory role for DAPK in mTORC1 signaling. J Biol Chem. 2009;284:334–344. | ||
Tanaka T, Bai T, Yukawa K. Death-associated protein kinase is essential for the survival of various types of uterine cancer cells. Int J Oncol. 2010;37:1017–1022. | ||
Tanaka T, Bai T, Yukawa K. Suppressed protein expression of the death-associated protein kinase enhances 5-fluorouracil-sensitivity but not etoposide-sensitivity in human endometrial adenocarcinoma cells. Oncol Rep. 2010;24:1401–1405. | ||
Tanaka T, Bai T, Yukawa K. Specific downregulation of death-associated protein kinase enhances Fas-mediated apoptosis in the human differentiated endometrial adenocarcinoma cell line, HHUA. Eur J Gynaecol Oncol. 2011;32:293–296. | ||
Yoo HJ, Byun HJ, Kim BR, Lee KH, Park SY, Rho SB. DAPk1 inhibits NF-kappaB activation through TNF-alpha and INF-gamma-induced apoptosis. Cell Signal. 2012;24:1471–1477. | ||
Wang WJ, Kuo JC, Ku W, et al. The tumor suppressor DAPK is reciprocally regulated by tyrosine kinase Src and phosphatase LAR. Mol Cell. 2007;27:701–716. | ||
Vaissiere T, Hung RJ, Zaridze D, et al. Quantitative analysis of DNA methylation profiles in lung cancer identifies aberrant DNA methylation of specific genes and its association with gender and cancer risk factors. Cancer Res. 2009;69:243–252. | ||
Ahrendt SA, Chow JT, Xu LH, et al. Molecular detection of tumor cells in bronchoalveolar lavage fluid from patients with early stage lung cancer. J Natl Cancer Inst. 1999;91:332–339. | ||
Soria JC, Rodriguez M, Liu DD, Lee JJ, Hong WK, Mao L. Aberrant promoter methylation of multiple genes in bronchial brush samples from former cigarette smokers. Cancer Res. 2002;62:351–355. | ||
Cang S, Lu Q, Ma Y, Liu D. Clinical advances in hypomethylating agents targeting epigenetic pathways. Curr Cancer Drug Targets. 2010; 10:539–545. | ||
Flotho C, Claus R, Batz C, et al. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia. 2009;23:1019–1028. | ||
Bender CM, Pao MM, Jones PA. Inhibition of DNA methylation by 5-aza-2′-deoxycytidine suppresses the growth of human tumor cell lines. Cancer Res. 1998;58:95–101. | ||
Momparler RL. Molecular, cellular and animal pharmacology of 5-aza-2′-deoxycytidine. Pharmacol Ther. 1985;30:287–299. | ||
Cheng JC, Matsen CB, Gonzales FA, et al. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J Natl Cancer Inst. 2003;95:399–409. | ||
Bauman J, Verschraegen C, Belinsky S, et al. A phase I study of 5-azacytidine and erlotinib in advanced solid tumor malignancies. Cancer Chemother Pharmacol. 2012;69:547–554. | ||
Schrump DS, Fischette MR, Nguyen DM, et al. Phase I study of decitabine-mediated gene expression in patients with cancers involving the lungs, esophagus, or pleura. Clin Cancer Res. 2006;12:5777–5785. | ||
Chu BF, Karpenko MJ, Liu Z, et al. Phase I study of 5-aza-2′-deoxycytidine in combination with valproic acid in non-small-cell lung cancer. Cancer Chemother Pharmacol. 2013;71:115–121. | ||
Momparler RL. Epigenetic therapy of non-small cell lung cancer using decitabine (5-aza-2′-deoxycytidine). Front Oncol. 2013;3:188. | ||
Issa JP, Garcia-Manero G, Huang X, et al. Results of phase 2 randomized study of low-dose decitabine with or without valproic acid in patients with myelodysplastic syndrome and acute myelogenous leukemia. Cancer. October 21, 2014. [Epub ahead of print]. | ||
He J, Xiu L, De Porre P, Dass R, Thomas X. Decitabine reduces transfusion dependence in older patients with acute myeloid leukemia: results from a post hoc analysis of a randomized phase III study. Leuk Lymphoma. November 3, 2014. [Epub ahead of print]. | ||
You BR, Park WH. Zebularine inhibits the growth of A549 lung cancer cells via cell cycle arrest and apoptosis. Mol Carcinog. 2014;53:847–857. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.