")
Back to Archived Journals » Advances in Genomics and Genetics » Volume 4
Clinical significance and implications of genetic and genomic studies in patients with osteoarthritis
Authors Muoh O, Malemud C, Askari A
Received 18 March 2014
Accepted for publication 3 June 2014
Published 10 November 2014 Volume 2014:4 Pages 193—206
DOI https://doi.org/10.2147/AGG.S64284
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Ogechi Muoh,1 Charles J Malemud,1,2 Ali D Askari1
1Department of Medicine, Division of Rheumatic Diseases, 2Department of Anatomy, Case Western Reserve University School of Medicine and University Hospitals Case Medical Center, Cleveland, Ohio, USA
Abstract: Idiopathic or primary osteoarthritis is a progressive musculoskeletal disease of diarthrodial synovial joints, whereas secondary osteoarthritis generally arises from an antecedent traumatic injury or abnormal synovial joint development. Presently, the medical therapy of osteoarthritis is limited, and focuses on alleviating the major clinical symptoms of osteoarthritis, which include pain, swelling, and reduced range of joint motion. The end-stage of the osteoarthritic process often requires joint replacement surgery. Although the pathogenetic components of human osteoarthritis remain to be completely elucidated, rodent and canine models of osteoarthritis have indicated that, at least in the early stages of development, osteoarthritis involves articular chondrocyte hypermetabolism, followed by an imbalance between chondrocyte anabolic and catabolic pathways, and limited cartilage repair. Mutations arising in several cartilage collagen isoforms, the proteoglycans aggrecan and asporin, cartilage oligomeric matrix protein, and matrilin-3 can be associated with precocious and age-related osteoarthritis, as well as chondrodysplasias. Whole genome scanning of blood cells from individuals with osteoarthritis in Iceland, the United Kingdom, and the United States indicated the strong likelihood of osteoarthritis gene susceptibility loci on chromosome regions, 7q34–7q36.3, 11p12–11q13.4, 6p21.1–6q15, 2q31.1–2q34, and 15q21.3–15q26.1. Genetic analyses of non-extracellular matrix proteins have suggested an association of osteoarthritis with single nucleotide polymorphisms in the genes encoding growth and differentiation factor-5, secreted Frizzle-related protein-3, deiodinase-2, and calmodulin-1. The continued elucidation of genetic markers of osteoarthritis may one day be employed to better define, for an individual, the probability of developing osteoarthritis, the likely rate of progression of osteoarthritis, the relative risk of requiring joint arthroplasty, and predict prosthetic joint loosening.
Keywords: genome-wide association studies, osteoarthritis, single nucleotide polymorphisms
Introduction
Osteoarthritis (OA) is the most commonly diagnosed arthritic condition in clinical practice. Advanced age and obesity are very strongly associated with OA. Half of the world’s population aged 50 and older have been diagnosed with OA. The most commonly affected synovial joints are the knee, hip, and hand.1 OA of the lower extremity is the most common cause of functional status limitation and disability2 in non-institutionalized elderly men and women, compared against other diseases. OA of the hip and knee is the most common reason for joint replacement surgery, and the cost of treatment and surgery only keeps growing as the world’s population ages and lives longer.3 The total cost for arthritis, including OA, in the United States has been estimated, and may exceed 2% of the United States gross domestic product.4
OA is also diagnosed in younger individuals under the age of 30. However, typically, this is due to mutations in extracellular matrix (ECM) protein genes that cause significant structural abnormalities and/or joint deformities.5
The prevalence of OA is difficult to ascertain, due to issues in differentiating between clinical and radiological OA, the specific joints being studied, and characteristics (eg, ethnicity, sex) of the study population. In population-based studies in 2005, it was estimated that among adults in the United States, nearly 27 million showed evidence of clinical OA, which indicated an increase in cases of OA from the 21 million cases reported in 1995.6 Obesity is considered to be a risk factor for knee OA, especially in females.7
It is commonly assumed that multiple factors, including, genetics, age, sex, metabolic status, obesity, and traumatic events can lead to OA. Thus, the development of OA is multifactorial, but the consequences of OA are destabilization of the normal coupling of anabolic and catabolic pathways for ECM proteins in articular cartilage and subchondral bone. This causes a skewing in homeostasis toward catabolism, and the pathology of OA eventually involves a disease process that affects the entire synovial joint.8
There are molecular and biomechanical changes that lead to osteophyte formation and the breakdown and loss of articular cartilage, which eventually lead to sclerosis of subchondral bone. Abnormal metabolism of osteoblast signals,9 particularly in the subchondral bone, which may result from altered local signals, can occur. Another consideration is enhanced bone remodeling that triggers articular cartilage damage.
An attempt to repair the damaged cartilage then leads to other biochemical adaptations in bone and cartilage, which may tip the balance from cartilage repair to further boney sclerosis and damage.10 Soft tissue structures in and around the joint are also affected, including the synovium, which may contain inflammatory infiltrates. The joint ligaments often become lax. Over time, muscles can lose function and become weakened. The primary reason for seeking medical treatment following a clinical diagnosis of OA is generally pain and loss of joint function. However, the underlying cause for the pain of OA is not entirely clear.11
OA can be differentiated into primary and secondary OA. Primary OA, the most common type, is typically seen in individuals in their late fifties. Secondary OA usually results from a previous underlying trauma or developmental abnormalities, which can cause precocious and, often, severe OA.
Structure and composition of articular cartilage
Articular cartilage is an aneural, avascular connective tissue comprised of chondrocytes that are embedded in an ECM. The ECM functions to allow the tissue to resist tensile and compressive forces, and to provide a lubricating surface, permitting low-frictional movement in the joint. These biomechanical properties are the result of the interactions between collagens, hyaluronic acid, sulfated proteoglycans, and a large number of accessory proteins found in the cartilage ECM. Type II collagen is the major collagenous component. However, other collagen isotypes, including Types III, VI, IX, XI, XII, and XIV also contribute to the mature cartilage ECM. Noncollagenous components include large amounts of the proteoglycan aggrecan, which binds non-covalently to hyaluronic acid, with its associated link protein, as well as to other collagen-binding proteoglycans, such as decorin, fibromodulin, lumican, versican, and accessory proteins such as proline/arginine-rich and leucine-rich repeat protein, fibronectin, lubricin, and cartilage oligomeric matrix protein (COMP).12 The structure and abundance of these ECM components change with aging.13 Importantly, mutations in the structural protein genes of the ECM help provide clues as to how the ECM proteins contribute to the pathogenesis of OA, and to possible therapeutic interventions.
Genetic analysis methodology for OA
Some of the investigational strategies used to define the role of genetics in OA include those from familial aggregation studies, twin studies, linkage analyses, and candidate gene and genome-wide association studies (GWAS). In order to identify genes and genetic variants involved in OA, these methods rely on risk factors, traits, or disease symptoms, like joint pain, cartilage loss on imaging, and joint replacement.14 Familial aggregation studies evaluate the risk ratio for a relative of an affected individual, compared against the population. In genetic epidemiology, GWAS is used to derive the statistical power available for a given condition to detect genetic linkage to a complex genetic disorder. Twin studies, which compare monozygote versus dizygote twins, help us to understand if the variance in the distribution of the trait under study might be attributable to genetic (rather than constitutional or environmental) factors that might be shared by individuals from the same family.15,16 These studies have shown that there is a strong genetic contribution to cartilage volume and progression of OA.17,18
Genetic linkage occurs when a gene locus involved in the trait of interest, and alleles at nearby markers, are jointly inherited. Therefore, genetic markers can be used as tools to track the inheritance pattern of a gene involved in a specific trait or disease. Genome-wide linkage scan studies19 have been performed on subjects with hip, knee, or hand OA in small families, or in twins of affected relatives. Such data have been collected in various countries, including the United States, the United Kingdom, and Iceland.20–26 The genome-wide linkage scans performed on these patients defined a large number of relatively broad genomic intervals that could harbor OA susceptibility genes on chromosomes 2, 4, 6, 7, 11, 16, 19, and the X chromosome. In fact, Lee et al,26 in a meta-analysis of three studies of OA whole-genome scans from 893 families with 3,000 affected individuals from Iceland, United Kingdom, and the United States, show the genetic regions, 7q34–7q36.3, 11p12–11q13.4, 6p21.1–6q15, 2q31.1–2q34, and 15q21.3–15q26.1 were the most likely ones to harbor OA susceptibility genes.
Mutations in extracellular matrix protein genes
Cartilage collagens and cartilage collagen mutations
Human articular cartilage contains at least five distinct collagen isoforms, with the Type II, IX, and XI collagen isoforms being considered “cartilage-specific”. Type II collagen provides the basic collagen framework for the ECM and, together with the Type IX and XI collagen isoforms, is cross-linked to form a copolymeric network, consisting of hybrid collagen fibrils, thus contributing to the framework of the cartilage ECM.27
Type II collagen comprises about 80%–90% of all collagen content in normal articular cartilage. Type II collagen is initially synthesized as pro-alpha chains that are assembled into a triple helical structure. Two forms of pro-collagen are found in cartilage: Type IIA (COL2A1) and Type IIB. These trimeric Type II collagen molecules are cross-linked to other collagens (ie, Types IX and XI) that form large collagen fibrils that bind to various ECM molecules. This provides the mechanism required by cartilage to resist tensile stress. Mutations in the Type II collagen gene can cause a spectrum of phenotypes, depending on where and what is the mutation in the collagen protein.28,29 The phenotypes that have been reported include familial OA as well as other spectrums comprising various Type II collagen mutations that can result in severe phenotypes, such as Stickler syndrome and several forms of chondrodysplasia. The most common amino acid substitution is at glycine residues in the triple helical domain.30
Type XI collagen is the second most abundant collagen in normal articular cartilage. It is a heterotrimeric molecule composed of three α-chains. The first two α-chains are encoded by the COL11A1 and COL11A2 genes, respectively, while the third α chain is coded by COL2A1. Type XI collagen is cross-linked with Type II collagen to form collagen fibrils that stabilize articular cartilage ECM. Similar to COL2A1, and depending on the type or location of the mutation, mutations in the Type XI collagen genes can cause early OA, Stickler syndrome, and chondrodysplasia.31
Type IX collagen is normally co-expressed with Type II collagen in hyaline cartilage. In adults, Type IX collagen makes up about 1% of the total collagen content in articular cartilage. Type IX collagen is covalently bound to the surface of large Type II collagen fibrils, and Type IX collagen is believed to constrain the lateral expansion of these fibrils.32 Missense mutations in the Type IX collagen genes have been associated with lumbar disk disease and multiple epiphyseal dysplasia, which can lead indirectly to the development of OA.33,34
Proteoglycans and proteoglycan mutations
The second major structural component of articular cartilage is the sulfated proteoglycans, of which aggrecan is the most prominent. These ECM proteins act predominantly to retain water in cartilage and to withstand compressive force during joint transition and loading. Aggrecan is a large chondroitin sulfate/keratan sulfate-containing proteoglycan that can bind to the polymeric non-sulfated glycosaminoglycan, hyaluronic acid (HA). Although HA is employed as an intra-articular therapy via joint injection for knee OA, its mechanism of action remains incompletely understood. It is believed that HA decreases OA-associated joint pain by increasing both the viscoelastic properties of synovial fluid and the lubrication of the articular surface, thus preventing the tissue from tearing as a result of friction generated during joint transition.35
The structure known as the proteoglycan aggregate serves to physically shield Type II collagen from proteolytic cleavage.36 Because of its overall high negative charge density, aggrecan attracts water into the cartilage ECM, providing the physiomechanical mechanism for permitting the cartilage to swell. Swelling gives the tissue its spring-like quality, helping it to withstand hydrostatic compressive forces that are applied to the joint during movement.
A missense mutation in the aggrecan gene produced a familial osteochondritis dissecans phenotype that clinically presents as multiple osteochondritic lesions in knees and/or hips and/or elbows, disproportionate short stature, and early development of OA.37,38
Fourteen years ago, a novel ECM protein, asporin,39 came to light, which was shown in genetic linkage studies to play a role in OA. Asporin, a small leucine-rich proteoglycan, is a non-collagenous ECM protein that is abundantly expressed in OA articular cartilage, and its expression is found to increase with progressive cartilage degeneration. Although the exact role of asporin is unknown, Nakajima et al40 have shown that asporin binds directly to transforming growth factor-β1 (TGF-β1) in vitro and inhibits TGF-β1-induced expression of ECM genes, as well as proliferation and differentiation of chondrocytes. These results strongly suggested that asporin acts as a negative regulator of TGF-β in cartilage, playing a critical role in the etiology and pathogenesis of OA.
Mutations in cartilage accessory proteins
Mutations in the two accessory ECM proteins, COMP and Matrilin 3 (MATN3), have also been implicated in the pathogenesis and, putatively, in the progression of OA and chondrodysplasias. However, at present, mutations in lubricin are far more associated with specific forms of chondrodysplasia than with OA.
With respect to COMP, Hecht et al41 reported that three COMP mutations, G427E, D469del, and D511Y, were produced by chondrocytes derived from individuals with pseudochondrodysplasia, and were associated with increased retention of COMP in the endoplasmic reticulum. In addition, these COMP mutations altered the secretion of Type IX collagen and MATN3, but not the secretion of aggrecan or Type II collagen. COMP has also proven to be a prominent and useful biomarker for assessing the progression of OA, where serum COMP levels were higher in aggressive forms of arthritis. COMP serum levels may even be useful for predicting the future progress of the disease.42 Most recently, Mu et al43 reported that direct sequencing of COMP in 70 subjects with early-onset OA revealed a c.2152C>T substitution in exon 18. Exon 18 encodes the C-terminal globular domain of COMP and, overall, the data suggested that COMP was an OA susceptibility gene. However, the role of COMP mutations and its potential as an OA susceptibility gene may be questionable in certain populations. Thus, genotype and allelic frequencies of COMP polymorphisms did not convey any increased relative risk for developing OA in a Japanese population.44
Matrilin (MATN) belongs to a family of oligomeric ECM proteins that are capable of binding to several types of collagen fibrils, to other noncollagenous proteins, and to aggrecan.45 A few distinct MATN3 polymorphisms have been shown to be associated with OA,24 as well as to some rare forms of chondrodysplasia. Two MATN3 mutations, pR116W and p.C299S, in particular, appear to be responsible for abnormal retention of MATN3 in chondrocyte endoplasmic reticulum, whereas a third MATN3 mutation, p.T298M, did not alter MATN3 secretion. In that regard, Otten et al46 showed that the p.T298M MATN3 mutation did not affect oligomerization of MATN3, nor was p.T298M altered by proteolytic processing via a disintegrin and metalloproteinase with thrombospondin motif (ADAMTS) 4 and 5. However, p.T298M did alter MATN3 conformational changes, which affected the in vitro fibril formation of Types II, IX, and XI collagen heterofibrils. Most noteworthy was the finding that the p.T298M MATN3 mutation did not change the affinity of MATN3 for these collagen isoforms, which suggested that MATN3 mutations affect the overall collagen self-assembly mechanism and, therefore, the integrity of collagen-containing cartilage ECM. The results of this study may also help to explain the association of two other MATN3 amino acid substitutions, T303M SNP547 and rs8176070 SNP6,48 with hand OA in a Dutch and a Chinese Han population, respectively. From the perspective of OA pathogenesis, it was also revealing that MATN3-null mice appeared to be highly predisposed to develop severe OA, compared against their wild-type littermates.49 In addition, the MATN3-null mice had significantly higher bone mineral densities than MATN1-null or wild-type mice.
Lubricin, a mucinous glycoprotein encoded by the PRG4 gene provides the mechanism by which the boundary lubrication properties of articular cartilage surfaces are maintained.50 Lubricin mutations or the absence of lubricin gene expression was found to be associated with camptodactyly-arthropathy-coxavara-pericarditis syndrome, synovial hyperplasia, and failed joints.51 However, it remains to be seen the extent to which lubricin mutations represent a relative risk and susceptibility factor for OA.
Mutations in ECM proteins that were identified in association with OA and, in other cases, with various types of chondrodysplasias, are summarized in Table 1.
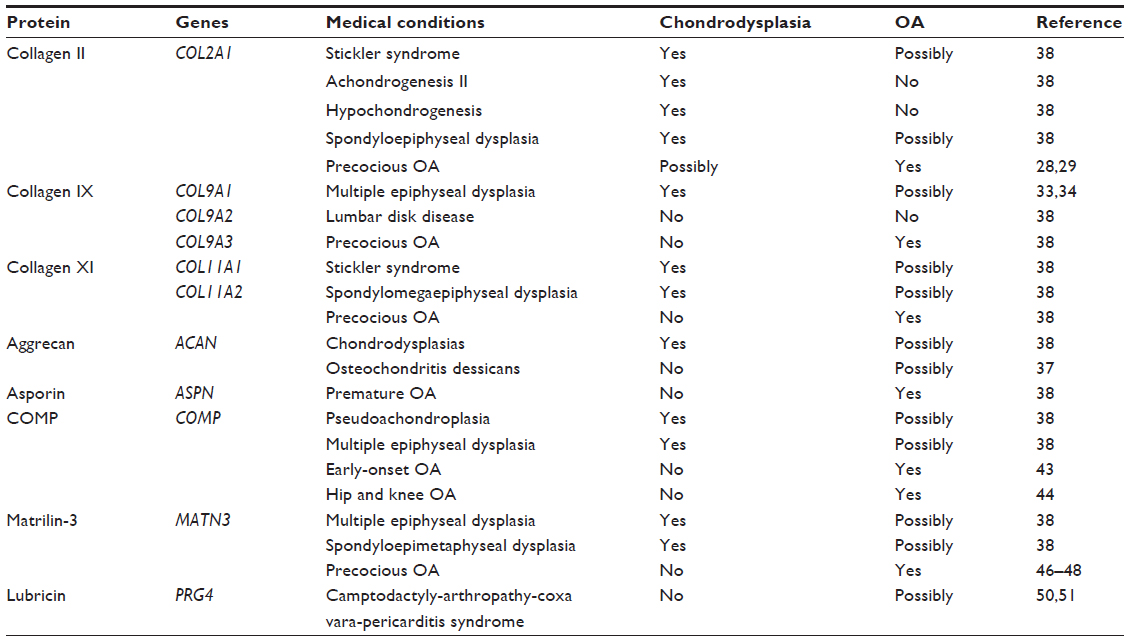 | Table 1 Cartilage ECM protein gene mutations: association with musculoskeletal diseases |
Genome-wide linkage studies
Studies of OA candidate genes have helped shed light on the role these genes might play in the pathogenesis of OA. However, many of the gene mutations that might be relevant to OA do not occur at a high enough frequency to be translated to a greater relative risk for developing OA in any population. So far, genetic analyses have been restricted, due to insufficient sample sizes and phenotype heterogeneity. Given this, investigators are now focusing on systematic investigations using GWAS to aid in identifying candidate genes from different patient populations. As with numerous other complex diseases, GWAS has recently been carried out to test the extent to which millions of single nucleotide polymorphisms (SNPs) associate with OA across the human genome.
There was significant association with OA and SNPs located in the growth and differentiation factor 5 (GDF5) and MCF.2 cell line derived transforming (MCF2L) genes, as well as a region on chromosome 7q22.52–55 In fact, novel genetic associations have been reported by the Arthritis Research UK Osteoarthritis Genetics (arcOGEN) consortium. Thus far, the largest GWAS project adding to the three previously established GWAS previously performed was conducted on 7,410 unrelated Northern European patients, who were prospectively and retrospectively selected for inclusion and compared against 11,009 unrelated controls; 80% have had total joint replacement of either the hip or knee. The signals identified were replicated in a second group of 7,473 cases and 42,938 controls, again from Northern Europe. The arcOGEN consortium identified five genome-wide significant loci for association with OA and three susceptibility loci just below this threshold. The strongest association was on chromosome 3 with rs6976 (odds ratio: 1.12; 95% confidence interval [CI]: 1.08–1.16; P=7.24×10−11), which was in perfect linkage disequilibrium with rs11177. Thus, the rs6976 SNP encodes a missense mutation within the nucleostemin-encoding gene, guanine nucleotide-binding protein-like 3. Of note, nucleostemin was elevated in chondrocytes from patients with OA ex vivo.
Other significant OA gene susceptibility loci were found on chromosome 9, close to astrotactin-2, chromosome 6, between filamin-A-interacting protein 1 and SUMO1/sentrin specific peptidase 6, chromosome 12, close to kelch domain containing 5 and parathyroid hormone-like hormone, and in another region of chromosome 12, close to carbohydrate (chondroitin 4) sulfotransferase 11. One of the signals, close to genome-wide significance, was within the fat mass and obesity-associated protein gene (also known as α-ketoglutarate-dependent dioxygenase), which is involved in regulating obesity, a strong risk factor for OA.56,57 Most of the identified OA gene susceptibility loci were associated with hip OA, rather than knee OA, supporting epidemiologic observations that hip OA is predominantly due to bone developmental abnormalities, whereas knee OA is more commonly associated with trauma and obesity. However, additional studies will be needed to identify the extent to which these gene SNPs are causally related to the pathogenesis of OA.
Non-extracellular matrix genes associated with susceptibility to OA
GDF5, secreted Frizzle-related protein 3 (FRZB), deiodinase 2 (DIO2), and calmodulin 1 (CALM1) are four non-ECM protein genes that have been identified by GWAS as potential candidate OA susceptibility genes58–62 (Figure 1). GDF5, FRZB, and DIO2 were reported to be associated with the development of hip and knee OA across various patient populations,63,64 whereas CALM1 was not.65–67 Several other genes of interest, purported to be involved in OA pathogenesis and progression, including, most notably, a disintegrin and metalloproteinase-12 (ADAM12), were found to be associated with subclinical OA as well as clinical knee OA in a population of male and female subjects in the United Kingdom.64 However, this was not confirmed in another GWAS analysis of subjects with knee OA from a Korean cohort.68
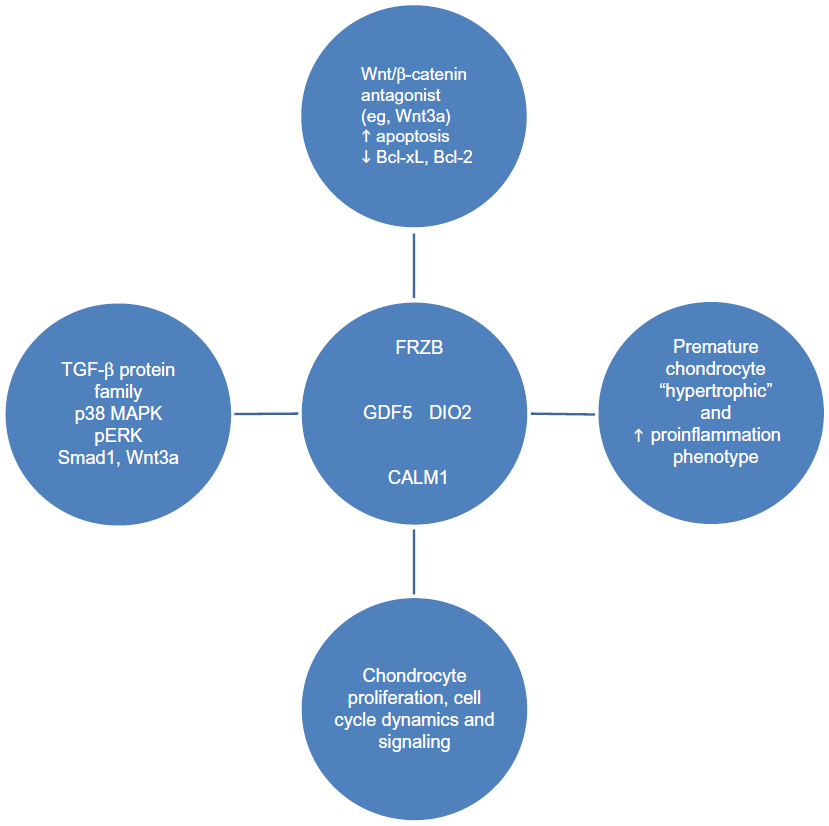 | Figure 1 Non-ECM genes identified as OA candidate genes. |
GDF5: functional implications for OA
GDF5 is a member of the transforming growth factor-β protein superfamily. GDF5 forms a heterodimeric complex after binding to bone morphogenetic protein receptor (BMPR) 1B and 2, or the actin-related protein 2a receptor. The interaction between GDF5 and GDFR was shown to result in the inhibition of phosphorylation of p38 kinase and of phosphorylated extracellular signal-regulated kinase. In contrast, GDF5 activated dominant/negative helix-loop-helix proteins, including inhibitor of differentiation 1 and inhibitor of differentiation 3, synthesized by human umbilical vein smooth muscle cells through the activation of the mothers against decapentaplegic homolog 1 (Smad1) pathway.69 Importantly, GDF5, along with bone morphogenetic protein 2 (BMP2), inhibited vascular smooth muscle cell and mouse embryonic fibroblast apoptosis70 by interacting with and activating BMPR2. This caused X-inhibitor of apoptosis protein (XIAP) to become more stable, through the inhibition of XIAP ubiquitination.71 In keeping with this finding and other in vitro results, ex vivo analyses of cardiomyocytes from GDF5-knockout mice showed that the cardiomyocytes from GDF5-knockout mice exhibited increased apoptosis, which was accompanied by decreased expression of the anti-apoptotic genes, Bcl-2 and Bcl-xL, when Bcl2 and Bcl-xL expression in the cardiomyocytes from the GDF5-knockout mice were compared against their wild-type counterparts.72 However, more recently, Ratnayake et al73 called into question the extent to which chondrocytes could respond in a positive manner to exogenous GDF5 in order to regulate gene responses associated with OA. Thus, Ratnayake et al73 compared the effects of the GDF5 variants, one of which contained the SNP rs143383 associated with OA susceptibility,61,74 against wild-type GDF5, on regulating intracellular Smad1 signaling, matrix metalloproteinase (MMP) 1 and 13, tissue inhibitor of metalloproteinase 1 (TIMP1), and COL2A1 gene expression by human OA chondrocytes.73 As would be expected, GDF5 activated Smad1. Furthermore, OA chondrocytes maintained in medium containing TGF-β1 downregulated MMP1 and MMP13 gene expression, while also consistently upregulating TIMP1 and COL2A1 genes. These results were consistent with the previously-reported role of TGF-β1 as a positive regulator of ECM protein production, including the synthesis of sulfated-proteoglycans.75 However, noteworthy was the finding that chondrocytes cultured in the presence of exogenous wild-type GDF5 or any of several GDF5 genetic variants tested under these culture conditions failed to consistently change the expression of MMP1, MMP13, TIMP1, and COL2A1 genes. These results suggested that any proposal to apply the use of exogenous GDF5 for the therapy of OA may have to take into account the possibility that OA chondrocytes no longer can respond to GDF5. This contention takes on added weight from the findings of Enochson et al,76 who showed that GDF5 inhibited the expression of MMP13 and ADAMTS4, while also stimulating aggrecan and the cartilage-specific transcription factor, SOX9, when non-arthritic human chondrocytes were cultured under “micromass” conditions, with or without the canonical Wnt signaling inhibitors Wnt3a and CHIR99021, or WAY262611, an inhibitor of dickkopf 1 homolog (DKK1) activity. Thus, these results showed that GDF5-mediated effects were accompanied by inhibition of Wnt3a signaling by upregulating DKK1 and FRZB genes, whereas GDF5 upregulation of DKK1 gene expression was responsible for downregulation of MMP13 gene expression.
The functional role of GDF5 as a regulator of chondrocyte gene expression has also been probed by employing microRNA (miR) technology.77,78 In that regard, Swingler et al78 showed that miR-455 directly targeted TGF-β-mediated signaling, including Smad2, whereas miR-21, which was increased in the cartilage of knee OA patients, compared against cartilage obtained from traumatic amputees, directly targeted GDF5, where miR-21 overexpression inhibited chondrogenesis in the human articular chondrocyte CH8 cell line.79 These results suggested that articular cartilage repair, which is significantly compromised in OA,80 may, in part, result from miR-21-mediated inhibition of GDF5. These findings also confirmed the results of other studies, which showed that overexpression of GDF5 resulted in enhanced osteogenic differentiation of human mesenchymal stem cells,81 as well as increased chondrogenesis and angiogenesis.82
Most relevant to any discussion of GDF5 as a major non-ECM protein for regulating musculoskeletal system development can be gleaned from results using Bp-J/+ mice deficient in GDF5. These mice were employed in several animal models of arthritis, including induction with collagenase or papain, medial meniscectomy, or treadmill running. All of these animal models resulted in synovial joint destabilization, gait abnormalities, and subchondral bone changes that were consistent with synovial joint damage.83 GDF5-deficient mice appeared phenotypically normal. No differences were detected among the three animal models with respect to the severity of OA, with one exception: GDF5-deficient mice showed increased synovial hypertrophy in the treadmill running animal model. Most strikingly, GDF5- (Bp-J/+) deficient mice demonstrated an abnormal gait pattern, with decreased subchondral bone density coupled to “distorted” collagen fiber arrangement. Taken together, these results suggested a potential protective effect of GDF5 expression in various mouse models of OA.
Is GDF5 an OA susceptibility gene across all studied populations?
The GDF5 rs143383 SNP was found to be associated with human OA and degenerative disk disease.84 In the original study results, Miyamoto et al85 reported that an SNP in the 5′ UTR of GDF5 (ie, +104T/C; rs143383) was associated with hip OA in two independent Japanese populations, and knee OA in Japanese and Han Chinese populations. These findings have since been confirmed and have genome-wide statistical significance for OA of the hip, hand,86 and knee.53,86
The rs143383 SNP was localized to the GDF5 core promoter where GDF5 promoter expression exhibited allelic differences and influenced chondrocyte transcriptional activity. Of note, the GDF5 susceptibility locus showed reduced GDF5 transcriptional activity. In a series of influential studies published by the research group directed by John Loughlin,87–89 an imbalance was consistently found in the GDF5 allele from tissues of OA patients undergoing hip or knee joint replacement surgery. In addition to SNP rs143383, Egli et al87 found a second GDF5 polymorphism in the 3′ UTR region, which also was shown to regulate the expression of SNP rs143383. Egli et al87 also reported finding a differential binding of the trans-acting factor, Deformed epidermal autoregulatory factor 1 (DEAF-1) to the two alleles of rs143383, the significance of which remains to be completely elucidated, although DEAF-1 appears to augment the synergistic activities of the Rel transcription factor and trans-acting T-cell-specific transcription factor, GATA-3, during the immune response. Most recently, Dodd et al88 used the Sanger method to “deep-sequence” GDF5, encompassing the protein-coding region of the two GDF5 exons, both untranslated regions and 100 bp of the proximal GDF5 promoter, in DNA obtained from 992 OA patients and 944 control subjects. Of the thirteen GDF5 variants detected, six were found to be “extremely” rare, with minor allelic frequencies. One of these alleles was the predicted transcription factor binding site in the GDF5 promoter, and the other seven variants were those previously known to exist. Dodd et al88 concluded that these GDF5 variants did not contain any novel sequences not previously reported that were likely to contribute to the “OA-association signal” represented by rs143383 SNP, nor did these sequences influence OA susceptibility independent of rs143383 SNP. Then, Reynard et al89 showed that the genetic effect of rs143383 SNP was regulated by the epigenome at the level of DNA methylation, which could influence the rate of penetrance for expression of the rs143383 allele.
Bos et al63 had previously claimed that GDF5 was an OA susceptibility gene across different patient populations. However, over the past 5 years, additional studies have been performed to determine the extent to which this result could be confirmed. In that regard, Valdes et al90 showed that the major GDF5 susceptibility allele at rs143383, but not the major allele of double von Willebrand factor A (DVWA) SNP rs7639618, was consistently associated with an elevated relative risk for developing knee OA in a United Kingdom population, whereas the data reported in the GDF5 meta-analysis conducted by Hao and Jin91 confirmed that the +104T/C polymorphism in the GDF5 5′ UTR region was associated with knee OA, irrespective of ethnicity. However, several recent studies, one performed in a Korean OA cohort68 and another in a Greek Caucasian population,92 could not confirm an association between the GDF5 +104T/C SNP and OA. Tawonsawatruk et al93 found such an association between the +104T/C SNP and knee OA in a Thai population. Overall, these genetic analyses of GDF5 polymorphisms pointed to the complexity in the relationship between the GDF5 rs143383 SNP, the influence of ethnic background, and the rs143383 gene susceptibility loci in these populations and the development of OA.
Wnt/β-catenin signaling and Frizzle- related protein: functional implications for OA
Wnt/β-catenin-mediated signal transductions fall into one of three categories: the canonical Wnt pathway that regulates gene transcription, the noncanonical planar cell polarity pathway that regulates cytoskeletal structure and cell shape, and the noncanonical Wnt/calcium pathway that regulates intracellular Ca2+ levels.94–96
All Wnt-mediated signaling is activated through the binding of a Wnt-protein ligand to a Frizzled family protein receptor. This ligand-receptor interaction then conveys a transduction signal to the intracellular phosphoprotein, dishevelled (Dsh). Importantly, Dsh phosphoproteins are evolutionally conserved, and these domains all share three similar structural homologies: an amino-terminal Dishevelled-aXin domain, a central post-synaptic density-95 domain, and a carboxy-terminal Dishevelled-EGL-10-Pleckstrin domain. Importantly, Habas and Dawid97 showed that it was the individual Dsh domains or combinations thereof that determined which of the three Wnt pathways were to become activated.
The Wnt/β-catenin protein family, in general,98 and, in particular, Wnt3a,99 Wnt-16, Wnt-2b, and Wnt-inducing signaling protein-1100 were shown to play critical roles in chondrogenesis, maintenance of cartilage homeostasis, and cartilage degeneration.101 Of note, Nalesso et al102 showed that Wnt3a activated the Wnt canonical pathway via β-catenin-mediated gene transcription, as well as the noncanonical Wnt/calcium pathway. Wnt3a promoted chondrocyte proliferation and the loss of COL2A1, aggrecan, and SOX9 gene expression. With respect to cartilage degeneration, inhibition of β-catenin signaling in chondrocytes derived from neonatal COL2A1-ICAT transgenic mice resulted in an increase in chondrocyte apoptosis.103 In that study, apoptosis was associated with a decrease in Bcl-2 and Bcl-XL expression, an increase in caspase-3 and -9 activities as well as cartilage destruction. In contrast, activation of β-catenin resulted in development of an “OA-phenotype” that was characterized by overexpression of ECM-degrading proteinases in adult mouse articular cartilage.104 It was further suggested that age-related changes in Wnt/β-catenin signaling may alter chondrocyte responses, with resultant precocious cartilage aging and development of OA.105
Secreted frizzled-related proteins (SFRPs) are well-characterized Wnt antagonists, and polymorphisms in FRZB-SFRP3 are associated with the development of OA106,107 and other chronic forms of inflammatory arthritis, including rheumatoid arthritis,108 Thus, FRZB-2, a member of the FRZB protein family, was strongly expressed in human OA chondrocytes where FRZB-2 immunoreactivity and TUNEL-positivity (an indicator of apoptosis) were co-localized.109 Interestingly, a comparison analysis of healthy human growth plate and articular cartilage from adolescent donors showed decreased Wnt signaling in the articular cartilage, which was shown to be partly due to increased expression of FRP and two other Wnt antagonists, Gremlin-1 and DKK-1.110,111 Thus, it was proposed that interfering with Wnt/β-catenin signaling using selective Wnt antagonists such as FRZB could constitute a novel therapeutic approach to improve cartilage repair and regeneration in OA.112 Importantly, recent evidence suggested that methylation of Wnt-regulated genes was a critical regulator of Wnt-mediated gene expression in osteoblasts isolated from human OA joints.113
FRZB: association with human OA?
Loughlin et al114 first established an association between a single SNP in FRZB, resulting in an Arg324Gly substitution at the C-terminus of the FRZB protein, and hip OA in females, which was independently confirmed in a second cohort of female hip OA patients. In addition, a haplotype encoding two highly-conserved Arg residues, namely, Arg200 (Arg200Trp) and Arg324 (Arg324Gly) was determined to constitute a strong risk factor for the development of primary hip OA. From a functional perspective, the FRP-3 Arg324Gly variant showed diminished Wnt-antagonistic activity in vitro. These results appeared to serve as an impetus to convincingly establish an association between FRZB polymorphisms and OA. However, in an analysis of hip OA, confirmed by radiographs, the minor Arg200Trp allele polymorphism was not statistically more common in OA than in the control group.115 Importantly, despite this lack of statistical significance, OA patients who were homozygous for the FRZB Arg200Trp allele had higher serum levels of FRP than the control group, who were homozygous for the major allele. In a cohort of Spanish OA patients, including patients with primary OA of the hip, knee, or hand,116 no significant differences in the allelic frequencies of Arg200Trp (ie, rs288326) or Arg324Trp (ie, rs7775) were found, compared against the control group of patients 55 years and older with no clinical evidence of OA. The only difference in allelic frequency reported in that analysis116 was that the Arg324Trp SNP “trended” toward a higher frequency in patients with clinical OA of several joints and, more specifically, in women in the hip replacement group, perhaps providing evidence of a link between Arg324Trp SNP and more progressive hip disease. In contrast, Snelling et al117 could not find any differential FRZB allelic expression in human chondrocytes obtained from subjects undergoing joint replacement who were heterozygous for one of the two FRZB SNPs. However, an analysis conducted by Valdes et al118 concluded that genetic variations in several genes, including ASPN, CALM1, COL2A1, COMP, and FRZB varied significantly between OA patient populations (ie, Japanese versus Caucasians) and sex, where these gene polymorphisms were significant only in the Caucasian population. Of note, a large-scale gene analysis to determine if a genetic association existed between FRZB and hand, knee, and hip OA patients failed to provide evidence of such an association between the FRZB rs7775 SNP and hand and knee OA, with only borderline significance (P=0.019) of rs7775 with hip OA. In a more recent analysis, Baker-Lepain et al119 showed that the rs288326 and rs7775 FRZB SNPs were associated with the shape of the proximal femur and, therefore, rs7775 FRZB SNP may be linked to hip shape and the influence of hip anatomy on OA.
Finally, several genes, including FRZB, were found to be differentially upregulated in bone tissue from patients with OA of the hip, which was confirmed by studies using bone cell cultures.120 However, the serum levels of FRZ and DKK-1 did not appear to differ from FRZ and DKK-1 serum levels in a control group when these individuals were compared against subjects with confirmed radiographic evidence of hip OA.121 In the same study, higher levels of serum DKK-1 were associated with a lowered relative risk for progression of hip OA, but only in women in the highest quartile of serum DKK-1 levels, compared against women in the lowest serum DKK-1 quartile.
To summarize, accumulated evidence points to a genetic association between the rs288326 and the rs7775 FRZB SNPs and OA. However, sex and ethnicity, as well the degree of OA severity and rate of OA progression to end-stage disease, appear to contribute, in part, to the variability in the results obtained in these genomic analyses.
DIO2: functional studies
Deiodinases are a class of homodimeric thioredoxin fold-containing selenoproteins involved in the activation and deactivation of thyroid hormones.122 The DIO2 is an initiator of thyroid hormone signaling, through its ability to activate the prohormone thyroxine (T4), converting it to the biologically-active T3 molecule.123
DIO2 has been identified as a modulator of chondrocyte function. For example, primary human chondrocytes were transfected with an siRNA specific for either DIO2 or the other major selenoproteins in this class; namely, glutathione peroxidase-1 (GPX1) and thioredoxin reductase-1 (TR1).124 The results of this study showed that basal, or IL-1β-induced, expression of cyclooxygenase-2 (COX-2) was increased in si-RNA/DIO2-transfected chondrocytes but not in siRNA/GPX1- or siRNA/TR1-transfected chondrocytes. Moreover, suppression of DIO2 also increased IL-1β-induced IL-1β and liver X receptor-α (LXR-α) but not LXR-β gene expression, suggesting that DIO2 mediated the expression of several critical proinflammatory genes expressed by human chondrocytes.
Nagase et al125 recently reported elevated DIO2 levels in articular cartilage of OA patients, compared against cartilage from normal subjects. The results of this study also showed that transgenic mice in which the DIO2 gene was specifically knocked out in articular cartilage, by transfecting with a bacterial artificial chromosome housing the entire rat COL2A1 gene and the human DIO2 gene construct, contained significantly higher levels of biomarkers characteristic of articular chondrocyte hypertrophy. Moreover, the COL2A1/DIO2-transgenic mice were more sensitive to knee destabilization-induced articular cartilage destruction than were their wild-type littermates. Thus, downregulation of DIO2 gene expression might be responsible for accelerating the onset of cartilage destruction, by regulating the transitioning of articular chondrocytes to the “hypertrophic phenotype”,126,127 although other genes of interest such as SOX9, Aggrecan, and Type I collagen, which are known to be associated with the development of OA, but not with chondrocyte hypertrophy, were also shown to be altered in both fibrillated and intact human cartilage.128
DIO2: association with OA?
Two studies have implicated DIO2 as a susceptibility gene in OA.129,130 In a 2008 DIO2 analysis, genotyping of tagged SNPs revealed a common coding variant, rs225014; Thr92Ala, in the DIO2 gene in OA patients from the United Kingdom and in Japanese female patients, the latter with confirmed symptomatic hip OA. Confirmation and replication by association of the minor rs225014 DIO2 allele and the common rs12885300 DIO2 allele was also obtained in females with advanced/symptomatic hip OA. In the second study, an elevated number of DIO2-positive cells were seen in OA cartilage, compared against cartilage from healthy donors. Furthermore, a statistically significant 1.3-fold higher presence for the rs225014 DIO2 ‘C’ allele was observed, relative to the DIO2 ‘T’ allele in 90.3% (28/31) of OA cartilage donors. Thus, these results confirmed the presence of an allelic imbalance in the DIO2 rs225014 ‘risk’ allele in OA, and established an association between this DIO2 genetic variant and the elevated relative risk for developing OA.
Calmodulin-1: functional studies and association with OA
Calmodulin-1, encoded by the CALM1 gene, is one of 20 members belonging to the calcium-modulating protein family. The calmodulin protein encoded by the CALM1 gene was initially identified by its localization in the cytosol, or as a component of cellular membranes where the calmodulin-1 protein was found oriented toward the cytosol and showed a high affinity for binding calcium. Calmodulin, in general, is comprised of 148 amino acids with four calcium-binding motifs. The varied functions of calmodulin-1 include its ability to regulate cell proliferation, cell cycle dynamics, and signal transduction.
CALM1 was first identified as a potential OA susceptibility gene by Mototani et al.59 They found a significant association between hip OA in a Japanese population and a CALM1 SNP, IVS3-293C>T, which was localized to the third intron of the CALM1 gene. CALM1 was expressed in cultured human chondrocytes and articular cartilage and, importantly, CALM1 was overexpressed in human OA cartilage. Linkage disequilibrium mapping identified five additional CALM1 SNPs with significant association, equivalent to IVS3-293C>T, with one of these SNPs, -16C>T, located in the CALM1 promoter region. Taken together, these initial data suggested that altered transcription of CALM1 was associated with OA susceptibility in this Japanese population. However, subsequent association studies in other OA populations have failed to confirm these initial findings in the Japanese cohort. Thus, no significant differences were found in the genotypic frequencies for the -16T/C CALM1 gene in Greek Caucasian OA subjects,67 in a Chinese Han population66 or in hip OA in Caucasians from the United Kingdom65. Although Poulou et al67 failed to find that hip OA susceptibility was linked to CALM1, additional data from this study suggested that the CALM1-16TT core promoter genotype in association with the ASPN D14 or D15 allele could together “influence” susceptibility for knee OA.
A clinical perspective on OA genetic susceptibility genes
There is now persuasive evidence from whole-genome scan studies that human chromosome regions including 7q34–7q36.3, 11p12–11q13.4, 6p21.1–6q15, 2q31.1–2q34, and 15q21.3–15q26.1 are most likely to harbor OA susceptibility genes. Importantly, a precocious form of OA was shown to arise from various Type II collagen point mutations. In addition, point or missense mutations in other cartilage collagen genes may also be the cause of altered cartilage homeostasis, chondrodysplasia, and OA. Most importantly, two SNPs in the Type XI collagen gene, COL11A1, (Table 1) was one of 199 candidate genes to be validated in association with OA from a large-scale meta-analysis of GWAS.131 SNPs in non-ECM proteins genes, most prominently, GDF5, FRZB, DIO2, and CALM1 have also been shown to be associated with an elevated relative risk and susceptibility to developing OA, with the proviso that not all populations of OA patients have been shown to harbor these OA susceptibility polymorphisms.
Despite the emergence of several genetic associations with OA and an improving understanding of the role that BMP-mediated signaling, “wingless”-type of signaling, cytokines, prostaglandins, pro- and anti-apoptotic molecules,132 angiogenesis factors, such as vascular endothelial growth factor,131 and cytoskeletal regulatory proteins (eg, leucine-rich repeats and calponin homology domain)133 play in OA,134 the clinical relevance of these novel findings to the diagnosis and medical therapy of OA remains to be further clarified. However, the fact that many of these signaling pathways are likely to be amenable to pharmacologic intervention, and can be applied to mechanisms that regulate pain relevant to OA,11 suggests that the study of genetic “markers” of OA may one day be employed to better define the rate of OA progression in an individual, or even the relative risk of requiring joint arthroplasty, or for predicting prosthetic joint loosening.
Disclosure
The authors have no conflicts of interest in this work.
References
Bijlsma JW, Berenbaum F, Lafeber FP. Osteoarthritis: an update with relevance for clinical practice. Lancet. 2011;377(9783):2115–2126. | |
Guccione AA, Felson DT, Anderson JJ, et al: The effects of specific medical conditions on the functional limitations of elders in the Framingham Study. Am J Public Health. 1994;84(3):351–358. | |
DeFrances CJ, Podgornik MN. 2004 National Hospital Discharge Survey. Adv Data. 2006;(371):1–19. | |
Yelin E. The economics of osteoarthritis. In: Brandt KD, Doherty M, Lohmander LS, editors. Osteoarthritis. New York: Oxford University Press; 1998. p. 23–30. | |
Li Y, Wei X, Zhou J, Wei L. The age-related changes in cartilage and osteoarthritis. Biomed Res Int. 2013;2013:916530. | |
Lawrence RC, Felson DT, Helmick CG, et al: Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58(1):26–35. | |
Felson DT, Lawrence RC, Dieppe PA, et al. Osteoarthritis: new insights. Part 1: the disease and its risk factors. Ann Intern Med. 2000;133(8):635–646. | |
Poole AR, Kojima T, Yasuda T, Mwale F, Kobayashi M, Laverty S. Composition and structure of articular cartilage: a template for tissue repair. Clin Orthop Relat Res. 2001;(Suppl 391):S26–S33. | |
Burr DB. The importance of subchondral bone in osteoarthrosis. Curr Opin Rheumatol. 1998;10(3):256–262. | |
Lajeunesse D. Altered subchondral osteoblast cellular metabolism in osteoarthritis: cytokines, eicosanoids, and growth factors. J Musculoskelet Neuronal Interact. 2002;2(6):504–506. | |
Dieppe PA, Lohmander LS. Pathogenesis and management of pain in osteoarthritis. Lancet. 2005;365(9463):965–973. | |
Poole AR. Cartilage in health and disease. In: Koopman W, editor. Arthritis and Allied Conditions. A Textbook of Rheumatology. 14th ed. New York: Lippincott Williams and Wilkins; 2001. p. 2260–2284. | |
Roughley PJ. Age-associated changes in cartilage matrix: implications for tissue repair. Clin Orthop Relat Res. 2001;(Suppl 391):S153–S160. | |
Chapman K, Valdes AM. Genetic factors in OA pathogenesis. Bone. 2012;51(2):258–264. | |
Risch N. Linkage strategies for genetically complex traits. I. Multilocus models. Am J Hum Genet. 1990;46(2):222–228. | |
Guo SW. Sibling recurrence risk ratio as a measure of genetic effect: caveat emptor! Am J Hum Genet. 2002;70(3):818–819. | |
Botha-Scheepers SA, Watt I, Slagboom E, et al. Influence of familial factors on radiologic disease progression over two years in siblings with osteoarthritis at multiple sites: a prospective longitudinal cohort study. Arthritis Rheum. 2007;57(4):626–632. | |
MacGregor AJ, Antoniades L, Matson M, Andrew T, Spector TD. The genetic contribution to radiographic hip osteoarthritis in women: results of a classic twin study. Arthritis Rheum. 2000;43(11):2410–2416. | |
Reynard LN, Loughlin J. The genetics and functional analysis of primary osteoarthritis susceptibility. Expert Rev Mol Med. 2013;15:e2. | |
Hunter DJ, Demissie S, Cupples LA, Aliabadi P, Felson DT. A genome scan for joint-specific hand osteoarthritis susceptibility: The Framingham Study. Arthritis Rheum. 2004;50(8):2489–2496. | |
Chapman K, Mustafa Z, Irven C, et al. Osteoarthritis-susceptibility locus on chromosome 11q, detected by linkage. Am J Hum Genet. 1999;65(1):167–174. | |
Loughlin J, Mustafa Z, Irven C, et al. Stratification analysis of an osteoarthritis genome screen-suggestive linkage to chromosomes 4, 6, and 16. Am J Hum Genet. 1999;65(6):1795–1798. | |
Livshits G, Kato BS, Zhai G, et al. Genomewide linkage scan of hand osteoarthritis in female twin pairs showing replication of quantitative trait loci on chromosomes 2 and 19. Ann Rheum Dis. 2007;66(5):623–627. | |
Stefansson SE, Jonsson H, Ingvarsson T, et al. Genomewide scan for hand osteoarthritis: a novel mutation in matrilin-3. Am J Hum Genet. 2003;72(6):1448–1459. | |
Ingvarsson T, Stefansson SE, Gulcher JR, et al. A large Icelandic family with early osteoarthritis of the hip associated with a susceptibility locus on chromosome 16p. Arthritis Rheum. 2001;44(11):2548–2555. | |
Lee YH, Rho YH, Choi SJ, Ji JD, Song GG. Osteoarthritis susceptibility loci defined by genome scan meta-analysis. Rheumatol Int. 2006;26(11):996–1000. | |
Eyre DR. The collagens of articular cartilage. Semin Arthritis Rheum. 1991;21(3 Suppl 2):2–11. | |
Eyre DR, Weis MA, Moskowitz RW. Cartilage expression of a type II collagen mutation in an inherited form of osteoarthritis associated with a mild chondrodysplasia. J Clin Invest. 1991;87(1):357–361. | |
Pun YL, Moskowitz RW, Lie S, et al. Clinical correlations of osteoarthritis associated with a single-base mutation (arginine 519 to cysteine) in type II procollagen gene. A newly defined pathogenesis. Arthritis Rheum. 1994;37(2):264–269. | |
Byers PH. Folding defects in fibrillar collagens. Philos Trans R Soc Lond B Biol Sci. 2001;356(1406):157–158. | |
Rodriguez RR, Seegmiller RE, Stark MR, Bridgewater LC. A type XI collagen mutation leads to increased degradation of type II collagen in articular cartilage. Osteoarthritis Cartilage. 2004;12(4):314–320. | |
Wu JJ, Woods PE, Eyre DR. Identification of cross-linking sites in bovine cartilage type IX collagen reveals an antiparallel type II-type IX molecular relationship and type IX to type IX bonding. J Biol Chem. 1992;267(32):23007–23014. | |
Näkki A, Videman T, Kujala UM, et al. Candidate gene association study of magnetic resonance imaging-based hip osteoarthritis (OA): evidence for COL9A2 gene as a common predisposing factor for hip OA and lumbar disc degeneration. J Rheumatol. 2011;38(4):747–752. | |
Jackson GC, Marcus-Soekarman D, Stolte-Dijkstra I, Verrips A, Taylor JA, Briggs MD. Type IX collagen gene mutations can result in multiple epiphyseal dysplasia that is associated with osteochondritis dissecans and a mild myopathy. Am J Med Genet A. 2010;152A(4):863–869. | |
Altman RD, Moskowitz R. Intraarticular sodium hyaluronate (Hyalgan) in the treatment of patients with osteoarthritis of the knee: a randomized clinical trial. Hyalgan Study Group. J Rheumatol. 1998;25(11):2203–2212. | |
Pratta MA, Yao W, Decicco C, et al. Aggrecan protects cartilage collagen from proteolytic cleavage. J Biol Chem. 2003;278(46):45539–45545. | |
Stattin EL, Wiklund F, Lindblom K, et al. A missense mutation in the aggrecan C-type lectin domain disrupts extracellular matrix interactions and causes dominant familial osteochondritis dissecans. Am J Hum Genet. 2010;86(2):126–137. | |
Jayasuriya CT, Chen Q. Cartilage extracellular matrix integrity and OA, In: Rothschild BM, editor. Principles of osteoarthritis – Its definition, character, derivation and modality-related recognition [Internet]. InTech; 2012. Available from: http://www.intechopen.com/books/principles-of-osteoarthritis-its-definition-character-derivation-and-modality-related-recognition/cartilage-extracellular-matrix-integrity-and-oa. | |
Lorenzo P, Aspberg A, Onnerfjord P, Bayliss MT, Neame PJ, Heinegard D. Identification and characterization of asporin. A novel member of the leucine-rich repeat protein family closely related to decorin and biglycan. J Biol Chem. 2001;276(15):12201–12211. | |
Nakajima M, Kizawa H, Saitoh M, Kou I, Miyazono K, Ikegawa S. Mechanisms for asporin function and regulation in articular cartilage. J Biol Chem. 2007;282(44):32185–32192. | |
Hecht JT, Hayes E, Haynes R, Cole WG. COMP mutations, chondrocyte function and cartilage matrix. Matrix Biol. 2005;23(8):525–533. | |
Posey KL, Hecht JT. The role of cartilage oligomeric matrix protein (COMP) in skeletal disease. Curr Drug Targets. 2008;9(10):869–877. | |
Mu SC, Lin YJ, Liu HC, et al. A mutation in cartilage oligomeric matrix protein (COMP) causes early-onset osteoarthritis in a large kindred study. Ann Hum Genet. 2011;75(5):575–583. | |
Mabuchi A, Ikeda T, Fukuda A, et al. Identification of sequence polymorphisms of the COMP (cartilage oligomeric matrix protein) gene and association study in osteoarthrosis of the knee and hip. J Hum Genet. 2001;46(8):456–462. | |
Klatt AR, Becker AK, Neascu CD, Paulsson M, Weganer R. The matrilins: modulators of extracellular matrix assembly. Int J Biochem Cell Biol. 2011;43(3):320–330. | |
Otten C, Hansen U, Talke A, Weganer R, Paulsson M, Zaucke F. A matrilin-3 mutation associated with osteoarthritis does affect collagen affinity but promotes the formation of wider cartilage collagen fibrils. Hum Mutat. 2010;31(3):254–263. | |
Min JL, Meulenbelt I, Riyazi N, et al. Association of matrilin-3 polymorphisms with spinal disc degeneration and osteoarthritis of the first carpometacarpal joint of the hand. Ann Rheum Dis. 2006;65(8):1060–1066. | |
Gu J, Rong J, Guan F, et al. MATN3 gene polymorphism is associated with osteoarthritis in Chinese Han population: a community-based case-control study. ScientificWorldJournal. 2012;2012:656084. | |
van der Weyden L, Wei L, Luo J, et al. Functional knockout of the matrilin-3 gene causes premature chondrocyte maturation to hypertrophy and increases bone mineral density and osteoarthritis. Am J Pathol. 2006;169(2):515–527. | |
Waller KA, Zhang LX, Elsaid KA, Fleming BC, Warman ML, Jay GD. Role of lubricin and boundary lubrication in the prevention of chondrocyte apoptosis. Proc Natl Acad Sci U S A. 2013;110(15):5852–5857. | |
Bao JP, Chen WP, Wu LD. Lubricin: a novel potential biotherapeutic approaches for the treatment of osteoarthritis. Mol Biol Rep. 2011;38(5):2879–2885. | |
Hochberg MC, Yerges-Armstrong L, Yau M, Mitchell BD. Genetic epidemiology of osteoarthritis: recent developments and future directions. Curr Opin Rheumatol. 2013;25(2):192–197. | |
Valdes AM, Evangelou E, Kerkhof HJ, et al. The GDF5 rs143383 polymorphism is associated with osteoarthritis of the knee with genome-wide statistical significance. Ann Rheum Dis. 2011;70(5):873–875. | |
Evangelou E, Valdes AM, Kerkhof HJ, et al. Meta-analysis of genome-wide association studies confirms a susceptibility locus for knee osteoarthritis on chromosome 7q22. Ann Rheum Dis. 2011;70(2):349–355. | |
Day-Williams AG, Southam L, Panoutsopoulou K, et al. A variant in MCF2L is associated with osteoarthritis. Am J Hum Genet. 2011; 89(3):446–450. | |
arcOGEN Consortium; arcOGEN Collaborators. Identification of new susceptibility loci for osteoarthritis (arcOGEN): a genome-wide association study. Lancet. 2012;380(9844):815–823. | |
Panoutsopoulou K, Southam L, Elliott KS, et al. Insights into the genetic architecture of osteoarthritis from stage 1 of the arcOGEN study. Ann Rheum Dis. 2011;70(5):864–867. | |
Loughlin J. Polymorphism in signal transduction is a major route through which osteoarthritis susceptibility is acting. Curr Opin Rheumatol. 2005;17(5):629–633. | |
Mototani H, Mabuchi A, Saito S, et al. A functional single nucleotide polymorphism in the core promoter region of CALM1 is associated with hip osteoarthritis in Japanese. Hum Mol Genet. 2005;14(8):1009–1017. | |
Ikegawa S. New gene associations in osteoarthritis: what do they provide, and where are we going? Curr Opin Rheumatol. 2007;19(5):429–434. | |
Dai J, Ikegawa S. Recent advances in association studies of osteoarthritis susceptibility genes. J Hum Genet. 2010;55(2):77–80. | |
Cornelis FM, Luyten FP, Lories RJ. Functional effects of susceptibility genes in osteoarthritis. Discov Med. 2011;12(63):129–139. | |
Bos SD, Slagboom PE, Meulenbelt I. New insights into osteoarthritis: early developmental features of an ageing-related disease. Curr Opin Rheumatol. 2008;20(5):553–559. | |
Valdes AM, Van Oene M, Hart DJ, et al. Reproducible genetic associations between candidate genes and clinical knee osteoarthritis in men and women. Arthritis Rheum. 2006;54(2):533–539. | |
Loughlin J, Sinsheimer JS, Carr A, Chapman K. The CALM1 core promoter polymorphism is not associated with hip osteoarthritis in a United Kingdom Caucasian population. Osteoarthritis Cartilage. 2006;14(3):295–298. | |
Shi D, Ni H, Dai J, et al. Lack of association between the CALM1 core promoter polymorphism (-16C/T) and susceptibility to knee osteoarthritis in a Chinese Han population. BMC Med Genet. 2008;9:91. | |
Poulou M, Kaliakatsos M, Tsezou A, Kanavakis E, Malizos KN, Tzetis M. Association of the CALM1 core promoter polymorphism with knee osteoarthritis in patients of Greek origin. Genet Test. 2008;12(2):263–265. | |
Shin MH, Lee SJ, Kee SJ, et al. Genetic association analysis of GDF5 and ADAM12 for knee osteoarthritis. Joint Bone Spine. 2012;79(5):488–491. | |
Chen X, Zankl A, Niroomand F, et al. Upregulation of ID protein by growth and differentiation factor 5 (GDF5) through a smad-dependent and MAPK-independent pathway in HUVSMC. J Mol Cell Cardiol. 2006;41(1):26–33. | |
Liu Z, Shen J, Pu K, et al. GDF5 and BMP2 inhibit apoptosis via activation of BMPR2 and subsequent stabilization of XIAP. Biochim Biophys Acta. 2009;1793(12):1819–1827. | |
Lewis AC, Malemud CJ. Correction of dysfunctional apoptosis in arthritis by pharmacologic interventions: Focus on altering the activity of inhibitor of apoptosis protein. In: Pandalai SG, editor. Recent Research Developments in Pharmacology. Kerala: Research Signpost; 2011. p. 69–84. | |
Zaidi SH, Huang Q, Momen A, Riazi A, Husain M. Growth differentiation factor 5 regulates cardiac repair after myocardial infarction. J Am Coll Cardiol. 2010;55(2):135–143. | |
Ratnayake M, Plöger F, Santibanez-Koref M, Loughlin J. Human chondrocytes respond discordantly to the protein encoded by the osteoarthritis susceptibility gene GDF5. PLoS One. 2014;9(1):e86590. | |
Dodd AW, Rodriguez-Fontenia C, Calaza M, et al. Deep sequencing of GDF5 reveals the absence of rare variants at this important osteoarthritis susceptibility locus. Osteoarthritis Cartilage. 2011;19(4):430–434. | |
Malemud CJ, Stevenson S, Mehraban F, Papay RS, Purchio AF, Goldberg VM. The proteoglycan synthesis repertoire of rabbit chondrocytes maintained in Type II collagen gels. Osteoarthritis Cartilage. 1994;2(1):29–42. | |
Enochson L, Stenberg J, Brittberg M, Lindahl A. GDF5 reduces MMP13 expression in human chondrocytes via DKK1 mediated canonical Wnt signaling inhibition. Osteoarthritis Cartilage. 2014;22(4):566–577. | |
Miyaki S, Nakasa T, Otsuki S, et al. MicroRNA-140 is expressed in differentiated human articular chondrocytes and modulates interleukin-1 responses. Arthritis Rheum. 2009;60(9):2723–2730. | |
Swingler TE, Wheeler G, Carmont V, et al. The expression and function of microRNAs in chondrogenesis and osteoarthritis. Arthritis Rheum. 2012;64(6):1909–1919. | |
Zhang Y, Jia J, Yang S, Liu X, Ye S, Tian H. MicroRNA-21 controls the development of osteoarthritis by targeting GDF-5 in chondrocytes. Exp Mol Med. 2014;46:e79. | |
Malemud CJ. Repair of injury to articular cartilage with chondrocyte progenitor cells. Rheumatol Curr Res. 2013;3(2):122. | |
Cheng X, Yang T, Meng W, Liu H, Zhang T, Shi R. Overexpression of GDF5 through an adenovirus vector stimulates osteogenesis of human mesenchymal stem cells in vitro and in vivo. Cells Tissues Organs. 2012;196(1):56–67. | |
Jin L, Li X. Growth differentiation factor 5 regulation in bone regeneration. Curr Pharm Des. 2013;19(19):3364–3373. | |
Daans M, Luyten FP, Lories RJ. GDF5 deficiency in mice is associated with instability-driven joint damage, gait and subchondral bone changes. Ann Rheum Dis. 2011;70(1):208–213. | |
Ikegawa S. The genetics of common degenerative skeletal disorders: osteoarthritis and degenerative disc disease. Annu Rev Genomics Hum Genet. 2013;14:245–256. | |
Miyamoto Y, Mabuchi A, Shi D, et al. A functional polymorphism in the 5′ UTR of GDF5 is associated with susceptibility to osteoarthritis. Nat Genet. 2007;39(4):529–533. | |
Evangelou E, Chapman K, Meulenbelt I, et al. Large-scale analysis of the association between GDF5 and FRZB variants and osteoarthritis of the hip, knee and hand. Arthritis Rheum. 2009;60(6):1710–1721. | |
Egli RJ, Southam L, Wilkins JM, et al. Functional analysis of the osteoarthritis susceptibility-associated GDF5 regulatory polymorphism. Arthritis Rheum. 2009;60(7):2055–2064. | |
Dodd AW, Syddall CM, Loughlin J. A rare variant in the osteoarthritis-associated locus GDF5 is functional and reveals a site that can be manipulated to modulate GDF5 expression. Eur J Hum Genet. 2013;21(5):517–521. | |
Reynard LN, Bui C, Canty-Laird EG, Young DA, Loughlin J. Expression of the osteoarthritis-associated gene GDF5 is modulated epigenetically by DNA methylation. Hum Mol Genet. 2011;20(17):3450–3460. | |
Valdes AM, Spector TD, Doherty S, Wheeler M, Hart DJ, Doherty M. Association of the DVWA and GDF5 polymorphisms with osteoarthritis in UK populations. Ann Rheum Dis. 2009;68(12):1916–1920. | |
Hao SW, Jin QH. Association between the +104T/C polymorphism in the 5′ UTR of GDF5 and susceptibility to knee osteoarthritis: a meta-analysis. Mol Med Rep. 2013;7(2):485–488. | |
Tsezou A, Satra M, Oikonomou P, Bargiotas K, Malizos KN. The growth differentiation factor 5 (GDF-5) core promoter polymorphism is not associated with knee osteoarthritis in the Greek population. J Orthop Res. 2008;26(1):136–140. | |
Tawonsawatruk T, Changthong T, Pingsuthiwong S, Trachoo O, Sura T, Wajanavisit W. A genetic association study between growth differentiation factor 5 (GDF5) polymorphism and knee osteoarthritis in Thai population. J Orthop Surg Res. 2011;6:47. | |
Logan CW, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. | |
Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis. 2008;4(2):68–75. | |
Nusse R, Varmus H. Three decades of Wnts: a personal perspective on how a scientific field developed. EMBO J. 2012;31(12):2670–2684. | |
Habas R, Dawid IB. Dishevelled and Wnt signaling: is the nucleus the final frontier? J Biol. 2005;4(1):2. | |
Schett G, Zwerina J, David JP. The role of Wnt proteins in arthritis. Nat Clin Pract Rheumatol. 2008;4(9):473–480. | |
Wang M, Shen J, Jin H, Im HJ, Sandy J, Chen D. Recent progress in understanding molecular mechanisms of cartilage degeneration during osteoarthritis. Ann N Y Acad Sci. 2011;1240:61–69. | |
Blom AB, Brockbank SM, van Lent PL, et al. Involvement of the Wnt signaling pathway in experimental and human osteoarthritis: prominent role of Wnt-induced signaling protein-1. Arthritis Rheum. 2009;60(2):501–512. | |
Pulsatelli L, Addimanda O, Brusi V, Pavloska B, Meliconi R. New findings in osteoarthritis pathogenesis: therapeutic implications. Ther Adv Chronic Dis. 2013;4(1):23–43. | |
Nalesso G, Sherwood J, Bertrand J, et al. WNT-3A modulates articular chondrocyte phenotype by activating both canonical and noncanonical pathways. J Cell Biol. 2011;193(3):551–564. | |
Zhu M, Chen M, Zuscik M, et al. Inhibition of β-catenin signaling in articular chondrocytes results in articular cartilage destruction. Arthritis Rheum. 2008;58(7):2053–2064. | |
Schroeppel JP, Crist JD, Anderson HC, Wang J. Molecular recognition of articular chondrocyte function and its significance in osteoarthritis. Histol Histopathol. 2011;26(3):377–394. | |
Pasold J, Osterberg A, Peters K, et al. Reduced expression of Sfrp1 during chondrogenesis and in articular chondrocytes correlates with osteoarthritis in STR/ort mice. Exp Cell Res. 2013;319(5):649–659. | |
Killock D. Osteoarthritis: Frzb knockout reveals the complexity of Wnt signaling in joint homeostasis. Nat Rev Rheumatol. 2012;8(3):123. | |
Thysen S, Cailotto F, Luyten FP, Lories RJ. A5.12 SFRPS in cartilage biology: more than just WNT antagonists. Ann Rheum Dis. 2014;73:A67–A68. | |
Lories RJ, Corr M, Lane NE. To Wnt or not to Wnt: the bone and joint health dilemma. Nat Rev Rheumatol. 2013;9(6):328–339. | |
James IE, Kumar S, Barnes MR, et al. FrzB-2: a human secreted frizzled-related protein with a potential role in chondrocyte apoptosis. Osteoarthritis Cartilage. 2000;8(6):452–463. | |
Leijten JC, Emons J, Sticht C, et al. Gremlin 1, frizzled-related protein and Dkk-1 are key regulators of human articular cartilage homeostasis. Arthritis Rheum. 2012;64(10):3302–3312. | |
Leijten JC, Bos SD, Landman EB, et al. GREM1, FRZB and DKK1 mRNA levels correlate with osteoarthritis and are regulated by osteoarthritis-associated factors. Arthritis Res Ther. 2013;15(5):R126. | |
Ma B, Landman EB, Miclea RL, et al. WNT signaling and cartilage: of mice and men. Calcif Tissue Res. 2013;92(5):399–411. | |
Garcia-Ibarbia C, Delgado-Calle J, Casafont I, et al. Contribution of genetic and epigenetic mechanisms to Wnt pathway activity in prevalent skeletal disorders. Gene. 2013;532(2):165–172. | |
Loughlin J, Dowling B, Chapman K, et al. Functional variants within the secreted frizzled-related protein 3 gene are associated with hip osteoarthritis in females. Proc Natl Acad Sci U S A. 2004;101(26):9757–9762. | |
Lane NE, Lian K, Nevitt MC, et al. Frizzled-related protein variants are risk factors for hip osteoarthritis. Arthritis Rheum. 2006;54(4):1246–1254. | |
Rodriguez-Lopez J, Pombo-Suarez M, Liz M, Gomez-Reino JJ, Gonzalez A. Further evidence of the role of frizzled-related protein gene polymorphisms in osteoarthritis. Ann Rheum Dis. 2007;66(8):1052–1055. | |
Snelling S, Ferreira A, Loughlin J. Allelic expression analysis suggests that cis-acting polymorphism of FRZB expression does not contribute to osteoarthritis susceptibility. Osteoarthritis Cartilage. 2007;15(1):90–92. | |
Valdes AM, Loughlin J, Oene MV, et al. Sex and ethnic differences in the association of ASPN, CALM1, COL2A1, COMP and FRZB with genetic susceptibility to osteoarthritis of the knee. Arthritis Rheum. 2007;56(1):137–146. | |
Baker-Lepain JC, Lynch JA, Parimi N, et al. Variant alleles of the Wnt antagonist FRZB are determinants of hip shape and modify the relationship between hip shape and osteoarthritis. Arthritis Rheum. 2012;64(5):1457–1465. | |
Velasco J, Zarrabeitia MT, Prieto JR, et al. Wnt pathway genes in osteoporosis and osteoarthritis: differential and genetic association study. Osteoporos Int. 2010;21(1):109–118. | |
Lane NE, Nevitt MC, Lui LY, et al. Wnt signaling antagonists are potential prognostic biomarkers for the progression of radiographic hip osteoarthritis in elderly Caucasian women. Arthritis Rheum. 2007;56(10):3319–3325. | |
Arrojo E, Drigo R, Bianco AC. Type II deiodinase at the crossroads of thyroid hormone action. Int J Biochem Cell Biol. 2011;43(10):1432–1441. | |
Gereben B, Zavacki AM, Ribich S, et al. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev. 2008;29(7):898–938. | |
Cheng AW, Bolognesi M, Kraus VB. DIO2 modifies inflammatory responses in chondrocytes. Osteoarthritis Cartilage. 2012;20(5):440–445. | |
Nagase H, Nagasawa Y, Tachida Y, et al. Deiodinase 2 upregulation demonstrated in osteoarthritis patients cartilage causes cartilage destruction in tissue-specific transgenic mice. Osteoarthritis Cartilage. 2013;21(3):514–523. | |
Van der Kraan PM, van den Berg WB. Chondrocyte hypertrophy and osteoarthritis: role in initiation and progression of cartilage degeneration. Osteoarthritis Cartilage. 2012;20(3):223–232. | |
Goldring MB. Insight into the function of DIO2, a susceptibility gene in human osteoarthritis, as an inducer of cartilage damage in a rat model: Is there a role for chondrocyte hypertrophy? Osteoarthritis Cartilage. 2013;21(5):643–645. | |
Brew CJ, Clegg PD, Boot-Handford RP, Andrew JG, Hardingham T. Gene expression in human chondrocytes in late osteoarthritis is changed in both fibrillated and intact cartilage without evidence of generalised chondrocyte hypertrophy. Ann Rheum Dis. 2010;69(1):234–240. | |
Meulenbelt I, Min JL, Bos S, et al. Identification of DIO2 as a new susceptibility locus for symptomatic osteoarthritis. Human Mol Genet. 2008;17(12):1867–1875. | |
Bos SD, Bovée JV, Duijnisveld BJ, et al. Increased type II deiodinase protein in OA-affected cartilage and allelic imbalance of OA risk polymorphism rs225014 at DIO2 in human joint tissues. Ann Rheum Dis. 2012;71(7):1254–1258. | |
Rodriguez-Fontenla C, Calaza M, Evangelou E, et al. Assessment of osteoarthritis candidate genes in a meta-analysis of nine genome-wide association studies. Arthritis Rheum. 2014;66(4):940–949. | |
Valdes AM, Spector TD. The clinical relevance of genetic susceptibility to osteoarthritis. Best Pract Res Clin Rheumatol. 2010;24(1):3–14. | |
Foussard H, Ferrer P, Valenti P, Polesello C, Carreno S, Payne F. LRCH proteins: a novel family of cytoskeletal regulators. PLoS One. 2010;5(8):e12257. | |
Spector TD, Reneland RH, Mah S, et al. Association between a variation in LRCH1 and knee osteoarthritis: a genome-wide single-nucleotide polymorphism association study using DNA pooling. Arthritis Rheum. 2006;54(2):524–532. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.