")
Back to Journals » Clinical Ophthalmology » Volume 10
Clinical presentation and visual status of retinitis pigmentosa patients: a multicenter study in southwestern Nigeria
Authors Onakpoya O, Adeoti C, Oluleye T, Ajayi I, Majengbasan T, Olorundare O
Received 6 March 2016
Accepted for publication 12 April 2016
Published 22 August 2016 Volume 2016:10 Pages 1579—1583
DOI https://doi.org/10.2147/OPTH.S107890
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Oluwatoyin Helen Onakpoya,1 Caroline Olufunlayo Adeoti,2 Tunji Sunday Oluleye,3 Iyiade Adeseye Ajayi,4 Timothy Majengbasan,4,5 Olayemi Kolawole Olorundare1
1Department of Ophthalmology, Obafemi Awolowo University Teaching Hospital, Ile-Ife, 2Department of Ophthalmology, Ladoke Akintola University of Technology Teaching Hospital, Osogbo, 3Department of Ophthalmology, University College Hospital, Ibadan, 4Department of Ophthalmology, University Teaching Hospital, Ado-Ekiti, 5Department of Ophthalmology, Federal Medical Centre, Ido-Ekiti, Nigeria
Background: To review the visual status and clinical presentation of patients with retinitis pigmentosa (RP).
Methodology: Multicenter, retrospective, and analytical review was conducted of the visual status and clinical characteristics of patients with RP at first presentation from January 2007 to December 2011. Main outcome measure was the World Health Organization’s visual status classification in relation to sex and age at presentation. Data analysis by SPSS (version 15) and statistical significance was assumed at P<0.05.
Results: One hundred and ninety-two eyes of 96 patients with mean age of 39.08±18.5 years and mode of 25 years constituted the study population; 55 (57.3%) were males and 41 (42.7%) females. Loss of vision 67 (69.8%) and night blindness 56 (58.3%) were the leading symptoms. Twenty-one (21.9%) patients had a positive family history, with RP present in their siblings 15 (71.4%), grandparents 11 (52.3%), and parents 4 (19.4%). Forty (41.7%) were blind at presentation and 23 (24%) were visually impaired. Blindness in six (15%) patients was secondary to glaucoma. Retinal vascular narrowing and retinal pigmentary changes of varying severity were present in all patients. Thirty-five (36.5%) had maculopathy, 36 (37.5%) refractive error, 19 (20%) lenticular opacities, and eleven (11.5%) had glaucoma. RP was typical in 85 patients (88.5%). Older patients had higher rates of blindness at presentation (P=0.005); blindness and visual impairment rate at presentation were higher in males than females (P=0.029).
Conclusion: Clinical presentation with advanced diseases, higher blindness rate in older patients, sex-related difference in blindness/visual impairment rates, as well as high glaucoma blindness in RP patients requires urgent attention in southwestern Nigeria.
Keywords: retinitis pigmentosa, blindness, glaucoma, visual impairment, Nigeria
Introduction
Retinitis pigmentosa (RP) is a group of hereditary retinal degenerative diseases affecting the photoreceptor cells and retinal pigment epithelium.1 The retinal dystrophy of RP causes visual impairment in all age groups.2 It is in fact one of the most frequent causes of blindness during working life in the industrialized nations.3
Globally, the prevalence of RP is about one in 4,000, with more than 1 million affected individuals worldwide.1 In Denmark, RP and optic neuropathy were the leading causes of blindness in individuals aged 20–64 years – each accounting for 29%.4 RP was the leading cause of visual disability in persons younger than 60 years of age in Kuwait.5
RP accounted for 0.69% of all new patients attending an eye outpatient department in Ibadan6 and 5.2% of all retinal diseases in Ile-Ife,7 both in southwestern Nigeria.
RP presents clinically with varied ocular symptoms; night blindness is usually the initial symptom followed by loss of visual field and visual acuity as the disease progresses.1,3,8 It evolves over several decades and final loss of central vision may not occur until the age of 60 years.1,9 Bilateral blindness of 30% and 50% in RP patients at presentation has been reported in single-center studies in sub-Saharan Africa.8,10 The typical variant of RP remains the most prevalent type globally; atypical RP and RP associated with systemic disorders do occur, although they are not as common as the typical variant.6,11
There is no RP register in Nigeria currently and the available population-based eye surveys are laden with the burden of causes of preventable blindness like cataract and glaucoma,12,13 both of which are highly prevalent in patients with RP compared with the general population.6 Considering that the burden of visual loss from RP occurs during working age, this study was carried out to determine the epidemiology, clinical presentation as well as the degree of visual loss from RP at their first clinic attendance.
Materials and methods
It was a multicenter, cross-sectional study involving five ophthalmic units/departments in tertiary hospitals all located in southwestern Nigeria: these are Obafemi Awolowo University Teaching Hospital, Ile-Ife; Ladoke Akintola University of Technology Teaching Hospital, Osogbo; University College Hospital, Ibadan; University Teaching Hospital, Ado-Ekiti; and Federal Medical Centre, Ido-Ekiti. All patients with a diagnosis of RP at first presentation from January 2007 to December 2011 constituted the study population. The study was performed according to the guidelines of the Declaration of Helsinki and approved by the Ethics and Research Committee of Ladoke Akintola University of Technology. Written informed patient consent was not sought as this was a retrospective study.
The pro forma for the study was developed by the authors based on the definition and clinical diagnostic criteria for RP, which are combinations of arteriolar narrowing/attenuation, retina pigments, optic atrophy, and night blindness. History obtained from the files of all the patients included presenting complaints, family history of RP and use of refractive spectacles. Eye examinations included visual acuity assessment, refraction, tonometry (applanation), slit lamp examination, and dilated funduscopy. One center had a fundus camera, so fundus photographs were available for a few patients. The age, sex, symptoms, visual acuity, intraocular pressure, retinal findings, and diagnosis on the first presentation to the eye outpatient departments were extracted from the patients’ records. The visual acuity was classified based on the World Health Organization’s category of vision14 as follows: visual acuity worse than 3/60 in the better eye was regarded as blind, from 3/60 to worse than 6/18 in the better eye was classified as visual impairment, while visual acuity of 6/18 and better in the better eye was classified as normal.
The main outcome measure was the World Health Organization’s visual status classification in relation to sex and age at presentation. The pattern of clinical presentation and causes of visual impairment and blindness were secondary outcome measures. Data analysis by SPSS, version 15 (SPSS Inc., Chicago, IL, USA) was carried out for univariate and multivariate analysis. Cross-tabulations were used to compare variables using chi square, and statistical significance was assumed at P<0.05.
Results
One hundred and ninety-two eyes of 96 patients constituted the study population, 55 (57.3%) of whom were males and 41 (42.7%) were females, giving a male to female ratio of 1.3:1. The mean age at presentation was 39.08±18.5 years, mode of 25 years. Majority (79.2%) of the patients were in the working age group, that is, young adults (16–44 years) and middle age (45–64 years) (Table 1).
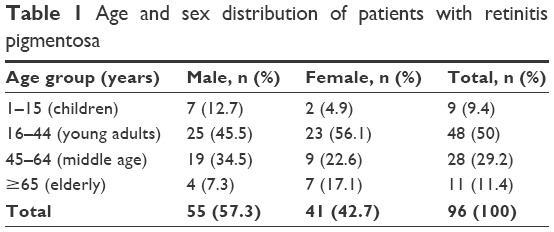 | Table 1 Age and sex distribution of patients with retinitis pigmentosa |
Of 96 patients, 19 (19.8%) had completed primary education, 29 (30.2%) secondary, 33 (34.4%) tertiary education, while 15 (15.6%) had no formal education. Loss of vision (69.8%) and night blindness (58.3%) were the leading presenting complaints. Positive family history of RP was elicited in 21 (21.9%) patients; present most commonly in siblings of 15 (71.4%) patients. Only one family member was involved in ten (47.6%) patients; two in nine (42.9%), and three in two (9.5%) patients. Refractive error was present in 36 (37.5%) patients, with myopic astigmatism being the most common (55.6%) type. The crystalline lenses were clear and in situ in both eyes of 77 (80.2%) patients and bilateral posterior subcapsular cataract in seven (7.3%) patients. Two patients (2.1%) had unilateral pseudophakia and one (1.04%) had unilateral couched cataractous lens in the vitreous; these three patients also had lenticular opacities in the fellow eye. All patients had retinal vascular narrowing and pigmentation. Bone spicule pigmentation in the mid-periphery was the most common variant seen in more than half of the patients while 35 (36.5%) had maculopathy (Table 2). Posterior vitreous detachment was present in seven (7.2%) and primary open angle glaucoma was seen in eleven (11.5%) patients. RP was of the typical variant in 85 (88.5%) while the atypical forms were sector RP in three (3.1%) patients. Usher syndrome was seen in two (2.1%), punctata albescens in one (1.05%), and Bardet–Biedl syndrome in one (1.05%) patients.
 | Table 2 Clinical characteristics of retinitis pigmentosa patients at presentation |
Forty (41.7%) patients were blind at first clinical presentation (Figure 1). Blindness and visual impairment at first clinical presentation were higher in males compared with female patients, P=0.030 (Figure 2). There was a statistically significant increase in the percentage of RP patients who presented with blindness with increasing age group (Figure 3). The mean age of RP patients based on the World Health Organization’s category of normal vision in the better eye was 32.31±15.9 years, visual impairment was 34.35±18.7 years while persons with blindness in the better eye had a mean age of 47.6±18.4 years (P=0.001). The causes of blindness were primarily RP in 31 (77.5%), glaucoma in six (15%), operable cataract in two (5%), and central corneal opacity in one (2.5%) RP blind patients.
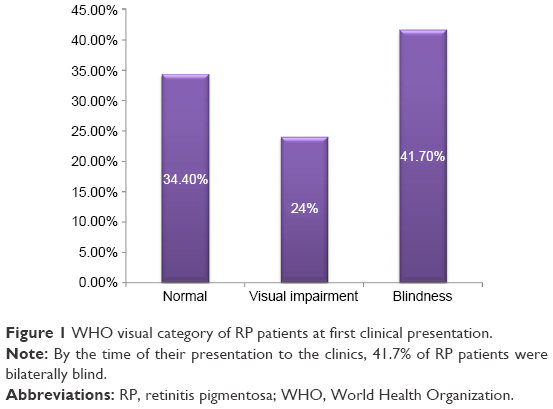 | Figure 1 WHO visual category of RP patients at first clinical presentation. |
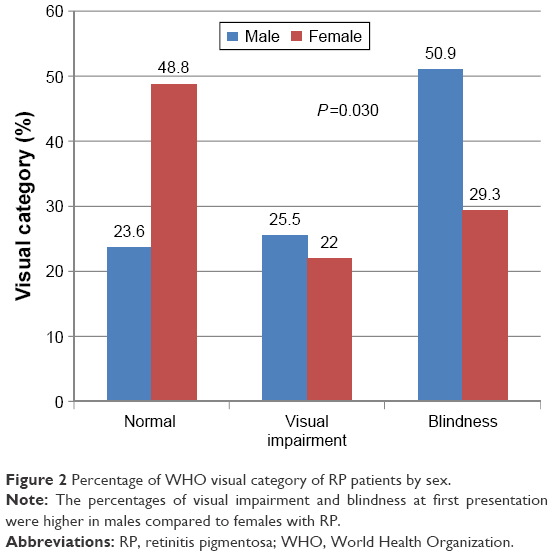 | Figure 2 Percentage of WHO visual category of RP patients by sex. |
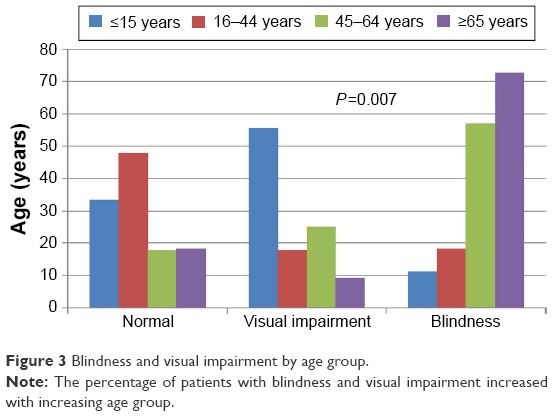 | Figure 3 Blindness and visual impairment by age group. |
Discussion
RP is not a very common disease.9 The mean age of 39 years in this study is similar to the 36.7 years reported in a single center study from southern Nigeria8 and the 35.1 years in Japan.15 Eballe et al,10 however, reported a higher mean age of 43.3 years in a Cameroonian hospital; late presentation for eye care was the reason reported for the high mean age in that study. The age of onset usually denotes the age of initial symptoms of RP; it may be influenced by the pattern of utilization of existing eye care facility, as well as individual differences in the level of awareness of their eye problems.1,8,10 The age of onset of symptoms remains an imprecise measure of RP severity because of variations in the awareness of individuals of their visual problems.1 The mode of inheritance as well as the variant may affect both the rate of disease progression and age at presentation.9,16 RP affects the working age group.4 Nearly 80% (79.2%) of the RP patients in the current study were aged between 16 and 64 years.
Reduction in vision was the leading (69.8%) symptom that prompted RP patients to seek medical consultation; this is similar to the previous reports.8,10 Reduction in vision was present in 85% and 90% of RP patients in previous hospital-based reports in Cameroon and Benin City, Nigeria, respectively.8,10 These reports in Cameroon and Benin City Nigeria are much higher than the 69.8% obtained in our series. Night blindness is the earliest symptom of RP.9 This was the second most common symptom in this study. Visual acuity could remain normal until the very advanced stage of RP; usually, night blindness is the earliest symptom, followed by visual field loss prior to visual acuity loss.1,9 Aside from late presentation, other nonclinical socio-environmental factors can affect the reported presence of night blindness. Presence of electrically illuminated nighttime environment sufficient for intended activity, residence in communities with very minimal nighttime activities, as well as age of onset being too young to appreciate and complain of night blindness could downplay the presence of symptoms.1,10
Visual impairment (24%) and blindness (41.7%) prevalence rates in RP patients studied are quite high. Grover et al17 reported a much lower blindness rate of 25% in the US,17 while 30% blindness rate was reported in Cameroon.10 Visual impairment and blindness have significant negative impact on the quality of life in any individual.18 Visual loss was categorized in this study based on visual acuity without consideration of visual field; thus underestimating visual impairment and blindness rate since RP has a profound effect on the visual field as the disease progresses.1,9,11 The economic impact of visual loss in RP patients is more disturbing, particularly as it occurs in individuals in the working age group and is a burden for individuals, families, and societies to bear.19 The prevalence of visual loss in this cohort increased with increasing age; RP is a progressive disease and visual loss tends to occur as the disease progresses.9,10,19 Cataract and glaucoma are the leading causes of blindness in Nigeria;12 glaucoma was the cause of blindness in 15% of the RP blind, this was second only to RP itself. The prevalence of glaucoma (11%) and lens opacity/pseudophakia (10.4%) among the RP patients studied is high. Previous reports have documented an increase in the prevalence of these disorders in RP patients.8,10,20,21 However, the glaucoma rate is higher than the 2.3% in People’s Republic of China21 and 7.5% in Cameroon.10 The usefulness of appropriate low vision devices for visually impaired RP patients cannot be overemphasized.9,22 RP was responsible for 16.6% of low vision patients in a new low vision aid center in southwestern Nigeria.23 More of such centers in this region will be beneficial.
Family history was documented in 21.9% of RP patients; this affected mainly siblings (71.4%). RP is a genetic disease with varying modes of inheritance, and family/genetic studies remain an important part of diagnosis and counseling. Detailed family tracing and examination were not done for the patients studied; this limitation as well as the absence of genetic studies prevents adequate reporting of the mode of inheritance. Large families and nonwillingness of relatives to attend ophthalmologic consultations due to various reasons are documented.10 This multicenter retrospective study is also limited by the absence of electrophysiological and visual field investigations largely due to their nonavailability in most parts of the region at the time of study.
In conclusion, severe visual morbidity is prevalent at first clinic presentation of RP patients in southwestern Nigeria. This is sometimes in addition to the more prevalent glaucoma and cataract. Education about the disease is necessary to enhance early presentation, prevent avoidable blindness, and enhance rehabilitation. Infrastructure for visual field analysis, electrophysiological tests, low vision aids, and genetic studies will enhance the local capacity for diagnosis and management of RP in the region.
Disclosure
The authors report no conflicts of interest in this work.
References
Harton DT, Berson EI, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. | ||
Wan M, Lin H, Bai Y, et al. Clinical evidence in concurrence of retinitis pigmentosa and glaucoma. Chin Med J. 2011;124:1270–1274. | ||
Parmegigiani F. Clinics, epidemiology and genetics of retinitis pigmentosa. Curr Genomics. 2011;2:236–237. | ||
Buch H, Vinding T, La Cour M, et al. Prevalence and causes of visual impairment and blindness among 9980 Scandinavian adults: the Copenhagen City Eye study. Ophthalmology. 2004;111:53–61. | ||
Al Merjan JI, Pandova MG, Al Ghanim M, et al. Registered blindness and low vision in Kuwait. Ophthalmic Epidemiol. 2005;12:251–257. | ||
Ashaye AO. Presumed hereditary retina degenerations: Ibadan experience. West Afr J Med. 2005;24:49–53. | ||
Onakpoya OH, Olateju SO, Ajayi IA. Retina diseases in a tertiary hospital: the need for establishment of a vitreoretina care unit. J Natl Med Assoc. 2008;100:1286–1289. | ||
Ukponmwan CU, Atamah A. Retinitis pigmentosa in Benin, Nigeria. East Afr Med J. 2004;81:254–257. | ||
Hammel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1:40. | ||
Eballe AO, Koki G, Emche CB, Bella LA, Kouam JM, Melong J. Blindness and visual impairment in retinitis pigmentosa: a Cameroonian hospital-based study. Clin Ophthalmol. 2010;4:661–665. | ||
Haim M. Epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol Scand Suppl. 2002;233:1–34. | ||
Abdull MM, Sivasubramaniam S, Murthy GVS, et al. Causes of blindness and visual impairment in Nigeria: the Nigeria national blindness and visual impairment survey. Invest Ophthalmol Vis Sci. 2009;50:4114–4118. | ||
Onakpoya OH, Adeoye AO, Akinsola FB, Adegbehingbe BO. Prevalence of blindness and visual impairment in Atakunmosa west local government area of southwestern Nigeria. Tanzan Health Res Bull. 2007;9:126–131. | ||
WHO. International Classification of Diseases and Related Problem. 10th Revision. Geneva: WHO; 1992;1:456–457. | ||
Tsujikawa M, WadaY, Sukegawa M, et al. Age at onset curves of retinitis pigmentosa. Arch Ophthalmol. 2008;126:337–340. | ||
Sandberg MA, Brockhurst RJ, Gaudio AR, et al. The association between visual acuity and central retinal thickness in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2005;46:3349–3354. | ||
Grover S, Fishman GA, Alexander KR, Anderson RJ, Derlacki DJ. Visual acuity impairment in patients with retinitis pigmentosa. Ophthalmology. 1996;103:1593–1600. | ||
Briensen S, Roberts H, Finger RP. The impact of visual impairment on health related quality of life in rural Africa. Ophthalmic Epidemiol. 2014;21(5):297–306. | ||
Marmor MF. Visual loss in retinitis pigmentosa. Am J Ophthalmol. 1990;89:692–698. | ||
Pruett RC. Retinitis pigmentosa: clinical observations and correlations. Trans Am Ophthalmol Soc. 1983;81:693–735. | ||
Peng DW. Retinitis pigmentosa associated with glaucoma. Zhongua Yan Ke Za Zhi. 1991;27:262–264. | ||
Mancil RM, Mancil GL, King E, et al. Improving nighttime mobility in persons with nightblindness caused by retinitis pigmentosa: A comparison of two low-vision mobility devices. J Rehabil Res Dev. 2005;42:471–486. | ||
Olusanya B, Onoja G, Ibraheem W, et al. Profile of patients presenting at a low vision clinic in a developing country. BMC Ophthalmol. 2012;12:31. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.