")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Clinical potential of vorapaxar in cardiovascular risk reduction in patients with atherosclerosis
Authors Diehl P, Bode C, Duerschmied D
Received 18 March 2015
Accepted for publication 28 May 2015
Published 3 August 2015 Volume 2015:11 Pages 1133—1138
DOI https://doi.org/10.2147/TCRM.S55469
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Philipp Diehl, Christoph Bode, Daniel Duerschmied
Department of Cardiology and Angiology I, Heart Center, University of Freiburg, Freiburg, Germany
Abstract: Vorapaxar (ZONTIVITY™, formerly known as SCH 530348) is a specific, orally active antagonist of the protease-activated receptor-1 (PAR-1) on platelets. It inhibits thrombin-induced platelet activation by binding to the ectodomain of PAR-1. After animal studies and Phase II studies showed that vorapaxar sufficiently inhibits platelet activation without significantly increasing bleeding complications, safety and efficacy of vorapaxar were assessed in two large multicenter trials in patients with coronary artery disease and atherosclerosis. The Thrombin-Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndromes (TRACER) trial investigated safety and efficacy of vorapaxar in patients with an acute coronary syndrome without ST-segment elevation. The Trial to Assess the Effects of Vorapaxar in Preventing Heart Attack and Stroke in Patients With Atherosclerosis-Thrombolysis In Myocardial Infarction 50 (TRA 2°P-TIMI 50) investigated atherothrombotic events in patients with stable atherosclerosis. Results of both studies suggested that vorapaxar given in addition to standard antiplatelet therapy can reduce atherothrombotic events, but increases the risk of mild and moderate bleeding complications. This review article summarizes the main results of TRACER and TRA 2°P-TIMI 50 and suggests patient cohorts that might benefit from treatment with vorapaxar in addition to standard antiplatelet therapy.
Keywords: vorapaxar, antiplatelet therapy, atherosclerosis, myocardial infarction
Protease activated receptors (PAR-1)
Platelet activation is a common phenomenon in several cardiovascular diseases and is of particular importance in patients with an acute coronary syndrome (ACS).1 Upon activation, platelets release several mediators (such as thromboxane A2, ADP, serotonin, plasminogen, PF4) that most often induce further platelet activation and aggregation, commonly resulting in platelet-rich thrombus formation, vessel occlusion, and ischemia of the downstream located tissue. One of the main platelet activators is thrombin, a serine protease of 36 kDa encoded in humans on the eleventh chromosome.2 Its function on the coagulation cascade is rather complex. However, it is evident that it catalyzes soluble fibrinogen to insoluble fibrin strands leading to platelet aggregation, and furthermore, cleaves a silencing ectodomain of the platelets protease activated receptors-1 (PAR-1). The unmasked new N-terminus of PAR-1 consecutively binds to an external binding site of PAR-1 and thereby induces receptor autoactivation (Figure 1). This interaction of the newly presented N-terminus to the binding site of its own receptor results in an increase of the cytosolic Ca2+ levels and a consecutive inhibition of the intracellular cyclic adenosine monophosphate (cAMP) concentration. This effect finally leads to activation of the platelet integrin glycoprotein IIb/IIIa with consecutive platelet aggregation and clot formation.
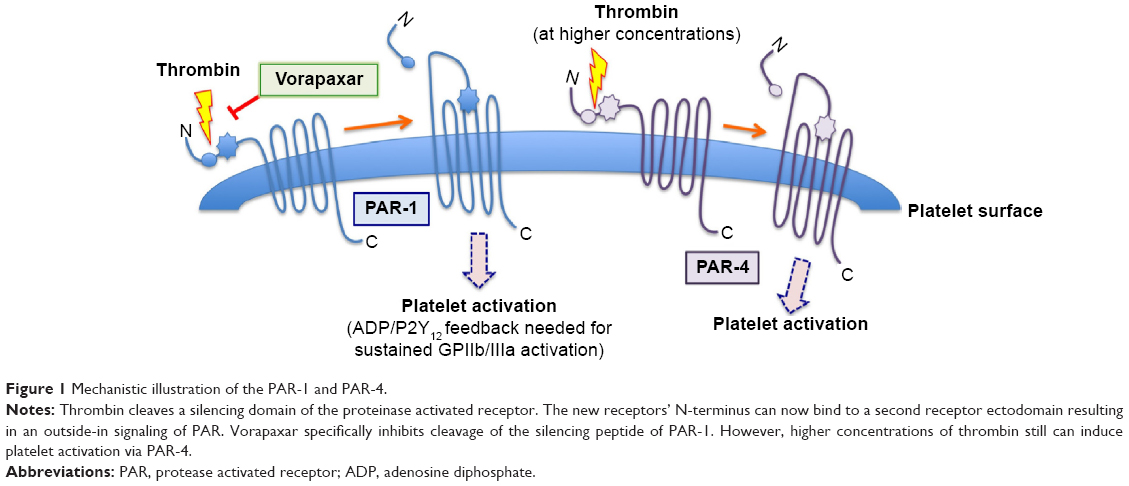 | Figure 1 Mechanistic illustration of the PAR-1 and PAR-4. |
PARs are a group of four different surface receptors (PAR-1–PAR-4), which are expressed on a variety of cells (platelets, endothelial cells, neurons).3 However, only the G-protein-coupled PAR-1 and PAR-4 are expressed on mature human platelets. At the end of the last century, it was found that PAR-1 has, in comparison to PAR-4, a particularly high affinity to thrombin (approximately 100-fold higher), and thus mediates the rapid thrombin-mediated platelet activation.4 Platelet activation, as can be found in several prothrombotic conditions, induces a massive increase of circulating thrombin resulting in platelet clot formation and often in infarction of the corresponding tissue. Hence, therapeutic inhibition of thrombin-mediated platelet activation was a new therapeutic approach in cardiovascular medicine. Supported by preclinical data showing that PAR-1 is mainly involved under circumstances of pathological platelet activation (such as atherothrombosis) and less in physiological hemostasis (to stop bleeding, requiring PAR-4 activation), the inhibition of PAR-1 was furthermore, believed to be associated with therapeutic antithrombotic effects, with a reduced risk for bleeding.5,6 These data were supported by a preclinical study in monkeys, which suggested that PAR-1 inhibition reduces the risk for thromboembolic complications without significantly affecting primary hemostasis.7
Vorapaxar: the first PAR-1 inhibitor that reached Phase III
Vorapaxar (ZONTIVITY™ [formerly known as SCH 530348] of Merck & Co., Kenilworth, NJ, USA) was the first in class antagonist of PAR-1 that reached clinical trials and was approved by the US Food and Drugs Administration (FDA) in 2014 for cardiovascular risk reduction. Vorapaxar is an ethyl carbamate based on the molecular structure of himbacine, which is an M20 alkaloid found in the bark of the Australian magnolia.8
Vorapaxar acts reversibly on the thrombin receptor PAR-1 and thereby inhibits thrombin-induced platelet activation. Other platelet activation pathways such as the thromboxane A2 or the ADP pathway are not impaired. Vorapaxar does not affect PAR-4, and high thrombin concentrations can thus still induce platelet clot formation. After oral intake, vorapaxar is hepatically metabolized to its major active agent M20 in a CYP450 dependant mechanism and peaks in serum after approximately 1 hour. Among all CYP450, CYP4503 A4 seems to have a major impact on hepatic vorapaxar metabolization.9 Hence, comedication with strong CYP450 inducers (such as phenytoin, carbamazepine) or inhibitors (such as clarithromycin, ketoconazole, nefazodone) or the presence of CYP-P4503A4 polymorphisms might influence the vorapaxar effect. Vorapaxar binds in blood to plasma proteins (>99% protein binding), preferentially to albumin, and is mainly hepatically cleared from plasma in a CYP3A4 and CYP2J2 manner before being excreted via feces (58%) and urine (25%). Due to the comparably long half-life time of vorapaxar (approximately 5–13 days) in relation to a platelet’s lifespan (approximately 8 days), vorapaxar affects platelet aggregation effectively irreversibly. Mild to moderate impairments of the liver function did not show significant changes of the vorapaxar effects. Nevertheless, vorapaxar should not be taken in case of severe hepatic insufficiency, as it might increase bleeding complications.10 Kosoglou et al11 investigated in a small clinical study the impact of patients with end-stage renal disease and hemodialysis on the vorapaxar bioavailability. They found that end-stage renal disease had no significant effect on pharmacokinetic profile of vorapaxar or its antiplatelet effect. However, besides metabolization and elimination rates it was recently published that blacks express more PAR-4 protein than Caucasians.12 Future clinical studies will need to investigate whether these differences are associated with altered antiplatelet effects.
Safety and efficacy of vorapaxar in patients with atherosclerosis
The safety of vorapaxar in patients with coronary artery disease (CAD) undergoing coronary angiography with nonurgent percutaneous coronary intervention (PCI) was first investigated in two Phase II studies.13,14 The results of these Phase II trials were promising as vorapaxar, given in addition to dual antiplatelet therapy, did not significantly increase bleeding complications, but reduced the risk of periprocedural myocardial infarction (MI). Driven by these results, vorapaxar was investigated in the Thrombin-Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndromes (TRACER) trail. TRACER investigated the safety profile of vorapaxar versus placebo in patients with an ACS but without ST-segment elevations.15 In this multicenter, double-blinded Phase III trial, 12,944 patients were randomized 1:1 into a vorapaxar group, in which patients obtained a loading dose of 40 mg followed by a maintenance dose of 2.5 mg daily for at least 1 year, or into a placebo cohort. Both, vorapaxar and placebo were given in addition to standard antiplatelet therapy. Exclusion criteria were, among others, a history of intracranial bleedings, any abnormal bleedings within the last 30 days, thrombocytopenia, and medication with oral anticoagulants such as warfarin or factor Xa inhibitors. The primary end point was defined as a composite of death due to cardiovascular diseases, MI, urgent coronary revascularization, or stroke. After a median follow-up time of 502 days (interquartile range from 349 to 667), 1,031 out of 6,473 patients from the vorapaxar group versus 1,102 out of 6,471 of the placebo group had reached the primary end point (Kaplan–Meier 2-year rate, 18.5% versus 19.9%; hazard ratio [HR], 0.92; 95% confidence interval [CI], 0.85–1.01; P=0.07). Death related to cardiovascular causes, MI, or stroke occurred less often in the vorapaxar group than in placebo controls (vorapaxar versus placebo: 14.7% versus 16.4%; HR, 0.89; 95% CI, 0.81–0.98; P=0.02). However, this beneficial effect of vorapaxar was associated with a significantly increased risk for moderate and severe bleedings (vorapaxar versus placebo: 7.2% versus 5.2%; HR, 1.35; 95% CI, 1.16–1.58; P<0.001), which outweighed the potential beneficial effect in reduction of atherothrombotic events at 2 years. Particularly, intracranial bleedings were highly significantly increased under vorapaxar (vorapaxar versus placebo: 1.1% versus 0.2%; HR, 3.39; 95% CI, 1.78–6.45; P<0.001). In TRACER patients with NSTE (nonST-segment elevation)-ACS and peripheral artery disease (PAD), there was a tendency of reduced ischemic end points and peripheral revascularization which, however, did not reach statistical significance.16
Platelet activation, particularly induced by thrombin, plays a key role in atherothrombotic events in patients with stable atherosclerosis.17 After it had been shown that vorapaxar reduces death due to cardiovascular causes in patients with unstable angina pectoris and nonST-elevation myocardial infarction (NSTEMI), safety and efficacy of vorapaxar in reducing atherothrombotic events in patients with atherosclerosis were assessed in the “Trial to Assess the Effects of Vorapaxar in Preventing Heart Attack and Stroke in Patients With Atherosclerosis-Thrombolysis In Myocardial Infarction 50” (TRA 2°P-TIMI 50).18 The hypothesis of TRA 2°P-TIMI 50 was that an inhibition of the PAR-1 in addition to standard platelet medication (eg, inhibition of the thromboxane A2 and the P2Y12 pathways) has a beneficial effect on the incidence of atherothrombotic events in patients with stable atherosclerosis and PAD. TRA 2°P-TIMI 50 was a multicenter, double-blinded, placebo-controlled Phase III trial in which 26,449 patients with stable CAD and prior MI, PAD or ischemic stroke were recruited.19 CAD or stroke patients were eligible for TRA 2°P-TIMI 50 if they had an MI or an ischemic stroke 2 weeks to 12 months before study inclusion. PAD patients were required to have intermittent claudication with an ankle-brachial index <0.85 or previous revascularization for limb ischemia. Patients with a planned revascularization, a history for bleeding complications, and a medication with vitamin K antagonists were excluded from the study. Patients eligible for TRA 2°P-TIMI 50 were randomized in a 1:1 ratio into a vorapaxar cohort (2.5 mg daily) or to placebo group. The primary end point was a composite of cardiovascular death, stroke, MI, or recurrent ischemia leading to urgent coronary revascularization. Bleeding complications were defined in accordance to the Global Use of Strategies to Open Occluded Coronary Arteries (GUSTO) and Thrombolysis in Myocardial Infarction (TIMI) classifications.20 Due to an excess of intracranial bleedings in patients treated with vorapaxar and a history of stroke, the safety monitoring board of TRA 2°P-TIMI 50 recommended discontinuation of vorapaxar in all patients with a previous stroke. Approximately 26,449 patients were enrolled in TRA 2°P-TIMI 50 and were randomly assigned to vorapaxar (13,225 patients) or placebo (13,244 patients). Approximately two-third of the patients were recruited to TRA 2°P-TIMI 50 because of prior MI, 18% due to ischemic stroke, and 14% because of PAD. The majority of patients with a history of MI were under thienopyridine therapy (eg, clopidogrel), 94% of all patients received acetylsalicylic acid (ASA). The primary end point was observed in 9.3% of the patients receiving vorapaxar versus 10.5% of the placebo group (HR, 0.87; 95% CI, 0.80–0.94; P<0.001). Furthermore, vorapaxar significantly reduced the risk for secondary end points (vorapaxar group versus placebo group: 11.2% versus 12.4%; HR, 0.88; 95% CI, 0.82–0.95; P=0.001). In patients with prior MI, compared to placebo controls, vorapaxar reduced the risk to reach the primary end point by 20%, that for MI by 21%, and that for ischemic stroke by 34%.
In patients with diabetes and prior MI, vorapaxar significantly reduced the risk to reach the primary end point (11.4% versus 14.3%; HR, 0.73; 95% CI, 0.60–0.89; P=0.002). Even if the incidence for minor and major GUSTO bleedings was increased, the net clinical outcome was improved in patients with diabetes and prior MI who were under vorapaxar treatment.21 The primary end point was not significantly different in patients who were qualified with PAD.22 But interestingly, rates for hospitalization due to acute limb ischemia and peripheral artery revascularization were significantly lower in the vorapaxar cohort compared to placebo.
However, the reduction of primary and secondary end points in patients under vorapaxar was associated with an increase in moderate to severe bleeding complications, as defined by the GUSTO criteria (vorapaxar versus placebo: 4.2% versus 2.5%, HR, 1.66; 95% CI, 1.43–1.93; P<0.001) and TIMI criteria. Intracranial bleedings appeared twice as often under vorapaxar than in the placebo group (vorapaxar versus placebo: 1.0% versus 0.5%; HR, 1.94; 95% CI, 1.39–2.70; P<0.001), but fatal bleeding complications were rare in both groups (vorapaxar versus placebo: 0.3% versus 0.2%; HR, 1.46; 95% CI, 0.82–2.58; P=0.19). In order to compare beneficial effects of vorapaxar with unwanted side effects, the net clinical outcome defined as the composite of cardiovascular death, MI, stroke, or moderate or severe GUSTO bleedings was calculated. In the vorapaxar group, 11.7% (1,315 patients) reached this end point compared to 12.1% (1,358 patients) in the control group (HR, 0.97; 95% CI, 0.90–1.04; P=0.40). Hence, vorapaxar given in addition to standard antiplatelet therapy reduces the risk for MI, stroke, and cardiovascular death in patients with stable atherosclerosis, and particularly in those with a history of MI. However, this risk reduction was associated with an increased risk for moderate and severe GUSTO bleedings.
Therefore, the FDA approval was primarily based on TRA-2P data demonstrating a reduction of the primary end point in post-MI or PAD patients without a history of stroke or transient ischemic attack (TIA). This antiischemic effect was pronounced in patients with low body weight (<60 kg), with diabetes, and <75 years of age. Of note, vorapaxar is not approved for ACS treatment (as tested in TRACER), but rather secondary prophylaxis (as tested in TRA 2P).
Subgroup analyses
Even if drug-eluting stents are used and antiplatelet medication is taken, stent thrombosis is a feared complication after stent implantation and is associated with clinically significant complications such as MI.23 In order to assess the effect of vorapaxar on event rates of stent thrombosis, a subanalysis of TRA 2°P-TIMI 50 was conducted.24 Stent thrombosis was defined in accordance to the Academic Research Consortium (ARC).25 Unexplained death within 30 days after stent implantation and MI without angiographic confirmation in the anatomical region of the previously implanted stent indicated possible stent thrombosis. Out of 26,449 patients of TRA 2°P-TIMI 50, 14,491 had at least one coronary stent. Approximately 97% of these patients received their coronary stents before study inclusion (eg, because of MI), whereas 3% obtained a coronary stent during the study follow-up period. Approximately 98% of all patients received ASA, 83% were treated with a dual antiplatelet therapy. During the observational period, 152 patients developed definite stent thrombosis, which in 92% occurred late (>30 days to <12 months after PCI) or very late (>12 months after PCI). In comparison to placebo, treatment with vorapaxar significantly reduced the rate of definite stent thrombosis (1.1% versus 1.4%; HR, 0.71; 95% CI, 0.51–0.98; P=0.037). This beneficial effect of vorapaxar on the rate of definite stent thrombosis was consistent through all subgroups such as STEMI patients, smoking habits, or stent type (bare metal stents versus drug-eluting stents), as well as through all time points.
In regard to primary end points, vorapaxar reduced the risk for MI, cardiovascular death, and ischemic stroke in patients with prior MI, whereas no significant increase of intracranial bleeding or fatal bleeding occurred.
However, an increase in moderate and severe bleedings was found. Hence, vorapaxar in addition to standard antiplatelet therapy reduces the risk for cardiovascular death or ischemic events in patients with MI, but is also associated with enhanced bleeding complications.
Conclusion and future outlook
Vorapaxar reduces major cardiovascular events in patients with a history of MI. Bleeding complications were unacceptably frequent only in patients with prior stroke or TIA. On the basis of these findings, primarily from large subgroup analyses of TRACER and TRA-2P, vorapaxar was approved by the FDA for secondary prophylaxis in patients with prior MI and PAD – without a history of stroke, TIA, or intracranial bleeding. In Europe, the Committee for Medicinal Products for Human Use (CHMP) recommended the European Medicines Agency (EMA) to approve vorapaxar in patients with prior MI in November 2014.
Vorapaxar provided net clinical benefit in patients at low risk for bleeding but high risk for ischemic events, because it especially prevented stent thrombosis after MI. It will therefore be a valuable tool enabling – for the first time – a concept of triple antiplatelet therapy in patients with particularly high ischemic risk. Its combination partners are ASA and clopidogrel, but not ticagrelor and prasugrel, because the newer ADP receptor antagonists have not been sufficiently evaluated in the vorapaxar trials (only 0.7% of patients in TRA-2P received prasugrel). Patients at high ischemic risk but low bleeding risk will hence have the choice between triple antiplatelet therapy with ASA, clopidogrel, and vorapaxar – and triple antithrombotic therapy with ASA, clopidogrel, and ultra-low-dose rivaroxaban for mild factor Xa inhibition.26,27 However, current European guidelines recommend prasugrel or ticagrelor as the first-line ADP receptor blocker after ACS.28
No studies have yet investigated safety and efficacy of vorapaxar in patients with STE MI. As these patients are in fact those with the most aggressive platelet activation and highest risk for stent thrombosis, triple antiplatelet therapy with vorapaxar may be particularly beneficial for patients after STEMI. Still, no trial investigating the effects of a PAR-1 inhibitor in patients with STEMI is currently registered at www.clinicaltrials.gov.
Finally, the optimal duration of dual antiplatelet therapy after PCI is not yet known, and much less is known about the optimal duration of triple or dual antiplatelet therapy with a PAR-1 antagonist. The median follow-up in TRACER was 502 days, and most patients received vorapaxar for much longer than 1 year. Although deviation of the Kaplan–Meier curves was evident within the first month, it is possible that shorter treatment courses allow optimization of patient management after PCI.
Disclosure
DD was investigator in the TRACER Ocular Safety substudy. CB was principal investigator in the TRACER trial and received speaker’s honoraries from Merck, Astra-Zeneca, and Sanofi. The authors report no other conflicts of interest in this work.
References
Heeschen C, Dimmeler S, Hamm CW, et al. Soluble CD40 ligand in acute coronary syndromes. N Engl J Med. 2003;348:1104–1111. | ||
Royle NJ, Irwin DM, Koschinsky ML, MacGillivray RT, Hamerton JL. Human genes encoding prothrombin and ceruloplasmin map to 11p11-q12 and 3q21-24, respectively. Somat Cell Mol Genet. 1987;13:285–292. | ||
Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. | ||
Andersen H, Greenberg DL, Fujikawa K, Xu W, Chung DW, Davie EW. Protease-activated receptor 1 is the primary mediator of thrombin-stimulated platelet procoagulant activity. Proc Natl Acad Sci U S A. 1999; 96:11189–11193. | ||
Cook JJ, Sitko GR, Bednar B, et al. An antibody against the exosite of the cloned thrombin receptor inhibits experimental arterial thrombosis in the African green monkey. Circulation. 1995;91:2961–2971. | ||
Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3:1800–1814. | ||
Chintala M, Strony J, Yang B, Kurowski S, Li Q. Sch 602539, a protease-activated receptor-1 antagonist, inhibits thrombosis alone and in combination with cangrelor in a folts model of arterial thrombosis in cynomolgus monkeys. Arterioscler Thromb Vasc Biol. 2010;30:2143–2149. | ||
Chackalamannil S, Wang Y, Greenlee WJ, et al. Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (sch 530348) with potent antiplatelet activity. J Med Chem. 2008;51:3061–3064. | ||
Ghosal A, Lu X, Penner N, et al. Identification of human liver cytochrome p450 enzymes involved in the metabolism of sch 530348 (vorapaxar), a potent oral thrombin protease-activated receptor 1 antagonist. Drug Metab Dispos. 2011;39:30–38. | ||
Zontivity™ (vorapaxar) tablets 2.08 mg*, for oral use *equivalent to 2.5 mg vorapaxar sulfate. Kenilworth, NJ: Merck Sharp & Dohme Corp (MSD); 2014. | ||
Kosoglou T, Kraft WK, Kumar B, et al. Pharmacokinetics and pharmacodynamics of the novel par-1 antagonist vorapaxar in patients with end-stage renal disease. Eur J Clin Pharmacol. 2012;68:1049–1056. | ||
Edelstein LC, Simon LM, Lindsay CR, et al. Common variants in the human platelet par4 thrombin receptor alter platelet function and differ by race. Blood. 2014;124:3450–3458. | ||
Becker RC, Moliterno DJ, Jennings LK, et al. Safety and tolerability of sch 530348 in patients undergoing non-urgent percutaneous coronary intervention: a randomised, double-blind, placebo-controlled phase ii study. Lancet. 2009;373:919–928. | ||
Goto S, Yamaguchi T, Ikeda Y, Kato K, Yamaguchi H, Jensen P. Safety and exploratory efficacy of the novel thrombin receptor (par-1) antagonist sch530348 for non-st-segment elevation acute coronary syndrome. J Atheroscler Thromb. 2010;17:156–164. | ||
Tricoci P, Huang Z, Held C, et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med. 2012;366:20–33. | ||
Jones WS, Tricoci P, Huang Z, et al. Vorapaxar in patients with peripheral artery disease and acute coronary syndrome: insights from thrombin receptor antagonist for clinical event reduction in acute coronary syndrome (tracer). Am Heart J. 2014;168:588–596. | ||
Jennings LK. Mechanisms of platelet activation: need for new strategies to protect against platelet-mediated atherothrombosis. Thromb Haemost. 2009;102:248–257. | ||
Morrow DA, Braunwald E, Bonaca MP, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012;366:1404–1413. | ||
Morrow DA, Scirica BM, Fox KA, et al. Evaluation of a novel antiplatelet agent for secondary prevention in patients with a history of atherosclerotic disease: design and rationale for the thrombin-receptor antagonist in secondary prevention of atherothrombotic ischemic events (tra 2 degrees p)-timi 50 trial. Am Heart J. 2009;158:335–341.e333. | ||
Mehran R, Rao SV, Bhatt DL, et al. Standardized bleeding definitions for cardiovascular clinical trials: a consensus report from the bleeding academic research consortium. Circulation. 2011;123:2736–2747. | ||
Cavender MA, Scirica BM, Bonaca MP, et al. Vorapaxar in patients with diabetes mellitus and previous myocardial infarction: findings from the thrombin receptor antagonist in secondary prevention of atherothrombotic ischemic events-timi 50 trial. Circulation. 2015;131:1047–1053. | ||
Bonaca MP, Scirica BM, Creager MA, et al. Vorapaxar in patients with peripheral artery disease: results from tra2{degrees}p-timi 50. Circulation. 2013;127:1522–1529, 1529e1–6. | ||
Iakovou I, Schmidt T, Bonizzoni E, et al. Incidence, predictors, and outcome of thrombosis after successful implantation of drug-eluting stents. JAMA. 2005;293:2126–2130. | ||
Bonaca MP, Scirica BM, Braunwald E, et al. Coronary stent thrombosis with vorapaxar versus placebo: results from the tra 2 degrees p-timi 50 trial. J Am Coll Cardiol. 2014;64:2309–2317. | ||
Mauri L, Hsieh WH, Massaro JM, Ho KK, D’Agostino R, Cutlip DE. Stent thrombosis in randomized clinical trials of drug-eluting stents. N Engl J Med. 2007;356:1020–1029. | ||
Mega JL, Braunwald E, Wiviott SD, et al. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med. 2012;366:9–19. | ||
Windecker S, Kolh P, Alfonso F, et al. 2014 esc/eacts guidelines on myocardial revascularization: the task force on myocardial revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS)developed with the special contribution of the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur Heart J. 2014;35:2541–2619. | ||
Steg PG, James SK, Atar D, et al. Esc guidelines for the management of acute myocardial infarction in patients presenting with st-segment elevation. Eur Heart J. 2012;33:2569–2619. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.