")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 7
Clinical potential of novel therapeutic targets in breast cancer: CDK4/6, Src, JAK/STAT, PARP, HDAC, and PI3K/AKT/mTOR pathways
Received 29 March 2014
Accepted for publication 8 May 2014
Published 6 August 2014 Volume 2014:7 Pages 203—215
DOI https://doi.org/10.2147/PGPM.S52762
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Sarah R Hosford,1 Todd W Miller1,2
1Department of Pharmacology and Toxicology, 2Comprehensive Breast Cancer Program, Norris Cotton Cancer Center, Geisel School of Medicine at Dartmouth, Lebanon, NH, USA
Abstract: Breast cancers expressing estrogen receptor α, progesterone receptor, or the human epidermal growth factor receptor 2 (HER2) proto-oncogene account for approximately 90% of cases, and treatment with antiestrogens and HER2-targeted agents has resulted in drastically improved survival in many of these patients. However, de novo or acquired resistance to antiestrogen and HER2-targeted therapies is common, and many tumors will recur or progress despite these treatments. Additionally, the remaining 10% of breast tumors are negative for estrogen receptor a, progesterone receptor, and HER2 (“triple-negative”), and a clinically proven tumor-specific drug target for this group has not yet been identified. Therefore, the identification of new therapeutic targets in breast cancer is of vital clinical importance. Preclinical studies elucidating the mechanisms driving resistance to standard therapies have identified promising targets including cyclin-dependent kinase 4/6, phosphoinositide 3-kinase, poly adenosine diphosphate–ribose polymerase, Src, and histone deacetylase. Herein, we discuss the clinical potential and status of new therapeutic targets in breast cancer.
Keywords: palbociclib, phosphoinositide 3-kinase, mammalian target of rapamycin
Introduction
Breast cancer is the most commonly diagnosed female cancer, and the second leading cause of cancer-related death in women in the United States with >232,000 new cases and 40,000 deaths projected in the year 2014.1 Clinical breast cancer subtypes are based on the expression of the hormone receptors (HRs) for estrogen (ER) and progesterone (PR), and overexpression/amplification of the human epidermal growth factor receptor 2 (HER2) (ERBB2) proto-oncogenic receptor, tyrosine kinase. Subtype drives the selection of appropriate therapies; HR+ tumors constitute ~75% of cases and are treated with antiestrogen therapies that block ER transcriptional activity (for example, aromatase inhibitors, tamoxifen, fulvestrant). HER2+ tumors comprise ~20% of cases, half of which are also HR-positive, and are managed with HER2-targeted agents (for example, trastuzumab, pertuzumab, lapatinib, trastuzumab emtansine). Tumors that lack the expression of ER, PR, and HER2 are termed “triple-negative breast cancers (TNBC)”; they comprise 5%–10% of cases, and are managed only with genotoxic chemotherapy.2 Breast tumors are also molecularly subtyped based on gene expression profiling into luminal A, luminal B, HER2-enriched, basal-like, claudin-low, and normal-like.3–5 Luminal A and B tumors tend to express ER, and basal and claudin-low tumors are often triple-negative.3,6
While tumor-targeted agents have been extremely effective in treating HR+ and HER2+ breast cancers, de novo or acquired drug resistance is common and many cancers will recur or progress.7,8 Alternatively, TNBC does not yet have an obvious tumor-specific receptor or pathway to target. Therefore, identifying new drug targets in breast cancer is a high clinical priority, and combining therapeutics to simultaneously target multiple oncogenic signaling pathways may be a key to overcoming/preventing resistance.
Targeting CDK4 and CDK6 in breast cancer
The role of CDK4/6 in the cell cycle
Phases of the cell cycle are tightly controlled by oscillating levels of cyclin proteins. In the G1 phase, cells grow and prepare to replicate deoxyribonucleic acid (DNA) in the S phase. In order for a cell to proliferate, it must first satisfy the requirements of restriction checkpoints in G1 before progressing into the S phase. If these checkpoints are not satisfied, the cell exits the cell cycle and enters a quiescent state (G0) and, under some circumstances, senescence (permanent cell cycle arrest). Alternatively, sufficient mitogenic signals will drive the production of cyclin D proteins that can associate with and activate their catalytic partners, cyclin-dependent kinase (CDK)4 or CDK6. CDK4 and CDK6 have overlapping and distinct functions,9 the most characterized of which is phosphorylation of the Rb (RB1) tumor suppressor. Once associated with cyclin D, CDK4/6 phosphorylates Rb at Ser780 and Ser795, resulting in Rb inactivation, thereby releasing E2F transcription factors to initiate the transcription of genes required for DNA replication. Progression through G1 is also limited by the abundance of cyclin-dependent kinase inhibitors (CKIs) such as p16INK4 proteins, which block the binding site of cyclin D on CDK4/6, and p27KIP1, which has broad specificity for the cyclin/CDK complexes.
Deregulation of the cell cycle in cancer
Aberrant cell cycle control is a hallmark of cancer,10 and multiple mechanisms contribute to deregulation of the G1-to-S checkpoint. Mitogen-activated pathways that drive the production of cyclin D1 may be stimulated by aberrant expression of growth factors or growth factor receptors, which can activate cells to produce cyclin D1 in an autocrine manner.11 These pathways may also be constitutively active due to the overexpression of downstream signaling molecules that drive the expression of cyclin D1, such as Ras proteins, which are frequently overexpressed in breast cancer and are associated with poor prognosis.12 Tumor cells have also been shown to frequently manipulate the expression of the cyclin and CDK genes (Figure 1). Many tumors alter the messenger ribonucleic acid (mRNA) and protein levels of key cell cycle regulatory components. For example, cyclin D1 mRNA is overexpressed in over 50% of breast cancers.13 Similarly, overexpression of CDK4/6 or the loss of CKIs is commonly seen in tumors.14 The gene encoding p16INK4A (CDKN2A) has more homozygous deletions than any other recessive cancer-related gene.15 It is important to note that deregulation of cell cycle proteins has been shown to differ according to breast cancer subtype. For example, amplification of cyclin D1 (CCND1) occurs in a much greater percentage of luminal B breast cancers than in luminal A (58% versus 29%, respectively).16 Similarly, the luminal B subtype is more often associated with a gain of CDK4 (25% of luminal B versus 14% of luminal A), and a loss of negative regulators including p16INK4A.16 In contrast, basal-like breast cancers do not typically display alterations in cyclin D1 or CDK4/6, but 20% of cases harbor mutations or lead to the homozygous loss of RB1, indicating that this breast cancer subtype may respond less frequently to treatment with CDK4/6 inhibitors.16 The relationship between cyclin D, CDK4/6, CKIs, and Rb allows cancer cells to gain a proliferative advantage by altering members of this pathway to enhance the inactivation of Rb. Therefore, it is not surprising that deregulation of at least one of these proteins is seen in nearly all human cancers.14,17
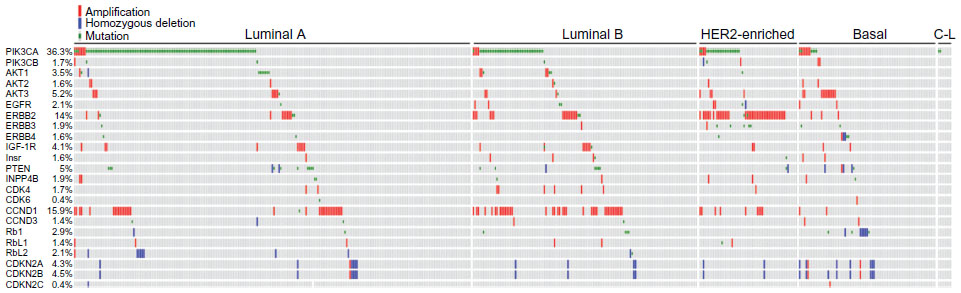 | Figure 1 Frequencies of genetic lesions identified in primary breast tumors. |
CDK4/6 inhibitors in clinical development
Until recently, nonspecific CDKIs have been the primary mechanism of therapeutically targeting kinases in the cell cycle.18 However, these therapeutics often elicit adverse effects in patients.18 The broad-spectrum CKI, flavopiridol, had promising preclinical results in multiple tumor cell types,19–21 but it exhibited adverse effects and high toxicity in early-phase clinical trials; furthermore, it did not meet expectations with regard to efficacy against most tumor types with the exception of leukemia.22–26 To limit toxicity and increase antitumor efficacy, there has been interest in the development of more specific CKIs. CDK4/6 are attractive targets due to their central role in the inactivation of Rb and their frequent deregulation in tumor cells. Palbociclib (PD-0332991) is the first highly selective inhibitor of CDK4/6 to be tested in humans. This drug exhibits an in vitro half maximal inhibitory concentration (IC50) of 10–15 nM for CDK4/6, compared to >5 μM for CDK2.27 Initial studies in cultured cancer cells showed a reduction in Rb phosphorylation within 4 hours of treatment with palbociclib, reaching a maximum at 16 hours posttreatment; Rb inhibition was completely reversed within 2 hours following drug removal.28 When tested against a panel of breast cancer cell lines, palbociclib preferentially inhibited growth of the ER+ cell lines, particularly among those with higher Rb and cyclin D levels, and lower p16INK4A expression.29 Palbociclib treatment also effectively suppressed the growth of ER+ human breast cancer xenografts in mice.30 The only known mechanism of resistance to CDK4/6 inhibition is the loss of Rb function.28
Despite immense progress in the treatment of ER+ breast cancer with the development of antiestrogens, approximately 50% of breast cancers will develop endocrine resistance and progress on antiestrogen therapies.31 One target gene of ER is cyclin D1 (CCND1), and antiestrogens have been shown to inhibit the cell cycle via downregulation of CDK activity and the subsequent phosphorylation of Rb to cause G0/G1 arrest.32–34 Therefore, it is not surprising that deregulation of cyclin D1 and Rb phosphorylation have been associated with antiestrogen resistance in in vitro and in vivo models.35–37 Given that tumors progressing on endocrine therapy are likely to have an intact cell cycle program, there is reason to investigate the therapies targeting CDK4/6 in endocrine-resistant tumors. Treatment of luminal ER+ breast cancer cells with palbociclib and tamoxifen elicits a synergistic effect.29 Furthermore, palbociclib treatment was able to partially restore sensitivity to tamoxifen in a tamoxifen-resistant cell line, and it abrogated growth of ER+ cells with acquired resistance to estrogen deprivation.29,30,38 Similarly, another CDK4/6 inhibitor now in clinical development (LEE011) has shown potent antitumor activity in preclinical models, including those resistant to the phosphoinositide 3-kinase (PI3K) inhibitor, BYL719 (described within the PI3K pathway inhibitors as single agents section).39 These promising preclinical data have led to numerous clinical trials of CDK4/6 inhibitors in various types of cancer.
Phase I trials of palbociclib tested different treatment schedules and doses in patients with Rb+ advanced solid tumors or non-Hodgkin’s lymphoma that were refractory to standard treatment. For both a 2-week-on, 1-week-off schedule (2:1), and a 3-week-on, 1-week-off schedule (3:1), neutropenia and thrombocytopenia were defined as the dose-limiting toxicities with maximum tolerated doses (MTDs) of 200 mg/day and 125 mg/day, respectively.40,41 With the 2:1 treatment schedule, three of 31 of patients exhibited stable disease for >10 cycles of treatment.40 Similarly, in the 3:1 treatment group, six of 37 patients maintained disease stabilization for >10 cycles of treatment; one patient who benefitted from 50 mg/day palbociclib had breast cancer with strong Rb positivity.41 Based on these results, a dose of 125 mg/day and a 3:1 schedule has been further pursued in Phase II trials of palbociclib (NCT01684215, NCT01536743, NCT01709370) (Table 1).
 | Table 1 Clinical development status of targeted therapeutics in breast cancer |
Based on the indications that palbociclib may be an effective treatment in combination with antiestrogens, and as a treatment for endocrine-resistant breast cancer, several clinical trials are ongoing to explore these findings in patients. A randomized, placebo-controlled, Phase II study to determine the benefit of adding palbociclib to the aromatase inhibitor, letrozole, as first-line therapy for advanced ER+/HER2− breast cancer in postmenopausal women (NCT00721409) has shown promising results thus far. The median progression-free survival (PFS) of the combination treatment was 26.1 months compared to 7.5 months for letrozole/placebo (hazard ratio=0.32; 95% confidence interval [CI]: 0.19–0.56; P<0.001). This study also established clinical benefit rates of 70% and 44% for treatment with letrozole/palbociclib and letrozole/placebo, respectively.42 In attempts to define biomarkers predictive of a response to palbociclib, this study analyzed the amplification of CCND1 and/or the loss of CDKN2A (p16INK4A) by fluorescence in situ hybridization. However, these alterations were not predictive of benefit from adding palbociclib, highlighting the difficulty in finding predictive biomarkers in the heterogeneous background of human tumors. The preliminary results of this trial led to palbociclib receiving “breakthrough therapy designation” from the United States Food and Drug Administration in April 2013, which allowed for the accelerated approval of drugs that treat a life-threatening disease and display significant improvement over current therapy. A Phase III trial of the same design (NCT01740427; PALOMA-2), a Phase III study of palbociclib/fulvestrant versus placebo/fulvestrant in patients with ER+/HER2– advanced disease that progressed on an antiestrogen (NCT01942135; PALOMA-3), and a Phase III study of palbociclib/antiestrogen versus placebo/antiestrogen as an adjuvant therapy in patients with early-stage ER+/HER2– disease (NCT01864746; PENELOPE-B) are underway.
Two other CDK4/6 inhibitors (LEE011 and LY2835219) have been tested in dose-finding Phase I studies.43 LEE011 is now being tested in a randomized Phase Ib/II study with the PI3K inhibitor BYL719 and letrozole (NCT01872260), and in a randomized Phase Ib/II study with the mammalian target of rapamycin (mTOR) complex 1 (mTORC1) inhibitor everolimus and the aromatase inhibitor exemestane (NCT01857193), both in patients with advanced ER+/HER2− breast cancer that progressed on an antiestrogen. LEE011 is also being tested with letrozole in a randomized presurgical study in patients with early-stage ER+/HER2− breast cancer to identify molecular changes in tumors and in a Phase III study to determine the efficacy of this combination (NCT01919229; MONALEESA-1, NCT01958021; MONALEESA-2). While palbociclib and LEE011 are being administered in a 3:1 schedule to ameliorate side effects such as neutropenia, LY2835219 is being administered on a continuous schedule with a lower incidence of neutropenia44 in a Phase Ib study in combination with several antiestrogens in patients with advanced ER+/HER2− antiestrogen-resistant disease (NCT02057133). The reason that these agents induce different adverse event profiles is unclear.
Potential of CDK4/6 inhibitors in combination with genotoxic agents
Traditional chemotherapeutic agents are thought to rely on the cell cycle to induce DNA damage that promotes apoptosis; therefore, the combination of these therapies with cell cycle inhibitors like palbociclib may not be a viable treatment strategy. In vitro data suggest that a concomitant treatment of chemotherapeutics and palbociclib may result in decreased efficacy as compared to single-agent treatments, and in some cases, combination treatment antagonizes chemotherapy-induced cell death.45–47 Palbociclib also antagonized mitotic catastrophe and subsequent cell death associated with paclitaxel, a microtubule-stabilizing agent.46
Although palbociclib does not appear to enhance tumor killing in combination with chemotherapeutics, there is potential in using CDK4/6 inhibitors transiently to synchronize cells before treating them with genotoxic agents to enhance the cytotoxic effect. Short-term treatment with palbociclib before or concurrent with paclitaxel suppressed cell growth more effectively than single agents or continuous treatment with both agents.46 A Phase I clinical study is ongoing to determine the MTD of palbociclib in combination with paclitaxel in patients with Rb+ advanced breast cancer (NCT01320592).
Targeting the PI3K/AKT/mTOR pathway in breast cancer
Pathway deregulation in breast cancer
PI3K/AKT/mTOR signaling regulates multiple cellular processes to promote cancer cell growth, survival, and metastasis.48–50 This is the most frequently aberrantly activated pathway in human breast cancer, with alterations in genes encoding the pathway components occurring in >80% of cases (Figure 1).51 The deregulation of class 1A PI3K signaling has also been associated with the development of resistance to a variety of cancer therapies, including antiestrogens, trastuzumab, radiation, and chemotherapy.52–54 However, mechanisms of PI3K pathway activation differ between breast cancer subtypes. Activating mutations in PIK3CA occur more frequently in luminal A, luminal B, and HER2+ breast cancers (45%, 29%, and 39%, respectively) as compared to basal-like breast cancers (9%).16 In contrast, phosphatase and tensin homolog (PTEN) expression is decreased in 67% of TNBCs, compared with 29%–44% in ER+ tumors and 22% in HER2+ tumors.55–58 The high frequency of PI3K pathway alteration in human breast tumors makes this pathway a promising target for therapeutics, and inhibitors of PI3K, AKT, and/or mTOR are in clinical development.
PI3K pathway inhibitors as single agents
PI3K inhibitors have exhibited clinical activity against advanced solid tumors and metastatic breast cancers.59,60 A Phase I study of the pan-PI3K inhibitor, BKM120, in patients with metastatic breast cancer reported partial responses and stable disease in 11% and 50% of patients, respectively.61 A Phase II trial is ongoing to investigate BKM120 for the treatment of metastatic TNBC (NCT01629615). Preliminary results of a first-in-human Phase I trial testing the PI3K/p110α isoform-specific inhibitor, BYL719, in advanced solid tumors that harbor activating mutations in p110α (PIK3CA) (NCT01219699) suggest a favorable safety profile and an MTD of 400 mg/day. Partial responses were observed in seven of 39 patients, including two with ER+ breast cancer. At a dose of 270 mg/day, the median PFS for 15 patients with ER+/HER2– metastatic breast cancer was 5.5 months.62 While the presence of PIK3CA mutations or PTEN loss is predictive of a response to PI3K and AKT inhibitors in cell lines,63–67 it remains to be determined whether these biomarkers remain predictive in human breast cancers.59,68,69 Phase I trials are ongoing to investigate BYL719 in combination with endocrine therapies including letrozole and exemestane in HR+ metastatic breast cancer (NCT01870505, NCT01791478).
The allosteric mTORC1 inhibitor, everolimus, also showed efficacy as a single agent in a Phase II placebo-controlled study in breast cancer patients with bone metastases. This study reported improved time-to-progression from 12.6 weeks in the placebo arm to 37 weeks in the everolimus arm (HR =0.464; 95% CI: 0.226–0.954; P=0.037, adjusted for endocrine therapy).70 Another Phase II trial is ongoing to test the benefit of everolimus after preoperative chemotherapy in patients with invasive breast cancer (NCT01088893).
PI3K pathway inhibitors for the treatment of ER+ breast cancer
There is ample evidence that HR+ tumors resistant to endocrine therapy exhibit increased PI3K/AKT/mTOR signaling.51,53,63 Accordingly, treatment of endocrine-resistant cell lines and xenografts with inhibitors of PI3K, AKT, and/or mTOR abrogates endocrine resistance.63,71–74 Therefore, treatment with inhibitors of the PI3K/ATK/mTOR pathway in combination with endocrine therapies may prevent an escape from endocrine dependence in these tumors, and restore sensitivity to endocrine agents in breast cancer.
The first PI3K/AKT/mTOR pathway-targeted therapeutic to enter routine clinical use for cancer is everolimus. A randomized Phase II trial investigating the clinical benefit of everolimus in combination with tamoxifen versus tamoxifen/placebo in patients with ER+/HER2− metastatic disease that had prior exposure to aromatase inhibitors reported an improved 6-month clinical benefit rate (61% versus 42%, exploratory P=0.045) and time-to-progression (8.6 months versus 4.5 months, exploratory P=0.002) in the combination arm compared to tamoxifen/placebo, corresponding to a reduction in the risk of progression (46% decrease) and the risk of death (55% decrease) with combination treatment.75 Similarly, the randomized Phase III Breast Cancer Trials of Oral Everolimus-2 (BOLERO-2) trial76 led to the approval of everolimus in combination with exemestane for the treatment of ER+/HER2− postmenopausal breast cancer patients with the disease that progressed during treatment with a nonsteroidal aromatase inhibitor. BOLERO-2 reported a median PFS of 11.0 months and 4.1 months for the combination treatment and exemestane/placebo, respectively (HR =0.38; 95% CI: 0.31–0.48; log-ranked P<0.0001).76 Sequencing of 182 genes to identify a lesion or lesions predictive of benefit from exemestane/everolimus did not reveal an obvious single biomarker, but patients with tumors harboring minimal combined lesions in PIK3CA, PTEN, CCND1, FGFR1, and FGFR2 derived greater benefit from the combination (HR =0.27).77 The benefit of continued endocrine therapy (with exemestane) is being tested in an ongoing randomized Phase III trial of everolimus alone versus everolimus/exemestane versus capecitabine in the same patient population (NCT01783444).
Preclinical and clinical data indicate that mTORC1 inhibition (for example, with everolimus) derepresses negative feedback that results in the upregulation of growth factor receptor, PI3K/AKT, and MEK/ERK signaling.78–80 Thus, targeting signaling nodes upstream of mTORC1 may be more effective. A Phase Ib/II trial testing the PI3K inhibitor, BKM120, with letrozole in patients with ER+/HER2− metastatic breast cancer is ongoing.81 Early results suggest that this drug combination is active with a tolerable safety profile. Unexpectedly, the mutation status of PIK3CA did not correlate with clinical benefit.81 BKM120 is being tested in Phase II and III trials in combination with fulvestrant in postmenopausal women with ER+/HER2− advanced disease with cancer that progressed on an aromatase inhibitor with or without an mTORC1 inhibitor (NCT01339442, NCT01610284 [BELLE-2], NCT01633060 [BELLE-3]). Similarly, the pan-PI3K inhibitor GDC-0941 and the PI3K/mTOR dual inhibitor GDC-0980 are being tested with fulvestrant in a randomized Phase II placebo-controlled study in patients with advanced disease resistant to aromatase inhibitors (NCT01437566). In addition, a Phase I trial is testing the efficacy of the AKT inhibitor, MK2206, in combination with anastrozole or fulvestrant, or with anastrozole plus fulvestrant in patients with metastatic ER+ disease (NCT01344031).
Targeting the PI3K pathway in HER2+ breast cancer
Activation of PI3K/AKT/mTOR signaling via a PIK3CA mutation in HER2+ breast cancer has been associated with resistance to trastuzumab and lapatinib, while the effects of PTEN loss remain controversial.82–86 In preclinical models, everolimus plus trastuzumab had greater antitumor effects than either treatment alone.78 Similarly, BKM120 was shown to synergize with trastuzumab in HER2+ breast cancer cell lines, and treatment with BKM120 partially restored sensitivity to trastuzumab in trastuzumab-resistant HER2+ xenografts.87 Recently, a Phase I/II trial88 was completed for patients with advanced HER2+ trastuzumab-resistant disease, showing that combined treatment with BKM120 and trastuzumab was well tolerated and elicited clinical benefit in 25% of patients. These results were comparable to those of a prior study89 examining the combination of everolimus and trastuzumab in a similar patient population, which reported a 35% rate of clinical benefit. Based on these encouraging results, a Phase II placebo-controlled trial is ongoing to evaluate the efficacy of BKM120 in combination with paclitaxel and trastuzumab in the neoadjuvant setting in patients with early-stage HER2+ breast cancer (NCT01816594, NeoPHOEBE). BKM120 is also being tested in combination with lapatinib in patients with advanced trastuzumab-resistant HER2+ breast cancer harboring lesions in PIK3CA or PTEN (NCT01589861, PIKHER2).
PI3K inhibitors in combination with genotoxic agents
Activation of PI3K/AKT signaling has been associated with resistance to DNA-damaging chemotherapeutics in vitro and in vivo,90 and several clinical trials are ongoing to explore the potential of combining PI3K inhibitors and genotoxic agents to prevent resistance. The benefit of BKM120 and paclitaxel as a first-line treatment for HER2-negative advanced breast cancer is being investigated in an ongoing placebo-controlled Phase II trial (NCT01572727). Similarly, GDC-0941 is being tested in combination with paclitaxel in a Phase II randomized study in patients with advanced disease (NCT01740336).
Inhibiting IGF-1R/InsR signaling in breast cancer
Treatment with antiestrogens and HER2-blocking agents has not only been shown to result in compensatory upregulation of PI3K/AKT/mTOR signaling; upstream receptor tyrosine kinases including insulin-like growth factor (IGF)-1 receptor (IGF-1R) are congruently upregulated.63,91,92 Furthermore, IGF-1R was found to be expressed in ~90% of breast cancers and associated with worse prognosis in ER+ breast cancer patients.93–95 As IGF-1R is a potent driver of PI3K signaling, preclinical and clinical studies explored the efficacy of IGF-1R-targeted agents in ER+ breast cancer. IGF-1R is highly homologous to the insulin receptor (InsR), and these proteins form homo- and heterodimers that confer ligand preferences for IGF-1 or insulin. Adenosine triphosphate (ATP)-competitive inhibitors target both receptors, while IGF-1R and IGF-1 (ligand-specific) antibodies are protein-specific. In ER+ models, treatment with an ATP-competitive IGF-1R/InsR inhibitor abrogated endocrine-resistant cell and tumor growth, while an IGF-1R antibody was less effective.96 Such effects are likely due, in part, to compensatory upregulation of InsR when IGF-1R is targeted.
A recent Phase I trial of everolimus combined with the monoclonal IGF-1R antibody, figitumumab, showed stable disease in 15 of 18 patients with advanced solid tumors, and partial response in one patient.97 Inhibition of PI3K signaling with BYL719 was shown to activate IGF-1R signaling in PI3KCA-mutant breast cancer cells and a xenograft model, while cotreatment of BYL719 and the IGF-1R antibody, ganitumab, induced tumor regression.98 A Phase Ib/II trial is ongoing to study the same drug combination in patients with PI3KCA-mutant breast cancers (NCT01708161).
There is extensive crosstalk between ER and IGF-1R signaling, and preclinical studies show that targeting both pathways may be beneficial.99,100 However, a Phase II placebo-controlled study101 testing the IGF-1R monoclonal antibody ganitumab (AMG-479) with exemestane or fulvestrant in postmenopausal women with HR+ metastatic disease did not result in improved PFS and negatively impacted overall survival. Likewise, the ATP-competitive IGF-1R/InsR inhibitor, OSI-906, demonstrated preclinical efficacy in tumor models96 and was being tested in a Phase II trial with letrozole ± the epidermal growth factor receptor inhibitor, erlotinib, in patients with ER+ metastatic breast cancer; however, this trial was terminated due to unacceptable toxicity and a lack of efficacy (NCT01205685). While initial trials of IGF-1R and IGF-1R/InsR inhibitors in ER+ disease have been disappointing, further study is necessary to determine the molecular markers that may predict benefit.
In vitro studies have demonstrated that IGF-1 plays a role in the development of resistance to chemotherapy in breast cancer.102,103 A Phase I trial of figitumumab in patients with advanced solid tumors showed clinical benefit, with ten of 15 patients experiencing stable disease at the maximum feasible dose of 20 mg/kg.104 A Phase II trial in small cell lung cancer showed that figitumumab significantly improved response to carboplatin and paclitaxel.105 While these results are promising, further study in breast cancer is needed to determine the best sequencing of IGF-1R inhibitors with chemotherapy to result in maximal tumor response.
IGF-1R signaling has been associated with resistance to HER2-targeted agents in preclinical studies.92,100,102,106 In vitro data have shown that the combination of trastuzumab and an anti-IGF1R antibody increases cytotoxicity compared to trastuzumab alone in breast cancer.106 A Phase II trial investigating the addition of the IGF-1R antibody, cixutumumab, to lapatinib and capecitabine in HER2+ breast cancer patients is ongoing (NCT00684983).
An alternative approach to inhibiting IGF-1R is the depletion of a ligand. BI 836845 is an antibody that binds and sequesters IGF-1 and IGF-2.107 This antibody is undergoing Phase I testing (NCT01403974, NCT02123823).
Anticancer effects of bone resorption inhibitors
Bone is a frequent site of metastasis in breast cancer patients. The bone microenvironment is enriched with growth factors to support cancer cell growth. Osteoclast-induced bone resorption leads to the increased release of growth factors, further enhancing proliferation and survival of cancer cells. Furthermore, breast cancer cells secrete growth factors and cytokines that contribute to osteoclast activity and inhibit osteoblast function, thereby maintaining a feed-forward cycle of bone resorption and cancer growth.108 Bisphosphonates potently inhibit osteoclast-mediated bone resorption, and these compounds have been used clinically to prevent complications related to bone metastasis. More recently, these compounds have been adopted as treatments to prevent bone loss associated with chemotherapy or endocrine therapy in breast cancer patients.109–111
Preclinical studies revealed that bisphosphonates have antitumor activity and prevent metastasis by inhibiting tumor cell invasion and promoting apoptosis.112–116 These observations prompted clinical investigations of bone resorption inhibitors as anticancer agents, which have produced mixed results. Clinical trials examining the bisphosphonate, zoledronic acid, showed improvements in disease-free and overall survival compared to standard therapy alone, both in chemotherapy and endocrine therapy combinations. However, the observed survival benefit was restricted to postmenopausal women, or premenopausal women who had suppressed estrogen levels due to treatment with aromatase inhibitors or ovarian suppression.117 Clinical trials investigating denosumab, an antibody against the osteoclast activator RANK ligand (RANKL), in the prevention of metastasis are ongoing (NCT00556374, NCT01077154).
Inhibiting PARP in triple-negative breast cancer
TNBCs are more aggressive and result in a worse prognosis when compared to other breast cancer subtypes.118,119 TNBCs are a heterogeneous subtype, and a single oncogenic driver has not been identified; therefore, tumor-targeted therapeutics are lacking. Poly(adenosine diphosphate–ribose) polymerase (PARP) is involved in the recognition and repair of DNA breaks, and PARP works in concert with other proteins including BRCA1 and BRCA2 to repair DNA damage. If DNA is irreparably damaged (for example, with genotoxic drugs), a cell will be forced to undergo apoptosis rather than replicate damaged DNA.120 Preclinical data demonstrate that deficiency in BRCA1/2 or other homologous recombination DNA repair proteins sensitizes cells to PARP inhibition.121 Therefore, as single agents, PARP inhibitors are likely to be more effective against tumors carrying mutations in genes encoding proteins involved in compensatory DNA repair mechanisms, such as BRCA1/2, as these mutations essentially prime the cells for DNA damage-induced apoptosis.122,123
There are two primary mechanisms of action of PARP inhibitors: catalytic PARP inhibition; and PARP–DNA trapping, where drugs trap PARP1 and PARP2 on single-stranded DNA breaks. Veliparib acts as a strong catalytic PARP inhibitor with a weak PARP–DNA trapping effect, while niraparib, olaparib, and rucaparib act through both mechanisms.124,125 A Phase I trial126 in breast cancer patients with germline BRCA1/2 mutations showed that the PARP inhibitor, olaparib, was effective as a monotherapy, achieving objective responses in 41% of patients. TNBCs have a gene expression signature similar to that of BRCA-deficient tumors.127 Furthermore, TNBCs frequently arise in carriers of BRCA1 mutations.127 It is therefore reasonable to suggest treating TNBC with PARP inhibitors in combination with genotoxic therapy to activate a synthetic lethal interaction of both PARP and BRCA deficiency in cancer cells.
In a recent Phase I trial128 of olaparib in combination with paclitaxel for metastatic TNBC, 37% of patients exhibited a partial response; however, this study was relatively small (n=19) and did not characterize the BRCA1/2 mutation status of patients. Rucaparib was well tolerated in a Phase I dose-escalation study129 in patients with advanced solid tumors; among 17 breast cancer patients, one BRCA1/2-mutant patient achieved a partial response, and four patients achieved stable disease for greater than 12 weeks. An exploratory analysis of BRCA1/2 mutation status in a Phase II study of veliparib in combination with temozolomide in metastatic TNBC reported a median PFS of 5.5 months in patients with known deleterious BRCA1/2 mutations versus 1.8 months in patients without these mutations.130 Thus, stratification based on BRCA1/2 status may be a useful biomarker to predict the response to PARP inhibitors. Clinical trials are investigating the use of PARP inhibitors in combination with genotoxic therapies in patients with BRCA1/2 mutations (NCT00494234, NCT01989546, NCT01074970, NCT01945775).
Histone deacetylase inhibitors for breast cancer
Preclinical studies have demonstrated that histone deacetylases (HDACs) modulate ER activity, and HDAC inhibitors reverse resistance to antiestrogen therapies in vitro.131–133 A Phase II trial134 in patients with advanced ER+ breast cancer resistant to aromatase inhibition reported that the HDAC inhibitor, entinostat, combined with exemestane improved PFS from 2.3 months to 4.3 months (HR =0.73; 95% CI: 0.50–1.07; P=0.055) and the median overall survival from 19.8 months to 28.1 months compared with exemestane/placebo (HR =0.59; 95% CI: 0.36–0.97; P=0.036). Similarly, Munster et al135 found in a Phase II trial in patients with ER+ endocrine-resistant metastatic disease that the combination of the HDAC inhibitor vorinostat and tamoxifen induced an objective response rate of 19%, a clinical benefit rate of 40%, and a median response duration of 10.3 months. A Phase II trial is ongoing in patients with chemotherapy-resistant advanced TNBC or ER+ disease to investigate combination treatment with entinostat and the DNA methyltransferase inhibitor, azacitidine (NCT01349959). Preliminary results of a Phase I trial136 in patients with trastuzumab-resistant HER2+ metastatic disease indicate that the HDAC inhibitor, panobinostat, with trastuzumab is well tolerated and shows promising antitumor activity, with two of 13 patients experiencing tumor reduction thus far (NCT00567879).
Targeting Src kinases in breast cancer
The Src family of nonreceptor tyrosine kinases is involved in a variety of functions (for example, proliferation, motility, and invasion) through its interaction with mediators including steroid HRs, integrins, G protein-coupled receptors, signal transducer and activator of transcription (STAT) family members, and several other receptors and intracellular proteins that may contributed to the malignant phenotype observed in cancer cells (as reviewed by Frame137). Gene expression profiling of primary and metastatic breast tumors indicates that a gene expression signature of Src pathway activation in the primary tumor is strongly predictive of bone metastasis,138 and high Src mRNA expression is correlated with decreased survival, making Src a potential target for therapeutic interventions.139–141 As a single agent, the antitumor activity of Src inhibitors has been disappointing in clinical trials for advanced breast cancer.142,143 However, in vitro studies have shown that Src inhibitors in combination with chemotherapy or endocrine therapy may enhance tumor cell death.144 In a Phase I trial,145 combination of the Src inhibitor dasatinib with paclitaxel in patients with metastatic breast cancer resulted in partial responses in four of 13 patients. Another Phase I trial146 reported that six of 25 advanced breast cancer patients exhibited partial responses, and 32% had stable disease following combination treatment with dasatinib plus capecitabine. The results of a randomized Phase II trial147 of dasatinib plus letrozole in HR+, HER2− postmenopausal metastatic breast cancer patients showed that the combination group had doubled PFS compared to the letrozole/placebo group (20.1 months versus 9.9 months, respectively; P=0.05). A Phase II trial148 investigating dasatinib plus exemestane compared with exemestane/placebo in advanced ER+ breast cancer patients that had progressed on an aromatase inhibitor reported a modest improvement in median PFS in the combination group (18.1 weeks with dasatinib/exemestane versus 16.1 weeks with exemestane/placebo; P=0.148), and the proportion of patients with clinical benefit was 30.6% in the combination group compared to 12.24% with exemestane/placebo. Overall, these findings support the use of Src inhibitors with chemotherapeutics and endocrine therapies to more effectively treat advanced breast cancers.
Targeting the JAK/STAT pathway in breast cancer
Janus kinases (JAKs) are mediators of cytokine and growth hormone signaling. Activated JAKs phosphorylate STAT proteins, leading to their nuclear translocation and the transcriptional regulation of genes that regulate cell proliferation, differentiation, and apoptosis.149,150 Mutations in JAK and STAT proteins have been extensively characterized in myeloproliferative disorders, and these mutations have been linked to hyperactivation of the JAK/STAT pathway and, consequently, unchecked cell proliferation.151 Comparable mutations have not been well studied in breast cancer. However STAT1, STAT3, and STAT5 are often constitutively phosphorylated, seemingly due to elevated levels of cytokines and receptors.152 Preclinical studies showed that the interleukin-6/JAK2/STAT3 pathway is preferentially activated in basal-like breast cancer cells, and inhibition of JAK2 with NBP-BSK805 hinders the growth of patient-derived breast tumor xenografts.153 There is also evidence supporting JAK2/STAT5 inhibitors as a means to overcome resistance to PI3K/mTOR inhibition in TNBC.154
The JAK1/JAK2 inhibitor, ruxolitinib, is approved for the treatment of myelofibrosis, and it is currently being tested as a single agent and in combination with paclitaxel in Phase II trials for inflammatory breast cancer and TNBC (NCT01562873, NCT02041429), and in advanced ER+ breast cancer in combination with exemestane (NCT01594216). A randomized, double-blind, placebo-controlled Phase II trial is also underway to investigate ruxolitinib in combination with capecitabine in metastatic breast cancer (NCT02120417). In patients with metastatic HER2+ breast cancer, an ongoing Phase II trial is testing ruxolitinib with trastuzumab (NCT02066532).
Conclusion
The high incidence of recurrence and progression on standard therapies highlights the importance of developing new therapeutics for breast cancer. Deregulation of the cyclin D–CDK4/6–Rb axis is frequently seen in antiestrogen-resistant tumors. Therefore, inhibitors of this pathway, including palbociclib, LEE011, and LY2835219, are promising as effective treatments for endocrine-resistant breast cancer. Activation of the PI3K/AKT/mTOR pathway has also been shown to promote drug resistance. The PI3K and mTOR inhibitors in clinical development have shown efficacy as single agents and in combination with antiestrogens or HER2-targeted agents. The search for a targeted therapeutic to treat TNBC has led to the development of PARP inhibitors, which appear to be considerably effective in a subgroup of TNBC patients harboring mutations in BRCA1/2. However, the TNBC subtype is heterogeneous, so further identification of targeted therapies for this group is needed. Immense progress has been made in identifying new targets in breast cancer, and future advances in therapy will likely include simultaneous or sequential targeting of multiple pathways to maximally inhibit tumor growth while thwarting the development of drug resistance.
Disclosure
TWM has received commercial research support from Piramal Life Sciences, Ltd. SRH reports no conflicts of interest in this work.
References
American Cancer Society. Breast Cancer: Facts and Figures 2013–2014. Atlanta, GA: American Cancer Society, Inc.; 2013. | |
Curtis C, Shah SP, Chin SF, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. | |
Sørlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. | |
Prat A, Parker JS, Karginova O, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12(5):R68. | |
Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. | |
Bertucci F, Finetti P, Cervera N, et al. How basal are triple-negative breast cancers? Int J Cancer. 2008;123(1):236–240. | |
Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit Rev Oncog. 2012;17(1):1–16. | |
Giuliano M, Schifp R, Osborne CK, Trivedi MV. Biological mechanisms and clinical implications of endocrine resistance in breast cancer. Breast. 2011;20:Suppl 3:42–49. | |
Anders L, Ke N, Hydbring P, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20(5):620–634. | |
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | |
Fleming TP, Matsui T, Molloy CJ, Robbins KC, Aaronson SA. Autocrine mechanism for v-sis transformation requires cell surface localization of internally activated growth factor receptors. Proc Natl Acad Sci U S A. 1989;86(20):8063–8067. | |
Clark GJ, Der CJ. Aberrant function of the Ras signal transduction pathway in human breast cancer. Breast Cancer Res Treat. 1995;35(1):133–144. | |
Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11(8):558–572. | |
Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer. 2001;1(3):222–231. | |
Bignell GR, Greenman CD, Davies H, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463(7283):893–898. | |
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. | |
Hall M, Peters G. Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv Cancer Res. 1996;68:67–108. | |
Dickson MA, Schwartz GK. Development of cell-cycle inhibitors for cancer therapy. Curr Oncol. 2009;16(2):36–43. | |
Blagosklonny MV. Flavopiridol, an inhibitor of transcription: implications, problems and solutions. Cell Cycle. 2004;3(12):1537–1542. | |
Byrd JC, Shinn C, Waselenko JK, et al. Flavopiridol induces apoptosis in chronic lymphocytic leukemia cells via activation of caspase-3 without evidence of bcl-2 modulation or dependence on functional p53. Blood. 1998;92(10):3804–3816. | |
Kitada S, Zapata JM, Andreeff M, Reed JC. Protein kinase inhibitors flavopiridol and 7-hydroxy-staurosporine down-regulate antiapoptosis proteins in B-cell chronic lymphocytic leukemia. Blood. 2000;96(2):393–397. | |
Shapiro GI, Supko JG, Patterson A, et al. A phase II trial of the cyclin-dependent kinase inhibitor flavopiridol in patients with previously untreated stage IV non-small cell lung cancer. Clin Cancer Res. 2001;7(6):1590–1599. | |
Schwartz GK, Ilson D, Saltz L, et al. Phase II study of the cyclin-dependent kinase inhibitor flavopiridol administered to patients with advanced gastric carcinoma. J Clin Oncol. 2001;19(7):1985–1992. | |
Aklilu M, Kindler HL, Donehower RC, Mani S, Vokes EE. Phase II study of flavopiridol in patients with advanced colorectal cancer. Ann Oncol. 2003;14(8):1270–1273. | |
Liu G, Gandara DR, Lara PN, et al. A Phase II trial of flavopiridol (NSC #649890) in patients with previously untreated metastatic androgen-independent prostate cancer. Clin Cancer Res. 2004;10(3):924–928. | |
Canavese M, Santo L, Raje N. Cyclin dependent kinases in cancer: potential for therapeutic intervention. Cancer Biol Ther. 2012;13(7):451–457. | |
Toogood PL, Harvey PJ, Repine JT, et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem. 2005;48(7):2388–2406. | |
Fry DW, Harvey PJ, Keller PR, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3(11):1427–1438. | |
Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77. | |
Miller TW, Balko JM, Fox EM, et al. ERα-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov. 2011;1(4):338–351. | |
Clarke R, Leonessa F, Welch JN, Skaar TC. Cellular and molecular pharmacology of antiestrogen action and resistance. Pharmacol Rev. 2001;53(1):25–71. | |
Watts CK, Brady A, Sarcevic B, deFazio A, Musgrove EA, Sutherland RL. Antiestrogen inhibition of cell cycle progression in breast cancer cells in associated with inhibition of cyclin-dependent kinase activity and decreased retinoblastoma protein phosphorylation. Mol Endocrinol. 1995;9(12):1804–1813. | |
Watts CK, Sweeney KJ, Warlters A, Musgrove EA, Sutherland RL. Antiestrogen regulation of cell cycle progression and cyclin D1 gene expression in MCF-7 human breast cancer cells. Breast Cancer Res Treat. 1994;31(1):95–105. | |
Carroll JS, Prall OW, Musgrove EA, Sutherland RL. A pure estrogen antagonist inhibits cyclin E-Cdk2 activity in MCF-7 breast cancer cells and induces accumulation of p130-E2F4 complexes characteristic of quiescence. J Biol Chem. 2000;275(49):38221–38229. | |
Wilcken NR, Prall OW, Musgrove EA, Sutherland RL. Inducible overexpression of cyclin D1 in breast cancer cells reverses the growth-inhibitory effects of antiestrogens. Clin Cancer Res. 1997;3(6):849–854. | |
Rudas M, Lehnert M, Huynh A, et al; Austrian Breast and Colorectal Cancer Study Group. Cyclin D1 expression in breast cancer patients receiving adjuvant tamoxifen-based therapy. Clin Cancer Res. 2008;14(6):1767–1774. | |
Bosco EE, Wang Y, Xu H, et al. The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J Clin Invest. 2007;117(1):218–228. | |
Thangavel C, Dean JL, Ertel A, et al. Therapeutically activating RB: reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr Relat Cancer. 2011;18(3):333–345. | |
Kim S, Loo A, Chopra R, et al. Abstract PR02: LEE011: an orally bioavailable, selective small molecule inhibitor of CDK4/6 – reactivating Rb in cancer. Mol Cancer Ther. 2013;12:PR02. | |
Schwartz GK, LoRusso PM, Dickson MA, et al. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br J Cancer. 2011;104(12):1862–1868. | |
Flaherty KT, Lorusso PM, Demichele A, et al. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012;18(2):568–576. | |
Finn RS, Crown JP, Boer K, et al. Results of a randomized phase 2 study of PD 0332991, a cyclin-dependent kinase (CDK) 4/6 inhibitor, in combination with letrozole vs letrozole alone for first-line treatment of ER+/HER2− advanced breast cancer(BC). 2012 Cancer Res. 2012; 72(Suppl 24):S1–S6. | |
Infante JR, Petronella W, Gerecitano J, et al. A phase I study of the single-agent CDK4/6 inhibitor LEE011 in pts with advanced solid tumors and lymphomas. J Clin Oncol. 2014;32(5s Suppl):Abstract 2528. | |
Shapiro G, Lee S, Tolcher AW, et al. A first-in-human phase I study of the CDK4/6 inhibitor, LY2835219, for patients with advanced cancer. J Clin Oncol. 2013;31Suppl:Abstract 2500. | |
McClendon AK, Dean JL, Rivadeneira DB, et al. CDK4/6 inhibition antagonizes the cytotoxic response to anthracycline therapy. Cell Cycle. 2012;11(14):2747–2755. | |
Dean JL, McClendon AK, Knudsen ES. Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J Biol Chem. 2012;287(34):29075–29087. | |
Roberts PJ, Bisi JE, Strum JC, et al. Multiple roles of cyclin- dependent kinase 4/6 inhibitors in cancer therapy. J Natl Cancer Inst. 2012;104(6):476–487. | |
Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. | |
Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–675. | |
Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9(6):667–676. | |
Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011;13(6):224. | |
Burris HA 3rd. Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother Pharmacol. 2013;71(4):829–842. | |
Miller TW, Balko JM, Arteaga CL. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J Clin Oncol. 2011;29(33):4452–4461. | |
Razis E, Bobos M, Kotoula V, et al. Evaluation of the association of PIK3CA mutations and PTEN loss with efficacy of trastuzumab therapy in metastatic breast cancer. Breast Cancer Res Treat. 2011;128(2):447–456. | |
Shoman N, Klassen S, McFadden A, Bickis MG, Torlakovic E, Chibbar R. Reduced PTEN expression predicts relapse in patients with breast carcinoma treated by tamoxifen. Mod Pathol. 2005;18(2):250–259. | |
Saal LH, Johansson P, Holm K, et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc Natl Acad Sci U S A. 2007;104(18):7564–7569. | |
Pérez-Tenorio G, Alkhori L, Olsson B, et al. PIK3CA mutations and PTEN loss correlate with similar prognostic factors and are not mutually exclusive in breast cancer. Clin Cancer Res. 2007;13(12):3577–3584. | |
Gonzalez-Angulo Am, Ferrer-Lozano J, Stemke-Hale K, et al. PI3K pathway mutations and PTEN levels in primary and metastatic breast cancer. Mol Cancer Ther. 2011;10(6):1093–1101. | |
Bendell JC, Rodon J, Burris HA, et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30(3):282–290. | |
Doi T, Ando Y, Bando H, et al. Phase I dose-escalation study of BKM120, an oral pan-class I PI3K inhibitor, in Japanese patients with advanced solid tumors. Mol Cancer Ther. 2011;10(11 Supplement 1): Abstract B159. | |
Rodon J, Bendell JC, Abdul RAR, et al. Safety profile and clinical activity of single-agent BKM120, a pan-class I PIK inhibitor, for the treatment of patients with metastatic breast carcinoma. Cancer Res. 2011;71(24 Suppl 3):Abstract P3-16-01. | |
Gonzalez-Angulo AM, Juric D, Argilés G, et al. Safety, pharmacokinetics, and preliminary activity of the α-specific PI3K inhibitor BYL719: results from the first-in-human study. J Clin Oncol. 2013; 31 Suppl:Abstract 2531. | |
Miller TW, Hennessy BT, González-Angulo AM, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest. 2010;120(7):2406–2413. | |
O’Brien C, Wallin JJ, Sampath D, et al. Predictive biomarkers of sensitivity to the phosphatidylinositol 3′ kinase inhibitor GDC-0941 in breast cancer preclinical models. Clin Cancer Res. 2010;16(14):3670–3683. | |
She QB, Chandarlapaty S, Ye Q, et al. Breast tumor cells with PI3K mutation or HER2 amplification are selectively addicted to Akt signaling. PLoS One. 2008;3(8):e3065. | |
Ihle NT, Lemos R Jr, Wipf P, et al. Mutations in the phosphatidylinositol- 3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009;69(1):143–150. | |
Wallin JJ, Edgar KA, Guan J, et al. GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol Cancer Ther. 2011;10(12):2426–2436. | |
Mayer IA, Balko JM, Kuba MG, et al. SU2C phase Ib study of pan-PI3K inhibitor BKM120 plus aromatase inhibitor letrozole in ER+/HER2− metastatic breast cancer (MBC). Cancer Res. 2011;71(24 Suppl 3): Abstract PD09-05. | |
Janku F, Wheler JJ, Westin SN, et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012;30(8):777–782. | |
Maass N, Harbeck N, Mundhenke C, et al; German Breast Group. Everolimus as treatment for breast cancer patients with bone metastases only: results of the phase II RADAR study. J Cancer Res Clin Oncol. 2013;139(12):2047–2056. | |
Cavazzoni A, Bonelli MA, Fumarola C, et al. Overcoming acquired resistance to letrozole by targeting the PI3K/AKT/mTOR pathway in breast cancer cell clones. Cancer Lett. 2012;323(1):77–87. | |
Ghayad SE, Cohen PA. Inhibitors of the PI3K/Akt/mTOR pathway: new hope for breast cancer patients. Recent Pat Anticancer Drug Discov. 2010;5(1):29–57. | |
Ghayad SE, Bieche I, Vendrell JA, et al. mTOR inhibition reverses acquired endocrine therapy resistance of breast cancer cells at the cell proliferation and gene-expression levels. Cancer Sci. 2008;99(10):1992–2003. | |
Vilquin P, Villedieu M, Grisard E, et al. Molecular characterization of anastrozole resistance in breast cancer: pivotal role of the Akt/mTOR pathway in the emergence of de novo or acquired resistance and importance of combining the allosteric Akt inhibitor MK-2206 with an aromatase inhibitor. Int J Cancer. 2013;133(7):1589–1602. | |
Bachelot T, Bourgier C, Cropet C, et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J Clin Oncol. 2012;30(22):2718–2724. | |
Yardley DA, Noguchi S, Pritchard KI, et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther. 2013;30(10):870–884. | |
Hortobagyi GN, Piccart-Gebhart MJ, Rugo HS, et al. Correlation of molecular alterations with efficacy of everolimus in hormone receptor-positive, HER2-negative advanced breast cancer: results from BOLERO-2. J Clin Oncol. 2013;31 Suppl:Abstract LBA509. | |
Miller TW, Forbes JT, Shah C, et al. Inhibition of mammalian target of rapamycin is required for optimal antitumor effect of HER2 inhibitors against HER2-overexpressing cancer cells. Clin Cancer Res. 2009;15(23):7266–7276. | |
O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66(3):1500–1508. | |
Carracedo A, Ma L, Teruya-Feldstein J, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118(9):3065–3074. | |
Mayer IA, Abramson VG, Balko JM, et al. SU2C phase Ib study of pan-PI3K inhibitor BKM120 with letrozole in ER+/HER2− metastatic breast cancer (MBC). J Clin Oncol. 2012;30 Suppl: Abstract 510. | |
Perez EA, Dueck AC, McCullough AE, et al. Impact of PTEN protein expression on benefit from adjuvant trastuzumab in early-stage human epidermal growth factor receptor 2-positive breast cancer in the North Central Cancer Treatment Group N9831 trial. J Clin Oncol. 2013;31(17):2115–2122. | |
Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12(4):395–402. | |
O’Brien NA, Browne BC, Chow L, et al. Activated phosphoinositide 3-kinase/AKT signaling confers resistance to trastuzumab but not lapatinib. Mol Cancer Ther. 2010;9(6):1489–1502. | |
Wang L, Zhang Q, Zhang J, et al. PI3K pathway activation results in low efficacy of both trastuzumab and lapatinib. BMC Cancer. 2011;11:248. | |
Esteva FJ, Guo H, Zhang S, et al. PTEN, PIK3CA, p-AKT, and p-p70S6K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am J Pathol. 2010;177(4):1647–1656. | |
O’Brien NA, McDonald K, Tong L, et al. PI3K/mTOR inhibition overcomes in vitro and in vivo trastuzumab resistance independent of feedback activation of pAKT. Cancer Res. 2012;72(24 Suppl 3): Abstract P4-08-01. | |
Saura C, Bendell J, Jerusalem G, et al. Phase Ib study of buparlisib plus trastuzumab in patients with HER2-positive advanced or metastatic breast cancer that has progressed on trastuzumab-based therapy. Clin Cancer Res. 2014;20(7):1935–1945. | |
Morrow PK, Wulf GM, Ensor J, et al. Phase I/II study of trastuzumab in combination with everolimus (RAD001) in patients with HER2-overexpressing metastatic breast cancer who progressed on trastuzumab-based therapy. J Clin Oncol. 2011;29(23):3126–3132. | |
West KA, Castillo SS, Dennis PA. Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resist Updat. 2002;5(6):234–248. | |
Massarweh S, Osborne CK, Creighton CJ, et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008;68(3):826–833. | |
Lu Y, Zi X, Zhao Y, Mascarenhas D, Pollak M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J Natl Cancer Inst. 2001;93(24):1852–1857. | |
Vadgama JV, Wu Y, Datta G, Khan H, Chillar R. Plasma insulin-like growth factor-I and serum IGF-binding protein 3 can be associated with the progression of breast cancer, and predict the risk of recurrence and the probability of survival in African-American and Hispanic women. Oncology. 1999;57(4):330–340. | |
Creighton CJ, Casa A, Lazard Z, et al. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J Clin Oncol. 2008;26(25):4078–4085. | |
Bonneterre J, Peyrat JP, Beuscart R, Demaille A. Prognostic significance of insulin-like growth factor 1 receptors in human breast cancer. Cancer Res. 1990;50(21):6931–6935. | |
Fox EM, Miller TW, Balko JM, et al. A kinome-wide screen identifies the insulin/IGF-I receptor pathway as a mechanism of escape from hormone dependence in breast cancer. Cancer Res. 2011;71(21):6773–6784. | |
Quek R, Wang Q, Morgan JA, et al. Combination mTOR and IGF-1R inhibition: phase I trial of everolimus and figitumumab in patients with advanced sarcomas and other solid tumors. Clin Cancer Res. 2011;17(4):871–879. | |
Cao AZ, Pinzon-Ortiz M, Chen Y, et al. Targeting PIK3CA mutant breast cancer with the combination of PIK3CA-specific inhibitor, BYL719, and IGF1-R antibody, ganitumab. J Clin Oncol. 2013; 31 Suppl:Abstract e13525. | |
Fagan DH, Yee D. Crosstalk between IGF1R and estrogen receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia. 2008;13(4):423–429. | |
Cohen BD, Baker DA, Soderstrom C, et al. Combination therapy enhances the inhibition of tumor growth with the fully human anti-type 1 insulin-like growth factor receptor monoclonal antibody CP-751,871. Clin Cancer Res. 2005;11(5):2063–2073. | |
Robertson JF, Ferrero JM, Bourgeois H, et al. Ganitumab with either exemestane or fulvestrant for postmenopausal women with advanced, hormone-receptor-positive breast cancer: a randomised, controlled, double-blind, phase 2 trial. Lancet Oncol. 2013;14(3):228–235. | |
Dunn SE, Hardman RA, Kari FW, Barrett JC. Insulin-like growth factor 1 (IGF-1) alters drug sensitivity of HBL100 human breast cancer cells by inhibition of apoptosis induced by diverse anticancer drugs. Cancer Res. 1997;57(13):2687–2693. | |
Gooch JL, Van Den Berg CL, Yee D. Insulin-like growth factor (IGF)-I rescues breast cancer cells from chemotherapy-induced cell death – proliferative and anti-apoptotic effects. Breast Cancer Res Treat. 1999;56(1):1–10. | |
Haluska P, Shaw H, Batzel GN, et al. Phase I dose escalation study of the anti-IGF-1R monoclonal antibody CP-751,871 in patients with refractory solid tumors. J Clin Oncol. 2007; 25(Suppl 18):Abstract 3586. | |
Karp DD, Paz-Ares LG, Novello S, et al. Phase II study of the anti-insulin-like growth factor type 1 receptor antibody CP-751,871 in combination with paclitaxel and carboplatin in previously untreated, locally advanced, or metastatic non-small-cell lung cancer. J Clin Oncol. 2009;27(15):2516–2522. | |
Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005;65(23):11118–11128. | |
Friedbichler K, Hofmann MH, Kroez M, et al. Pharmacodynamic and antineoplastic activity of BI 836845, a fully human IGF ligand-neutralizing antibody, and mechanistic rationale for combination with rapamycin. Mol Cancer Ther. 2014;13(2):399-409. | |
Chen YC, Sosnoski DM, Mastro AM. Breast cancer metastasis to the bone: mechanisms of bone loss. Breast Cancer Res. 2010;12(6):215. | |
Hershman DL, McMahon DJ, Crew KD, et al. Zoledronic acid prevents bone loss in premenopausal women undergoing adjuvant chemotherapy for early-stage breast cancer. J Clin Oncol. 2008;26(29):4739–4745. | |
Gnant M, Mlineritsch B, Luschin-Ebengreuth G, et al; Austrian Breast and Colorectal Cancer Study Group (ABCSG). Adjuvant endocrine therapy plus zoledronic acid in premenopausal women with early-stage breast cancer: 5-year follow-up of the ABCSG-12 bone-mineral density substudy. Lancet Oncol. 2008;9(9):840–849. | |
Shapiro CL, Halabi S, Hars V, et al. Zoledronic acid preserves bone mineral density in premenopausal women who develop ovarian failure due to adjuvant chemotherapy: final results from CALGB trial 79809. Eur J Cancer. 2011;47(5):683–689. | |
Green JR. Bisphosphonates: preclinical review. Oncologist. 2004; 9 Suppl 4:3–13. | |
Winter MC, Holen I, Coleman RE. Exploring the anti-tumour activity of bisphosphonates in early breast cancer. Cancer Treat Rev. 2008;34(5):453–475. | |
Daubiné F, Le Gall C, Gasser J, Green J, Clézardin P. Antitumor effects of clinical dosing regimens of bisphosphonates in experimental breast cancer bone metastasis. J Natl Cancer Inst. 2007;99(4):322–330. | |
Ottewell PD, Lefley DV, Cross SS, Evans CA, Coleman RE, Holen I. Sustained inhibition of tumor growth and prolonged survival following sequential administration of doxorubicin and zoledronic acid in a breast cancer model. Int J Cancer. 2010;126(2):522–532. | |
Hiraga T, Williams PJ, Ueda A, Tamura D, Yoneda T. Zoledronic acid inhibits visceral metastases in the 4T1/luc mouse breast cancer model. Clin Cancer Res. 2004;10(13):4559–4567. | |
Gnant M. Zoledronic acid in the treatment of early-stage breast cancer: is there a final verdict? Curr Oncol Rep. 2012;14(1):35–43. | |
Dent R, Trudeau M, Pritchard KI, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15 Pt 1):4429–4434. | |
Kennecke H, Yerushalmi R, Woods R, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol. 2010;28(20):3271–3277. | |
Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006;12(9):440–450. | |
McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66(16):8109–8115. | |
Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. | |
Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. | |
Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–5599. | |
Murai J, Huang SY, Renaud A, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13(2):433–443. | |
Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–244. | |
Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–819. | |
Dent RA, Lindeman GJ, Clemons M, et al. Phase I trial of the oral PARP inhibitor olaparib in combination with paclitaxel for first- or second-line treatment of patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2013;15(5):R88. | |
Kristeleit RS, Shapiro G, LoRusso P, et al. A phase I dose-escalation and PK study of continuous oral rucaparib in patients with advanced solid tumors. J Clin Oncol. 2013;31 Suppl: Abstract 2585. | |
Isakoff SJ, Overmoyer B, Tung NM, et al. A phase II trial expansion cohort of the PARP inhibitor veliparib (ABT888) and temozolomide in BRCA1/2 associated metastatic breast cancer. Cancer Res. 2011; 71(24 Suppl 3):P3-16-05. | |
Fan J, Yin WJ, Lu JS, et al. ER alpha negative breast cancer cells restore response to endocrine therapy by combination treatment with both HDAC inhibitor and DNMT inhibitor. J Cancer Res Clin Oncol. 2008;134(8):883–890. | |
Jang ER, Lim SJ, Lee ES, et al. The histone deacetylase inhibitor trichostatin A sensitizes estrogen receptor alpha-negative breast cancer cells to tamoxifen. Oncogene. 2004;23(9):1724–1736. | |
Thomas S, Thurn KT, Biçaku E, Marchion DC, Münster PN. Addition of a histone deacetylase inhibitor redirects tamoxifen-treated breast cancer cells into apoptosis, which is opposed by the induction of autophagy. Breast Cancer Res Treat. 2011;130(2):437–447. | |
Yardley DA, Ismail-Khan RR, Melichar B, et al. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol. 2013;31(17):2128–2135. | |
Munster PN, Thurn KT, Thomas S, et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer. 2011;104(12):1828–1835. | |
Conte P, Campone M, Pronzato P, et al. Phase I trial of panobinostat (LBH589) in combination with trastuzumab in pretreated HER2-positive metastatic breast cancer (mBC): preliminary safety and tolerability results. J Clin Oncol. 2009;27(15s Suppl): Abstract 1081. | |
Frame MC. Src in cancer: deregulation and consequences for cell behavior. Biochim Biophys Acta. 2002;1602(2):114–130. | |
Zhang XH, Wang Q, Gerald W, et al. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell. 2009;16(1):67–78. | |
Elsberger B, Tan BA, Mitchell TJ, et al. Is expression or activation of Src kinase associated with cancer-specific survival in ER-, PR- and HER2-negative breast cancer patients? Am J Pathol. 2009;175(4):1389–1397. | |
Elsberger B, Tan BA, Mallon EA, Brunton VG, Edwards J. Is there an association with phosphorylation and dephosphorylation of Src kinase at tyrosine 530 and breast cancer patient disease-specific survival. Br J Cancer. 2010;103(12):1831–1834. | |
Morgan L, Gee J, Pumford S, et al. Elevated Src kinase activity attenuates Tamoxifen response in vitro and is associated with poor prognosis clinically. Cancer Biol Ther. 2009;8(16):1550–1558. | |
Herold CI, Chadaram V, Peterson BL, et al. Phase II trial of dasatinib in patients with metastatic breast cancer using real-time pharmacodynamic tissue biomarkers of Src inhibition to escalate dosing. Clin Cancer Res. 2011;17(18):6061–6070. | |
Finn RS, Bengala C, Ibrahim N, et al. Dasatinib as a single agent in triple-negative breast cancer: results of an open-label phase 2 study. Clin Cancer Res. 2011;17(21):6905–6913. | |
Fedele P, Calvani N, Marino A, et al. Targeted agents to reverse resistance to endocrine therapy in metastatic breast cancer: where are we now and where are we going? Crit Rev Oncol Hematol. 2012;84(2):243–251. | |
Fornier MN, Morris PG, Abbruzzi A, et al. A phase I study of dasatinib and weekly paclitaxel for metastatic breast cancer. Ann Oncol. 2011;22(12):2575–2581. | |
Somlo G, Atzori F, Strauss LC, et al. Dasatinib plus capecitabine for advanced breast cancer: safety and efficacy in phase I study CA180004. Clin Cancer Res. 2013;19(7):1884–1893. | |
Paul D, Vukelja SJ, Holmes FA, et al. Letrozole plus dasatinib improves progression-free survival (PFS) in hormone receptor-positive, HER2-negative postmenopausal metastatic breast cancer (MBC) patients receiving first-line aromatase inhibitor (AI) therapy. Cancer Res. 2013;73:Abstract S3-07. | |
Bristol-Myers Squibb. Safety and efficacy of exemestane plus dasatinib versus placebo for advanced ER+ breast cancer. Available from: http://clinicaltrials.gov/show/NCT00767520. NLM identifier: NCT00767520. Accessed March 14, 2014. | |
Parganas E, Wang D, Stravopodis D, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93(3):385–395. | |
Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7(9):673–683. | |
Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. | |
Sansone P, Bromberg J. Targeting the interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol. 2012;30(9):1005–1014. | |
Marotta LL, Almendro V, Marusyk A, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24− stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121(7):2723–2735. | |
Britschgi A, Andraos R, Brinkhaus H, et al. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: a rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell. 2012;22(6):796–811. | |
Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. | |
Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.