")
Back to Journals » Clinical and Experimental Gastroenterology » Volume 7
Clinical potential of naloxegol in the management of opioid-induced bowel dysfunction
Authors Poulsen JL, Brock C , Olesen AE , Nilsson M, Drewes AM
Received 11 June 2014
Accepted for publication 21 July 2014
Published 19 September 2014 Volume 2014:7 Pages 345—358
DOI https://doi.org/10.2147/CEG.S52097
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Jakob Lykke Poulsen,1 Christina Brock,1,2 Anne Estrup Olesen,1,2 Matias Nilsson,1 Asbjørn Mohr Drewes1,3
1Mech-Sense, Department of Gastroenterology and Hepatology, Aalborg University Hospital, Aalborg, Denmark; 2Department of Drug Design and Pharmacology, University of Copenhagen, Copenhagen, Denmark; 3Department of Clinical Medicine, Aalborg University, Aalborg, Denmark
Abstract: Opioid-induced bowel dysfunction (OIBD) is a burdensome condition which limits the therapeutic benefit of analgesia. It affects the entire gastrointestinal tract, predominantly by activating opioid receptors in the enteric nervous system, resulting in a wide range of symptoms, such as reflux, bloating, abdominal cramping, hard, dry stools, and incomplete evacuation. The majority of studies evaluating OIBD focus on constipation experienced in approximately 60% of patients. Nevertheless, other presentations of OIBD seem to be equally frequent. Furthermore, laxative treatment is often insufficient, which in many patients results in decreased quality of life and discontinuation of opioid treatment. Novel mechanism-based pharmacological approaches targeting the gastrointestinal opioid receptors have been marketed recently and even more are in the pipeline. One strategy is prolonged release formulation of the opioid antagonist naloxone (which has limited systemic absorption) and oxycodone in a combined tablet. Another approach is peripherally acting, µ-opioid receptor antagonists (PAMORAs) that selectively target µ-opioid receptors in the gastrointestinal tract. However, in Europe the only PAMORA approved for OIBD is the subcutaneously administered methylnaltrexone. Alvimopan is an oral PAMORA, but only approved in the US for postoperative ileus in hospitalized patients. Finally, naloxegol is a novel, oral PAMORA expected to be approved soon. In this review, the prevalence and pathophysiology of OIBD is presented. As PAMORAs seem to be a promising approach, their potential effect is reviewed with special focus on naloxegol's pharmacological properties, data on safety, efficacy, and patient-focused perspectives. In conclusion, as naloxegol is administered orally once daily, has proven efficacious compared to placebo, has an acceptable safety profile, and can be used as add-on to existing pain treatment, it is a welcoming addition to the targeted treatment possibilities for OIBD.
Keywords: opioids, gut, dysfunction, constipation, naloxegol, opioid antagonists
Introduction
Opioids have been used for thousands of years for the treatment of diarrhea and moderate to severe pain of both benign and malignant origin. Opioids are currently the most commonly prescribed medications to treat severe pain in the Western world and it has been estimated that up to 90% of US patients presenting to specialized pain centers are treated with opioids.1
Most opioids are opioid receptor agonists and mainly exert their analgesic effect within the central nervous system (CNS), although it is well known that opioid receptors are synthesized in the dorsal root ganglia and then transported to both central and peripheral nerve terminals.2,3
Unfortunately, the clinical use is limited by the development of tolerance. Moreover, adverse effects including sedation, respiratory depression, nausea, and gastrointestinal (GI)-related symptoms may also indirectly interfere with opioid efficiency. Some adverse effects are restricted to the peripheral action of opioids. In the GI tract, they mainly exert their action on the enteric nervous system (ENS), where they bind to opioid receptors in the myenteric and submucosal plexuses. Here they cause dysmotility, decreased fluid secretion, and sphincter dysfunction. In concert, these synergistic effects lead to opioid-induced bowel dysfunction (OIBD).4 Hence, OIBD is a pharmacologically induced condition manifested with different symptoms such as dry mouth, gastroesophageal reflux, vomiting, bloating, abdominal pain, anorexia, hard stools, constipation, and incomplete evacuation.5,6 Unfortunately, tolerance to OIBD does not evolve and consequently several studies have confirmed a high prevalence of OIBD in pain patients.5,7 Regrettably, the negative impact of OIBD is often overlooked and it is one of the most common reasons for patients to avoid or discontinue opioid use.8,9
Typical treatment strategies to alleviate OIBD rely on the use of different laxatives, in combination with non-pharmacological strategies, such as increased dietary fiber and fluid intake, encouraging exercise, bio-feedback, etc. However, these strategies do not address the underlying pathophysiology of OIBD.4 Another approach is to use opioids with additional effects, such as tapentadol, which has a noradrenergic reuptake inhibitory action that results in an additional analgesic effect. Consequently the dose can be reduced, resulting in less opioid receptors (including those in the gut) being blocked, thereby improving the adverse effect profile.10,11 An alternative treatment targeting the pathophysiology of OIBD is the use of opioid antagonists. For the antagonist not to interfere with the analgesic effect, the antagonistic effect must be restricted to peripheral opioid receptors. One strategy has been the administration of opioid antagonists with limited systemic absorption, as seen in the combined prolonged-release oxycodone and naloxone tablet. Another strategy has been the development of peripherally acting μ-opioid receptor antagonists (PAMORAs) that selectively target μ-receptors in the GI-tract. Currently only two PAMORAs are marketed – the subcutaneously administered methylnaltrexone and the oral tablet alvimopan. However, their clinical use is limited by various factors that will be addressed in the section “Peripherally acting μ-opioid receptor antagonists”.
Naloxegol is a novel PAMORA. Approval for marketing has been filed in the US and Europe, and a final positive decision from the US Food and Drug Administration is expected by September 2014. It is an oral polymer conjugate of naloxone, incorporating a polyethylene glycol (PEG) moiety that limits its capacity to cross the blood–brain barrier.12
In the present review, clinical presentation and prevalence of OIDB is presented as background information, along with GI physiology and the relation to opioid pharmacology. Pathophysiology of OIBD and potential methods for assessment are covered in order to understand the potential impact of PAMORAs on OIBD symptoms. Lastly, available treatment possibilities for OIBD are presented and, due to the expected release, special focus on the safety, efficacy, and tolerability profile of naloxegol is presented and discussed, along with patient-focused perspectives for evaluation of the clinical potential of naloxegol in the management of OIBD. Although methylnaltrexone originally was designed to shorten the duration of postoperative ileus, and the indication of alvimopan is postoperative ileus under specific conditions, the scope of the review is bowel dysfunction brought on by the administration of exogenous opioids, and not surgical/anesthesia-induced bowel dysfunction.
Symptoms and prevalence of OIBD
Recently, the prevalence of GI symptoms in patients treated with opioids for non-malignant pain was examined and constipation was found in 47% of patients, gastroesophageal reflux related symptoms in 33%, nausea in 27%, and vomiting in 9%. Furthermore, chronic abdominal pain was reported by 58%.13 Similar results appeared in a population-based survey, where increased frequency of constipation-related symptoms (including straining, hard stools, and infrequent bowel movements) were reported, along with other GI symptoms, such as nausea, heartburn, abdominal pain, and bloating.14 Another prospective survey of incidence, prevalence, and severity of morphine adverse effects during repeated individualized dosing for chronic cancer pain found that 95% of all patients reported dry mouth, 88% reported sedation and constipation, and <50% reported nausea.15
The prevalence of constipation is probably the most studied adverse effect in opioid treated patients. However, prevalence rates vary significantly ranging from 15% to 81% in non-cancer patients.16–20 The highest rate (81%) of constipation – despite concomitant use of laxatives – was reported in a multinational internet-based survey of 322 chronic pain patients taking daily oral opioids.17 Along this line, constipation was frequently reported (57% of patients) in a population-based survey of 2,055 patients treated with opioids and laxatives for chronic non-cancer pain.18 These numbers should be seen in light of the prevalence of chronic constipation in the general population, which has been estimated to affect – depending on the definition used – between 2% and 27% of the adult population with an average around 15%.21–23 However, conflicting clinical scenarios have also been reported where diarrhea-related symptoms (including urgency, loose bowel movements, and frequent bowel movements) appeared to be increased in patients treated with opioids, most likely a result of overflow diarrhea.24 Of note, constipation is often defined as the number of spontaneous bowel movements (SBMs), and in fact this is only ranked number five in patients suffering from constipation, where symptoms such as straining and gas are far more prevalent. This strongly emphasizes the limitations of subjective clinical symptoms such as constipation in clinical trials.25 Furthermore, other risk factors for constipation in pain patients, such as reduced mobility, age, low-fiber diet, sex, and different drugs, may also contribute to the collective GI symptomatology.22
Collectively, these findings underline the diversity of GI-related adverse effects associated with short term and chronic (often defined as >90 days) opioid use. While constipation may present the dominant symptom in most cases, it is vastly important to keep in mind the multifaceted pathophysiology and symptomatology of OIBD when evaluating patients in the clinic.
Gastrointestinal physiology and opioid pharmacology
The clinical potential of naloxegol in the management of OIBD must be seen in light of the underlying mechanisms of opioid actions by combining knowledge of opioid agonist and antagonist pharmacology together with GI anatomy and physiology.
Visceral afferents
Visceral afferents run predominantly with sympathetic nerves to the CNS; however some afferents join parasympathetic and parallel pathways. They mediate conscious sensation and modulate local functions and reflexes such as gut motility, secretion, mucosal transport, and blood flow.26 Motor neurons in ENS coordinate these functions by transducing neural input originating from local sensory neurons, although some also receive inputs from the CNS via autonomic (both sympathetic and parasympathetic) pathways.27
The enteric nervous system
The GI innervation is established via the ENS and sensory neurons (extrinsic afferents). The ENS is situated within the entire gut wall from esophagus to anus, and forms an integrative nervous network regulating motor neurons and effector systems. Two major divisions of ENS motor neurons exist:1) musculomotor neurons innervating the muscularis externa and the muscularis mucosae; and 2) secretomotor neurons innervating the intestinal secretory glands.28 The myenteric plexus and the submucosal plexus control and regulate these delicate balances.
The myenteric plexus is situated between the longitudinal and circular muscle layers in the gut wall and is primarily responsible for controlling motor activity,26,29–31 whereas the submucosal plexus controls secretory and absorptive activities.26,32
Opioid pharmacology
Opioid receptor sub-types exist: μ-receptors, δ-receptors, κ-receptors, and the opioid receptor like-1.33 μ-, δ-, and κ-subtype receptors have been identified in the GI tract of rodents and humans, but the relative distribution differs throughout the GI tract depending on the specific region and histological layer of the gut, and between species. The primary mediator of opioid analgesic effects in the CNS is the μ-receptor (μ1), which is also present in the GI- tract (μ2). In the human GI tract, μ-receptors are predominantly localized on myenteric and submucosal neurons and on immune cells in the lamina propria.7,31
Endogenous ligands (eg, enkephalins, endorphins, and dynorphins) and most clinically applied opioids can activate μ-receptors,34 which transduce their signals through interaction with guanine nucleotide binding proteins.35 The comprehensive intracellular signaling involves numerous steps that ultimately result in inhibition of adenylyl cyclase, which converts adenosine triphosphate to cyclic adenosine monophosphate (AMP). Consequently, opioids decrease the formation of cyclic AMP,36 which otherwise would have activated several target molecules to regulate cellular functions. The overall effect is the reduced release of neurotransmitters and decreased neuronal activity.
Pathophysiology of OIBD
The diverse clinical presentations of OIBD are a net result of disturbances in three essential GI functions: gut motility, secretion, and sphincter function. In the following, the pathophysiological background for OIBD is described. As objective measures are important to provide credible and reliable evaluation of new treatments, suggestions for assessment methods are provided for each of the three functions.
Gut motility
The gut motility is controlled by neurotransmitter secretion from the enteric neurons and excitation–contraction coupling in the circular smooth muscles. Among others, the neurotransmitters include acetylcholine, serotonin, vasoactive intestinal peptide, and nitric oxide. Acetylcholine-release activates the cholinergic excitatory motor neurons situated in the longitudinal smooth muscles, in contrast to nitric oxide and vasoactive intestinal peptide, which control the inhibition of the non-cholinergic inhibitory motor neurons in the circular smooth muscles. Hence, the coordination of the contractile and propulsive gut motility is determined by a balance between acetylcholine and nitric oxide/vasoactive intestinal peptide release.37,38 Because opioids inhibit neurotransmitter release, administration will directly disrupt this balance, resulting in abnormal coordination of motility.39
Assessment of gut motility
Assessment of gut motility has traditionally been carried out by radio-opaque marker studies and radionuclide studies; however no golden standard exists.40 Moreover, most of these techniques are only capable of estimating total transit time or in one segment (gastric, small bowel, colonic) per examination. As OIBD affects the entire GI tract, objective assessments of interventions should include segmental transit times, ie, gastric emptying, small bowel transit, colonic transit, and total transit. This can be carried out by recently introduced wireless motility capsule systems, such as the SmartPill®, which samples and transmits intraluminal pH, pressure, and temperature, allowing measurement of segmental transit times comparable to scintigraphic measures, but is carried out in a single examination.41–43 Another option is the motility tracking system that monitors the progress of a magnetic pill through the GI tract in real time. Besides segmental transit times, it also provides information about the position, direction, velocity, and amplitude of bowel movements, and thus disturbed propulsive dynamics of OIBD can be evaluated.44,45 The principle of the motility tracking system is illustrated in Figure 1.
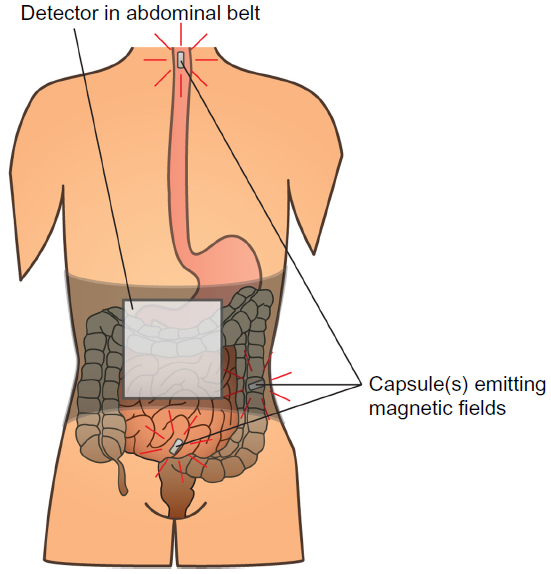 | Figure 1 Principle of the motility tracking system for evaluation of gut motility. |
Gut secretion
Intestinal fluid secretion is crucial to the establishment of an ideal environment favoring effective digestion, nutrient absorption, and propulsion. The GI tract secretes approximately 8–9 L of fluid on a daily basis (approximately 2 L saliva, 2.5 L gastric juice, 0.5–1 L bile, 1.5 L pancreatic juice, and 1.5–2 L in the small intestine). Since fluid cannot be actively secreted into the gut, this process is dependent on an osmotic gradient within the enterocyte.46
Chloride secretion is the major determinant of mucosal hydration. Chloride secretion can be either reduced or increased, such as in cystic fibrosis and secretory diarrhea. Enterocyte secretion is controlled by a variety of endocrine, paracrine, autocrine, neuronal, and immunological effectors. The classical basic and well-characterized second messengers that modulate intestinal ion transport are the cyclic nucleotides cyclic AMP, cyclic guanosine monophosphate, and free cytosolic Ca2+.47 In many cells expressing opioid receptors, adenyl cyclase (which converts adenosine triphosphate to cyclic AMP) inhibition has been demonstrated and consequently opioids directly decrease the formation of cyclic AMP.36 Cyclic AMP production is also indirectly increased by vasoactive intestinal peptide release.28,48 Furthermore, as gut motility is also dependent on volume inside the lumen via local stretch reflexes, decreased secretion (and volume) will also result in the slowing of motility.
Assessment of secretion
Assessing the secretory capacity of human intestinal tissue in vivo is difficult. Animal experiments often rely on the removal of the intestine; a critical methodological limitation for human trials.49 As animal data cannot uncritically be transferred to the human situation, other methods are used. One of these is the assessment of intestinal secretion with stool electrolyte and osmolality assays. However, these methods are mainly focused on differentiating the etiology of secretory and osmotic diarrhea (eg, congenital chloridorrhea versus lactose intolerance).50,51 A possible and commonly used way of circumventing these problems is the Ussing chamber. The Ussing chamber relies on fixing viable tissue between two chambers, respectively simulating gut lumen and blood supply. Conventionally, relatively large tissue samples have been required, acquired from surgical resections.52 Recently, however, a novel approach has been developed which relies on small tissue samples, such as endoscopically acquired mucosa biopsies.53 By measuring the osmotic gradient between the two chambers, an indirect measure of the ion transport is obtained. Because intestinal secretion largely depends on the ion flux across the gut epithelium, the Ussing chamber is an obvious choice for the assessment of intestinal secretory capacity.54–56 Figure 2 illustrates the principle of the Ussing chamber.
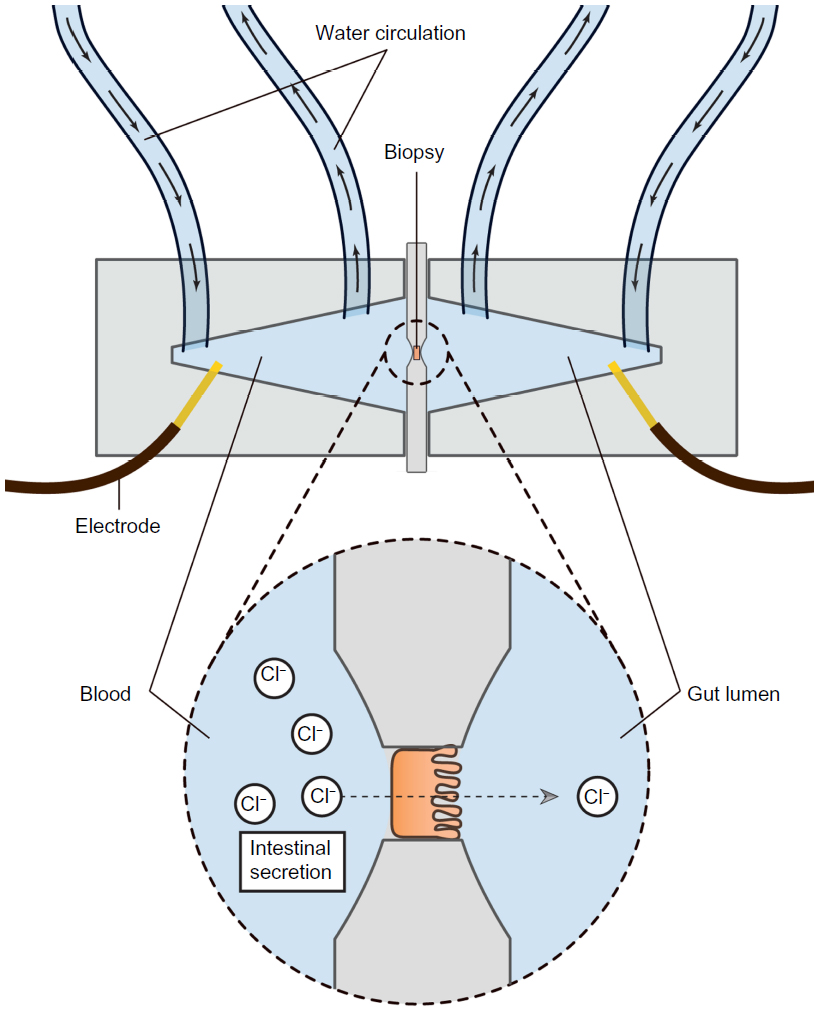 | Figure 2 Schematic representation of the Ussing chamber measuring gut secretion. |
Sphincter function
There are six sphincters throughout the GI tract; upper esophageal sphincters control food entry into the esophagus and the lower esophageal sphincter prevents reflux of gastric contents. At the distal end of the stomach the pyloric sphincter controls the passage of chyme into the duodenum. The ileocecal sphincter regulates propulsive movements between the small and large intestine, whereas the internal and external anal sphincters control defecation. Finally, the sphincter of Oddi is placed at the junction of the bile duct, pancreatic duct, and duodenum, controlling flow of bile and pancreatic juices into the duodenum and preventing reflux of duodenal content into the ducts.57 Thus, each of the sphincters play an important role for normal transit, and any sphincter dyscoordination will lead to bowel dysfunction.
Opioids may induce esophageal muscular dysfunction which leads to decreased lower esophageal sphincter pressure, and thereby increased incidence of gastroesophageal reflux.58 However, the opposite has also been reported where incomplete lower esophageal sphincter relaxation leads to achalasia-like symptoms.59,60
Opioid-induced sphincter of Oddi dysfunction is a well-known adverse effect of opioid administration, characterized by spasm of the sphincter of Oddi and consequently increased intrabiliary duct pressure.61–64 The patients present with upper abdominal pain, clinically resembling a gallbladder attack, and in some cases also dilation of the bile ducts and with or without a dilated pancreatic duct on abdominal ultrasound.65
Although anal sphincter dysfunction in OIBD has not been systematically evaluated, one-third of patients in a recent study of opioid-treated patients reported the sensation of anal blockage.13 This is in line with studies indicating that opioids not only inhibit detection of stool in the upper anal canal, but also relaxation of the internal anal sphincter.66,67
Assessment of sphincter function
Sphincter (dys)function and dyscoordination is complex and very hard to assess. Anorectal manometry is the most common technique for assessment of anorectal dysfunction such as impaired rectal contraction, paradoxical anal contraction, impaired anal relaxation, or a combination of these mechanisms. Other possible tests are defecography or the balloon expulsion test, which provides an assessment of the patient’s ability to defecate.40 However, in biomechanical terms, the overall ability of a sphincter to distend or open is related to many factors, including muscle tone, passive viscoelasticity, mechanoreceptor-mediated reflexes and perception, none of which can be measured with the previous mentioned methods.68 To overcome problems like this, methods such as the functional lumen imaging probe technique (FLIP) have been developed to study GI sphincters.69 The main advantage is that it can distinguish between an open and closed sphincter and record the distensibility during challenge tests (ie, squeezing, expulsion), whereas manometry only shows the difference between a toned and a relaxed sphincter.70,71 Thus, the FLIP may provide useful information in the evaluation of anorectal dysfunction in OIBD patients. Figure 3 illustrates the principle of the FLIP system.
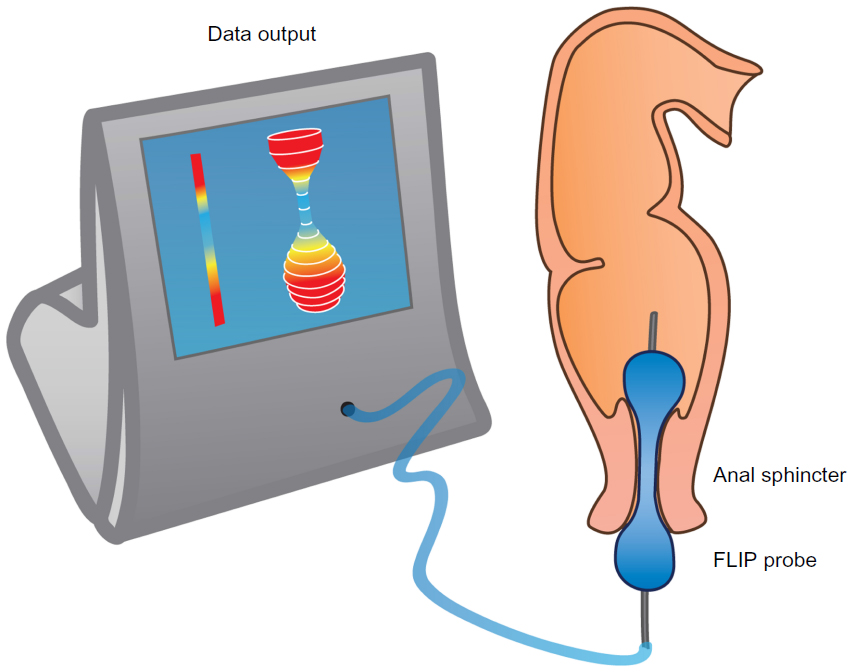 | Figure 3 The FLIP probe. |
Treatment of OIBD
Treatment of OIBD is a challenge, and the current recommended strategies involve the use of different laxatives in combination with non-pharmacological strategies, such as increased dietary fiber and fluid intake, encouraging exercise, etc. However, these interventions are rarely sufficient to increase the number of bowel movements, while other presentations of OIBD may persist. In the following, a short overview of the current treatment possibilities and pharmacological approaches in the pipeline are presented. Table 1 illustrates pharmacological mechanisms, clinical symptoms, and the potential effect of laxatives and PAMORAs, as they represent the traditional treatment and the most promising mechanism-based treatment, respectively.
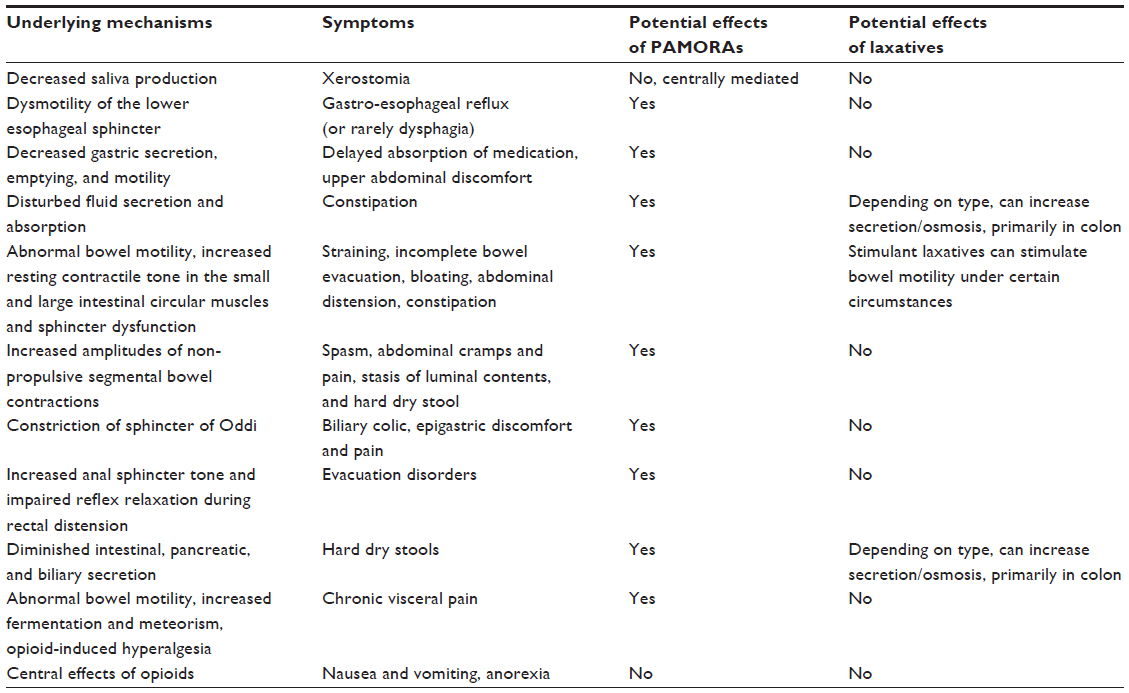 | Table 1 Underlying mechanisms, symptoms, and the potential effects of PAMORAs and laxatives, respectively |
Laxatives
Generally laxatives are recommended as first line treatment in all patients to whom opioids are prescribed.72,73 Laxatives can be divided into different sub-groups, including osmotic agents (magnesium, lactulose, polyethylene glycol), stimulants (bisacodyl, senna), bulking agents (methylcellulose, psyllium), and stool softeners (anionic surfactants). Although traditional laxatives have proven useful in inducing bowel movements, they are temporary, quick fixes and often come with undesirable side effects, and hence their efficacy is, overall, insufficient.17 For further details about mechanisms and recommendations for laxatives, see Rao,40 and Lembo and Camilleri.74
Opioid antagonists
Opioid antagonists target the underlying pathophysiology of OIBD – blockage of the μ-opioid receptors in the gut. It is mainly the pharmacokinetic properties that separate them, rendering them more or less suitable for treatment of OIBD. In the following, their effect on the three pathophysiological mechanisms in OIBD is described and subsequently the different alternatives of opioid antagonists available for treatment of OIBD are presented.
Effect on gut motility
Opioid antagonists will alleviate the opioid-induced increased resting tone in the circular muscle layer. Moreover, opioid antagonists will enhance tonic inhibition of the muscle tone. Consequently, they will act towards normalization of the tone in the circular muscle layer and prevent opioid-induced rhythmic contractions, with propulsive phasic contractions. Taken together, the outcome of opioid antagonism treatment is diminished segmental contraction along with a normalization of peristaltic propulsion, and consequently decreased transit time. Indirectly this will also counteract the increased passive absorption of fluids as seen due to stasis of luminal contents during opioid treatment.39 This will likely reduce typical OIBD symptoms, including constipation, gut spasm, and abdominal cramps.38,75,76
Effect on gut secretion
Opioid antagonists will counteract the opioid-induced decreased cyclic AMP formation and consequently tend to normalize chloride secretion. When opioids act on μ-receptors within the ENS, it in return increases activity in the sympathetic nervous system, leading to inhibited vasoactive intestinal peptide release, and hence decreasing gut secretion in an indirect manner.26,32 This effect is also counterbalanced by opioid antagonists. Collectively, the patient will experience less dry and softer stools.37,39,77
Effect on sphincter function
Opioid antagonists will – at least in theory – normalize sphincter coordination. Opioid antagonists can prevent opioid-induced sphincter of Oddi dysfunction and thus acute biliary-like type of pain attacks.78 Finally, opioid antagonists may diminish opioid-induced dysfunction of the anal sphincter, which has been associated with straining, hemorrhoids, and/or incomplete evacuation.4 The sphincter has, however, two components (internal with visceral innervation and external with somatic, voluntary innervation), and this complicates the evaluation of the effect of opioids and their antagonists.
Prolonged release naloxone – an opioid antagonist with limited systemic absorption
Naloxone is a competitive opioid receptor antagonist, with high affinity to opioid receptors. It is widely used to treat opioid overdose and is administered intravenously or as intramuscular injection. In this formulation it reverses both centrally and peripherally mediated effects of opioids. However, naloxone has a very high first-pass metabolism, thus when administered orally means bioavailability is less than 2%.79 Even though first-pass metabolism of naloxone is close to 100%, the part that passes may cross the blood–brain barrier and cause withdrawal symptoms and reversal of analgesia. This might explain why naloxone in oral formulation is not marketed, despite the fact that it has been shown to improve opioid-induced constipation (OIC).80 To avoid this, a combined oral prolonged release formulation of oxycodone and prolonged release naloxone in a 2:1 ratio has been developed. The aim of this formulation has been to counteract OIC through the local antagonistic effect of naloxone in the gut wall, while maintaining analgesia due to the low bioavailability of oral prolonged release naloxone. Studies have shown promising analgesic efficacy as well as improvement in OIBD-related symptoms.81–83 However, as naloxone is primarily metabolized in the liver, there is a risk of greater bioavailability in patients with hepatic impairment,84,85 which complicates treatment. The fixed combined doses, high costs, and maximum recommended daily dose (which may not be sufficient to relieve the pain) may, however, limit its use. Furthermore, the fixed combination to oxycodone makes opioid rotation difficult.
Peripherally acting μ-opioid receptor antagonists
Another approach is methylnaltrexone-bromide, a drug originally designed to shorten the length of postoperative ileus.86 It is a PAMORA and a derivative of the opioid antagonist naltrexone. Due to its ammonium group, the drug does not pass the blood–brain barrier.72 It has been shown to relieve OIC and induce laxation,87,88 and so far methylnaltrexone is the only approved PAMORA for the treatment of OIBD. However, it is only available in subcutaneous formulation and only approved in palliative care in patients with advanced illness. Hence it is of limited benefit for the general OIBD population.
Alvimopan is another orally administered PAMORA that does not cross the blood–brain barrier at clinically relevant doses, and does not reverse analgesia or cause opioid withdrawal symptoms.89–91 Alvimopan has been shown to increase spontaneous bowel movements,90 however cardiovascular safety concerns (increased risk of myocardial infarction) halted further development. Yet the Food and Drug Administration approved alvimopan for postoperative ileus following partial small or large bowel resection with primary anastomosis in hospitalized patients, and it is only registered in the US, hence it is also of little benefit for the general OIBD population.
Other PAMORAs in earlier stages of development are ADL-5945 and ADL-7445 (Cubist®), and TD-1211 (Theravance®). ADL-5945 and ADL-7445 have proven tolerable and effective in producing SBMs in Phase I trials, but to the authors’ knowledge no Phase II data have been published, even though a Phase II study was announced in 2010.72 The TD-1211 has shown to be well-tolerated, and have a linear pharmacokinetic profile, but is still being evaluated in Phase II trials.4,72
Naloxegol is another investigational PAMORA, which has shown promising results and expected to be approved for marketing in September 2014. Therefore a special focus on its pharmacological properties, safety, and efficacy is given separately in the next section.
Naloxegol – PEGylated naloxone
Naloxegol is a PEGylated naloxone molecule. The result of covalently attaching a PEG moiety to a therapeutically useful molecule is what has come to be known as PEGylation. The effect of the PEGylation is determined by several factors, including the pharmacological properties of the parent molecule, the number of PEG chains attached to the molecule, the molecular weight and structure of the PEGs, and their location on the parent molecule, as well as the chemical processes used to attach the PEG.92 PEGylation is used to alter the functional and structural properties of the molecule. These include:1) reduction of immunogenicity by shielding antigenic epitopes; 2) reduce renal clearance due to increased molecule size; 3) improve drug solubility; 4) prolong plasma half-life; 5) reduce toxicity; and 6) alter biodistribution.
Naloxone possesses the potent μ-receptor antagonism desirable for a PAMORA. However, blood–brain barrier penetration and the extensive first-pass metabolism complicate its use. In order for naloxone to be clinically applicable, increased bioavailability and particularly peripheral restriction is warranted. These features have been successfully achieved in naloxegol by PEGylation of naloxone.12 Figure 4 illustrates the pharmacological principle of naloxegol, normal GI physiology (in regards to motility, secretion, and sphincter function), OIBD pathophysiology, and the pharmacodynamics of naloxegol.
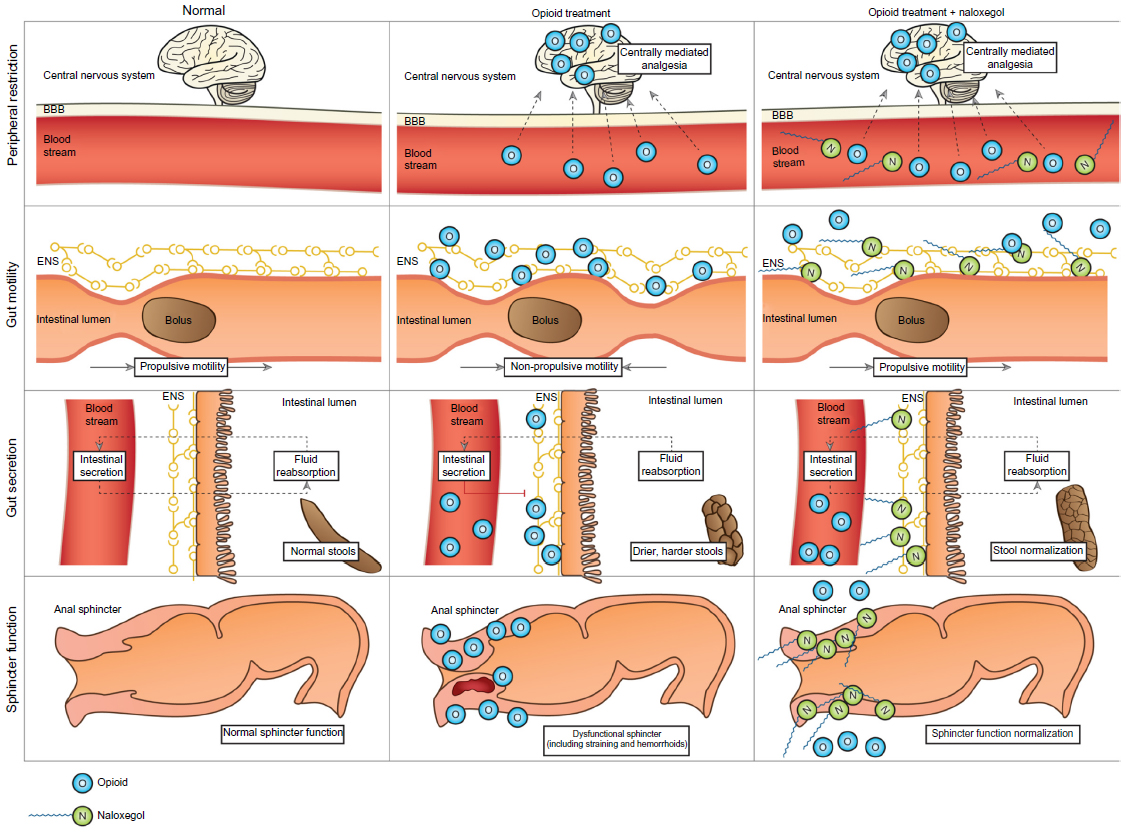 | Figure 4 Pharmacological principle of naloxegol under normal conditions (left column), during opioid treatment (middle) and opioid and naloxegol treatment (right). |
PEGs are also used as osmotic laxatives (eg, macrogol 3350/4000), where they act as non-metabolized, non-absorbable, osmotic agents, forming hydrogen bonds with water in the intestinal lumen. Conversely fecal volume increases, which in turn enhances gut motility through activation of the neuromuscular pathways.93 As described in the next section, naloxegol is absorbed rapidly and so far no evidence points toward a secondary effect as an osmotic laxative, although it has not been evaluated specifically and cannot be ruled out. However, the lower molecular weight of naloxegol versus osmotic laxatives based on PEGs, 651 g/mol versus 3,350/4,000 g/mol respectively,94,95 supports better absorptive properties of naloxegol.
Pharmacokinetics
In vitro naloxegol displays decreased receptor binding affinity compared to naloxone, likely due to the structural modification of the PEGylation. However compared to opioids, naloxegol displays higher receptor binding affinity and hence will displace, eg, morphine from μ-opioid receptors in the myenteric and submucosal plexuses. As noted, peripheral restriction is of utmost importance, and it has been shown that blood–brain barrier permeation of naloxegol is 15-fold lower than naloxone, and similar to atenolol, which is considered as having no appreciable blood–brain barrier permeation.96 This peripheral restriction was emphasized when the effect of naloxegol versus naloxone was investigated in morphine-treated rats. Hence, whilst naloxone and naloxegol completely reversed the opioid effects within the GI system, the centrally mediated opioid analgesia was only markedly decreased during naloxone administration.96
Clinical studies of oral naloxegol exploring a dose range of 8–1,000 mg found rapid absorption with time to peak concentration of 1.5 hours, and plasma concentrations of naloxegol were orders of magnitude greater than those of oral naloxone.96–98
Safety, tolerability, and efficacy
In preclinical and clinical studies, naloxegol has been examined for potential drug–drug interactions, toxicology, and drug accumulation.96,99–102 Concomitant administration of drugs interacting with CYP3A4 and/or P-glycoprotein transporters can potentially affect plasma concentration of naloxegol. Renal impairment and mild or moderate hepatic impairment appear to have minimal impact on the pharmacokinetics of naloxegol.100,101 Otherwise there were no reports of adverse events (AEs), toxicology, or drug accumulation.
In an early clinical study, the investigators examined the efficacy of naloxegol on morphine-induced delay in oral–cecal transit time using the lactulose hydrogen breath GI motility test.103 Naloxegol significantly reduced morphine-induced prolongation with 61% relative to baseline following NKTR-118 (early code-name for naloxegol) with morphine, compared to placebo with morphine.
In a Phase II trial, 207 patients with OIC on a stable opioid regime of 30–1,000 mg/day oral morphine equivalent units were randomized to receive 4 weeks double-blind placebo or naloxegol (5, 25, or 50 mg) once daily.12 The efficacy of naloxegol was evaluated by median change from baseline in SBMs per week after week 1 of drug administration. In both the 25 and 50 mg cohorts a significant increase was observed. The median change from baseline in SBMs per week across the 4-week study period was also significantly greater in these two treatment groups versus placebo. Similarly, the time to first laxation was significantly shorter in these treatment groups. For the 5 mg treatment group there were no significant changes in any of the efficacy endpoints. In the 50 mg cohort, the incidence of treatment-emergent AEs was higher than in the placebo group. Most frequently reported AEs were transient GI complaints, typically resolving within the first week, and often reported as mild or moderate. No clinical relevant changes in serum chemistry, hematology, or electrocardiogram were revealed, confirming the results from two Phase I studies.97,98 There was no significant reduction of opioid-mediated analgesia. A significant change in the median Clinical Opiate Withdrawal Score was observed in the 50 mg group day 1 of week 1. However, this difference was in the GI component and when excluded, there was no difference between placebo and naloxegol.
The largest studies evaluating the safety and efficacy of naloxegol are two identical Phase III, randomized, double blind, 12-week studies referred to as KODIAC-04 and KODIAC-05. Data from these studies have recently been published.104 In total, 1,352 patients with OIC participated in the two studies. They were randomized to naloxegol (12.5 and 25 mg) versus placebo for 12 weeks. The primary endpoint was the efficacy of naloxegol assessed as percentage of responders during the 12 weeks (intent-to-treat population), defined as having ≥three spontaneous bowel movements per week with ≥three spontaneous bowel movements per week increase over baseline for ≥9 weeks and ≥three of the last four weeks of treatment. It was met with in the 25 mg groups in both studies and for the 12.5 mg dose in KODIAC-04. Furthermore, the investigators evaluated the percentage of responders in a subpopulation of patients with an inadequate response to laxatives before enrollment. Interestingly, the response rates were even higher, ie, the number needed to treat was lower than in the intention-to-treat group. Pain scores and daily opioid use were unaffected by naloxegol treatment and assessment of opioid-mediated central analgesia was unaltered by treatment with naloxegol. AEs were reported more frequently in the 25 mg group, but most were GI related and mild in severity.
Patient-focused perspectives
Objective assessments of the different pathophysiological mechanisms of OIBD described earlier provide credible and reliable evaluations of new treatments. However, due to the multifaceted symptomatology of OIBD (Table 1), focusing only on, eg, SBMs or transit times will not only miss other GI-related adverse effects, but also important aspects like patient-focused perspectives as subjective severity and impact on quality-of-life. This is underlined by the fact that many opioid-treated patients report normal stool frequency, but still experience symptoms of OIBD.17 Thus, a combination of objective and subjective assessment methods is mandatory when evaluating the treatment effect of a drug on OIBD. A detailed discussion about tools for evaluating OIBD is beyond the scope of this review, but reviewed elsewhere.105
The most comprehensive subjective assessment of naloxegol treatment has been carried out by Webster et al.12 For both the 25 and 50 mg group mean scores for physical discomfort, dissatisfaction was lower at multiple time points, reflecting increased quality of life. Additionally, the 25 mg cohort experienced statistically significant improvement in physical functioning, mental health, social functioning, and vitality. In the Phase III study only simple subjective measures were carried out, including severity of straining measured on a 5-point scale and stool consistency assessed on the Bristol stool scale. Improvement of both endpoints was observed in the 25 mg group in both studies and in the 12.5 mg group in the KODIAC-5 study, just as the primary endpoint.
Patient-focused perspectives also include acceptability and adherence to treatment, which are not easily measured. However, the oral formulation is obviously an advantage compared to the subcutaneously administered methylnaltrexone. Likewise, as methylnaltrexone is only approved for palliative care in patients with advanced illness, it remains limited to OIBD symptom management in a small subgroup of OIBD patients. Lastly, it is an advantage that naloxegol can be used as an add-on to existing pain therapy, as many pain patients are on a stable and satisfactory analgesic regime, but suffer from OIBD symptoms.
Conclusion
OIBD is a challenge in pain therapy and is often neglected by clinicians. Importantly, opioids affect the whole gut, and even when focusing on constipation, bowel movements are often not the most important symptom for the patient. The traditional treatment with laxatives is of limited efficacy and adverse effects are frequent. Therefore new treatment modalities, such as prolonged release naloxone and PAMORAs, are promising as they target the underlying pathophysiology of OIBD. Naloxegol is a novel, oral PAMORA, which has proven efficacious compared to placebo, has an acceptable safety profile, and can be used as an add-on to existing pain treatment. This makes it a welcome addition to the targeted treatment possibilities for OIBD. However, further evaluations of these new treatment modalities are warranted in order to justify their clinical use. First, this should be done as comparative studies between an adequate laxative treatment and the new treatments modalities. Secondly, head-to-head comparison of the different new modalities is needed to evaluate whether one is superior to another. Thirdly, these studies should include a comprehensive combination of objective and subjective assessments in order to encompass the multifaceted presentation of OIBD.
Disclosure
Asbjørn Mohr Drewes has received unrestricted research grants from Mundipharma, AstraZeneca, Grünenthal, Lundbeck and Pfizer and served as a Consultant/Advisory Board member for Mundipharma, Grünenthal, AstraZeneca, Almirall and Shire. The authors report no other conflicts of interest in this work.
References
Trescot AM, Glaser SE, Hansen H, Benyamin R, Patel S, Manchikanti L. Effectiveness of opioids in the treatment of chronic non-cancer pain. Pain Physician. 2008;11(Suppl 2):S181–S200. | |
Ji RR, Zhang Q, Law PY, Low HH, Elde R, Hökfelt T. Expression of mu-, delta-, and kappa-opioid receptor-like immunoreactivities in rat dorsal root ganglia after carrageenan-induced inflammation. J Neurosci. 1995;15(12):8156–8166. | |
Laduron PM. Axonal transport of opiate receptors in capsaicin- sensitive neurones. Brain Res. 1984;294(1):157–160. | |
Brock C, Olesen SS, Olesen AE, Frøkjaer JB, Andresen T, Drewes AM. Opioid-induced bowel dysfunction: pathophysiology and management. Drugs. 2012;72(14):1847–1865. | |
Pappagallo M. Incidence, prevalence, and management of opioid bowel dysfunction. Am J Surg. 2001;182(Suppl 5A):11S–18S. | |
Looström H, Akerman S, Ericson D, Tobin G, Götrick B. Tramadol-induced oral dryness and pilocarpine treatment: effects on total protein and IgA. Arch Oral Biol. 2011;56(4):395–400. | |
Kurz A, Sessler DI. Opioid-induced bowel dysfunction: pathophysiology and potential new therapies. Drugs. 2003;63(7):649–671. | |
Thorpe DM. Management of opioid-induced constipation. Curr Pain Headache Rep. 2001;5(3):237–240. | |
Tamayo AC, Diaz-Zuluaga PA. Management of opioid-induced bowel dysfunction in cancer patients. Support Care Cancer. 2004;12(9):613–618. | |
Afilalo M, Morlion B. Efficacy of tapentadol ER for managing moderate to severe chronic pain. Pain Physician. 2013;16(1):27–40. | |
Niesters M, Proto PL, Aarts L, Sarton EY, Drewes AM, Dahan A. Tapentadol potentiates descending pain inhibition in chronic pain patients with diabetic polyneuropathy. Br J Anaesth. 2014;113(1):148–156. | |
Webster L, Dhar S, Eldon M, Masuoka L, Lappalainen J, Sostek M. A phase 2, double-blind, randomized, placebo-controlled, dose-escalation study to evaluate the efficacy, safety, and tolerability of naloxegol in patients with opioid-induced constipation. Pain. 2013;154(9):1542–1550. | |
Tuteja AK, Biskupiak J, Stoddard GJ, Lipman AG. Opioid-induced bowel disorders and narcotic bowel syndrome in patients with chronic non-cancer pain. Neurogastroenterol Motil. 2010;22(4):424–430. | |
Choung RS, Locke GR 3rd, Zinsmeister AR, Schleck CD, Talley NJ. Opioid bowel dysfunction and narcotic bowel syndrome: a population-based study. Am J Gastroenterol. 2009;104(5):1199–1204. | |
Glare P, Walsh D, Sheehan D. The adverse effects of morphine: a prospective survey of common symptoms during repeated dosing for chronic cancer pain. Am J Hosp Palliat Care. 2006;23(3):229–235. | |
Allan L, Hays H, Jensen NH, et al. Randomised crossover trial of transdermal fentanyl and sustained release oral morphine for treating chronic non-cancer pain. BMJ. 2001;322(7295):1154–1158. | |
Bell TJ, Panchal SJ, Miaskowski C, Bolge SC, Milanova T, Williamson R. The prevalence, severity, and impact of opioid-induced bowel dysfunction: results of a US and European Patient Survey (PROBE 1). Pain Med. 2009;10(1):35–42. | |
Cook SF, Lanza L, Zhou X, et al. Gastrointestinal side effects in chronic opioid users: results from a population-based survey. Aliment Pharmacol Ther. 2008;27(12):1224–1232. | |
Kalso E, Edwards JE, Moore RA, McQuay HJ. Opioids in chronic non-cancer pain: systematic review of efficacy and safety. Pain. 2004;112(3):372–380. | |
Moore RA, McQuay HJ. Prevalence of opioid adverse events in chronic non-malignant pain: systematic review of randomised trials of oral opioids. Arthritis Res Ther. 2005;7(5):R1046–R1051. | |
Sanchez MI, Bercik P. Epidemiology and burden of chronic constipation. Can J Gastroenterol. 2011;25 Suppl B:11B–15B. | |
Wirz S, Nadstawek J, Elsen C, Junker U, Wartenberg HC. Laxative management in ambulatory cancer patients on opioid therapy: a prospective, open-label investigation of polyethylene glycol, sodium picosulphate and lactulose. Eur J Cancer Care (Engl). 2012;21(1):131–140. | |
Higgins PD, Johanson JF. Epidemiology of constipation in North America: a systematic review. Am J Gastroenterol. 2004;99(4):750–759. | |
Bril S, Shoham Y, Marcus J. The “mystery” of opioid-induced diarrhea. Pain Res Manag. 2011;16(3):197–199. | |
Johanson JF, Kralstein J. Chronic constipation: a survey of the patient perspective. Aliment Pharmacol Ther. 2007;25(5):599–608. | |
Furness J, Costa M. The Enteric Nervous System. Edinburgh: Churchill Livingstone; 1987. | |
Aziz Q, Thompson DG. Brain-gut axis in health and disease. Gastroenterology. 1998;114(3):559–578. | |
Wood JD. Enteric nervous system: sensory physiology, diarrhea and constipation. Curr Opin Gastroenterol. 2010;26(2):102–108. | |
Furness JB, Kunze WA, Bertrand PP, Clerc N, Bornstein JC. Intrinsic primary afferent neurons of the intestine. Prog Neurobiol. 1998;54(1):1–18. | |
Holzer P. Treatment of opioid-induced gut dysfunction. Expert Opin Investig Drugs. 2007;16(2):181–194. | |
Sternini C, Patierno S, Selmer IS, Kirchgessner A. The opioid system in the gastrointestinal tract. Neurogastroenterol Motil. 2004; 16 Suppl 2:3–16. | |
Huizinga JD, Lammers WJ. Gut peristalsis is governed by a multitude of cooperating mechanisms. Am J Physiol Gastrointest Liver Physiol. 2009;296(1):G1–G8. | |
Fioravanti B, Vanderah TW. The ORL-1 receptor system: are there opportunities for antagonists in pain therapy? Curr Top Med Chem. 2008;8(16):1442–1451. | |
Greenwood-Van Meerveld B, Gardner CJ, Little PJ, Hicks GA, Dehaven-Hudkins DL. Preclinical studies of opioids and opioid antagonists on gastrointestinal function. Neurogastroenterol Motil. 2004;16 Suppl 2:46–53. | |
Jordan B, Devi LA. Molecular mechanisms of opioid receptor signal transduction. Br J Anaesth. 1998;81(1):12–19. | |
Sharma SK, Nirenberg M, Klee WA. Morphine receptors as regulators of adenylate cyclase activity. Proc Natl Acad Sci U S A. 1975;72(2):590–594. | |
Wood JD, Galligan JJ. Function of opioids in the enteric nervous system. Neurogastroenterol Motil. 2004;16 Suppl 2:17–28. | |
Sarna SK, Otterson MF. Small intestinal amyogenesia and dysmyogenesia induced by morphine and loperamide. Am J Physiol. 1990;258(2 Pt 1):G282–G289. | |
Thomas J. Opioid-induced bowel dysfunction. J Pain Symptom Manage. 2008;35(1):103–113. | |
Rao SS. Constipation: evaluation and treatment of colonic and anorectal motility disorders. Gastroenterol Clin North Am. 2007;36(3):687–711. | |
Parkman HP. Assessment of gastric emptying and small-bowel motility: scintigraphy, breath tests, manometry, and SmartPill. Gastrointest Endosc Clin N Am. 2009;19(1):49–55. | |
Maqbool S, Parkman HP, Friedenberg FK. Wireless capsule motility: comparison of the SmartPill GI monitoring system with scintigraphy for measuring whole gut transit. Dig Dis Sci. 2009;54(10):2167–2174. | |
Zhang WQ, Yan GZ, Ye DD, Chen CW. Simultaneous assessment of the intraluminal pressure and transit time of the colon using a telemetry technique. Physiol Meas. 2007;28(2):141–148. | |
Stathopoulos E, Schlageter V, Meyrat B, Ribaupierre Y, Kucera P. Magnetic pill tracking: a novel non-invasive tool for investigation of human digestive motility. Neurogastroenterol Motil. 2005;17(1):148–154. | |
Hiroz P, Schlageter V, Givel JC, Kucera P. Colonic movements in healthy subjects as monitored by a Magnet Tracking System. Neurogastroenterol Motil. 2009;21(8):838–e57. | |
Murek M, Kopic S, Geibel J. Evidence for intestinal chloride secretion. Exp Physiol. 2010;95(4):471–478. | |
Barrett KE, Keely SJ. Chloride secretion by the intestinal epithelium: molecular basis and regulatory aspects. Annu Rev Physiol. 2000;62:535–572. | |
Kunzelmann K, Mall M. Electrolyte transport in the mammalian colon: mechanisms and implications for disease. Physiol Rev. 2002;82(1):245–289. | |
Li Y, Xiang YY, Lu WY, Liu C, Li J. A novel role of intestine epithelial GABAergic signaling in regulating intestinal fluid secretion. Am J Physiol Gastrointest Liver Physiol. 2012;303(4):G453–G460. | |
Thomas PD, Forbes A, Green J, et al. Guidelines for the investigation of chronic diarrhoea, 2nd ed. Gut. 2003;52 Suppl 5:v1–v15. | |
Eherer AJ, Fordtran JS. Fecal osmotic gap and pH in experimental diarrhea of various causes. Gastroenterology. 1992;103(2):545–551. | |
van de Kerkhof EG, Ungell AL, Sjöberg AK, et al. Innovative methods to study human intestinal drug metabolism in vitro: precision-cut slices compared with ussing chamber preparations. Drug Metab Dispos. 2006;34(11):1893–1902. | |
Larsen R, Mertz-Nielsen A, Hansen MB, Poulsen SS, Bindslev N. Novel modified Ussing chamber for the study of absorption and secretion in human endoscopic biopsies. Acta Physiol Scand. 2001;173(2):213–222. | |
Osbak PS, Bindslev N, Poulsen SS, Kaltoft N, Tilotta MC, Hansen MB. Colonic epithelial ion transport is not affected in patients with diverticulosis. BMC Gastroenterol. 2007;7:37. | |
Sun X, Wang X, Wang GD, et al. Lubiprostone reverses the inhibitory action of morphine on mucosal secretion in human small intestine. Dig Dis Sci. 2011;56(2):330–338. | |
Kaltoft N, Tilotta MC, Witte AB, et al. Prostaglandin E2-induced colonic secretion in patients with and without colorectal neoplasia. BMC Gastroenterol. 2010;10:9. | |
Toouli J. Sphincter of Oddi: Function, dysfunction, and its management. J Gastroenterol Hepatol. 2009;24 Suppl 3:S57–S62. | |
Penagini R, Bartesaghi B, Zannini P, Negri G, Bianchi PA. Lower oesophageal sphincter hypersensitivity to opioid receptor stimulation in patients with idiopathic achalasia. Gut. 1993;34(1):16–20. | |
Kraichely RE, Arora AS, Murray JA. Opiate-induced oesophageal dysmotility. Aliment Pharmacol Ther. 2010;31(5):601–606. | |
Thorén T, Carlsson E, Sandmark S, Wattwil M. Effects of thoracic epidural analgesia with morphine or bupivacaine on lower oesophageal motility – an experimental study in man. Acta Anaesthesiol Scand. 1988;32(5):391–394. | |
Helm JF, Venu RP, Geenen JE, et al. Effects of morphine on the human sphincter of Oddi. Gut. 1988;29(10):1402–1407. | |
Thompson DR. Narcotic analgesic effects on the sphincter of Oddi: a review of the data and therapeutic implications in treating pancreatitis. Am J Gastroenterol. 2001;96(4):1266–1272. | |
Wu SD, Zhang ZH, Jin JZ, et al. Effects of narcotic analgesic drugs on human Oddi’s sphincter motility. World J Gastroenterol. 2004;10(19):2901–2904. | |
Coelho JC, Senninger N, Runkel N, Herfarth C, Messmer K. Effect of analgesic drugs on the electromyographic activity of the gastrointestinal tract and sphincter of Oddi and on biliary pressure. Ann Surg. 1986;204(1):53–58. | |
Sharma SS. Sphincter of Oddi dysfunction in patients addicted to opium: an unrecognized entity. Gastrointest Endosc. 2002;55(3):427–430. | |
Bouvier M, Kirschner G, Gonella J. Actions of morphine and enkephalins on the internal anal sphincter of the cat: relevance for the physiological role of opiates. J Auton Nerv Syst. 1986;16(3):219–232. | |
Burleigh DE, D’Mello A. Neural and pharmacologic factors affecting motility of the internal anal sphincter. Gastroenterology. 1983;84(2):409–417. | |
Gregersen H. Biomechanics of the Gastrointestinal Tract. New Perspectives in Motility. Research and Diagnostics. London: Springer-Verlag; 2002:93–114. | |
McMahon BP, Frøkjaer JB, Liao D, Kunwald P, Drewes AM, Gregersen H. A new technique for evaluating sphincter function in visceral organs: application of the functional lumen imaging probe (FLIP) for the evaluation of the oesophago-gastric junction. Physiol Meas. 2005;26(5):823–836. | |
McMahon BP, Drewes AM, Gregersen H. Functional oesophago-gastric junction imaging. World J Gastroenterol. 2006;12(18):2818–2824. | |
Alqudah MM, Gregersen H, Drewes AM, McMahon BP. Evaluation of anal sphincter resistance and distensibility in healthy controls using EndoFLIP ©. Neurogastroenterol Motil. 2012;24(12):e591–e599. | |
Diego L, Atayee R, Helmons P, Hsiao G, von Gunten CF. Novel opioid antagonists for opioid-induced bowel dysfunction. Expert Opin Investig Drugs. 2011;20(8):1047–1056. | |
Herndon CM, Jackson KC 2nd, Hallin PA. Management of opioid-induced gastrointestinal effects in patients receiving palliative care. Pharmacotherapy. 2002;22(2):240–250. | |
Lembo A, Camilleri M. Chronic constipation. N Engl J Med. 2003;349(14):1360–1368. | |
Frantzides CT, Cowles V, Salaymeh B, Tekin E, Condon RE. Morphine effects on human colonic myoelectric activity in the postoperative period. Am J Surg. 1992;163(1):144–148; discussion 148–149. | |
Telford GL, Condon RE, Szurszewski JH. Opioid receptors and the initiation of migrating myoelectric complexes in dogs. Am J Physiol. 1989;256(1 Pt 1):G72–G77. | |
De Luca A, Coupar IM. Insights into opioid action in the intestinal tract. Pharmacol Ther. 1996;69(2):103–115. | |
Torres D, Parrinello G, Trapanese C, Licata G. Sudden severe abdominal pain after a single low dose of paracetamol/codein in a cholecystectomized patient: learning from a case report. Am J Ther. 17(4):e133–e134. | |
Smith K, Hopp M, Mundin G, et al. Low absolute bioavailability of oral naloxone in healthy subjects. Int J Clin Pharmacol Ther. 2012;50(5):360–367. | |
Meissner W, Schmidt U, Hartmann M, Kath R, Reinhart K. Oral naloxone reverses opioid-associated constipation. Pain. 2000;84(1):105–109. | |
Vondrackova D, Leyendecker P, Meissner W, et al. Analgesic efficacy and safety of oxycodone in combination with naloxone as prolonged release tablets in patients with moderate to severe chronic pain. J Pain. 2008;9(12):1144–1154. | |
Burness CB, Keating GM. Oxycodone/Naloxone prolonged-release: a review of its use in the management of chronic pain while counteracting opioid-induced constipation. Drugs. 2014;74(3):353–375. | |
Leppert W. The place of oxycodone/naloxone in chronic pain management. Contemp Oncol (Pozn). 2013;17(2):128–133. | |
Sykes NP. An investigation of the ability of oral naloxone to correct opioid-related constipation in patients with advanced cancer. Palliat Med. 1996;10(2):135–144. | |
Leppert W. New treatment possibilities for opioid-induced bowel dysfunction. Pain. 2013;154(9):1491–1492. | |
Kraft MD. Methylnaltrexone, a new peripherally acting mu-opioid receptor antagonist being evaluated for the treatment of postoperative ileus. Expert Opin Investig Drugs. 2008;17(9):1365–1377. | |
Portenoy RK, Thomas J, Moehl Boatwright ML, et al. Subcutaneous methylnaltrexone for the treatment of opioid-induced constipation in patients with advanced illness: a double-blind, randomized, parallel group, dose-ranging study. J Pain Symptom Manage. 2008;35(5):458–468. | |
Thomas J, Karver S, Cooney GA, et al. Methylnaltrexone for opioid-induced constipation in advanced illness. N Engl J Med. 2008;358(22):2332–2343. | |
Schmidt WK. Alvimopan* (ADL 8-2698) is a novel peripheral opioid antagonist. Am J Surg. 2001;182(Suppl 5A):27S–38S. | |
Paulson DM, Kennedy DT, Donovick RA, et al. Alvimopan: an oral, peripherally acting, mu-opioid receptor antagonist for the treatment of opioid-induced bowel dysfunction – a 21-day treatment-randomized clinical trial. J Pain. 2005;6(3):184–192. | |
Camilleri M. Alvimopan, a selective peripherally acting mu-opioid antagonist. Neurogastroenterol Motil. 2005;17(2):157–165. | |
Roberts MJ, Bentley MD, Harris JM. Chemistry for peptide and protein PEGylation. Adv Drug Deliv Rev. 2002;54(4):459–476. | |
Belsey JD, Geraint M, Dixon TA. Systematic review and meta analysis: polyethylene glycol in adults with non-organic constipation. Int J Clin Pract. 2010;64(7):944–955. | |
The PubChem Project– [homepage on the Internet]. Naloxegol. National Center for Biotechnology Information. Available at: http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=56959087#itabs-2d. Accessed July 10, 2014. | |
Wirz S, Klaschik E. Management of constipation in palliative care patients undergoing opioid therapy: is polyethylene glycol an option? Am J Hosp Palliat Care. 2005;22(5):375–381. | |
Eldon M, Song D, Neumann T. Oral NKTR-118 (oral PEG-naloxol), a PEGylated derivative of naloxone; demonstration of selective peripheral opioid antagonism after oral administration. Poster presented at: The American Academy of Pain Management 18th Annual Clinical Meeting; September 27–30, 2007; Las Vegas, NV. | |
Gottfridsson C, Carlson G, Lappalainen J, Sostek M. Evaluation of the effect of Naloxegol on cardiac repolarization: a randomized, placebo- and positive-controlled crossover thorough QT/QTc study in healthy volunteers. Clin Ther. 2013;35(12):1876–1883. | |
Paaschen H van, Sahner D, Marcantonio A, Eldon MA. Results from a phase 1, double-blind, randomized, placebo-controlled. Multiple-dose study evaluating the safety, tolerability, and pharmacokinetics of oral doses of NKTR-118 (PEG-Naloxol). J Pain. 2008; 9(4, Supplement 2):28. Available from http://www.nektar.com/pdf/pipeline/NKTR-118/NKTR-118_Phase_1_APS_Poster_20080507.pdf. Accessed August 19, 2014. | |
Odinecs A, Song Y, Harite S, Lee MG, Kugler AR, Eldon MA. NKTR-118, an oral peripheral opioid antagonist, has low potential for drug-drug interactions. Poster presented at: The American College of Clinical Pharmacology 38th Annual Meeting; September 13–15, 2009; San Antonio, TX. | |
Bui K, She F, Sostek M. The effects of mild or moderate hepatic impairment on the pharmacokinetics, safety, and tolerability of naloxegol. J Clin Pharmacol. Epub 2014 Jun 19. | |
Bui K, She F, Sostek M. The effects of renal impairment on the pharmacokinetics, safety, and tolerability of naloxegol. J Clin Pharmacol. Epub 2014 Jun 19. | |
US Food and Drug Administration. Naloxegol NDA 204-760 Briefing Document for the Anesthetic and Analgesic Drug Products Advisory Committee (AADPAC). Washington DC: US Food and Drug Administration. Available at: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AnestheticAndAnalgesicDrugProductsAdvisoryCommittee/UCM400209.pdf. Accessed August 14, 2014. | |
Neumann T, Paaschen H Van, Marcantonio A, Song D, Kugler AR, Eldon MA. Evaluation of PEG-Naloxol (NKTR-118) as an oral peripheral opioid antagonist in healthy male subjects: a double-blind, placebo-controlled, dose escalation crossover study. Poster presented at: The American College of Clinical Pharmacology 36th Annual Meeting; September 9–11, 2007; San Francisco, CA. | |
Chey WD, Webster L, Sostek M, Lappalainen J, Barker PN, Tack J. Naloxegol for opioid-induced constipation in patients with noncancer pain. N Engl J Med. 2014;370(25):2387–2396. | |
Olesen AE, Drewes AM. Validated tools for evaluating opioid-induced bowel dysfunction. Adv Ther. 2011;28(4):279–294. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.