")
Back to Journals » ImmunoTargets and Therapy » Volume 3
Clinical potential of apremilast in the treatment of psoriatic arthritis
Authors Cauli A, Porru G, Piga M , Vacca A, Dessole G, Mathieu A
Received 17 December 2013
Accepted for publication 13 March 2014
Published 9 June 2014 Volume 2014:3 Pages 91—96
DOI https://doi.org/10.2147/ITT.S40199
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Alberto Cauli, Giovanni Porru, Matteo Piga, Alessandra Vacca, Grazia Dessole, Alessandro Mathieu
Rheumatology Unit, Department of Medical Sciences, Policlinico of University of Cagliari, Monserrato, Italy
Abstract: Psoriatic arthritis (PsA) is a frequent chronic inflammatory disease characterized by joint and skin involvement, and by typical extra-articular manifestations. Although the pathogenesis of PsA is still under investigation, the available evidence suggests the importance of the patient's genetic background, microbial or environmental triggers, and an imbalance in the adaptive and acquired immune system, resulting in the production of inflammatory mediators. New therapeutic approaches have been proposed, among them the use of modulators of intracellular signals and gene transcription such as PDE4-inhibiting compounds, which are able to modulate the activity of transcription factors such as CREB and NF-κB and therefore the synthesis of inflammatory mediators, resulting in immunoregulation. This paper summarizes the mechanism of action of apremilast, a PDE4 inhibitor, and the clinical data available on its clinical efficacy and safety profile in the treatment of PsA patients.
Keywords: psoriatic arthritis, apremilast, therapy
Introduction
Psoriatic arthritis (PsA) is a frequent inflammatory disease that affects both peripheral and axial joints, enthuses, and the skin (psoriatic dermatitis, Ps). It is usually seronegative for rheumatoid factor and it has been included in the spectrum of the spondyloarthropathies (SpA). According to the traditional Moll and Wright classification,1 PsA clinical subsets, often overlapping, can be classified as follows: peripheral joint arthritis (polyarticular, oligoarticular, distal, or mutilans) and inflammatory axial disease. Enthesitis, dactylitis, and uveitis are common features that are shared with the other spondyloarthropathies such as ankylosing spondylitis (AS), reactive arthritis (ReA), and SpA associated with inflammatory bowel disease (IBD).2
PsA pathogenesis is believed to be deeply influenced by genetic susceptibility,3–5 immune system imbalance, and environmental triggers. It is mainly mediated by T cells6 interacting with antigen-presenting cells (APC) and other cells of the inflammatory milieu,7 giving rise to the inflammatory cascade that leads to joint damage and repair mechanisms. In this context, increasing evidence has recently suggested the importance of a new subset of T lymphocyte, named Th17 according to its signature cytokine.8
The initial treatments of PsA are non-steroidal anti-inflammatory drugs (NSAIDs), steroid intraarticular injections if a single joint is affected, shortly followed by one or more disease-modifying anti-rheumatic agents (DMARDs) in peripheral joint disease subsets. In clinical practice, and according to international and national guidelines,9,10 the most widely used DMARDs are methotrexate (effective both in skin and joint manifestations), sulfasalazine, leflunomide, and cyclosporine A.11–15
In “non-responders” and in the axial subset, anti-TNFα agents are indicated in order to suppress inflammation and stop structural changes. It is recommended not to delay the switch from a traditional DMARD to the “biologic treatment” when needed in order to avoid erosive changes and joint damage.9,10
However, a proportion of patients does not respond or develop side effects to traditional or biologic DMARDs; therefore, alternative drugs with different mechanisms of action are needed. Among the new molecules which have been recently proposed to the clinical community, apremilast is the most promising and attractive because of its novel mechanism of action.
Cyclic adenosine monophosphate and phosphodiesterase-4 in the inflammatory milieu
Biologic DMARDs available for chronic arthritis specifically target a single cytokine, receptor, or surface antigen in order to influence the inflammatory cascade and milieu. Another approach is to act at an earlier point, interfering with intracellular signaling that controls gene expression of cytokines and inflammatory mediators involved in the disease mechanisms.
Second intracellular messengers, such as cyclic adenosine monophosphate (cAMP), are involved in responses to various stimuli (endogenous and exogenous) in almost all types of cells, such as lymphocytes, monocytes, APCs, and various cells involved in immune responses. The intracellular concentrations of cAMP represent the balance of formed and degraded cAMP by means of the activity of the adenylyl cyclases (mainly activated via G protein-coupled receptors [GPCRs]) and the family of phosphodiesterases (PDEs), some of them expressed in a tissue-specific distribution.14 Phosphodiesterase-4 (PDE4) is well-expressed in dendritic cells, T cells, macrophages, and monocytes.16–18 PDE4 is also expressed in keratinocytes, smooth muscle cells, vascular endothelium, and chondrocytes.17,19,20 It is noteworthy that PDE4 is also expressed in the central nervous system, and particularly in the area postrema which controls the emetic reflex21 (see also side effects of inhibitor compounds).
The reduction in intracellular concentration of cAMP, by means of PDE4, induces the secretion of pro-inflammatory cytokines and decreases the synthesis of anti-inflammatory mediators. On the other hand, inhibition of PDE4 determines the increase of cAMP and the inhibition of the secretion of cytokines, such as tumor necrosis factor-α (TNF-α), interferon gamma (IFN-γ), and interleukin-1 (IL-2) from immune cells,22 and the increase in cytokines such as IL-10, which are characterized by anti-inflammatory properties.23 On the basis of the important role of PDE4 in regulating the balance of inflammatory cytokines, specific molecules have been investigated as potential therapeutic drugs capable of modulating this enzymatic activity; among these, apremilast has emerged as the most successful one and has been proposed for the treatment of psoriasis and PsA.
Apremilast: mechanism of action (MOA)
Apremilast specifically inhibits the enzymatic activity of PDE4, and therefore influences the expression of several pro- and anti-inflammatory cytokines.24 Pro-inflammatory signals, such as those derived by toll-like receptors in monocytes and dendritic cells, activate transcription factor nuclear factor-kappa B (NF-kB) and the expression of pro-inflammatory cytokines such as IL-23, tumor necrosis factor-α (TNF-α), and interferon gamma (IFN-γ). Moreover, GPCRs trigger the activation of adenylyl cyclase, resulting in an increased concentration of cAMP.16,17,22–24
In monocytes and dendritic cells, cAMP is degraded to AMP mainly by PDE4. PDE4 inhibition by apremilast increases intracellular cAMP levels, which regulates the activation of protein kinase A (PKA). PKA activation induces the phosphorylation of transcription factors such as cAMP responsive element-binding protein (CREB), cAMP responsive element modulator (CREM), and activating transcription factor 1 (ATF-1). These transcription factors also bind to CRE sites within promoters of IL-10, increasing IL-10 expression. Transcriptional co-activators such as CREB-binding protein (CBP) or the homologous protein p300 are also involved in these processes, resulting in inhibition of NF-kB transcriptional activity, and reduced expression of IL-23, TNF-α, and IFN-γ. The decreased production of inflammatory mediators reduces cellular infiltration in the target tissue and the activation and proliferation of keratinocytes and synoviocytes in the psoriatic skin and synovium. This may reduce epidermal thickening in psoriasis and decrease synovitis and articular damage in the affected joints. Direct effects of apremilast on keratinocytes and synoviocytes are also under investigation (see Figure 1).16,17,22–24
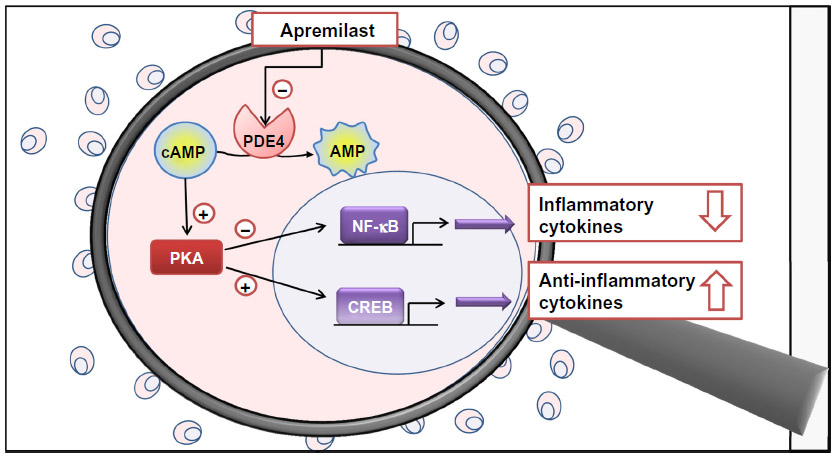 | Figure 1 Magnifying glass on the mechanism of action of apremilast, simplified and schematic representation. In monocytes and dendritic cells, cAMP is degraded to AMP mainly by PDE4. PDE4 inhibition by apremilast increases intracellular cAMP levels and determines the activation of PKA. PKA activation induces the phosphorylation of transcription factors such as CREB. These transcription factors also bind to sites within promoters of IL-10, increasing IL-10 expression. Co-activators are also involved resulting in the inhibition of NF-kB transcriptional activity and expression of IL-23, TNF-α, and IFN-γ. The decreased production of inflammatory mediators reduces cellular infiltration in the target tissue and the activation and proliferation of keratinocytes and synoviocytes in the psoriatic skin and synovium. |
Efficacy of apremilast in PsA clinical studies
Following early evidence of the therapeutic potential of apremilast in the treatment of PsA, Schett et al performed a Phase II randomized, double-blind, placebo-controlled clinical trial in order to study the efficacy and safety of apremilast in active PsA.25 Two hundred and four patients with active PsA, defined as having at least three swollen joints and at least three tender joints at the time of recruitment, were involved in the study. All patients had discontinued immunosuppressant drugs, including traditional and biologic DMARDs (with the exception of methotrexate) and phototherapy in patients with extensive skin involvement, according to a definite scheduled washout period, before being treated with apremilast. Stable therapy with methotrexate, corticosteroids, and/or nonsteroidal anti-inflammatory drugs (NSAIDs) was permitted. Recruited patients were randomized in three arms to receive placebo, apremilast 20 mg twice daily (BID), or apremilast 40 mg once daily (QD) for 12 weeks. Following the first 12 weeks, all patients could exit the trial, enter a 4-week observational phase or enter into a 12-week treatment extension phase. Patients originally randomized to receive placebo rather than active drug were re-randomized to apremilast 20 mg BID or 40 mg QD treatment arms. The primary endpoint was clinical response as measured by ACR 20 (American College of Rheumatology 20% improvement response) at week 12, while secondary endpoints were: evaluation of PsA response criteria (PsARC), disease activity score28 (DAS28), ACR 50, ACR 70, Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F), Health Assessment Questionnaire-Disability Index (HAQ-DI), pain visual analog scale (VAS), 36-Item Short-Form Health Survey (SF-36), and assessment of the incidence of side effects and general safety issues.25 One hundred and sixty-five patients completed the treatment phase at week 12 (out of 204). Almost half of the patients (43.5%) receiving apremilast 20 mg BID (P<0.001) and 35.8% of patients receiving 40 mg QD (P=0.002) reached an ACR 20 response compared with 11.8% in the placebo group. At week 24 (end of the treatment extension phase), more than 40% of all treatment groups fulfilled the ACR 20 response criteria. Reported adverse effects were mild or moderate and included diarrhea, headache, nausea, fatigue, and nasopharyngitis. On the basis of these results, the authors concluded that apremilast, at the dosage of 20 mg twice per day or 40 mg once a day was effective and well-tolerated in the treatment of active PsA.25
On the basis of the promising results of this Phase II study, four Phase III studies (psoriatic arthritis long-term assessment of clinical efficacy [PALACE]) were designed and conducted in order to assess the long-term clinical efficacy and safety of apremilast. In all these trials, around 500 patients were randomized into three arms to receive apremilast 30 mg BID, apremilast 20 mg BID, or placebo. After 24 weeks of treatment, patients might enter, on a voluntary basis, an extension phase where patients in the placebo arms were re-randomized to one of the apremilast active drug arms. In the extension phases, the recruited patients are planned to be followed for 2 or 4.5 additional years, in the PALACE 4 trial and PALACE 1, 2, 3 trials, respectively. The primary endpoint of the PALACE studies is fulfillment of ACR 20 response criteria. Secondary endpoints include ACR 50, ACR 70, Health-Related Quality of Life questionnaire, and assessment of treatment safety. Treatment with methotrexate, leflunomide, or sulfasalazine was allowed in PALACE 1, 2, and 3, while patients in PALACE 4 were DMARD therapy-naïve. The results of these trials have been presented at international meetings and published only in abstract form.26–30 The data publicly available in October 2013 support the efficacy of apremilast in the long term (52 weeks), showing that in patients who received apremilast from baseline, an ACR 20 response was achieved by 63% and 54.6% in the apremilast 20 mg and 30 mg arms, respectively. At week 52, PASI-75 (Psoriasis Area and Severity Index score) was achieved by 25% and 37% of patients who received apremilast 20 mg and 30 mg BID, respectively, compared with baseline (PALACE 1).27 Preliminary results from PALACE 2, 3, and 4 studies31–33 are shown in Table 1.
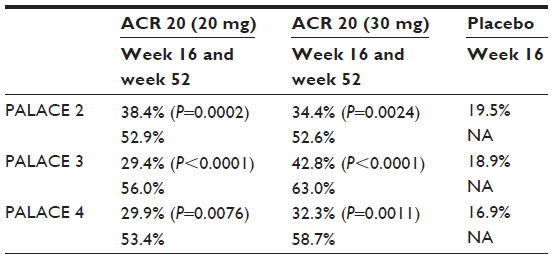 | Table 1 Detailed preliminary results of ongoing randomized double-blind controlled trials PALACE 2, 3, and 4 at primary endpoint (week 16, placebo-controlled) and long-term (52 weeks) data, which include patients originally randomized to apremilast and completing 52 weeks of study |
Long-term data also confirm the efficacy of apremilast in treating typical manifestations of PsA such as enthesitis and dactylitis. Pooled results from three Phase III RCT were analyzed by Gladman et al28 and presented at the ACR 2013 annual meeting, demonstrating that in patients randomized to apremilast and completing 52 weeks of study, a MASES (Maastricht Ankylosing Spondylitis Enthesitis Score) score of 0, indicating no pain at any of the entheses assessed, was achieved by 41.4% and 37.4% of patients treated with 20 or 30 mg BID, respectively.34 At week 52, dactylitis count decreased to 0 in 66.9% and 65.9% of patients (apremilast 20 or 30 mg, respectively). The authors concluded that apremilast confirmed its efficacy in the treatment of PsA, including improvements in enthesitis and dactylitis.
Further evidence of the efficacy of apremilast in the treatment of psoriatic skin dermatitis comes from a Phase II randomized study, performed by Papp et al: patients treated with apremilast at 10 mg, 20 mg, and 30 mg BID reached PASI-75 in a dose-dependent fashion (11%, 29%, and 41%, respectively, compared to 6% in the placebo group).35 Moreover, Gottlieb et al demonstrated a reduction in epidermal thickness, T cells, and CD11c cell infiltration in patients with psoriasis only, following treatment with apremilast.36
Safety profile of apremilast
The overall safety and tolerability of apremilast was well assessed in a pooled analysis of PALACE 1, 2, and 3 performed by Mease et al.29,30 No new safety findings were identified compared with previous reports from early time points assessment. The most commonly reported side effects were diarrhea (14.3%), nausea (12.6%), and headache (10.1%), which were dose-dependent, as well as upper respiratory tract infection (10.3%) and nasopharyngitis (7.4%). The majority of adverse effects were mild/moderate and discontinuation of treatment due to side effects was low (7.5% and 8.3% in patients receiving apremilast 20 or 30 mg BID, respectively) and occurred mainly in the first 24 weeks of treatment. Nausea and diarrhea were predominantly mild and occurred mainly in the first 2 weeks of treatment. The majority of cases resolved within 4 weeks despite continued therapy. Remarkably, incidence rates of serious infections, major adverse cardiac events, or occurrence of malignancies were comparable to the incidence in the placebo group. There were no clinically relevant changes in laboratory markers, suggesting that laboratory monitoring may be not indicated. Of note, no novel or reactivation cases of tuberculosis (TB) were reported in the apremilast treatment groups and TB screening was not required per protocol. In summary, apremilast demonstrated a satisfactory safety profile and was generally well-tolerated for up to 52 weeks with no concerns in the long-term exposure. The good safety profile and tolerability of apremilast is also confirmed in other clinical studies, including those performed in patients with skin psoriasis without joint involvement.35–38
Conclusion
The precise pathogenesis of PsA is still under investigation, but the evidence so far available supports an important role of the individual’s genetic background, environmental triggers, and an imbalance in the adaptive and acquired immune system resulting in the production of inflammatory mediators. New therapeutic approaches have been proposed, among them the use of modulators of intracellular signals and gene transcription such as PDE4-inhibiting compounds, being PDE4 involved in degrading cAMP and therefore altering the activity of transcription factors such as CREB and NF-kB. The reduction of the inflammatory mediators like TNF-α and IL-23, and the increase of anti-inflammatory mediators like IL-10 have proved beneficial in animal models and clinical trials. The data available on the PDE4 inhibitor apremilast, summarized in this review, support its role as an immunoregulator, as well as its clinical efficacy, good tolerability, and safety profile in the treatment of PsA.
Disclosure
The authors report no conflicts of interest in this work.
References
Namey TC. In: Wright V, Moll JM, editors. Seronegative Polyarthritis. Amsterdam and New York: North Holland Publishing Co; 1976. | |
Gladman DD. Psoriatic arthritis. In: Harris ED, Budd RC, Firestein GS, et al, editors. Kelly’s Textbook of Rheumatology. 7th ed. Philadelphia, PA: WB Saunders Co; 2005:1155–1164. | |
O’Rielly DD, Rahman P. Genetics of susceptibility and treatment response in psoriatic arthritis. Nat Rev Rheumatol. 2011;7:718–732. | |
Mameli A, Cauli A, Taccari E, et al. Association of MICA alleles with psoriatic arthritis and its clinical forms. A multicenter Italian study. Clin Exp Rheumatol. 2008;26:649–652. | |
Paladini F, Taccari E, Fiorillo MT, et al. Distribution of HLA-B27 subtypes in Sardinia and continental Italy and their association with spondylarthropathies. Arthritis Rheum. 2005;52:3319–3321. | |
Pitzalis C, Cauli A, Pipitone N, et al. Cutaneous lymphocyte antigen-positive T lymphocytes preferentially migrate to the skin but not to the joint in psoriatic arthritis. Arthritis Rheum. 1996;39:137–145. | |
Cauli A, Pitzalis C, Yanni G, Hawad M, Panayi GS. CD1 positive antigen presenting cells in psoriatic and rheumatoid arthritis. Rheumatology. 2000;6:666–673. | |
Cauli A, Mathieu A. Th17 and interleukin 23 in the pathogenesis of psoriatic arthritis and spondyloarthritis. J Rheumatol Suppl. 2012;89(7):15–18. | |
Ritchlin CT, Kavanaugh A, Gladman DD, et al. Treatment recommendations for psoriatic arthritis. Annals Rheu Dis. 2009;68:1387–1394. | |
Salvarani C, Pipitone N, Marchesoni A, et al. Recommendations for the use of biologic therapy in the treatment of psoriatic arthritis: update from the Italian Society for Rheumatology. Clin Exp Rheumatol. 2011;29:S28–S41. | |
Clegg DO, Reda DJ, Mejias E, et al. Comparison of sulfasalazine and placebo in the treatment of psoriatic arthritis. A Department of Veterans Affairs Cooperative Study. Arthritis Rheum. 1996;39:2013–2020. | |
Kaltwasser JP, Nash P, Gladman D, et al. Efficacy and safety of leflunomide in the treatment of psoriatic arthritis and psoriasis: a multinational, double-blind, randomized, placebo-controlled clinical trial. Arthritis Rheum. 2004;50:1939–1950. | |
Olivieri I, Salvarani C, Cantini F, et al. Therapy with cyclosporine in psoriatic arthritis. Semin Arthritis Rheum. 1997;27:36–43. | |
Salvarani C, Macchioni P, Olivieri I, et al. A comparison of cyclosporine, sulfasalazine, and symptomatic therapy in the treatment of psoriatic arthritis. J Rheumatol. 2001;28:2274–2282. | |
Kingsley GH, Kowalczyk A, Taylor H, et al. A randomized placebo-controlled trial of methotrexate in psoriatic arthritis. Rheumatology. 2012;51:1368–1377. | |
Conti M, Richter W, Mehats C, Livera G, Park JY, Jin C. Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. J Biol Chem. 2003;278:5493–5496. | |
Houslay MD, Schafer P, Zhang KY. Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov Today. 2005;10:1503–1519. | |
Shepherd MC, Baillie GS, Stirling DI, Houslay MD. Remodelling of the PDE4 cAMP phosphodiesterase isoform profile upon monocyte-macrophage differentiation of human U937 cells. Br J Pharmacol. 2004;142:339–351. | |
Heystek HC, Thierry A-C, Soulard P, Moulon C. Phosphodiesterase 4 inhibitors reduce human dendritic cell inflammatory cytokine production and Th1-polarizing capacity. Int Immunol. 2003;15:827–835. | |
Tenor H, Hedbom E, Hauselmann HJ, Schudt C, Hatzelmann A. Phosphodiesterase isoenzyme families in human osteoarthritis chondrocytes – functional importance of phosphodiesterase 4. Br J Pharmacol. 2002;135:609–618. | |
Mori F, Perez-Torres S, De Caro R, et al. The human area postrema and other nuclei related to the emetic reflex express cAMP phosphodiesterases 4B and 4D. J Chem Neuroanat. 2010;40:36–42. | |
Claveau D, Chen SL, O’Keefe S, et al. Preferential inhibition of T helper 1, but not T helper 2, cytokines in vitro by L-826,141 [4-[2-(3,4-Bisdifluromethoxyphenyl)-2-[4-(1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl)-phenyl]-ethyl]3-methylpyridine-1-oxide], a potent and selective phosphodiesterase 4 inhibitor. J Pharmacol Exp Ther. 2004;310:752–760. | |
Eigler A, Siegmund B, Emmerich U, Bauman KH, Hartmann G, Endres S. Anti-inflammatory activity of cAMP-elevating agents: enhancement of IL-10 synthesis and concurrent suppression of TNF production. J Leukoc Biol. 1998;63:101–107. | |
Schafer PH, Parton A, Gandhi AK, et al. Apremilast, a cAMP phosphodiesterase-4 inhibitor, demonstrates anti-inflammatory activity in vitro and in a model of psoriasis. Br J Pharmacol. 2010;159:842–855. | |
Schett G, Wollenhaupt J, Papp K, et al. Oral apremilast in the treatment of active psoriatic arthritis: results of a multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2012;64:3156–3167. | |
Kavanaugh A, Mease PJ, Gomez-Reino JJ, et al. Apremilast, an oral phosphodiesterase 4 inhibitor, in patients with psoriatic arthritis: results of a phase 3, randomized, controlled trial. Proceedings of the Annual Scientific Meeting of the American College of Rheumatology (ACR/ARHP). November 9–14, 2012; Washington, DC, USA: abst L13. | |
Kavanaugh A, Mease PJ, Adebajo AO, et al. Long-term (52-week) results of a phase 3, randomized, controlled trial of apremilast, an oral phosphodiesterase 4 inhibitor, in patients with psoriatic arthritis. Proceedings of the 14th Annual European League Against Rheumatism (EULAR); June 12–15, 2013; Madrid, Spain: abst LB0001. | |
Gladman DD, Mease PJ, Kavanaugh A, Adebajo AO, Gomez-Reino JJ, Wollenhaupt J. Apremilast, an oral phosphodiesterase 4 inhibitor, is associated with long-term (52-week) improvements in enthesitis and dactylitis in patients with psoriatic arthritis: pooled results from three phase 3, randomized, controlled trials. Proceedings of the annual scientific meeting of the American College of Rheumatology (ACR/ARHP). October 25–30, 2013; San Diego, CA, USA: abst 0816. | |
Mease PJ, Kavanaugh A, Adebajo AO, et al. Apremilast: pooled safety analysis of three Phase 3, randomized, controlled trials in patients with psoriatic arthritis. Proceedings of the annual scientific meeting of the European League Against Rheumatism (EULAR). June 12–15, 2013; Madrid, Spain: abst SAT0299. | |
Mease PJ, Kavanaugh A, Gladman DD, et al. Long-term safety and tolerability of Apremilast, an oral phosphodiesterase 4 inhibitor, in patients with psoriatic arthritis: pooled safety analysis of three phase 3, randomized, controlled trials. Proceedings of the annual scientific meeting of the American College of Rheumatology (ACR/ARHP). October 25–30, 2013; San Diego, CA, USA: abst 310. | |
Cutolo M, Myerson GE, Fleischmann RM, et al. Long term (52 week) results of a phase 3, randomized, controlled trial of apremilast, an oral phosphodiesterase 4 inhibitor, in patients with psoriatic arthritis (PALACE 2). Proceedings of the annual scientific meeting of the American College of Rheumatology (ACR/ARHP). October 25–30, 2013; San Diego, CA, USA: abst 815. | |
Edwards CJ, Blanco FJ, Crowley J, Hu C, Stevens RM, Birbara CA. Long term (52 week) results of a phase 3, randomized, controlled trial of apremilast, an oral phosphodiesterase 4 inhibitor, in patients with psoriatic arthritis and current skin involvement (PALACE 3). Proceedings of the annual scientific meeting of the American College of Rheumatology (ACR/ARHP). October 25–30, 2013; San Diego, CA, USA: abst 311. | |
Wells AF, Edwards CJ, Adebajio AO, et al. Apremilast in the treatment of DMARD-Naïve psoriatic arthritis patients: results of a phase 3 randomized, controlled trial (PALACE 4). Proceedings of the annual scientific meeting of the American College of Rheumatology (ACR/ARHP). October 25–30, 2013; San Diego, CA, USA: abst L4. | |
Heuft-Dorenbosch L, Spoorenberg A, van Tubergen A, Landewé R, van der Tempel H, Mielants H, Dougados M, van der Heijde D. Assessment of enthesitis in ankylosing spondylitis. Ann Rheum Dis. 2003;62(2):127–132. | |
Papp K, Cather JC, Rosoph L, et al. Efficacy of apremilast in the treatment of moderate to severe psoriasis: a randomised controlled trial. Lancet. 2012;380:738–746. | |
Gottlieb AB, Strober B, Krueger JG, et al. An open-label, single-arm pilot study in patients with severe plaque-type psoriasis treated with an oral anti-inflammatory agent, apremilast. Curr Med Res Opin. 2008;24:1529–1538. | |
Papp KA, Kaufmann R, Thaçi D, Hu C, Sutherland D, Rohane P. Efficacy and safety of apremilast in subjects with moderate to severe plaque psoriasis: results from a phase II, multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison study. J Eur Acad Dermatol Venereol. 2013;27(3):e376–e383. | |
Palfreeman AC, McNamee KE, McCann FE. New developments in the management of psoriasis and psoriatic arthritis: a focus on apremilast. Drug Design Develop Ther. 2013;7:201–210. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.