")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Clinical pharmacology study of cariprazine (MP-214) in patients with schizophrenia (12-week treatment)
Authors Nakamura T, Kubota T, Iwakaji A, Imada M, Kapas M, Morio Y
Received 25 August 2015
Accepted for publication 10 November 2015
Published 14 January 2016 Volume 2016:10 Pages 327—338
DOI https://doi.org/10.2147/DDDT.S95100
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Tadakatsu Nakamura,1 Tomoko Kubota,1 Atsushi Iwakaji,1 Masayoshi Imada,1 Margit Kapás,2 Yasunori Morio1
1Sohyaku. Innovative Research Division, Mitsubishi Tanabe Pharma Corporation, Tokyo, Japan; 2Developmental Drug Metabolism and Pharmacokinetics, Gedeon Richter Plc, Budapest, Hungary
Purpose: Cariprazine is a potent dopamine D3-preferring D3/D2 receptor partial agonist in development for the treatment of schizophrenia, bipolar mania, and depression. Pharmacokinetics of cariprazine and the two clinically relevant metabolites (desmethyl- and didesmethyl-cariprazine) was evaluated in a clinical pharmacology study.
Methods: This was a multicenter, randomized, open-label, parallel-group, fixed-dose (3, 6, or 9 mg/day) study of 28-week duration (≤4-week observation, 12-week open-label treatment, and 12-week follow-up). Once-daily cariprazine was administered to 38 adult patients with schizophrenia. The pharmacokinetics of cariprazine, metabolites, and total active moieties (sum of cariprazine and two metabolites) was evaluated; efficacy and safety were also assessed.
Results: Steady state was reached within 1–2 weeks for cariprazine and desmethyl-cariprazine, 4 weeks for didesmethyl-cariprazine, and 3 weeks for total active moieties. Cariprazine and desmethyl-cariprazine levels decreased >90% within 1 week after the last dose, didesmethyl-cariprazine decreased ~50% at 1 week, and total active moieties decreased ~90% within 4 weeks. Terminal half-lives of cariprazine, desmethyl-cariprazine, and didesmethyl-cariprazine ranged from 31.6 to 68.4, 29.7 to 37.5, and 314 to 446 hours, respectively. Effective half-life (calculated from time to steady state) of total active moieties was ~1 week. Incidence of treatment-emergent adverse events was 97.4%; 15.8% of patients discontinued due to adverse events. No abnormal laboratory values or major differences from baseline in extrapyramidal symptoms were observed.
Conclusion: Cariprazine and its active metabolites reached steady state within 4 weeks, and exposure was dose proportional over the range of 3–9 mg/day. Once-daily cariprazine was generally well tolerated in adult patients with schizophrenia.
Keywords: schizophrenia, cariprazine, pharmacokinetics, desmethyl-cariprazine, didesmethyl-cariprazine
Introduction
Schizophrenia is a complex disorder associated with positive, negative, and mood symptoms as well as neurocognitive impairment.1 The mainstay of treatment for schizophrenia are antipsychotics, which act to modulate neurotransmitter function in order to decrease the frequency and severity of psychotic episodes.2 First-generation or typical antipsychotics are efficacious in reducing the positive symptoms of schizophrenia but have shown limited efficacy for treating negative symptoms or cognitive impairment; additionally, typical antipsychotics were associated with substantial risk of extrapyramidal symptoms (EPS).3 Second-generation or atypical antipsychotics are thought to have a lower risk of EPS and better, though still limited, efficacy in negative symptoms and cognitive deficits.3,4
All clinically effective antipsychotics show some affinity for the dopamine D2 receptor,5 but their affinity to other receptors may play an important role in their clinical efficacy and safety profiles. Identifying new molecular targets for the treatment of schizophrenia is crucial to optimally manage this debilitating disorder. The dopamine D3 receptor has emerged as a potential target for antipsychotics.6 In rodent models, selective antagonism of D3 receptors eliminated D2 receptor agonist-induced catalepsy,7,8 did not produce motor adverse events (AEs),9 and improved learning and memory.10,11 Furthermore, targeted disruption of D3 receptors in mice results in heightened sensitivity to antidepressants compared with wild-type littermates.12 Additional lines of evidence in animal studies, such as facilitation of nesting behavior and social contact,13 suggest that D3 receptor blockade could counter negative symptoms associated with schizophrenia. An antipsychotic with high affinity for both D3 and D2 receptors may offer a treatment option that is effective in resolving positive, negative, and mood symptoms and improving cognitive deficits associated with schizophrenia.14,15
Cariprazine is a potent D3/D2 receptor partial agonist with preferential binding to D3 receptors. It has been recently approved for the treatment of schizophrenia and manic or mixed episodes associated with bipolar I disorder by the US Food and Drug Administration. Cariprazine has an almost tenfold greater affinity for D3 than D2 receptors in vitro,16 with high and balanced occupancy of both receptor subtypes in rats and humans16,17 at pharmacologically effective doses. In animal models, cariprazine was associated with D3 receptor-dependent procognitive and antidepressant-like effects, suggesting utility in treating these schizophrenia symptom domains.18,19 Cariprazine has demonstrated efficacy, safety, and tolerability in three positive Phase II/III randomized, placebo-controlled trials in patients with acute exacerbation of schizophrenia.20–22 The D3-binding preference of the drug may offer a unique treatment option for patients with persistent and predominant negative symptoms. Cariprazine has indeed shown clinical potential for treating negative symptoms based on results from a prospectively defined schizophrenia study of patients with persistent and predominant negative symptoms.23
The metabolism of cariprazine is via the CYP3A4 and to a lesser extent by CYP2D6 pathways.24 Two clinically relevant metabolites of cariprazine, desmethyl-cariprazine, and didesmethyl-cariprazine, have similar pharmacological activity as cariprazine, though didesmethyl-cariprazine has a much longer terminal half-life.24 Studies in healthy volunteers and patients with schizophrenia showed that cariprazine and its metabolites provide dose-proportional systemic exposure with up to 1 month of treatment.24–26 However, the pharmacokinetics of didesmethyl-cariprazine, which has a long terminal half-life (t1/2,z), was not well defined in earlier studies because of shorter study duration.
Understanding the steady-state pharmacokinetics of a drug is important to guide long-term treatment of patients with schizophrenia. Pharmacokinetic data, particularly half-life, may be used to predict the time to steady state. For a drug with multi-exponential disposition, the effective half-life (t1/2,eff) value, determined from the time to reach steady state, can better predict the accumulation and elimination of the drug than the terminal half-life (t1/2,z) associated with a deep compartment.
Reported here are results from a clinical pharmacology study designed to evaluate the steady-state pharmacokinetics of cariprazine, desmethyl-cariprazine, and didesmethyl-cariprazine following 12 weeks of once-daily oral cariprazine administration in Japanese patients with schizophrenia.
Materials and methods
This study was conducted at 20 sites in Japan from May 7, 2012 to August 23, 2013, in compliance with guidelines for Good Clinical Practice, Pharmaceutical Affairs Law, and Declaration of Helsinki. The study protocol was approved by institutional review boards at each study site. Prior to study start, written informed consent was obtained from all participants.
Study design
Japanese patients with schizophrenia were evaluated in a 28-week, multicenter, randomized, open-label, parallel-group, fixed-dose clinical pharmacology study. The study consisted of a screening visit, an observation period for up to 4 weeks, a 12-week treatment period, and a 12-week follow-up period. If study drug was discontinued during the treatment period, the 12-week follow-up period started from the day of drug discontinuation. The treatment and follow-up periods were set as 12 weeks in order to evaluate time to steady state of the plasma concentrations and elimination of cariprazine and its metabolites.
Patients were randomized (1:1:1) to one of three parallel therapeutic doses of cariprazine: 3, 6, or 9 mg/day. Cariprazine treatment was initiated at the 1.5 mg/day dose, and then the dose increased by 1.5 mg/day until the target therapeutic dose was achieved. All cariprazine doses were administered once daily after breakfast.
Study participants
Participants in this study were male or female Japanese patients between 20 and 64 years of age with a diagnosis of schizophrenia for ≥1 year based on Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision criteria. It was required that patients have a Positive and Negative Syndrome Scale (PANSS) total score ≤120 and normal physical examinations, vital signs, clinical laboratory tests, and electrocardiogram (ECG) finding or abnormal results that were judged to be not clinically significant.
Patients with any mental disorder other than schizophrenia were excluded from the study. Additional exclusion criteria included substance abuse or dependence within 3 months prior to consent (excluding nicotine or caffeine dependence), therapy-resistant schizophrenia (ie, poor response to two or more antipsychotics during the past 2 years), history of hypersensitivity to any drugs, suicide attempt or suicidal ideation, body mass index ≤17.5 or >35 kg/m2, pregnancy or breast-feeding, unstable or decompensated cardiovascular disease, gastric bypass, or other conditions that may have an effect on drug absorption. Patients with human immunodeficiency virus or hepatitis virus infection, or any significant/unstable medical condition that might interfere with study participation were excluded, as were patients who required prohibited concomitant medications (eg, psychotropic drugs, CYP3A4 inducers/inhibitors, CYP2D6 inhibitors). Allowed concomitant medications included hypnotics (eg, zolpidem or chloral hydrate), antiparkinson drugs, and lorazepam. Treatment-related exclusions included electroconvulsive therapy or extended-release antipsychotics within 3 months prior to consent, clozapine within 1 year prior to consent, or cariprazine treatment in the past.
Assessments
Pharmacokinetics
Blood samples were collected prior to the first dose and at 3, 4, 6, 8, 24, and 48 hours after the first dose, as well as pre-dose on the last day of each week during the 12-week treatment period (ie, every 7 days starting from day 7) with the exception of weeks 9 and 11. Additionally, for weeks 2 and 4, blood samples were collected before administration and 3–4 hours post-dose. Blood samples were also taken prior to the last dose and at 3, 4, 6, 8, and 24 hours after the last dose. During the follow-up period, blood samples were collected on days 1, 2, and 3, and on the last day of weeks 1, 2, 4, 6, 8, and 12.
Blood samples were collected from the vein into vacuum collection tubes with the anticoagulant K2EDTA (dipotassium ethylenediaminetetraacetic acid), and centrifuged using a refrigerated centrifuge within 30 minutes after blood withdrawal (4°C, 1,800× g, 15 minutes); plasma was then separated, aliquoted, and quickly frozen at ≤−20°C. Plasma samples were analyzed for cariprazine and its metabolites, desmethyl-cariprazine, and didesmethyl-cariprazine, using a previously validated liquid chromatography–tandem mass spectrometry technique with a lower limit of quantification of 0.02 ng/mL.27
Pharmacokinetic parameters, calculated from plasma concentration–time data using noncompartmental analyses, included area under the plasma concentration–time curve from time zero to 24 hours (AUC0–24), maximum plasma concentration (Cmax), apparent terminal elimination rate constant (kel), time to Cmax (tmax), and terminal elimination half-life (t1/2,z). The time to reach steady state and dose proportionality were also evaluated. Time to reach steady state was defined as the first time point where the pre-dose plasma concentration relative to the week 12 value was ≥90%. Dose proportionality of cariprazine, desmethyl-cariprazine, and didesmethyl-cariprazine on the last day of treatment was assessed using a power model: log(Y) = α + β × log(X) + error, where Y is the pharmacokinetic variable, α is the y-intercept, β is the slope, and X is dosage. When the confidence interval (CI) included 1, this confirmed that there is dose proportionality of Cmax and AUC0–24. The t1/2,eff was also assessed for cariprazine, desmethyl-cariprazine, didesmethyl-cariprazine, and total active moieties; t1/2,eff was determined based on time to reach steady state divided by 3.32, where 3.32 is the number of half-lives required to reach 90% of steady state assuming a one-compartment model.
Efficacy assessments
The primary objective of this study was to evaluate the pharmacokinetics of cariprazine and its metabolites; as such, efficacy assessments were collected but were not planned for inferential analyses due to the small sample size of the study. Efficacy assessments were collected at baseline, week 6, and week 12 of the study (or at study discontinuation). The PANSS scale is a clinician-administered assessment that evaluates both positive and negative symptoms in patients with schizophrenia.28 The Clinical Global Impressions-Severity (CGI-S) scale and the Clinical Global Impressions-Improvement (CGI-I) scale are clinician-administered assessments that evaluate overall severity of disease (CGI-S) and improvement or worsening of disease from baseline (CGI-I).29
Safety and tolerability
Safety and tolerability were assessed by AE monitoring, vital sign and clinical laboratory evaluations, ECG parameters, physical and ophthalmologic examinations, and results of the Columbia-Suicide Severity Rating Scale. AEs were monitored throughout the treatment and follow-up periods, with additional monitoring after the follow-up period if recovery from an AE was not observed at study completion. The occurrence of EPS was assessed using the Drug-Induced Extrapyramidal Symptoms Scale (DIEPSS), Abnormal Involuntary Movement Scale (AIMS), and Barnes Akathisia Rating Scale (BARS).
Statistical analysis
The pharmacokinetic analysis population, defined as all patients who received one or more doses of cariprazine following randomization, was used to assess cariprazine pharmacokinetics. Descriptive statistics were calculated for each dose group and at selected blood sampling times for the plasma concentrations and pharmacokinetic parameters of cariprazine, desmethyl-cariprazine, and didesmethyl-cariprazine.
The safety analysis population comprised all randomized patients who received one or more doses of cariprazine and for whom safety data were available. The incidence of AEs, adverse drug reactions, and serious adverse events (SAEs) was calculated for each dose group. All safety measures were analyzed using descriptive statistics. The efficacy analysis population comprised all randomized schizophrenia patients who received one or more doses of cariprazine and had a baseline and one or more postbaseline efficacy assessments. Descriptive statistics were calculated for PANSS total score, PANSS positive score, PANSS negative score, CGI-S score, and CGI-I score. Change from baseline for each efficacy parameter was analyzed using analysis of covariance with treatment group as fixed effect and baseline of the corresponding parameter. Least squares mean and corresponding 95% CIs were calculated. In addition, last observation carried forward was used for missing values of each efficacy parameter.
Results
Patient disposition and demographic characteristics
Of the 38 enrolled individuals, 24 (63.2%) completed the study, with completion rates similar among cariprazine dose groups (Figure 1). A total of six (15.8%) patients discontinued the study due to AEs, seven (18.4%) withdrew consent, and one (2.6%) was found to be ineligible for the study.
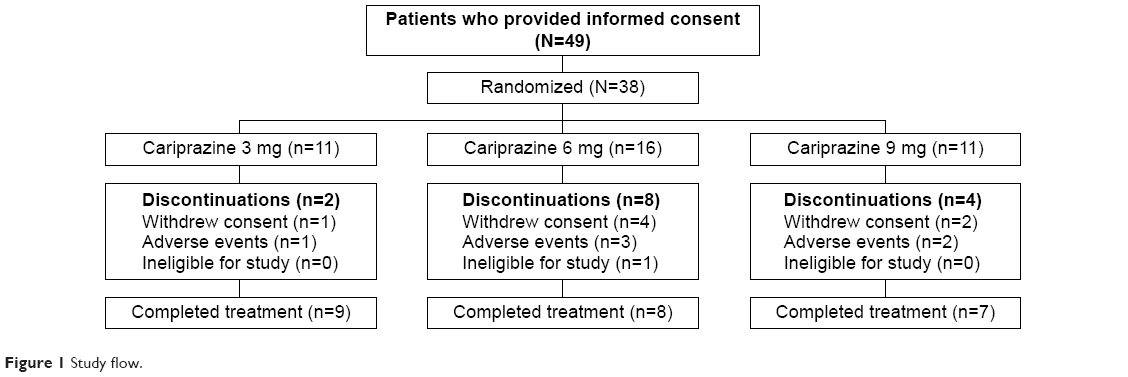 | Figure 1 Study flow. |
Baseline demographics and disease characteristics were similar among treatment groups (Table 1). Compliance to cariprazine treatment among patients in all dose groups was 95.6% for the 12-week treatment period.
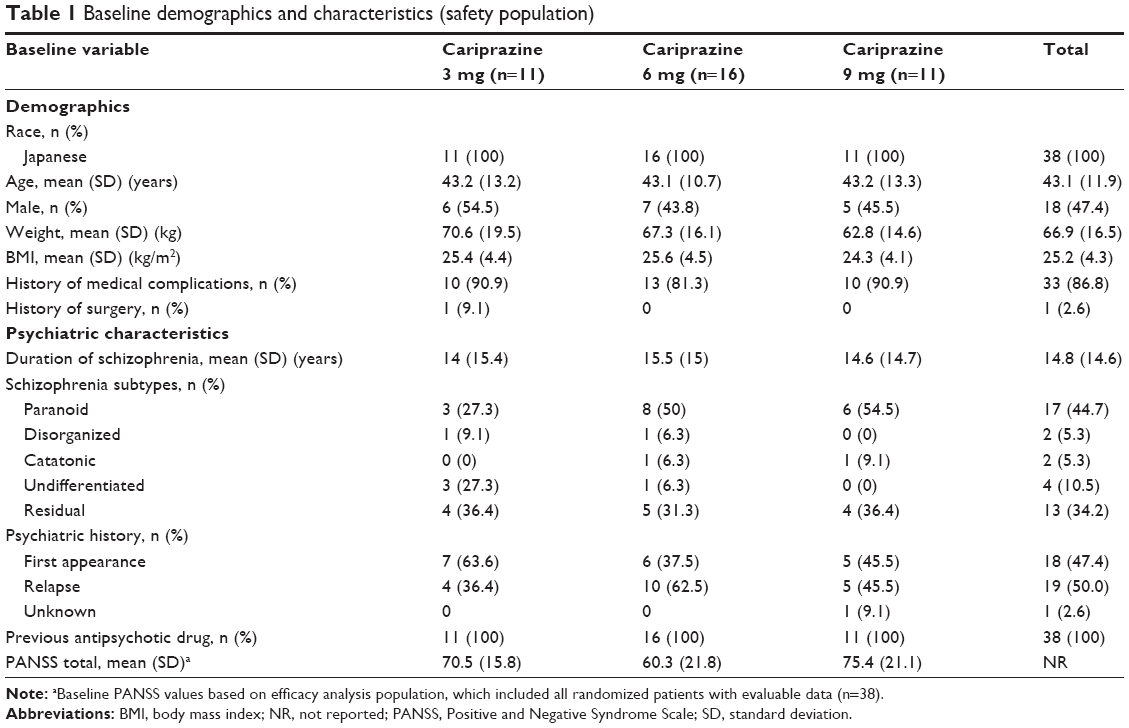 | Table 1 Baseline demographics and characteristics (safety population) |
Pharmacokinetic analyses
Cariprazine, desmethyl-cariprazine, and didesmethyl-cariprazine, and total active moieties all reached steady state following repeated administration over the 12-week treatment period (Figure 2). Trough concentrations of cariprazine, desmethyl-cariprazine, and didesmethyl-cariprazine increased by repetitive administration and stabilized thereafter. The ratios calculated from plasma trough concentrations compared to those on the last day of treatment are presented in Table 2. For cariprazine, the mean trough concentration ratios reached ≥90% for the first time on day 3 of treatment in 3 mg group (mean [95% CI]: 1.907 [−1.006 to 4.819]), and at week 1 of treatment in the 6 and 9 mg groups (0.917 [0.719–1.116] and 0.927 [0.713–1.142], respectively). For desmethyl-cariprazine, the mean trough concentration ratios reached ≥90% for the first time at week 2 of treatment for the 3 and 9 mg groups (1.003 [0.791–1.216] and 1.025 [0.852–1.197], respectively), and at week 1 in the 6 mg group (0.960 [0.764–1.156]). For didesmethyl-cariprazine, the mean trough concentration ratios reached ≥90% for the first time at week 2 of treatment in 3 mg group (0.985 [0.085–1.886]), and at week 4 in the 6 and 9 mg groups (0.954 [0.802–1.107] and 0.932 [0.751–1.113], respectively). The results across the doses are consistent and do not show any dose-dependency for the moieties. Specifically, steady state was reached in each dose group within 1–2 weeks for cariprazine and desmethyl-cariprazine, within 4 weeks for didesmethyl-cariprazine, and within 3 weeks for the total active moieties.
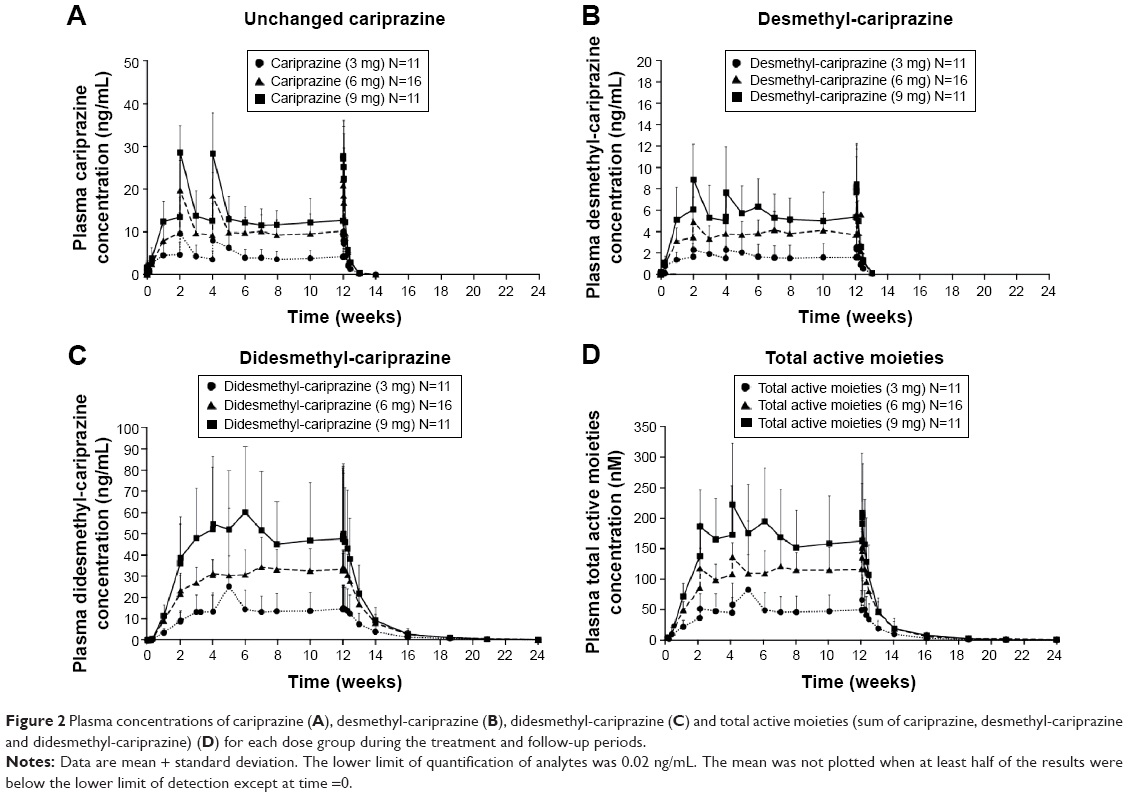 | Figure 2 Plasma concentrations of cariprazine (A), desmethyl-cariprazine (B), didesmethyl-cariprazine (C) and total active moieties (sum of cariprazine, desmethyl-cariprazine and didesmethyl-cariprazine) (D) for each dose group during the treatment and follow-up periods. |
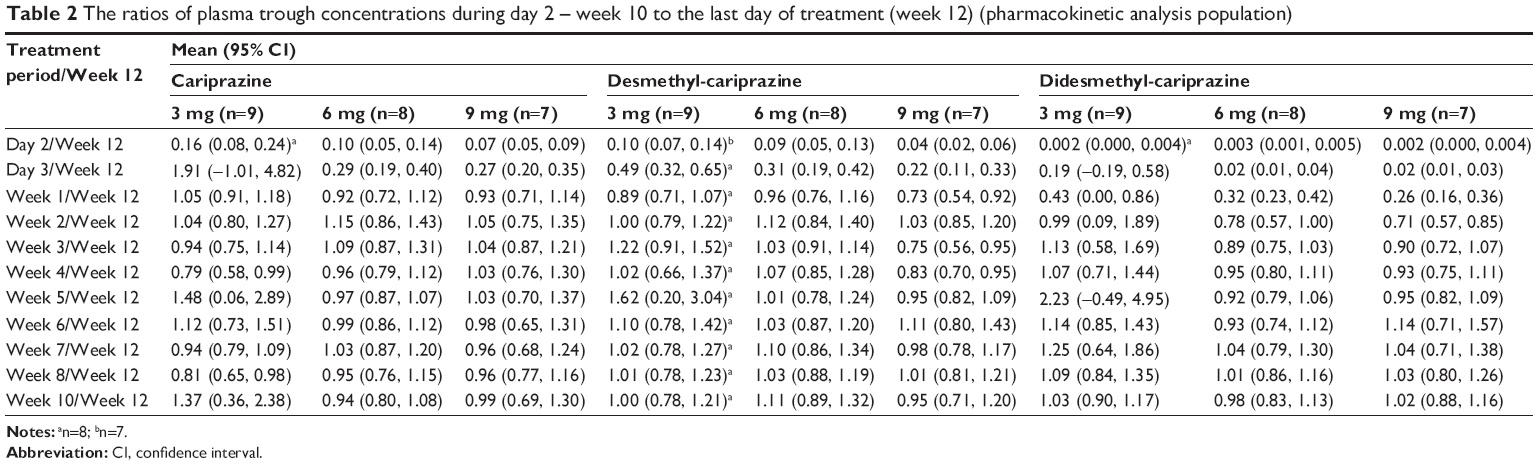 | Table 2 The ratios of plasma trough concentrations during day 2 – week 10 to the last day of treatment (week 12) (pharmacokinetic analysis population) |
Pharmacokinetic parameters of cariprazine, desmethyl-cariprazine, and didesmethyl-cariprazine for each dose group measured on the first and last days of the 12-week treatment period are presented in Table 3. At steady state, didesmethyl-cariprazine is the prominent moiety, with exposure (AUC0–24) about two- to threefold higher than for cariprazine. Steady-state exposure of desmethyl-cariprazine is ~30%–40% of cariprazine exposure. The t1/2,z for cariprazine, desmethyl-cariprazine, and didesmethyl-cariprazine in the dose groups ranged from 31.6 to 68.4, 29.7 to 37.5, and 313.6 to 446.0 hours, respectively. Despite the long t1/2,z, plasma concentrations of cariprazine and desmethyl-cariprazine decreased >90% within 1 week after the last dose, while didesmethyl-cariprazine and total active moieties decreased ~50% at 1 week and ~90% within 4 weeks after the last dose, respectively. The calculated t1/2,eff was ~2 days for cariprazine and desmethyl-cariprazine, 8.4 days for didesmethyl-cariprazine, and 1 week for total active moieties.
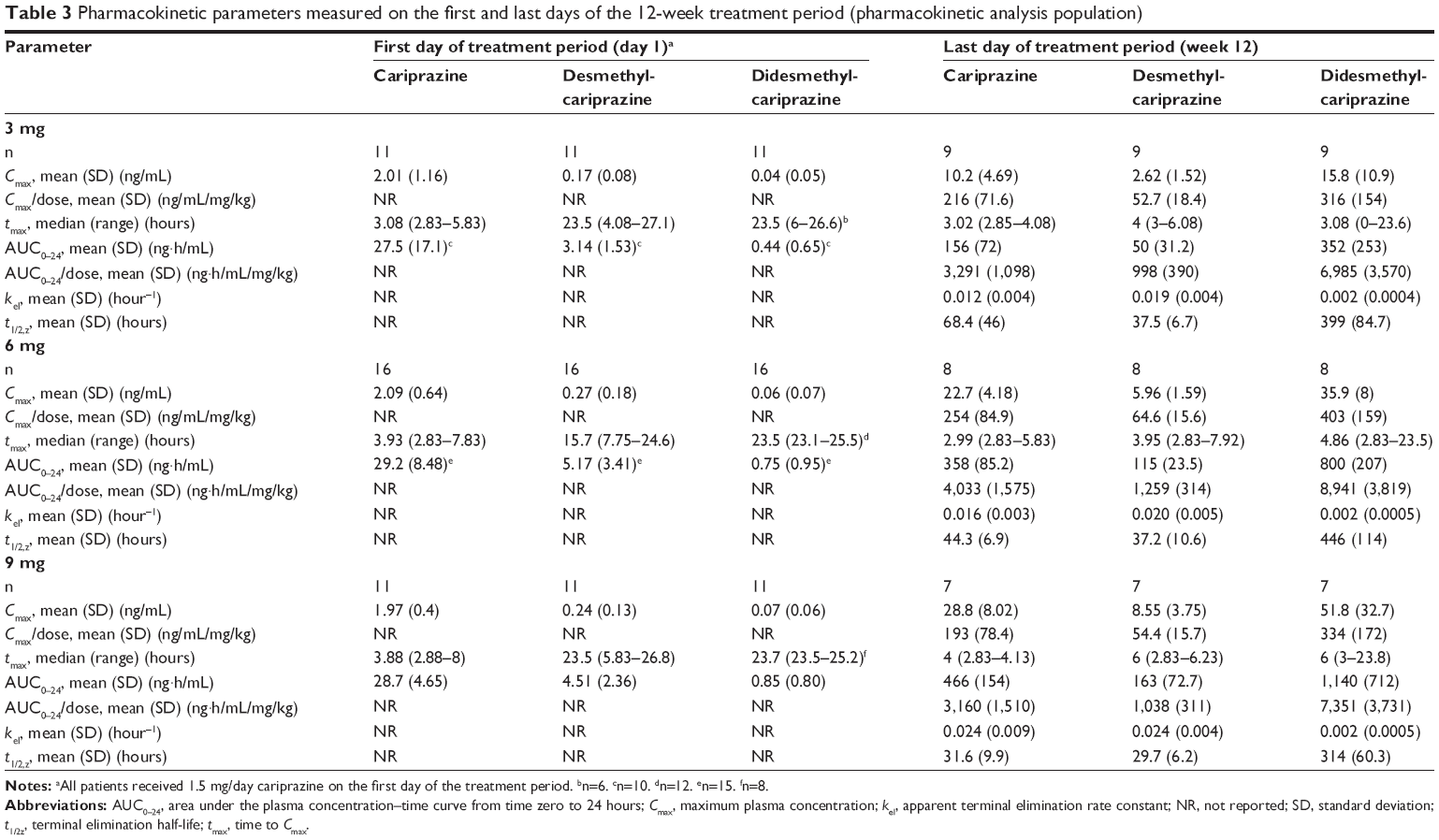 | Table 3 Pharmacokinetic parameters measured on the first and last days of the 12-week treatment period (pharmacokinetic analysis population) |
For cariprazine, the estimated slope β (two-sided 95% CI) of AUC0–24 and Cmax was 1.1 (0.7–1.4) and 1.0 (0.7–1.3), respectively. The estimated slope β (95% CI) of AUC0–24 and Cmax for desmethyl-cariprazine was 1.2 (0.8–1.6) and 1.2 (0.8–1.6), respectively. The estimated slope β (95% CI) of AUC0–24 and Cmax for didesmethyl-cariprazine was 1.4 (0.6–2.2) and 1.3 (0.6–2.1), respectively. Dose proportionality was confirmed for cariprazine, desmethyl-cariprazine, and didesmethyl-cariprazine within the range of 3–9 mg/day, since the two-sided 95% CIs for the estimated value of β contained 1 for both AUC0–24 and Cmax. Dose-adjusted values of Cmax and AUC0–24 for cariprazine, desmethyl-cariprazine, and didesmethyl-cariprazine evaluated on the last day of treatment are presented in Table 3. Dose-adjusted values of AUC0–24 and Cmax were also similar between doses for cariprazine, desmethyl-cariprazine, and didesmethyl-cariprazine.
Safety and tolerability outcomes
Exposure
All 38 patients who enrolled in the study were evaluable for safety analyses. The mean duration of treatment was 61.8 days, with the most frequent duration of treatment ranging from 71 to 84 days for 50% of patients.
Adverse events
A total of 37 (97.4%) patients reported at least one treatment-emergent AE, and 31 (81.6%) patients experienced at least one adverse drug reaction, defined as AEs for which causal relationship with cariprazine was determined to be “reasonably possible” (Table 4). AEs observed in this study were similar to those reported in previous studies.20–22 No patients in the 3 or 9 mg dose groups experienced any SAEs; two patients in the 6 mg group reported the SAEs of exacerbation of schizophrenia.
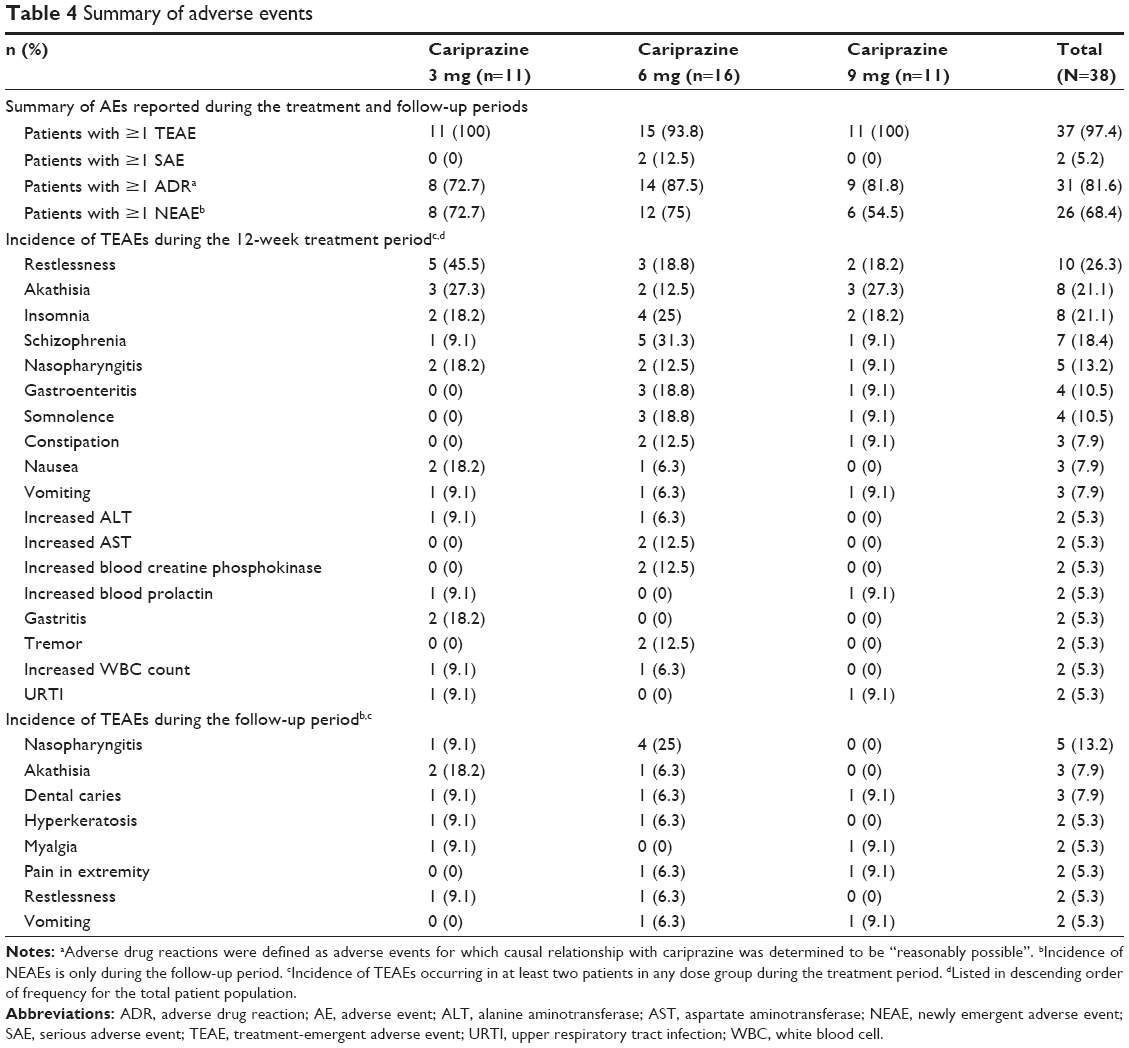 | Table 4 Summary of adverse events |
AEs leading to study discontinuation included exacerbation of schizophrenia (n=3), vomiting (n=1), abnormal hepatic function (n=1), cold sweat (n=1), chills (n=1), and abnormal liver function test (n=1). In each case, the AE was deemed as having a “reasonable possibility” of being related to cariprazine treatment. Neuroleptic malignant syndrome and syncope, classified as significant AEs based on the pharmacological action of cariprazine, were not observed; one case of cataract (also a significant AE) deemed mild in severity was reported. No deaths were reported during the study.
Laboratory parameters, vital signs, ECG, and physical examinations
For most laboratory parameters, changes from baseline were small, and no major differences were observed between dose groups. A change in mean values from baseline to end of study for aspartate aminotransferase and alanine aminotransferase levels was greater for the 9 versus 3 and 6 mg dose groups; however, median values showed no major differences among the three dose groups. The mean change from baseline in creatine kinase levels at the end of the study was higher in the 6 mg group than 3 and 9 mg dose groups. For all dose groups, there was considerable interpatient variability in creatine kinase levels, and no major differences in median values were observed. Mean serum prolactin values declined during treatment and were within the reference range at the end of the study. There was an increase in fasting insulin levels across all dose groups from baseline to the end of study, though all levels were within reference ranges. No significant changes were observed for additional metabolic parameters (eg, glucose, triglycerides, and high-density lipoprotein or low-density lipoprotein cholesterol).
No clinically relevant changes from baseline or differences between dose groups were observed for vital sign tests, ECG, ophthalmologic, or physical examination results. In all dose groups, there was a decrease in weight from baseline. Columbia-Suicide Severity Rating Scale assessments revealed that seven (18.4%) patients across all dose groups exhibited suicidal ideation at least once during the treatment period, though no patients displayed suicidal behavior.
Extrapyramidal symptoms
EPS included treatment-emergent parkinsonism (6 mg, n=3) or treatment-emergent akathisia (3 mg, n=1; 6 mg, n=2; 9 mg, n=2). Additional assessments of EPS revealed no major differences from baseline to the end of study. There were no major variations in DIEPSS scores from baseline to the end of study across dose groups. AIMS scores were elevated for patients in the 6 mg dose group at weeks 6 and 12, though these scores declined back to baseline levels by the end of the study. No major variation in AIMS scores was observed for the 3 or 9 mg dose groups. Overall, there was a slight increase in BARS scores from baseline in all dose groups at the end of the study (change from baseline [mean ± SD]: 3 mg, 0.5±1.1; 6 mg, 0.4±1.1; 9 mg, 0.3±0.6).
Efficacy assessments
PANSS scores at baseline were relatively low compared with baseline PANSS scores in the Phase II/III acute exacerbation studies. Changes from baseline in PANSS total, positive, and negative scores were small during this study (Table 5). Additionally, there was very little change in CGI-S and CGI-I scores for all dose groups (Table 5).
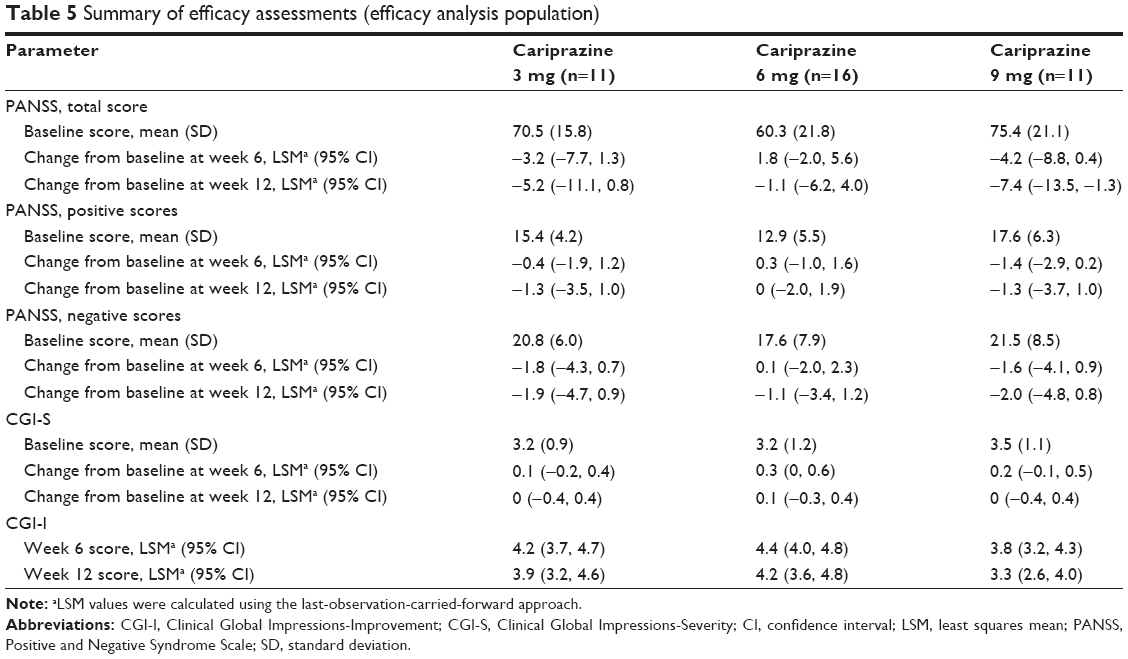 | Table 5 Summary of efficacy assessments (efficacy analysis population) |
Discussion
The primary objective of this study was to evaluate the steady-state pharmacokinetics of cariprazine, desmethyl-cariprazine, didesmethyl-cariprazine, and total active moieties. Prior studies assessing the pharmacokinetics of cariprazine and its metabolites were shorter in length, and insufficient information was collected about steady-state levels, particularly for didesmethyl-cariprazine, which is known to have a prolonged half-life. This study confirmed that cariprazine, desmethyl-cariprazine, didesmethyl-cariprazine, and total active moieties reached steady-state levels within 4 weeks of daily cariprazine administration and were eliminated almost completely in the 12 weeks following the last cariprazine dose. Furthermore, exposure for each of cariprazine and its metabolites was dose proportional over the range of 3–9 mg/day.
While t1/2,z for cariprazine ranged from 1.3 to 2.9 days, the t1/2,eff of the parent compound was 2 days, and the t1/2,eff calculated for the total active moieties, which takes into account cariprazine and the two metabolites, was ~1 week. The t1/2,eff value for the total cariprazine concentrations is considered the best predictor for estimating plasma accumulation and elimination of this drug as it captures the pharmacokinetics of all three active moieties.
The impact of race on the pharmacokinetics of cariprazine, desmethyl-cariprazine, and didesmethyl-cariprazine was evaluated in a single-dose study of 1 mg cariprazine administered to Japanese, South Korean, Taiwanese, and Caucasian healthy subjects. Differences in Cmax and AUC between races were ~≤50% (unpublished). As the impact of race did not appear to be substantial, the pharmacokinetic data gathered from this study are considered representative worldwide, not only for the Japanese population.
The AEs observed during this study were similar to those reported previously.20–22 Of the significant AEs (neuroleptic malignant syndrome, syncope, and cataract), there was only one case of cataract that was considered possibly related to cariprazine treatment and was deemed as being mild in intensity.
Occupancy of D2 receptors is considered the common mechanism of action of all antipsychotics and is thought to be associated with risk of EPS.30 Atypical antipsychotics, which may have greater affinities for other receptors or different activity (ie, partial agonism) at D2 receptors, may reduce the risk of EPS.5 In this study, drug-induced parkinsonism was observed in three patients who received 6 mg/day cariprazine. Additionally, across all dose groups, akathisia was observed in 21% of patients during cariprazine treatment. The DIEPSS, AIMS, and BARS assessments of EPS revealed no variation in DIEPSS and AIMS scores from baseline to the end of study, with a small increase in BARS score at the end of the study for all dose groups. Overall, these assessments suggested that the risk of EPS was relatively small with once-daily cariprazine 3–9 mg/day for 12 weeks.
Though many atypical antipsychotics may have reduced risk of EPS compared with typical antipsychotics, some have a propensity to induce weight gain and metabolic changes, which increase the risk of cardiovascular complications and type 2 diabetes.5 This may make treatment especially challenging in patients with schizophrenia, who tend to have high rates of comorbid medical conditions.31 In this study, clinically relevant weight gain was not observed in any dose group. Moreover, there was a mean decrease in weight, though the AE “weight decreased” was not reported for any patients. An increase in fasting insulin levels from baseline to end of study was observed in all dose groups, though levels were all within reference ranges. No significant changes were observed from baseline to end of study for glucose and lipid levels (ie, triglycerides and total high-density lipoprotein and low-density lipoprotein cholesterol).
Maintenance of efficacy with cariprazine has been demonstrated in a recent positive randomized, placebo-controlled, relapse prevention study.32 The current study supports the maintenance of efficacy reported in the relapse prevention study. In the current 12-week study, PANSS total, positive, and negative scores all improved during the study, though the extent of change throughout the treatment period for patients was small, likely due to the low mean PANSS scores for patients at baseline (3 mg, 70.5; 6 mg, 60.3; 9 mg, 75.4). Importantly, patients did not worsen during treatment suggesting that cariprazine maintained efficacy through 12 weeks of treatment. Additionally, as all patients in the three dose groups had taken an antipsychotic drug prior to study start, residual effects of prior therapy may have been sustained during cariprazine treatment and captured in the assessments of efficacy reported here. This study did not have a placebo control group, and the efficacy results must be interpreted accordingly.
As is the case with all registered clinical trials for schizophrenia, careful selection of patients ensured that all trial participants could provide written informed consent and had no significant comorbid psychiatric or medical conditions. This factor must be considered when interpreting results for the heterogeneous population of patients seen in clinical practice, particularly those who are difficult-to-treat, have comorbid diseases, and who may require higher doses. The small sample size and lack of a placebo or active control in this study must also be considered when interpreting these results.
With its unique pharmacological properties, cariprazine may be a favorable treatment option for patients with schizophrenia. The pharmacokinetic profile determined from this study supports the proposed dosing regimen for cariprazine, which includes up-titration for the first 4 days to the defined target dose followed by once-daily dosing. This study also further supports the safety and tolerability of once-daily cariprazine (3–9 mg) during 12 weeks of treatment and 12 weeks of follow-up in patients with schizophrenia.
Acknowledgments
The authors thank the participants of this study, as well as members of the MP-214A11 study group. This study was funded by Mitsubishi Tanabe Pharma Corporation (Osaka). Writing assistance and editorial support for the preparation of this manuscript was provided by Sharanya Ford, PhD, and Adam Ruth, PhD, of Prescott Medical Communications Group, Chicago, IL, a contractor of Forest Research Institute, an affiliate of Actavis, Inc.
Disclosure
Tadakatsu Nakamura, Tomoko Kubota, Atsushi Iwakaji, Masayoshi Imada, and Yasunori Morio acknowledge a potential conflict of interest as employees of Mitsubishi Tanabe Pharma Corporation, Tokyo, Japan. Margit Kapás acknowledges a potential conflict of interest as an employee of Gedeon Richter Plc, Budapest, Hungary.
References
Tandon R, Nasrallah HA, Keshavan MS. Schizophrenia, “just the facts” 4. Clinical features and conceptualization. Schizophr Res. 2009;110(1–3):1–23. | ||
Haller CS, Padmanabhan JL, Lizano P, Torous J, Keshavan M. Recent advances in understanding schizophrenia. F1000Prime Rep. 2014;6:57. | ||
Tandon R, Nasrallah HA, Keshavan MS. Schizophrenia, “just the facts” 5. Treatment and prevention. Past, present, and future. Schizophr Res. 2010;122(1–3):1–23. | ||
Kapur S, Mamo D. Half a century of antipsychotics and still a central role for dopamine D2 receptors. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27(7):1081–1090. | ||
Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209–1223. | ||
Gross G, Wicke K, Drescher KU. Dopamine D(3) receptor antagonism – still a therapeutic option for the treatment of schizophrenia. Naunyn Schmiedebergs Arch Pharmacol. 2013;386(2):155–166. | ||
Millan MJ, Gressier H, Brocco M. The dopamine D3 receptor antagonist, (+)-S 14297, blocks the cataleptic properties of haloperidol in rats. Eur J Pharmacol. 1997;321(3):R7–R9. | ||
Gyertyán I, Sághy K, Laszy J, et al. Subnanomolar dopamine D3 receptor antagonism coupled to moderate D2 affinity results in favourable antipsychotic-like activity in rodent models: II. Behavioural characterisation of RG-15. Naunyn Schmiedebergs Arch Pharmacol. 2008;378(5):529–539. | ||
Millan MJ, Dekeyne A, Rivet JM, Dubuffet T, Lavielle G, Brocco M. S33084, a novel, potent, selective, and competitive antagonist at dopamine D(3)-receptors: II. Functional and behavioral profile compared with GR218,231 and L741,626. J Pharmacol Exp Ther. 2000;293(3):1063–1073. | ||
Laszy J, Laszlovszky I, Gyertyán I. Dopamine D3 receptor antagonists improve the learning performance in memory-impaired rats. Psychopharmacology (Berl). 2005;179(3):567–575. | ||
Millan MJ, Di Cara B, Dekeyne A, et al. Selective blockade of dopamine D(3) versus D(2) receptors enhances frontocortical cholinergic transmission and social memory in rats: a parallel neurochemical and behavioural analysis. J Neurochem. 2007;100(4):1047–1061. | ||
Leggio GM, Micale V, Drago F. Increased sensitivity to antidepressants of D3 dopamine receptor-deficient mice in the forced swim test (FST). Eur Neuropsychopharmacol. 2008;18(4):271–277. | ||
Gross G, Drescher K. The role of dopamine D(3) receptors in antipsychotic activity and cognitive functions. Handb Exp Pharmacol. 2012;(213):167–210. | ||
Kiss B, Laszlovszky I, Horváth A, et al. Subnanomolar dopamine D3 receptor antagonism coupled to moderate D2 affinity results in favourable antipsychotic-like activity in rodent models: I. Neurochemical characterisation of RG-15. Naunyn Schmiedebergs Arch Pharmacol. 2008;378(5):515–528. | ||
Joyce JN, Millan MJ. Dopamine D3 receptor antagonists as therapeutic agents. Drug Discov Today. 2005;10(13):917–925. | ||
Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328–340. | ||
Kiss B, Horti F, Bobok A. Cariprazine, a D3/D2 dopamine receptor partial agonist antipsychotic, displays greater D3 receptor occupancy in vivo compared with other antipsychotics. Schizophr Res. 2012;136(Suppl 1):S190. | ||
Papp M, Gruca P, Lason-Tyburkiewicz M, Adham N, Kiss B, Gyertyán I. Attenuation of anhedonia by cariprazine in the chronic mild stress model of depression. Behav Pharmacol. 2014;25(5–6):567–574. | ||
Zimnisky R, Chang G, Gyertyán I, Kiss B, Adham N, Schmauss C. Cariprazine, a dopamine D(3)-receptor-preferring partial agonist, blocks phencyclidine-induced impairments of working memory, attention set-shifting, and recognition memory in the mouse. Psychopharmacology (Berl). 2013;226(1):91–100. | ||
Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2–3):450–457. | ||
Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367–373. | ||
Durgam S, Cutler AJ, Lu K, et al. Cariprazine in acute exacerbation of schizophrenia: a fixed-dose, phase III randomized, double-blind, placebo- and active-controlled trial. J Clin Psychiatry. In press. | ||
Debelle M, Németh G, Szalai E, et al. Cariprazine as monotherapy for the treatment of schizophrenia patients with predominant negative symptoms: a double-blind, active controlled trial. Eur Neuropsychopharmacol. 2015;25(Suppl 2):S510. | ||
Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193–206. | ||
Mészáros GP, Kapás M, Borsos M, et al. Pharmacokinetics of RGH-188, a new dopamine D3/D2 antagonist/partial agonist atypical antipsychotic, in healthy subjects. Eur Neuropsychopharmacol. 2007;17(Suppl 4):S451–S452. | ||
Ereshefsky L, Andor G, Gage A, et al. Phase I study of RGH-188 in schizophrenic patients. Poster presented at: the 161st Annual Meeting of the American Psychiatric Association; May 3–8; 2008; Washington, DC. | ||
Mészáros GP, Ágai-Csongor E, Kapás M. Sensitive LC-MS/MS methods for the quantification of RGH-188 and its active metabolites, desmethyl- and didesmethyl-RGH-188 in human plasma and urine. J Pharm Biomed Anal. 2008;48(2):388–397. | ||
Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13(2):261–276. | ||
Guy W, editor. The clinician global severity and impression scales. In: ECDEU Assessment Manual for Psychopharmacology. Rockville, MD: National Institute of Mental Health; 1976:218–222. DHEW Publication No 76-338. | ||
Pierre JM. Extrapyramidal symptoms with atypical antipsychotics: incidence, prevention and management. Drug Saf. 2005;28(3):191–208. | ||
Weber NS, Cowan DN, Millikan AM, Niebuhr DW. Psychiatric and general medical conditions comorbid with schizophrenia in the National Hospital Discharge Survey. Psychiatr Serv. 2009;60(8):1059–1067. | ||
Durgam S, Earley W, Li R, et al. Long-term cariprazine treatment for the prevention of relapse in patients with schizophrenia: a double-blind, placebo-controlled trial. Eur Neuropsychopharmacol. 2015;25(Suppl 2):S512–S513. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.