")
Back to Journals » Cancer Management and Research » Volume 13
Clinical Management of Anemia in Patients with Myelodysplastic Syndromes: An Update on Emerging Therapeutic Options
Authors Lewis R, Bewersdorf JP , Zeidan AM
Received 14 November 2020
Accepted for publication 13 January 2021
Published 25 January 2021 Volume 2021:13 Pages 645—657
DOI https://doi.org/10.2147/CMAR.S240600
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Beicheng Sun
Russell Lewis, Jan Philipp Bewersdorf, Amer M Zeidan
Department of Medicine, Section of Hematology, Yale University, New Haven, CT, USA
Correspondence: Amer M Zeidan
Department of Medicine, Section of Hematology, Smilow Cancer Center at Yale New Haven Hospital, 333 Cedar Street, PO Box 208028, New Haven, CT 06520-8028, USA
Email [email protected]
Abstract: For the majority of patients with lower-risk myelodysplastic syndrome (LR-MDS), one of the primary clinical goals is to alleviate the symptoms associated with the resultant cytopenias and to minimize the transfusion burden. While supportive red blood cell (RBC) transfusions and erythropoiesis-stimulating agents (ESAs) may lead to clinical improvement, frequent transfusions are often complicated by iron overload and decreased quality of life; furthermore, most patients either do not respond to ESAs or will eventually develop resistance. As such, there is a great need for further therapeutic options in the management of anemia related to MDS. Several additional therapeutics are now available in select patients with LR-MDS and symptomatic anemia including luspatercept, lenalidomide, and immunosuppressive therapy. Furthermore, several novel agents are currently in development to address this area of clinical need such as imetelstat and roxadustat. In this article, we review the currently available therapeutic options for symptomatic anemia in LR-MDS as well as review the therapeutic agents in development.
Keywords: myelodysplastic syndrome, erythropoiesis-stimulating agents, novel agents, clinical trials
Introduction
Myelodysplastic syndromes (MDS) are a heterogeneous group of hematologic clonal malignancies characterized by dysfunctional hematopoiesis, cytopenias, and an increased risk of progression to acute myeloid leukemia. They occur most frequently in older adults, with a median age of diagnosis of 70 years, and so management is often complicated by the presence of comorbidities and an inability to tolerate intensive treatments.1 The presentation of disease is heterogeneous, but patients often manifest with symptoms related to cytopenias such as fatigue, infections, or hemorrhagic complications. A diagnostic evaluation of MDS in a patient with unexplained persistent cytopenia(s) currently requires a bone marrow biopsy and aspiration to detect dysplasia and assess marrow cellularity. Cytogenetic testing is a standard of care and there is often a need to exclude other causes of cytopenias.2 Cytogenetic assessment has implications for prognosis as well as treatment, as is the case with del (5q) and the use of lenalidomide.1,3 There is an increasing awareness of the importance of molecular testing in the context of workup and diagnosis of MDS, though their full integration remains a work in progress.4
The 2016 World Health Organization (WHO) classification system for MDS differentiates between several subtypes based on the number of dysplastic lineages and cytopenias, the presence of ring sideroblasts, the presence of del (5q), and the presence of excess blasts.5 MDS has historically been risk stratified by use of the International Prognostic Scoring System (IPSS) for MDS, and more recently with the revised-IPSS (IPSS-R).6–8 These tools categorize disease risk based on cytogenetic abnormalities, the degree of cytopenias, and the percentage of bone marrow blasts. The IPSS-R subdivides patients into five groups (very low-, low-, intermediate-, high-, very high-risk) that differ in terms of survival and risk of leukemic transformation. This is clinically relevant as the treatment approach differs between higher-risk and lower-risk patient subgroups.9–11 However, it has been well recognized that some patients with lower-risk (LR)-MDS using IPSS or IPSS-R do not fare well.12 Therefore, inherent limitations of the current prognostic tools should be recognized in the process of making clinical decision for individual patients.13–15 The integration of molecular assessment in risk stratification tools will likely increase their precision and utility in clinical practice.
In patients with higher-risk MDS (HR-MDS) the goals of treatment are to alter the natural history of the disease, decrease to risk of leukemic progression, and improve survival. Allogeneic hematopoietic cell transplant is the recommended treatment option in such patients if they are candidates for the procedure.10,16 However, most patients are not transplant candidates, and so they are most often treated with the hypomethylating agents azacitidine or decitabine.9,10,17–19 Targeted therapies are now under investigation for HR-MDS patients such as the IDH1 and IDH2 inhibitors which are being evaluated in clinical trials with ivosidenib and enasidenib, respectively.20,21 Furthermore, there is a promising pipeline of novel agents under active investigation in HR-MDS.22,23
For patients with LR-MDS, the goals of treatment are to improve quality of life by managing the underlying cytopenias and their side effects. The majority of patients are anemic at presentation, and this represents a major clinical challenge.3 Treatment options in this group of patients include active surveillance, red blood cell (RBC) transfusions, erythropoiesis-stimulating agents (ESAs), lenalidomide in patients with del (5q), hypomethylating agents (HMAs), luspatercept in patients with ring sideroblasts, and immunosuppressive agents in a select group of patients (see Table 1).17,24–26 While these modalities have made significant improvements in minimizing transfusion requirements and improving quality of life for LR-MDS patients, most patients either are or eventually become refractory to treatment and become transfusion dependent. As such, there is significant interest in developing novel agents to manage anemia in these patients. In this article, we review the current approach and therapeutics used to manage anemia in LR-MDS as well as discuss novel agents under investigation (see Table 2).
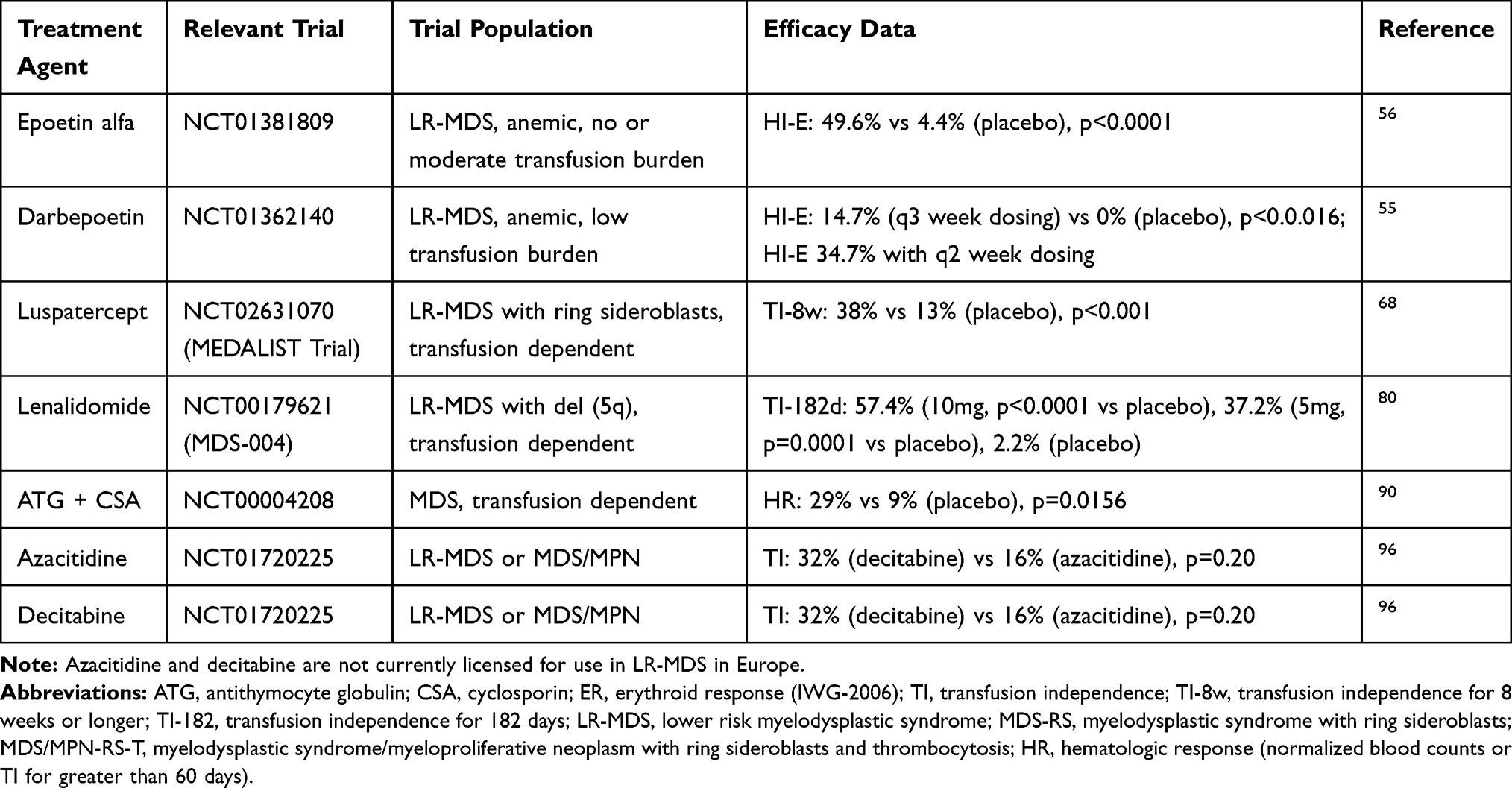 |
Table 1 Currently Available Agents for the Treatment of Anemia in LR-MDS |
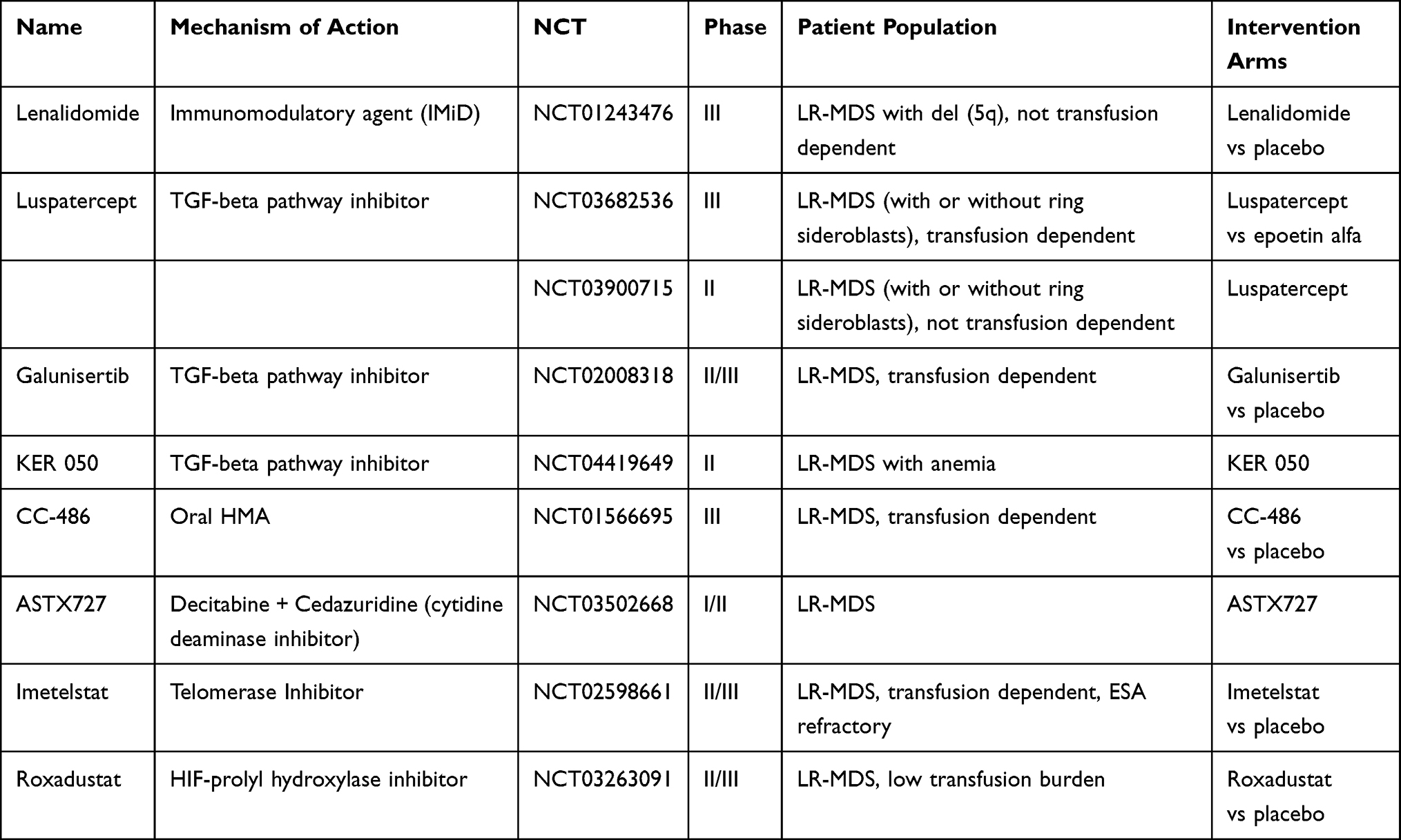 |
Table 2 Investigational Agents for the Management of Anemia in LR-MDS |
Current Treatment Paradigm
Anemia Evaluation
The initial evaluation of anemia in MDS should seek to identify alternative etiologies for the anemia such as iron deficiency, nutrient deficiencies, hypothyroidism, renal disease, or gastrointestinal bleeding.27 Copper deficiency should be considered, especially in patients with a history of gastrointestinal surgery or zinc supplementation, as this can lead to hematologic abnormalities that are very similar to MDS and which may resolve completely with supplementation alone.28,29 It is also important to assess for symptoms, as asymptomatic low-risk patients without any significant cytopenias can undergo active surveillance alone with deferred treatment.30
Supportive Care and RBC Transfusions
The majority of patients with MDS will either have symptomatic anemia at diagnosis or develop symptoms during the course of their disease requiring RBC transfusion support.31,32 Transfusion dependence is, however, associated with a significant decrease in quality of life, health-care costs, and complications related to iron overload.31,33,34 There is no uniform consensus on transfusion targets in MDS in the absence of other comorbid conditions (such as coronary artery disease, heart failure, or stroke); however, the National Comprehensive Cancer Network (NCCN) recommends using the minimum number of RBC transfusions to alleviate symptoms and restore the patient to safe hemoglobin levels.10 A retrospective study of patients with MDS and anemia found that hemoglobin levels <9g/dL in males or <8 g/dL in females were associated with decreased overall survival (OS) and increased rates of cardiac death and non-leukemic death.35 However, there is no clear consensus on the lowest safe threshold to forgo RBC transfusions among MDS patients.36,37
Although transfusions may alleviate symptoms of anemia, chronic iron overload from excessive transfusions can have a detrimental impact on cardiac and hepatic function.38,39 Furthermore, retrospective data have suggested that transfusional iron overload may be associated with increased mortality.40–42 This highlights the importance of monitoring serum ferritin levels as well as end-organ function for all patients receiving frequent transfusions. Additionally, iron chelation therapy has been studied in the transfusion-dependent LR-MDS patient population, and retrospective data suggest that it may improve overall survival, prolong leukemia-free survival, and decrease cardiac event-free survival.43–47 The use of deferasirox in heavily transfusion-dependent LR-MDS was recently evaluated in the randomized placebo-controlled TELESTO trial, and deferasirox was found to significantly increase event-free survival (3.9 years vs 3.0 years; HR 0.64 [CI, 0.42–0.96]), defined as time to first non-fatal event (related to cardiac or hepatic dysfunction, or progression to AML) or death.48 However, this trial had several limitations such as significant under-accrual which limited the ability to assess for OS difference. Furthermore, the difference in event-free survival seems to have been driven primarily by heart failure hospitalizations and worsening cardiac function while AML progression and OS were similar. Currently, the NCCN recommends consideration of deferasirox in low and intermediate-risk MDS patients who receive more than 20 RBC transfusions, are anticipated to have ongoing transfusions, or who have a ferritin above 2500 ng/mL.10
Erythropoiesis-Simulating Agents (ESAs)
For patients with low- to intermediate-risk MDS with symptomatic anemia and low EPO levels (<500 mU/mL), ESAs are generally an appropriate frontline treatment option, either with or without granulocyte-colony stimulating factor (G-CSF). Both recombinant human erythropoietin (rHu EPO) and darbepoetin, a longer acting and highly glycosylated form, are acceptable options, though darbepoetin-alfa has a more convenient dosing schedule.49
Earlier trial data in anemic patients with LR-MDS showed that erythropoietin (EPO) led to an erythroid hematologic improvement (HI-E) response rate of 36%, with HI-E defined as improvement of hemoglobin by at least 1 g/dl or reduction in transfusion support by >50% (IWG 2000 criteria).50,51 Although ESA use was not associated with improvements in OS or leukemic progression, those patients who did respond to ESAs had improved survival compared with nonresponders (5.5 vs 2.3 years median OS, p=0.004).50 Another study of MDS patients treated with EPO plus G-CSF showed similar HI-E (39%) but did demonstrate a treatment-associated improvement in OS (Hazard ratio [HR] 0.61 [CI, 0.44 to 0.83]) and a median duration of response of 23 months.52 The initial studies of darbepoetin showed erythroid response rates of 40–71% in patients with low- to intermediate-risk MDS.53,54 More recent Phase 3 data in low-risk MDS has shown HI-E response rates of 46% in patients treated with EPO, and 35% in patients treated with darbepoetin (by IWG 2006 criteria).55,56
Although there is not a standardized regimen or dosing for use of ESAs, the NCCN panel recommends their use for symptomatic anemia in MDS with a target hemoglobin of 10–12 g/dL.10 Furthermore, the American Society of Clinical Oncology (ASCO) and American Society of Hematology (ASH) guidelines for management of cancer-related anemia recommend consideration of ESAs in patients with lower risk MDS and serum erythropoietin levels (sEPO) below 500 IU/L.57 This is based on numerous clinical trials, which have shown increased efficacy of ESAs in lower-risk MDS patients with lower sEPO levels; in studies using an sEPO cutoff of 500 IU/L, those with lower sEPO levels had a 48–55% response rate to ESAs versus 10–16% in those with higher sEPO levels.58 However, data suggest poor compliance with these guidelines in use of ESAs in the community.25,59,60 Factors predictive of response to ESAs include low EPO levels, low baseline transfusion requirements, higher baseline hemoglobin, fewer bone marrow blasts, and normal cytogenetics.53,58 Unfortunately, most patients who initially respond to ESAs will eventually become treatment refractory.
It remains controversial whether or not the addition of G-CSF to ESAs improves erythroid response rates. In randomized trials comparing ESAs alone versus ESAs in combination with G-CSF, HI-E rates improved by about 35% in patients receiving combination therapy; however, the applicability of these studies to current practice is not clear because they used low-/standard dose ESA, rather than full-dose ESA as is currently recommended.61,62 In fact, a meta-analysis concluded that full-dose ESA produced higher rates of HI-E (64.5%) than either standard dose ESA alone (49%, p<0.001) or ESA in combination with G-CSF (50.6%, p=0.007).63 Although there have been additional studies investigating the benefit of G-CSF in this setting, these trials have been limited by issues of inadequate EPO dosing as well as the use of sequential drug administration designs, in which G-CSF is administered after a minor response or no response to ESA; this is a suboptimal design approach as it makes it difficult to differentiate a delayed hematologic response to ESA from an actual response to the addition of G-CSF. Based on this data and these trial limitations, a recent systemic review concluded that there is insufficient evidence to strongly recommend a benefit of additional G-CSF over full-dose ESA alone.64
Luspatercept
Luspatercept is an erythroid maturation agent (EMA) that is structurally formed as a recombinant fusion protein that acts as an activin receptor ligand trap inhibiting signaling via the transforming growth factor-β (TGF-β) pathway.24 It consists of an extracellular domain of activin receptor type IIB fused to a human immunoglobulin G1 Fc domain. Increased activation of the TGF-β has been associated with ineffective erythropoiesis in MDS and increased downstream SMAD2 and SMAD3 signaling.24 Use of luspatercept in murine models has demonstrated decreased SMAD2 and SMAD3 signaling, reduced erythroid hyperplasia, improved erythropoiesis, and improved anemia (see Figure 1).65,66
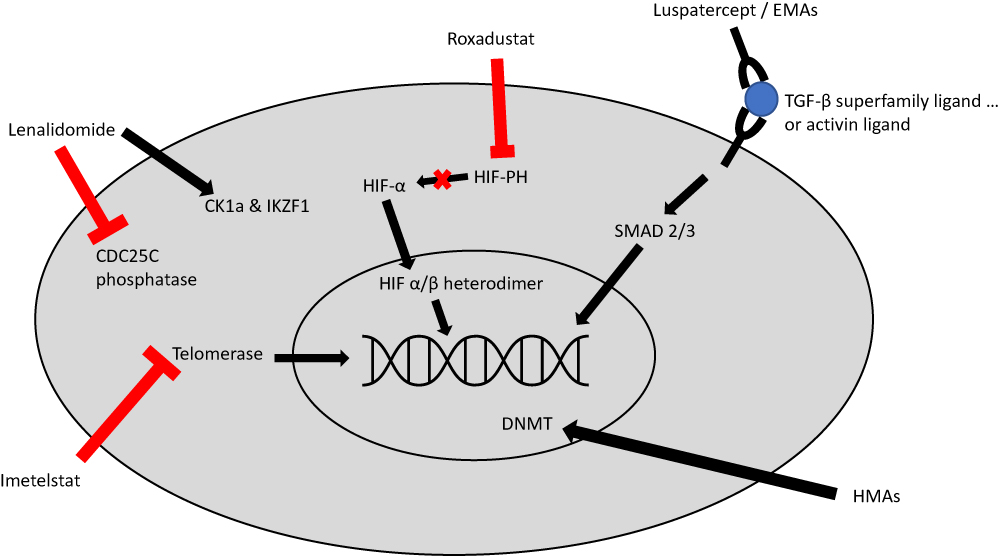 |
Figure 1 Mechanism of action for select agents used or under investigation for the management of LR-MDS, Imetelstat directly inhibits telomerase, thus preventing telomerase from adding telomere repeat sequences to the 3ʹ-end of telomeres. Roxadustat inhibits HIF-PH, thus leading to decreased HIF-alpha degradation and increased HIF-alpha signaling. Luspatercept and similar erythropoiesis maturation agents (EMAs) act as a ligand trap which prevents TGF-beta activation and leads to decreased downstream SMAD2 and SMAD3 signaling. Lenalidomide has a complex mechanism of action; it has a direct antiproliferative effect via inhibition of CDC25C phosphatase which leads to cell cycle arrest; it also leads to ubiquitination of CK1a and IKZF1 which leads to apoptosis. Hypomethylating agents (HMA) lead to increased DNA methylation by causing degradation of DNA methyltransferase (DNMT). This leads to decreased inactivation of tumor suppressor genes. Abbreviations: Hypomethylating agents, (HMA); lead to increased DNA methylation by causing degradation of DNA methyltransferase, (DNMT); this leads to decreased inactivation of tumor suppressor genes. |
The Phase 2 PACE-MDS trial studied the use of luspatercept in anemic patients with IPSS low- or intermediate-risk MDS or non-proliferative chronic myelomonocytic leukemia (CMML).67 The primary end point was HI-E (by IWG 2006 criteria), defined as a hemoglobin improvement of 1.5 g/dL or higher for 14 days in low transfusion burden patients, or a reduction in RBC transfusion requirement by 4 or more units over an 8-week period or a 50% reduction from baseline. Among patients treated with higher doses of luspatercept, HI-E response was seen in 63% and transfusion independence was achieved in 38%. Subgroup analysis revealed that the presence of ring sideroblasts, SF3B1 mutation, and positive spliceosome mutational status was associated with increased response rates.67
These results led to the randomized, double-blind, placebo-controlled, phase 3 MEDALIST trial, which investigated luspatercept in patients with IPSS-R defined very low- to intermediate-risk MDS with ring sideroblasts who were transfusion dependent and who were refractory to or unlikely to respond to ESAs. A total of 229 patients were enrolled, and 153 were assigned to receive luspatercept (1.0 to 1.75 mg/kg). Thirty-eight percent of patients in the luspatercept group achieved transfusion independence for 8 weeks or longer, compared with 13% in the placebo group (p<0.001). The median duration of the longest period of transfusion independence was 30.6 weeks in the luspatercept group vs 13.6 weeks in the placebo group. The drug was well tolerated; common adverse effects included fatigue, diarrhea, asthenia, nausea, and dizziness; however, the majority of these were grade 1 or grade 2.68 Based on these encouraging results, luspatercept was approved by the FDA in April 2020 for the treatment of anemia in patients with very low- to intermediate-risk MDS with ring sideroblasts (MDS-RS) who have failed an ESA and require at least 2 or more RBC units over 8 weeks.69
Lenalidomide
Lenalidomide is an immunomodulatory (IMiD) agent with a complex mechanism of action (see Figure 1), which has shown significant benefit in patients with MDS who harbor a deletion in the long arm of chromosome 5, del (5q).70–72 5q deletions are relatively common in MDS, occurring in 5–15% of patients,73–75 and they predict a poor response to ESAs.76,77
Earlier trials evaluating lenalidomide in transfusion-dependent low- or intermediate-risk MDS patients with del (5q) demonstrated a high rate of response, with 76% of patients having reduced transfusion requirements and 67% having transfusion independence.78 Furthermore, 73% had a cytogenetic response, and 45% had complete cytogenetic remission. The time to response was quick (median 4.6 weeks) and the median hemoglobin improvement was 5.4 g/dl; encouragingly the duration of response was prolonged (median duration of transfusion independence not reached). Neutropenia and thrombocytopenia were the most common grade 3 or 4 adverse effects (occurring in 55% and 44% of patients, respectively). Subsequent Phase 2/3 trials have shown similar results.79–81 The MDS-004 study in low- to intermediate-risk MDS with del (5q) and transfusion dependence showed high rates of transfusion independence with lenalidomide 10 mg (57% vs 2% with placebo, p<0.0001) and high rates of cytogenetic response (57% vs 0%, p<0.0001); although lenalidomide did not improve survival, being a responder to lenalidomide did predict increased OS.80
In patients without del (5q), lenalidomide has shown activity, though the results have been more subdued. Phase 2 data in low- or intermediate-risk MDS without del (5q) showed a 43% overall hematologic improvement with lenalidomide and a 26% rate of transfusion independence (TI); median duration of TI was 41 weeks.82 In a phase 3 trial enrolling patients with ESA refractory non-del (5q) LR-MDS, lenalidomide plus erythropoietin beta was shown to have higher rates of erythroid response than lenalidomide alone (39.4% vs 23.1%, respectively, p=0.044).83 Currently, lenalidomide is only approved for lower-risk transfusion-dependent MDS with del (5q); however, it is often used outside this indication.
Of note, TP53 mutations are present in about a fifth of LR-MDS patients with del (5q), and are associated with an increased risk of leukemic progression.84 Prior studies have shown that these patients are less sensitive to lenalidomide.85
Retrospective data have evaluated the use of lenalidomide in non-transfusion dependent MDS patients; no clear OS benefit was found compared to risk match-controls, though these patients did appear to have an increased risk of thromboembolic event (10.8% vs 6.0%, p=0.04) compared to transfusion-dependent patients.86 However, the question of lenalidomide’s value in non-transfusion dependent patients with LR-MDS with del (5q) is currently being investigated in a phase 3 trial (NCT01243476).
The issue of impact of sequencing of therapies on response has been studied in retrospective analyses which suggested that use of lenalidomide before a HMA might be a better strategy than the reverse order.87,88 One study evaluated 63 patients with LR-MDS who had failed ESA and received both lenalidomide and azacitidine as second- and third-line therapy; patients had a significantly higher HI-E response rate to lenalidomide when they received it prior to azacitidine (38% vs 12%, p=0.04), whereas response rates to azacitidine were similar irrespective of sequencing (38% lenalidomide first vs 35% azacitidine first, p=0.69). Notably, sequencing of lenalidomide and azacitidine had no impact on OS or leukemic progression.87 In a retrospective analysis of US payer claims database, patients who received lenalidomide first followed by a HMA had a longer median time to insurance disenrollment (22.4 vs 16.1 months, p<0.001), which was used as a proxy for survival, compared to patients who received a hypomethylating agent prior to lenalidomide.88
High-dose lenalidomide has been used in more advanced MDS states but therapy was poorly tolerated with minimal activity.89
Immunosuppressive Therapy (IST)
Immunologic dysregulation has been observed in patients with MDS and may play a direct role in impaired hematopoiesis. Prior studies have observed oligoclonal T-cell expansion in patients with MDS as well as suppression of hematopoietic progenitors by cytotoxic T-cells.90 This provides a rationale for the observed efficacy of immunosuppressive agents in a subset of patients with hypoplastic MDS.
A 2011 phase 3 study in patients with transfusion-dependent MDS evaluated the efficacy of horse antithymocyte globulin (ATG) and cyclosporin (CSA) versus best supportive care (BSC). The rate of hematologic response (defined as normalization of blood counts or transfusion independence for greater than 60 days) was significantly higher in the ATG+CSA group, at 29% versus 9% for BSC (p=0.0156), though there was no difference in OS or rates of leukemic transformation.90 Prior studies have shown that the response rate to ATG combined with CSA is higher than it is with either agent on its own, suggesting some degree of synergy.91 Also, it is notably that the effects of IST have been found to be most beneficial in a subset of patients with HLA-DR15 positivity, marrow hypoplasia, normal cytogenetics, low-risk disease, and those with PNH clone; however, these predictors of response are controversial, as a recent large retrospective center study found only marrow hypocellularity to be predictive of response to IST.90,92,93 Though its role is not clearly defined, the NCCN recommends consideration of IST in MDS patients with symptomatic anemia, elevated EPO (>500), and good probability of responding to IST (defined by patients <60 years old with no more than 5% bone marrow blasts, or those with hypocellular marrow, PNH clone positivity, or STAT-3 mutant cytotoxic T-cell clones).10
Hypomethylating Agents
Hypomethylating agents (HMA) are thought to function by leading to decreased methylation at DNA promotor regions, thereby leading to re-expression of tumor suppressor genes (see Figure 1).94 The HMAs azacitidine and decitabine are standard treatment options in HR-MDS, though they have shown modest efficacy in LR-MDS as well. In a Phase II study of ESA refractory LR-MDS, patients who received azacitidine had a 16% rate of transfusion independence after 6 cycles, and 35% had an erythroid response (IWG 2000).95 Another phase II study randomly assigned LR-MDS patients to receive either decitabine or azacitidine; after a median follow-up of 20 months, patients who received decitabine had slightly better rates of transfusion independence (32% vs 16%, p=0.20) and rates of cytogenetic response (61% vs 25%, p=0.02).96 Real-life analyses have confirmed similar rates of TI with HMAs.97 HMAs remain a treatment consideration, particularly in younger patients with aggressive features and those who have failed other treatment options. However, HMAs are not currently licensed for use in LR-MDS in Europe.
Emerging Therapeutics for Management of Anemia in LR-MDS
Erythropoiesis Maturation Agents (EMAs)
While luspatercept represents the only FDA-approved therapeutic in MDS that functions via promotion of erythropoiesis maturation, there are several other drugs that also work via TGF-β pathway inhibition and these have been and currently are under investigation for LR-MDS.
Sotatercept and galunisertib are both TGF-β pathway inhibitors that lead to downstream SMAD2 and SMAD3 signaling and stimulate hematopoiesis.3,98,99 Although these agents had promising phase 2 trial results in the management of transfusion-dependent LR-MDS, further clinical development has been halted for both.100,101 Sotatercept was halted because the manufacturer chose to pursue luspatercept instead for further clinical development.30 Development of galunisertib was discontinued by the manufacturer in early 2020 for unspecified reasons.
KER-050 is a ligand trap composed of the activin receptor type IIA fused to the human immunoglobulin G1 Fc domain. It has had promising results in murine models, and is now being developed for the treatment of cytopenias in patients with MDS and myelofibrosis (MF).102 It is currently in a phase 2 clinical trial that is evaluating the drug’s effect on very low- to intermediate-risk MDS patients with anemia (NCT04419649).
Novel Hypomethylating Agents
ASTX727 is a novel oral medication that combines decitabine with cedazuridine, which is an inhibitor of cytidine deaminase in the gastrointestinal tract and liver to prevent decitabine’s breakdown and inactivation. In a recent phase 2 study, 80 patients with intermediate-1/2- or high-risk MDS or chronic myelomonocytic leukemia (CMML) were randomized to receive ASTX727 or IV decitabine, with crossover occurring in cycle 2.103 Clinical responses were observed in 48 patients (60%) including 17 (21%) with complete response. The study showed that the efficacy, side effect profile, and systemic decitabine exposure of ASTX727 are similar to that of IV decitabine, and it was approved by the FDA in July 2020 for IPSS intermediate-1, intermediate-2, and high-risk MDS. However, a phase I/II trial is ongoing with oral cedazuridine/decitabine at a lower dose in LR-MDS (NCT03502668).
Azacitidine has also been developed as an oral agent, CC-486, and this formulation was recently approved by the FDA for maintenance therapy in AML patients in first remission after induction therapy, based on the results of the phase 3 QUAZAR AML-001 study.104 In this trial, CC-486 maintenance therapy after induction chemotherapy for AML led to significant improvement in median OS compared to placebo (24.7 months vs 14.8 months, p=0.0009). More recently, CC-486 was studied in a Phase III trial to assess its efficacy and safety in transfusion-dependent LR-MDS (NCT01566695).105 In this trial, significantly more patients in the CC-486 arm achieved the primary endpoint of RBC-TI for >56 days compared to placebo (31% vs 11%, p=0.0002). Furthermore, rates of hematologic platelet improvement (HI-P) were greater in the CC-486 arm (p=0.0003), though rates of HI-E were comparable (p=0.12). While the sample size (n=215) was not powered for interim OS analysis, no difference in OS was seen between CC-486 and placebo (17.3 vs 16.7 months, p=0.88). Furthermore, while there was no difference in overall death rate between the two arms, there were an increased number of early deaths in the CC-486 arm, mostly related to infection, which led to the decision to close enrollment early.
Imetelstat
Telomeres play an important role in maintaining normal hematopoiesis, and telomere shortening and dysregulation of telomerase is thought to play a role in the development of MDS and AML. Prior studies have shown that patients with MDS have shorter telomere lengths and that shorter telomere length is independently associated with poor survival.106,107 While most normal human cells only express telomerase transiently and thus develop telomere shortening with each cell division, cancer cells often express high levels of telomerase allowing for significant proliferation.108 In fact, MDS patients with increased levels of telomerase activity (TA) have worse survival compared to those with low TA levels.109
Imetelstat is a potent telomerase inhibitor (see Figure 1), which has been studied in early phase trials for several malignancies and which has already shown promising results in the treatment of myeloproliferative neoplasms (MPNs). In a phase 2 trial of patients with previously treated essential thrombocythemia, imetelstat induced a complete hematologic response in 16 of 18 patients treated.110 A phase 2 trial in patients with myelofibrosis showed a complete or partial response to imetelstat in 21% of the patients, though with significant grade 4 thrombocytopenia (18%), grade 4 neutropenia (12%), and grade 3 anemia (30%).111 The IMerge phase 2/3 trial (NCT02598661) is currently recruiting patients, and it is investigating the use of imetelstat in patients with transfusion-dependent lower-risk MDS refractory to ESAs. Preliminary trial results, with 38 patients enrolled, showed an 8-week transfusion independence rate of 45% with a median TI duration of 8.5 months; TI rates did not differ based on baseline sEPO levels. HI-E was achieved in 68% of patients. Furthermore, five out of six patients (83%) with intermediate- or poor-risk cytogenetics achieved 8-week TI.112 While these results are preliminary, they are encouraging.
Hypoxia-Inducible Factor Prolyl Hydroxylase (HIF-PH) Inhibitors – Roxadustat
Hypoxia-inducible factors (HIFs) are transcription factors that mediate the cellular response to hypoxic conditions via upregulation of several genes that are key to erythropoiesis including those for erythropoietin, the EPO receptor, and proteins involved in iron metabolism.113 In normoxic conditions, HIF is degraded by hydroxylation of prolyl residues of the HIF-α subunit by HIF prolyl hydroxylases.114 Thus, inhibition of HIF prolyl hydroxylase represents an attractive target to prevent HIF degradation and thus promote erythropoiesis. In fact, several HIF prolyl hydroxylase inhibitors have been developed for the treatment of anemia, including daprodustat, desidustat, roxadustat, molidustat, and vadadustat.115–119 While these agents have been primarily studied in the setting of anemia of CKD, Roxadustat is also being investigated in patients with MDS.
Roxadustat is an oral HIF-PH inhibitor (see Figure 1). Prior phase 2 studies in patients with anemia of chronic kidney disease (CKD) have shown that roxadustat increases hemoglobin levels, increases erythropoietin, and decreases hepcidin levels.115 Phase 3 data in patients with anemia of CKD treated with roxadustat, showed increases in hemoglobin levels by 1.9 g/dL after 8 weeks and also showed significant reductions in hepcidin levels. Common adverse effects included hyperkalemia (16%) and metabolic acidosis (12%), though serious adverse events were rare (9% overall).113 The drug is now approved for the treatment of CKD with or without dialysis in China, and a new drug application has been filed with the FDA in early 2020 seeking regulatory approval in the United States. Roxadustat is currently being studied (NCT03263091) in a phase 2/3 trial in patients with very low- to intermediate-risk MDS and low transfusion burden, defined as 1–4 RBC units per 8 weeks; patients also needed to have EPO levels<400 mIU/mL and to have not received an ESA within 8 weeks.120 Early results from the open-label dose-finding portion of the study have been published. Of the 24 recruited patients, 38% achieved transfusion independence for at least 8 weeks and 17% (4/24) remained TI for at least 20 consecutive weeks. Fifty-eight percent achieved at least a 50% reduction in RBC units required per 8-week period. There was an acceptable side effect profile.120 Based on these results, the randomized placebo-controlled portion of the trial is currently enrolling.
Enrollment in clinical trials remains very improvement to continue to advance therapeutic options for patients with MDS.121
Conclusion
For patients with lower-risk MDS, the upfront treatment of choice for the majority of patients is going to involve supportive care and erythropoiesis-stimulating agents. Certain subsets of patients will have additional treatment options available that offer meaningful clinical efficacy – in particular patients with del (5q) benefit from lenalidomide and those with ring sideroblasts benefit from luspatercept. Hypomethylating agents and IST are also an option for these patients in certain situation (though HMAs are not currently licenced for use in LR-MDS in Europe). However, the development of ESA and treatment refractory anemia is a common and frustrating clinical scenario. It is encouraging that there are several promising new therapeutic options in active clinical trial development, including EMAs, telomerase inhibitors, and HIF-PH inhibitors.
Funding
Amer Zeidan is a Leukemia and Lymphoma Society Scholar in Clinical Research and was also supported by an NCI’s Cancer Clinical Investigator Team Leadership Award (CCITLA). Research reported in this publication was in part supported by the National Cancer Institute of the National Institutes of Health under Award Number P30 CA016359. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure
A.M.Z. received research funding (institutional) from Celgene/BMS, AbbVie, Astex, Pfizer, Medimmune/AstraZeneca, Boehringer-Ingelheim, Trovagene/Cardiff oncology, Incyte, Takeda, Novartis, Aprea, and ADC Therapeutics. A.M.Z participated in advisory boards, and/or had a consultancy with and received honoraria from AbbVie, Otsuka, Pfizer, Celgene/BMS, Jazz, Incyte, Agios, Boehringer-Ingelheim, Novartis, Acceleron, Astellas, Daiichi Sankyo, Cardinal Health, Taiho, Seattle Genetics, BeyondSpring, Trovagene/Cardiff Oncology, Takeda, Ionis, Amgen, Janssen, Epizyme, Syndax, and Tyme. A.M.Z served on clinical trial committees for Novartis, AbbVie, Geron and Celgene/BMS. A.M.Z received travel support for meetings from Pfizer, Novartis, and Trovagene/Cardiff Oncology. None of these relationships were related to the development of this manuscript. R.L. and J.P.B. have no conflicts of interest.
References
1. Cazzola M. Myelodysplastic syndromes. N Engl J Med. 2020;383(14):1358–1374. doi:10.1056/NEJMra1904794
2. Zeidan AM, Faltas B, Douglas Smith B, Gore S. Myelodysplastic syndromes: what do hospitalists need to know? J Hosp Med. 2013;8(6):351–357. doi:10.1002/jhm.2049
3. Platzbecker U. Treatment of MDS. Blood. 2019;133(10):1096–1107. doi:10.1182/blood-2018-10-844696
4. Nazha A, Sekeres MA, Gore SD, Zeidan AM. Molecular testing in myelodysplastic syndromes for the practicing oncologist: will the progress fulfill the promise? Oncologist. 2015;20(9):1069–1076. doi:10.1634/theoncologist.2015-0067
5. Hong M, The HG. 2016 Revision to the world health organization classification of myelodysplastic syndromes. J Transl Int Med. 2017;5(3):139–143. doi:10.1515/jtim-2017-0002
6. Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89(6):2079–2088. doi:10.1182/blood.V89.6.2079
7. Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454–2465. doi:10.1182/blood-2012-03-420489
8. Zeidan AM, Gore SD, Komrokji RS. Higher-risk myelodysplastic syndromes with del (5q): is sequential azacitidine-lenalidomide combination the way to go? Expert Rev Hematol. 2013;6(3):251–254. doi:10.1586/ehm.13.30
9. Malcovati L, Hellström-Lindberg E, Bowen D, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood. 2013;122(17):2943–2964.
10. NCCN. Myelodysplastic Syndromes (Version 2.2020). Available from: https://www.nccn.org/professionals/physician_gls/pdf/mds.pdf.
11. Pfeilstöcker M, Tuechler H, Sanz G, et al. Time-dependent changes in mortality and transformation risk in MDS. Blood. 2016;128(7):902–910. doi:10.1182/blood-2016-02-700054
12. Zeidan AM, Sekeres MA, Wang XF, et al. Comparing the prognostic value of risk stratifying models for patients with lower-risk myelodysplastic syndromes: is one model better? Am J Hematol. 2015;90(11):1036–1040. doi:10.1002/ajh.24173
13. Zeidan AM, Al Ali N, Barnard J, et al. Comparison of clinical outcomes and prognostic utility of risk stratification tools in patients with therapy-related vs de novo myelodysplastic syndromes: a report on behalf of the MDS Clinical Research Consortium. Leukemia. 2017;31(6):1391–1397. doi:10.1038/leu.2017.33
14. Zeidan AM, Sekeres MA, Barnard J, Steensma DP, Komrokji R. Therapy-related myelodysplastic syndromes-specific risk stratification: are we putting the cart before the horse? Leukemia. 2017;31(11):2539–2541. doi:10.1038/leu.2017.238
15. Zeidan AM, Sekeres MA, Garcia-Manero G, et al. Comparison of risk stratification tools in predicting outcomes of patients with higher-risk myelodysplastic syndromes treated with azanucleosides. Leukemia. 2016;30(3):649–657. doi:10.1038/leu.2015.283
16. Zeidan AM, Stahl M, Hu X, et al. Long-term survival of older patients with MDS treated with HMA therapy without subsequent stem cell transplantation. Blood. 2018;131(7):818–821. doi:10.1182/blood-2017-10-811729
17. Abou Zahr A, Saad Aldin E, Barbarotta L, Podoltsev N, Zeidan AM. The clinical use of DNA methyltransferase inhibitors in myelodysplastic syndromes. Expert Rev Anticancer Ther. 2015;15(9):1019–1036. doi:10.1586/14737140.2015.1061936
18. Lee EJ, Zeidan AM. Genome sequencing in myelodysplastic syndromes: can molecular mutations predict benefit from hypomethylating agent therapy? Expert Rev Hematol. 2015;8(2):155–158. doi:10.1586/17474086.2015.1016905
19. Fenaux P, Mufti GJ, Santini V, et al. Azacitidine (AZA) treatment prolongs Overall Survival (OS) in higher-risk MDS patients compared with Conventional Care Regimens (CCR): results of the AZA-001 Phase III Study. Blood. 2007;110(11):817. doi:10.1182/blood.V110.11.817.817
20. Richard-Carpentier G, DeZern AE, Takahashi K, et al. Preliminary results from the phase II study of the IDH2-inhibitor enasidenib in patients with high-risk IDH2-mutated Myelodysplastic Syndromes (MDS). Blood. 2019;134(Supplement_1):678. doi:10.1182/blood-2019-130501
21. DiNardo CD, Watts JM, Stein EM, et al. Ivosidenib (AG-120) induced durable remissions and transfusion independence in patients with IDH1-mutant relapsed or refractory myelodysplastic syndrome: results from a phase 1 dose escalation and expansion study. Blood. 2018;132(Supplement 1):1812. doi:10.1182/blood-2018-99-111264
22. Bewersdorf JP, Zeidan AM. Following in the footsteps of acute myeloid leukemia: are we witnessing the start of a therapeutic revolution for higher-risk myelodysplastic syndromes? Leuk Lymphoma. 2020;61(10):2295–2312. doi:10.1080/10428194.2020.1761968
23. Abou Zahr A, Bernabe Ramirez C, Wozney J, Prebet T, Zeidan AM. New Insights into the Pathogenesis of MDS and the rational therapeutic opportunities. Expert Rev Hematol. 2016;9(4):377–388. doi:10.1586/17474086.2016.1135047
24. Bewersdorf JP, Zeidan AM. Transforming growth factor (TGF)-β pathway as a therapeutic target in lower risk myelodysplastic syndromes. Leukemia. 2019;33(6):1303–1312. doi:10.1038/s41375-019-0448-2
25. Hendrick F, Davidoff AJ, Zeidan AM, Gore SD, Baer MR. Effect of erythropoiesis-stimulating agent policy decisions on off-label use in myelodysplastic syndromes. Medicare Medicaid Res Rev. 2014;4:4. doi:10.5600/mmrr.004.04.a02
26. Shallis RM, Chokr N, Stahl M, Pine AB, Zeidan AM. Immunosuppressive therapy in myelodysplastic syndromes: a borrowed therapy in search of the right place. Expert Rev Hematol. 2018;11(9):715–726. doi:10.1080/17474086.2018.1503049
27. Shallis RM, Xu ML, Podoltsev NA, et al. Be careful of the masquerades: differentiating secondary myelodysplasia from myelodysplastic syndromes in clinical practice. Ann Hematol. 2018;97(12):2333–2343. doi:10.1007/s00277-018-3474-7
28. Fong T, Vij R, Vijayan A, DiPersio J, Blinder M. Copper deficiency: an important consideration in the differential diagnosis of myelodysplastic syndrome. Haematologica. 2007;92(10):1429–1430. doi:10.3324/haematol.11314
29. Gregg XT, Reddy V, Prchal JT. Copper deficiency masquerading as myelodysplastic syndrome. Blood. 2002;100(4):1493–1495. doi:10.1182/blood-2002-01-0256
30. Bewersdorf JP, Zeidan AM. Evolving therapies for lower-risk myelodysplastic syndromes. Ann Hematol. 2020;99(4):677–692. doi:10.1007/s00277-020-03963-1
31. Gupta P, LeRoy SC, Luikart SD, Bateman A, Morrison VA. Long-term blood product transfusion support for patients with myelodysplastic syndromes (MDS): cost analysis and complications. Leuk Res. 1999;23(10):953–959. doi:10.1016/S0145-2126(99)00113-7
32. Zeidan AM, Gore SD, Padron E, Komrokji RS. Current state of prognostication and risk stratification in myelodysplastic syndromes. Curr Opin Hematol. 2015;22(2):146–154. doi:10.1097/MOH.0000000000000110
33. Gattermann N. Iron overload in myelodysplastic syndromes (MDS). Int J Hematol. 2018;107(1):55–63. doi:10.1007/s12185-017-2367-1
34. Koutsavlis I. Transfusion thresholds, quality of life, and current approaches in myelodysplastic syndromes. Anemia. 2016;2016:8494738. doi:10.1155/2016/8494738
35. Malcovati L, Della Porta MG, Strupp C, et al. Impact of the degree of anemia on the outcome of patients with myelodysplastic syndrome and its integration into the WHO classification-based prognostic scoring system (WPSS). Haematologica. 2011;96(10):1433–1440. doi:10.3324/haematol.2011.044602
36. Abel GA, Klepin HD, Magnavita ES, et al. Feasibility of peri-transfusion quality of life assessment for patients with myelodysplastic syndromes. Blood. 2020;136(Supplement 1):25–26.
37. Tanasijevic AM, Revette A, Klepin HD, et al. Consensus minimum hemoglobin level above which patients with myelodysplastic syndromes can safely forgo transfusions. Leuk Lymphoma. 2020;1–5.
38. Schafer AI, Cheron RG, Dluhy R, et al. Clinical consequences of acquired transfusional iron overload in adults. N Engl J Med. 1981;304(6):319–324. doi:10.1056/NEJM198102053040603
39. Jensen PD, Jensen FT, Christensen T, Nielsen JL, Ellegaard J. Relationship between hepatocellular injury and transfusional iron overload prior to and during iron chelation with desferrioxamine: a study in adult patients with acquired anemias. Blood. 2003;101(1):91–96. doi:10.1182/blood-2002-06-1704
40. Malcovati L. Impact of transfusion dependency and secondary iron overload on the survival of patients with myelodysplastic syndromes. Leuk Res. 2007;31(Suppl 3):S2–6. doi:10.1016/S0145-2126(07)70459-9
41. Zeidan AM, Griffiths EA. To chelate or not to chelate in MDS: that is the question! Blood Rev. 2018;32(5):368–377. doi:10.1016/j.blre.2018.03.002
42. Zeidan AM, Pullarkat VA, Komrokji RS. Overcoming barriers to treating iron overload in patients with lower-risk myelodysplastic syndrome. Crit Rev Oncol Hematol. 2017;117:57–66. doi:10.1016/j.critrevonc.2017.07.002
43. Leitch HA, Parmar A, Wells RA, et al. Overall survival in lower IPSS risk MDS by receipt of iron chelation therapy, adjusting for patient-related factors and measuring from time of first red blood cell transfusion dependence: an MDS-CAN analysis. Br J Haematol. 2017;179(1):83–97. doi:10.1111/bjh.14825
44. Remacha ÁF, Arrizabalaga B, Villegas A, et al. Evolution of iron overload in patients with low-risk myelodysplastic syndrome: iron chelation therapy and organ complications. Ann Hematol. 2015;94(5):779–787. doi:10.1007/s00277-014-2274-y
45. Mitchell M, Gore SD, Zeidan AM. Iron chelation therapy in myelodysplastic syndromes: where do we stand? Expert Rev Hematol. 2013;6(4):397–410. doi:10.1586/17474086.2013.814456
46. Zeidan AM, Giri S, DeVeaux M, Ballas SK, Duong VH. Systematic review and meta-analysis of the effect of iron chelation therapy on overall survival and disease progression in patients with lower-risk myelodysplastic syndromes. Ann Hematol. 2019;98(2):339–350. doi:10.1007/s00277-018-3539-7
47. Zeidan AM, Hendrick F, Friedmann E, et al. Deferasirox therapy is associated with reduced mortality risk in a medicare population with myelodysplastic syndromes. J Comp Eff Res. 2015;4(4):327–340. doi:10.2217/cer.15.20
48. Angelucci E, Li J, Greenberg P, et al. Iron chelation in transfusion-dependent patients with low- to intermediate-1-risk myelodysplastic syndromes: a randomized trial. Ann Intern Med. 2020;172(8):513–522. doi:10.7326/M19-0916
49. Duong VH, Baer MR, Hendrick F, et al. Variations in erythropoiesis-stimulating agent administration in transfusion-dependent myelodysplastic syndromes impact response. Leuk Res. 2015;39(6):586–591. doi:10.1016/j.leukres.2015.03.013
50. Greenberg PL, Sun Z, Miller KB, et al. Treatment of myelodysplastic syndrome patients with erythropoietin with or without granulocyte colony-stimulating factor: results of a prospective randomized phase 3 trial by the Eastern Cooperative Oncology Group (E1996). Blood. 2009;114(12):2393–2400. doi:10.1182/blood-2009-03-211797
51. Cheson BD, Bennett JM, Kantarjian H, et al. Report of an international working group to standardize response criteria for myelodysplastic syndromes. Blood. 2000;96(12):3671–3674.
52. Jädersten M, Malcovati L, Dybedal I, et al. Erythropoietin and granulocyte-colony stimulating factor treatment associated with improved survival in myelodysplastic syndrome. J Clin Oncol. 2008;26(21):3607–3613. doi:10.1200/JCO.2007.15.4906
53. Musto P, Lanza F, Balleari E, et al. Darbepoetin alpha for the treatment of anaemia in low-intermediate risk myelodysplastic syndromes. Br J Haematol. 2005;128(2):204–209. doi:10.1111/j.1365-2141.2004.05288.x
54. Mannone L, Gardin C, Quarre MC, et al. High-dose darbepoetin alpha in the treatment of anaemia of lower risk myelodysplastic syndrome results of a phase II study. Br J Haematol. 2006;133(5):513–519. doi:10.1111/j.1365-2141.2006.06070.x
55. Platzbecker U, Symeonidis A, Oliva EN, et al. A phase 3 randomized placebo-controlled trial of darbepoetin alfa in patients with anemia and lower-risk myelodysplastic syndromes. Leukemia. 2017;31(9):1944–1950. doi:10.1038/leu.2017.192
56. Fenaux P, Santini V, Spiriti MAA, et al. A phase 3 randomized, placebo-controlled study assessing the efficacy and safety of epoetin-α in anemic patients with low-risk MDS. Leukemia. 2018;32(12):2648–2658. doi:10.1038/s41375-018-0118-9
57. Bohlius J, Bohlke K, Castelli R, et al. Management of cancer-associated anemia with erythropoiesis-stimulating agents: ASCO/ASH clinical practice guideline update. J Clin Oncol. 2019;37(15):1336–1351. doi:10.1200/JCO.18.02142
58. Park S, Kelaidi C, Meunier M, Casadevall N, Gerds AT, Platzbecker U. The prognostic value of serum erythropoietin in patients with lower-risk myelodysplastic syndromes: a review of the literature and expert opinion. Ann Hematol. 2020;99(1):7–19. doi:10.1007/s00277-019-03799-4
59. Davidoff AJ, Hendrick FB, Zeidan AM, et al. Patient cost sharing and receipt of erythropoiesis-stimulating agents through medicare part D. J Oncol Pract. 2015;11(2):e190–198. doi:10.1200/JOP.2014.001527
60. Davidoff AJ, Weiss SR, Baer MR, et al. Patterns of erythropoiesis-stimulating agent use among Medicare beneficiaries with myelodysplastic syndromes and consistency with clinical guidelines. Leuk Res. 2013;37(6):675–680. doi:10.1016/j.leukres.2013.02.021
61. Nair V, Mishra DK, Sharma A, et al. Erythropoetin (EPO) and granulocyte colony stimulating factor (GCS-F) based therapy in patients with low risk mds: a single centre experience from India. Blood. 2006;108(11):4869. doi:10.1182/blood.V108.11.4869.4869
62. Balleari E, Rossi E, Clavio M, et al. Erythropoietin plus granulocyte colony-stimulating factor is better than erythropoietin alone to treat anemia in low-risk myelodysplastic syndromes: results from a randomized single-centre study. Ann Hematol. 2006;85(3):174–180. doi:10.1007/s00277-005-0044-6
63. Mundle S, Lefebvre P, Vekeman F, Duh MS, Rastogi R, Moyo V. An assessment of erythroid response to epoetin alpha as a single agent versus in combination with granulocyte- or granulocyte-macrophage-colony-stimulating factor in myelodysplastic syndromes using a meta-analysis approach. Cancer. 2009;115(4):706–715. doi:10.1002/cncr.24090
64. Affentranger L, Bohlius J, Hallal M, Bonadies N. Efficacy of granulocyte colony stimulating factor in combination with erythropoiesis stimulating agents for treatment of anemia in patients with lower risk myelodysplastic syndromes: A systematic review. Crit Rev Oncol Hematol. 2019;136:37–47. doi:10.1016/j.critrevonc.2019.01.021
65. Suragani RN, Cadena SM, Cawley SM, et al. Transforming growth factor-β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med. 2014;20(4):408–414. doi:10.1038/nm.3512
66. Suragani RN, Cawley SM, Li R, et al. Modified activin receptor IIB ligand trap mitigates ineffective erythropoiesis and disease complications in murine β-thalassemia. Blood. 2014;123(25):3864–3872. doi:10.1182/blood-2013-06-511238
67. Platzbecker U, Germing U, Gotze KS, et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017;18(10):1338–1347. doi:10.1016/S1470-2045(17)30615-0
68. Fenaux P, Platzbecker U, Mufti GJ, et al. Luspatercept in patients with lower-risk myelodysplastic syndromes. N Engl J Med. 2020;382(2):140–151. doi:10.1056/NEJMoa1908892
69. Administration FD. FDA approves luspatercept-aamt for anemia in adults with MDS; 2020. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-luspatercept-aamt-anemia-adults-mds.
70. Stahl M, Zeidan AM. Lenalidomide use in myelodysplastic syndromes: insights into the biologic mechanisms and clinical applications. Cancer. 2017;123(10):1703–1713.
71. Shallis RM, Zeidan AM. Lenalidomide in non-deletion 5q lower-risk myelodysplastic syndromes: a glass quarter full or three quarters empty? Leuk Lymphoma. 2018;59(9):2015–2017. doi:10.1080/10428194.2018.1430797
72. Stahl M, Zeidan AM. Management of lower-risk myelodysplastic syndromes without del5q: current approach and future trends. Expert Rev Hematol. 2017;10(4):345–364. doi:10.1080/17474086.2017.1297704
73. Bejar R, Levine R, Ebert BL. Unraveling the molecular pathophysiology of myelodysplastic syndromes. J clin oncol. 2011;29(5):504–515. doi:10.1200/JCO.2010.31.1175
74. Pozdnyakova O, Miron PM, Tang G, et al. Cytogenetic abnormalities in a series of 1029 patients with primary myelodysplastic syndromes: a report from the US with a focus on some undefined single chromosomal abnormalities. Cancer. 2008;113(12):3331–3340.
75. Solé E, Sanz, et al. Incidence, characterization and prognostic significance of chromosomal abnormalities in 640 patients with primary myelodysplastic syndromes. Br J Haematol. 2000;108(2):346–356. doi:10.1046/j.1365-2141.2000.01868.x
76. Kelaidi C, Park S, Brechignac S, et al. Treatment of myelodysplastic syndromes with 5q deletion before the lenalidomide era; the GFM experience with EPO and thalidomide. Leuk Res. 2008;32(7):1049–1053. doi:10.1016/j.leukres.2007.11.037
77. Park S, Grabar S, Kelaidi C, et al. Predictive factors of response and survival in myelodysplastic syndrome treated with erythropoietin and G-CSF: the GFM experience. Blood. 2008;111(2):574–582. doi:10.1182/blood-2007-06-096370
78. List A, Dewald G, Bennett J, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355(14):1456–1465. doi:10.1056/NEJMoa061292
79. Fenaux P, Giagounidis A, Selleslag D, et al. A randomized phase 3 study of lenalidomide versus placebo in RBC transfusion-dependent patients with Low-/Intermediate-1-risk myelodysplastic syndromes with del5q. Blood. 2011;118(14):3765–3776. doi:10.1182/blood-2011-01-330126
80. Giagounidis A, Mufti GJ, Mittelman M, et al. Outcomes in RBC transfusion-dependent patients with Low-/Intermediate-1-risk myelodysplastic syndromes with isolated deletion 5q treated with lenalidomide: a subset analysis from the MDS-004 study. Eur J Haematol. 2014;93(5):429–438. doi:10.1111/ejh.12380
81. Schuler E, Giagounidis A, Haase D, et al. Results of a multicenter prospective phase II trial investigating the safety and efficacy of lenalidomide in patients with myelodysplastic syndromes with isolated del (5q) (LE-MON 5). Leukemia. 2016;30(7):1580–1582. doi:10.1038/leu.2015.340
82. Raza A, Reeves JA, Feldman EJ, et al. Phase 2 study of lenalidomide in transfusion-dependent, low-risk, and intermediate-1 risk myelodysplastic syndromes with karyotypes other than deletion 5q. Blood. 2008;111(1):86–93. doi:10.1182/blood-2007-01-068833
83. Toma A, Kosmider O, Chevret S, et al. Lenalidomide with or without erythropoietin in transfusion-dependent erythropoiesis-stimulating agent-refractory lower-risk MDS without 5q deletion. Leukemia. 2016;30(4):897–905. doi:10.1038/leu.2015.296
84. Jädersten M, Saft L, Smith A, et al. TP53 mutations in low-risk myelodysplastic syndromes with del (5q) predict disease progression. J Clin Oncol. 2011;29(15):1971–1979. doi:10.1200/JCO.2010.31.8576
85. Mossner M, Jann JC, Nowak D, et al. Prevalence, clonal dynamics and clinical impact of TP53 mutations in patients with myelodysplastic syndrome with isolated deletion (5q) treated with lenalidomide: results from a prospective multicenter study of the german MDS study group (GMDS). Leukemia. 2016;30(9):1956–1959. doi:10.1038/leu.2016.111
86. Brunner AM, Weng S, Cronin A, et al. Impact of lenalidomide use among non-transfusion dependent patients with myelodysplastic syndromes. Am J Hematol. 2018;93(9):1119–1126. doi:10.1002/ajh.25166
87. Zeidan AM, Al Ali NH, Padron E, Lancet J, List A, Komrokji RS. Lenalidomide treatment for lower risk nondeletion 5q myelodysplastic syndromes patients yields higher response rates when used before azacitidine. Clin Lymphoma Myeloma Leuk. 2015;15(11):705–710. doi:10.1016/j.clml.2015.08.083
88. Zeidan AM, Klink AJ, McGuire M, Feinberg B. Treatment sequence of lenalidomide and hypomethylating agents and the impact on clinical outcomes for patients with myelodysplastic syndromes. Leuk Lymphoma. 2019;60(8):2050–2055. doi:10.1080/10428194.2018.1551538
89. Zeidan AM, Smith BD, Carraway HE, Gojo I, DeZern A, Gore SD. A phase 2 trial of high dose lenalidomide in patients with relapsed/refractory higher-risk myelodysplastic syndromes and acute myeloid leukaemia with trilineage dysplasia. Br J Haematol. 2017;176(2):241–247. doi:10.1111/bjh.14407
90. Passweg JR, Giagounidis AA, Simcock M, et al. Immunosuppressive therapy for patients with myelodysplastic syndrome: a prospective randomized multicenter phase III trial comparing antithymocyte globulin plus cyclosporine with best supportive care–SAKK 33/99. J Clin Oncol. 2011;29(3):303–309.
91. Sloand EM, Wu CO, Greenberg P, Young N, Barrett J. Factors affecting response and survival in patients with myelodysplasia treated with immunosuppressive therapy. J Clin Oncol. 2008;26(15):2505–2511. doi:10.1200/JCO.2007.11.9214
92. Molldrem JJ, Caples M, Mavroudis D, Plante M, Young NS, Barrett AJ. Antithymocyte globulin for patients with myelodysplastic syndrome. Br J Haematol. 1997;99(3):699–705. doi:10.1046/j.1365-2141.1997.4423249.x
93. Stahl M, DeVeaux M, de Witte T, et al. The use of immunosuppressive therapy in MDS: clinical outcomes and their predictors in a large international patient cohort. Blood Advances. 2018;2(14):1765–1772. doi:10.1182/bloodadvances.2018019414
94. Christman JK. 5-Azacytidine and 5-aza-2ʹ-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21(35):5483–5495. doi:10.1038/sj.onc.1205699
95. Thépot S, Ben Abdelali R, Chevret S, et al. A randomized phase II trial of azacitidine ± epoetin-β in lower-risk myelodysplastic syndromes resistant to erythropoietic stimulating agents. Haematologica. 2016;101(8):918–925. doi:10.3324/haematol.2015.140988
96. Jabbour E, Short NJ, Montalban-Bravo G, et al. Randomized phase 2 study of low-dose decitabine vs low-dose azacitidine in lower-risk MDS and MDS/MPN. Blood. 2017;130(13):1514–1522. doi:10.1182/blood-2017-06-788497
97. Zeidan AM, Zhu W, Stahl M, et al. RBC transfusion independence among lower risk MDS patients receiving hypomethylating agents: a population-level analysis. Leuk Lymphoma. 2019;60(13):3181–3187. doi:10.1080/10428194.2019.1622700
98. Carrancio S, Markovics J, Wong P, et al. An activin receptor IIA ligand trap promotes erythropoiesis resulting in a rapid induction of red blood cells and haemoglobin. Br J Haematol. 2014;165(6):870–882. doi:10.1111/bjh.12838
99. Herbertz S, Sawyer JS, Stauber AJ, et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther. 2015;9:4479–4499. doi:10.2147/DDDT.S86621
100. Komrokji R, Garcia-Manero G, Ades L, et al. Sotatercept with long-term extension for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes: a phase 2, dose-ranging trial. Lancet Haematol. 2018;5(2):e63–e72. doi:10.1016/S2352-3026(18)30002-4
101. Santini V, Valcárcel D, Platzbecker U, et al. Phase II study of the ALK5 inhibitor galunisertib in very low-, low-, and intermediate-risk myelodysplastic syndromes. Clin Cancer Res. 2019;25(23):6976–6985. doi:10.1158/1078-0432.CCR-19-1338
102. Feigenson M, Nathan R, Materna C, et al. Ker-050, a novel inhibitor of Tgfβ superfamily signaling, induces red blood cell production by promoting multiple stages of erythroid differentiation. Blood. 2020;136(Supplement 1):34.
103. Garcia-Manero G, Griffiths EA, Steensma DP, et al. Oral cedazuridine/decitabine for MDS and CMML: a phase 2 pharmacokinetic/pharmacodynamic randomized crossover study. Blood. 2020;136(6):674–683. doi:10.1182/blood.2019004143
104. Wei AH, Döhner H, Pocock C, et al. The QUAZAR AML-001 maintenance trial: results of a phase iii international, randomized, double-blind, placebo-controlled study of CC-486 (oral formulation of azacitidine) in patients with Acute Myeloid Leukemia (AML) in first remission. Blood. 2019;134(Supplement_2):
105. Guillermo Garcia-Manero VS, Almeida A, Platzbecker U, et al. Aristoteles Giagounidis. Abstract: A phase III placebo-controlled trial of CC-486 in patients with red blood cell transfusion-dependent (RBC-TD) anemia and thrombocytopenia due to IPSS lower-risk myelodysplastic syndromes (LR-MDS). EHA Library. 2020.
106. Lange K, Holm L, Vang Nielsen K, et al. Telomere shortening and chromosomal instability in myelodysplastic syndromes. Genes Chromosomes Cancer. 2010;49(3):260–269. doi:10.1002/gcc.20737
107. Park HS, Choi J, See CJ, et al. Dysregulation of Telomere Lengths and Telomerase Activity in Myelodysplastic Syndrome. Ann Lab Med. 2017;37(3):195–203. doi:10.3343/alm.2017.37.3.195
108. Röth A, Harley CB, Baerlocher GM. Imetelstat (GRN163L)–telomerase-based cancer therapy. Recent Results Cancer Res. 2010;184:221–234.
109. Gürkan E, Tanriverdi K, Başlamişli F. Telomerase activity in myelodysplastic syndromes. Leuk Res. 2005;29(10):1131–1139. doi:10.1016/j.leukres.2005.03.006
110. Baerlocher GM, Oppliger Leibundgut E, Ottmann OG, et al. Telomerase inhibitor imetelstat in patients with essential thrombocythemia. N Engl J Med. 2015;373(10):920–928. doi:10.1056/NEJMoa1503479
111. Tefferi A, Lasho TL, Begna KH, et al. A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N Engl J Med. 2015;373(10):908–919. doi:10.1056/NEJMoa1310523
112. Fenaux PSD, Van Eygen K, Raza A, et al. Abstract: treatment with imetelstat provides durable transfusion independence (TI) in heavily transfused non-del (5q) lower risk MDS (LR-MDS) relapsed/refractory (R/R) to erythropoiesis stimulating agents (ESAS). EHA Library. 2019.
113. Chen N, Hao C, Peng X, et al. Roxadustat for anemia in patients with kidney disease not receiving dialysis. N Engl J Med. 2019;381(11):1001–1010. doi:10.1056/NEJMoa1813599
114. Yang Y, Yu X, Zhang Y, et al. Hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat (FG-4592) protects against cisplatin-induced acute kidney injury. Clin Sci (Lond). 2018;132(7):825–838. doi:10.1042/CS20171625
115. Besarab A, Provenzano R, Hertel J, et al. Randomized placebo-controlled dose-ranging and pharmacodynamics study of roxadustat (FG-4592) to treat anemia in nondialysis-dependent chronic kidney disease (NDD-CKD) patients. Nephrol Dial Transplant. 2015;30(10):1665–1673. doi:10.1093/ndt/gfv302
116. Pergola PE, Spinowitz BS, Hartman CS, Maroni BJ, Haase VH. Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis-dependent chronic kidney disease. Kidney Int. 2016;90(5):1115–1122. doi:10.1016/j.kint.2016.07.019
117. Ariazi JL, Duffy KJ, Adams DF, et al. Discovery and preclinical characterization of GSK1278863 (daprodustat), a small molecule hypoxia inducible factor-prolyl hydroxylase inhibitor for Anemia. J Pharmacol Exp Ther. 2017;363(3):336–347. doi:10.1124/jpet.117.242503
118. Kansagra KA, Parmar D, Jani RH, et al. Phase I clinical study of ZYAN1, A novel Prolyl-Hydroxylase (PHD) inhibitor to evaluate the safety, tolerability, and pharmacokinetics following oral administration in healthy volunteers. Clin Pharmacokinet. 2018;57(1):87–102. doi:10.1007/s40262-017-0551-3
119. Flamme I, Oehme F, Ellinghaus P, Jeske M, Keldenich J, Thuss U. Mimicking hypoxia to treat anemia: HIF-stabilizer BAY 85-3934 (Molidustat) stimulates erythropoietin production without hypertensive effects. PLoS One. 2014;9(11):e111838. doi:10.1371/journal.pone.0111838
120. Henry DH, Glaspy J, Harrup RA, et al. Roxadustat (FG4592; ASP1517; AZD9941) in the treatment of anemia in patients with Lower Risk Myelodysplastic Syndrome (LR-MDS) and Low Red Blood Cell (RBC) Transfusion Burden (LTB). Blood. 2019;134(Supplement_1):843. doi:10.1182/blood-2019-128714
121. Zeidan AM, Stahl M, Sekeres MA, Steensma DP, Komrokji RS, Gore SD. A call for action: increasing enrollment of untreated patients with higher-risk myelodysplastic syndromes in first-line clinical trials. Cancer. 2017;123(19):3662–3672. doi:10.1002/cncr.30903
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.