")
Back to Journals » Patient Preference and Adherence » Volume 10
Clinical importance of achieving biochemical control with medical therapy in adult patients with acromegaly
Authors Christofides E
Received 11 December 2015
Accepted for publication 24 March 2016
Published 13 July 2016 Volume 2016:10 Pages 1217—1225
DOI https://doi.org/10.2147/PPA.S102302
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Johnny Chen
Elena A Christofides
Endocrinology Associates, Inc., Columbus, OH, USA
Abstract: In acromegaly, achieving biochemical control (growth hormone [GH] level <1.0 ng/mL and age- and sex-normalized levels of insulin-like growth factor 1 [IGF-1]) through timely diagnosis and appropriate treatment provides an opportunity to improve patient outcomes. Diagnosis of acromegaly is challenging because it is rooted in observing subtle clinical manifestations, and it is typical for acromegaly to evolve for up to 10 years before it is recognized. This results in chronic exposure to elevated levels of GH and IGF-1 and delay in patients receiving appropriate treatment, which consequently increases mortality risk. In this review, the clinical impact of elevated GH and IGF-1 levels, the effectiveness of current therapies, and the potential role of novel treatments for acromegaly will be discussed. Clinical burden of acromegaly and benefits associated with management of GH and IGF-1 levels will be reviewed. Major treatment paradigms in acromegaly include surgery, medical therapy, and radiotherapy. With medical therapies, such as somatostatin analogs, dopamine agonists, and GH receptor antagonists, a substantial proportion of patients achieve reduced GH and normalized IGF-1 levels. In addition, signs and symptoms, quality of life, and comorbidities have also been reported to improve to varying degrees in patients who achieve biochemical control. Currently, there are several innovative therapies in development to improve patient outcomes, patient use, and access. Timely biochemical control of acromegaly ensures that the patient can ultimately improve morbidity and mortality from this disease and its extensive consequences.
Keywords: disease burden, growth hormone, insulin-like growth factor 1, medical therapy, pituitary
Introduction
Acromegaly is a rare and chronic disease characterized most often by hypersecretion of growth hormone (GH) from benign somatotrophic adenomas.1 Hypersecretion of GH stimulates excess production of insulin-like growth factor 1 (IGF-1) from the liver and systemic tissues.2,3 Earlier estimates of the acromegaly annual incidence rate of three to four cases per million per year and the prevalence rate of ~70 total cases per million4 were recently reexamined.5 Recent estimates describe a similar overall incidence and prevalence between 2008 and 2012, with an overall incidence at eleven per million person-years and an overall prevalence at 78 cases per million each year. Much higher prevalence of up to 480 cases per million has been reported, although, in that study, 2,270 patients with type 2 diabetes mellitus or glucose intolerance were screened, three were found to have acromegaly, and calculations were made based on the prevalence of type 2 diabetes mellitus and glucose intolerance in the general population and in patients with acromegaly.6 The disease occurs at all ages and is equally distributed between sexes.4 Although there were no differences in incidence or prevalence noted between males and females, increasing age was associated with increased incidence and prevalence.5 Incidence rates were estimated to be between three and eight cases per million person-years in children 0 year to 17 years of age and nine to 18 cases per million person-years in adults >65 years of age. Prevalence rates were estimated to range from 29 to 37 cases per million person-years in children and from 148 to 182 cases per million person-years in adults. The average delay in diagnosing acromegaly appears to be between 4 years and 10 years after the onset of symptoms in adults, as determined from duration of symptoms and alterations in appearance confirmed via photographs.4,5,7
The clinical manifestations of acromegaly result from the pleiotropic effects of increased levels of GH and IGF-1 on many organs, which lead to a multisystem disease associated with various comorbidities, premature mortality, and physical disfigurement.2,3,7,8 The classic signs and symptoms of acromegaly include acral soft-tissue changes, widening teeth spacing, and brow ridge prominence. More commonly consistent patient complaints include excessive perspiration, soft-tissue swelling, large-joint osteoarthritis, and carpal tunnel syndrome.2,7,9 Importantly, patients with acromegaly also develop a significant number of additional comorbidities, which consist of cardiovascular disease (eg, hypertension and cardiomyopathy), insulin resistance, respiratory complications (eg, sleep apnea), and neoplasia (eg, colon polyps).7 Overall, although there have been recent improvements, acromegaly is associated with an increased mortality rate, with GH and IGF-1 levels being important determinants.10,11 Patient age at the time of surgery and treatment with somatostatin analogs (SSAs) is associated with lower mortality. In one study published in 2004, the mean age (± standard deviation [SD]) of patients at initial diagnosis of acromegaly was 42 (±13) years; the mean duration of follow-up (±SD) was 13.4 (±9.9) years; and the mean age (±SD) at death was 61 (±12.8) years.12 The main causes of death were cardiovascular, cerebrovascular, and cancer. Although baseline GH levels were similar between surviving patients and those who had died, surviving patients had significantly lower GH levels at the time of the last follow-up visit (P<0.0001). In general, outcomes have improved with time.13 A recent meta-analysis found that later year of publication was an important efficacy determinant for acromegaly outcomes with treatment.
Currently, there are three treatment options available: surgery, medical therapy, and radiotherapy. The goals of acromegaly treatment include shrinkage or removal of the tumor, safeguarding normal pituitary function, improving symptoms caused by excess GH and IGF-1 levels, including comorbidities and a decrease in mortality risk, and achieving biochemical control.13 According to the most recent acromegaly clinical guidelines from the Endocrine Society (ENDO), biochemical control is defined as achieving a random GH level <1.0 ng/mL and an age-normalized serum IGF-1 level.3 Achieving these recommended biochemical target goals has been shown to correlate with reduced mortality risk and improvement in clinical symptoms and outcomes in patients with acromegaly. This review discusses the clinical evidence and guideline recommendations for current medical therapies used to achieve biochemical control, the importance of biochemical control in patients with acromegaly, and the potential role of investigational medical therapies in the treatment of patients with acromegaly.
Medical therapies for biochemical control
Transsphenoidal surgery is recommended as the primary therapy in most patients with acromegaly.3 Remission had been defined as achieving a normal IGF-1 level and a GH level <1.0 ng/mL during a glucose load (oral glucose tolerance test [OGTT]).14 Advances in the sensitivity of assays led to revising recommendations to achieve normal IGF-1, GH <0.4 ng/mL during glucose load, or a random GH <1.0 ng/mL.14,15 A lack of consensus in the field for particular criteria has led to a lack of consistent remission rate reporting.14–16 What is clear is that although surgical success is reported in many patients, some do not achieve adequate control of disease after surgery; this lack of biochemical control is characterized by persistently elevated GH and IGF-1 levels. Thus, in these patients with persistent or recurrent disease, additional therapeutic intervention such as medical therapy is needed to improve patient outcomes.
Medical therapy is recommended in patients who are poor surgical candidates and in those who did not achieve biochemical remission with surgery.3 Medical therapy has also been considered for use preoperatively to improve surgical outcomes. When using a criterion of GH nadir <1.0 ng/mL during OGTT, pretreatment with medical therapy for 3–6 months increased surgical cure rates from 15% to 18% in the control groups receiving surgery alone and to 38% to 42% in the groups receiving medical treatment and surgery.17–19
Three different medical therapy approaches can be taken to address control of GH and IGF-1, including stimulation of somatostatin receptors (SSA), inhibition of GH receptors (GH receptor antagonist [GHRA]), and stimulation of dopamine receptors (dopamine agonist [DA]; Figure 1).
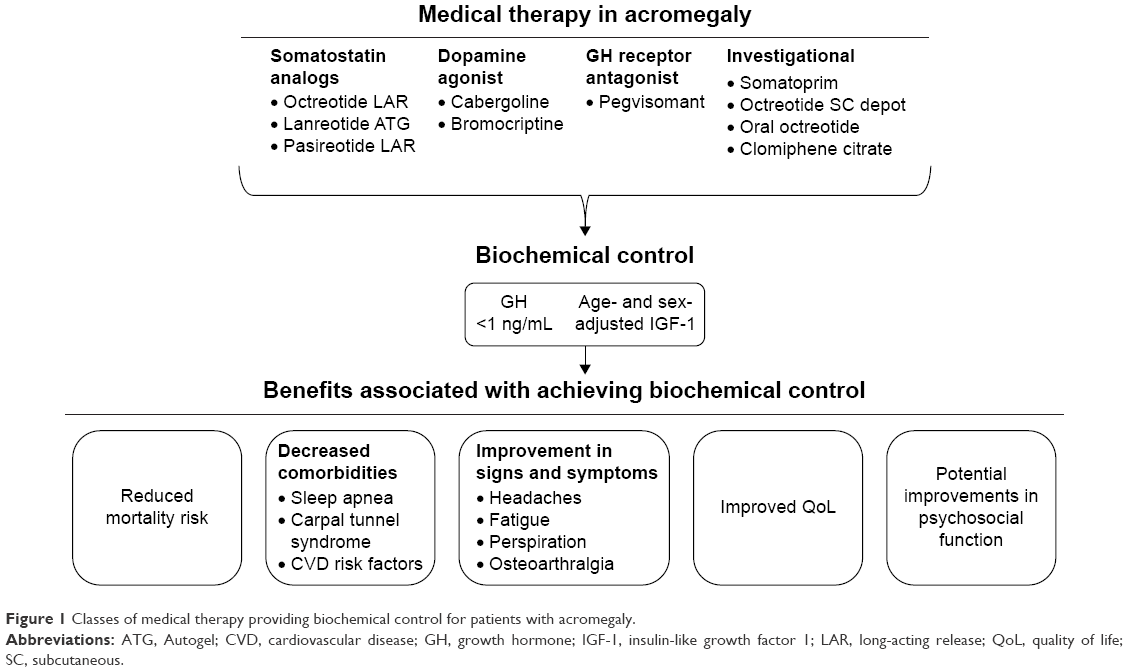 | Figure 1 Classes of medical therapy providing biochemical control for patients with acromegaly. |
Somatostatin analogs
Somatostatin, also known as GH-inhibiting hormone, inhibits the release of GH from the pituitary.20 SSAs, which are synthetic compounds that mimic activity of endogenous somatostatin receptor (sst) ligand, are considered a mainstay of acromegaly therapy.13,20 There are five subtypes of ssts, with sst2 and sst5 being the predominantly expressed ssts in GH-secreting pituitary adenomas.2,7,21 Several SSAs are currently approved for the treatment of acromegaly, including octreotide long-acting release (LAR), octreotide subcutaneous (SC), lanreotide Autogel® (ATG), and pasireotide LAR. Octreotide and lanreotide are considered first-generation SSAs, whereas pasireotide is a next-generation, multireceptor-targeted SSA.3,22
In the ENDO clinical guidelines, medical therapy is recommended in patients with persistent disease following surgery.3 The results of a meta-analysis in acromegaly showed that approximately one-half of patients achieved control of GH and/or IGF-1 levels with either octreotide or lanreotide, with longer treatment duration having an appreciable effect on GH levels.23 In addition to improved GH and/or IGF-1 levels, up to 97% of patients treated with octreotide or lanreotide gained control of tumor growth.23,24 It has also been reported that tumor shrinkage occurred more frequently in patients treated with octreotide LAR as primary therapy than as secondary therapy; however, cautious interpretation is warranted as patients in these two different treatment groups were not matched for baseline tumor size.23
The benefits associated with preoperative SSA therapy remain unclear.25,26 Although several small studies suggested a better surgical outcome in patients with invasive macroadenomas who were pretreated with SSAs, other larger studies have not reported a significant difference.26 Thus, recent ENDO clinical guidelines recommend that preoperative therapy should not be used to improve biochemical control after surgery.3
Pasireotide LAR is a next-generation SSA that has demonstrated efficacy in patients with acromegaly and is indicated for the treatment of patients with acromegaly who have had inadequate response to surgery and/or in whom surgery is not an option.23 Unlike octreotide or lanreotide, which primarily exert their effects through binding to sst2, pasireotide binds with high affinity to sst5.27 In a Phase III study, 31.3% of medically naïve patients with acromegaly achieved biochemical control after 12 months of treatment with pasireotide LAR compared with 19.2% of patients treated with octreotide LAR.28 Tumor volume shrinkage and improvements in symptoms and quality of life (QoL) were similar between treatment groups. In another Phase III study in patients with inadequately controlled acromegaly despite treatment with octreotide LAR or lanreotide ATG, 15% and 20% of patients achieved biochemical control with pasireotide LAR 40 mg and 60 mg, respectively, compared with 0% of patients who continued treatment with octreotide LAR or lanreotide ATG.29 Tumor volume reduction (>25%) was also greater in those treated with pasireotide LAR (40 mg, 18.5%; 60 mg, 10.8%) than in those who continued treatment with the active control (1.5%). In addition, more patients in the pasireotide LAR group had improvements in symptom severity scores than those in the active control group. As demonstrated in both Phase III studies, pasireotide LAR has a safety profile that was similar to that of the other SSAs used, except for a higher frequency of hyperglycemia.28,29
GH receptor antagonists
GHRAs block binding of endogenous GH to its receptor and consequently inhibit the secretion of IGF-1 from the liver.7,30 Because GHRAs inhibit the action of GH but not its secretion, GH concentrations should not be used to evaluate treatment efficacy.7 Therefore, IGF-1 should be used as a surrogate marker to assess treatment outcomes. Pegvisomant is indicated for patients with acromegaly who have had an inadequate response to surgery or radiotherapy or for whom these therapies are not appropriate.31 ENDO guidelines recommend that pegvisomant be used in patients with moderate-to-severe acromegaly who have inadequately responded to SSAs.3 In a 12-week study, a greater number of patients with acromegaly achieved normalized IGF-1 levels with pegvisomant in a dose-dependent manner (10 mg, 54%; 15 mg, 81%; 20 mg, 89%) compared with placebo (10%).30 Furthermore, improved scores for signs and symptoms, including soft-tissue swelling, excessive perspiration, fatigue, and ring size, were also observed with pegvisomant compared with placebo. A long-term study of pegvisomant treatment for 5 years demonstrated that IGF-1 levels were normalized in 63.2% of patients with acromegaly.32 In contrast to the pituitary-targeted SSAs, pegvisomant does not have tumor suppressive effects.22 In fact, tumor growth has been reported in 3% of patients treated with pegvisomant, although whether this is related to the drug or to the natural history of the tumor remains to be further elucidated.32,33 Therefore, GHRAs provide an alternate option for achieving control of IGF-1, especially when control is not achieved with an SSA.
Dopamine agonists
Dopamine is a catecholamine neurotransmitter that binds to dopamine receptors expressed in cells of the anterior pituitary gland.34 Bromocriptine and cabergoline are currently available DAs for the treatment of acromegaly.35 Cabergoline, which is limited to use in patients with acromegaly who have modest elevations in IGF-1 levels and mild signs and symptoms of GH excess,3 has a longer half-life and higher selectivity for D2 receptors than bromocriptine. Although cabergoline as a single-agent therapy has modest efficacy in acromegaly, it can often serve as an effective combination therapy with SSAs for achieving full biochemical normalization.
Monitoring success of therapy
Up to 45% of patients do not achieve biochemical control with SSA monotherapy.13 Reasons for lack of response can include diminished expression or mutation of the ssts, specific genetic mutations, or alterations to intracellular signaling pathways.36 Reduction in GH levels in patients with acromegaly in response to a selective sst2 antagonist was directly correlated with expression of sst2, suggesting that receptor density is directly correlated with response.37 In addition, expression of mutated sst5 was negatively correlated with SSA effect, implying that a loss of functional receptor translates to a loss of response.21,37 Alterations to intracellular signaling pathways downstream of sst2 also affect the ability of the patient to respond to SSA therapy. Patients with genetic-mutated aryl hydrocarbon receptor-interacting protein (AIP) gene are resistant to SSA treatment,38 whereas mutations in IGF-binding protein 3 are not correlated with response to therapy.39 Therefore, given that there are various alterations that may affect SSA-mediated biochemical control, close monitoring of response to medical therapy is critical to ensure appropriate medical management.
Recommendations for monitoring successful biochemical control vary depending on the medical treatment option chosen (Figure 2). Guidelines recommend monitoring serum GH and IGF-1 levels when either an SSA or a cabergoline is used, whereas IGF-1 levels and an MRI are recommended with pegvisomant administration.3 Lack of suppression of GH to <1 ng/mL following an OGTT is recommended for diagnostic confirmation of acromegaly in the presence of equivocal serum IGF-1 levels and following surgery. However, assessing GH suppression after an OGTT in patients with acromegaly who have type 2 diabetes mellitus is not recommended, as the OGTT is unreliable.40 Interpretation of test results is not always straightforward because of biological and assay variability. Levels of IGF-1 have to be interpreted in the context of age. Not only is GH secretion episodic, but there is also considerable variability among GH assays.3 Even within one testing center using the same assay, GH levels can vary, making it a challenge to define a value that clearly defines normal. Therefore, to understand biochemical control, it is important to maintain the same GH assay for the same patient throughout management.
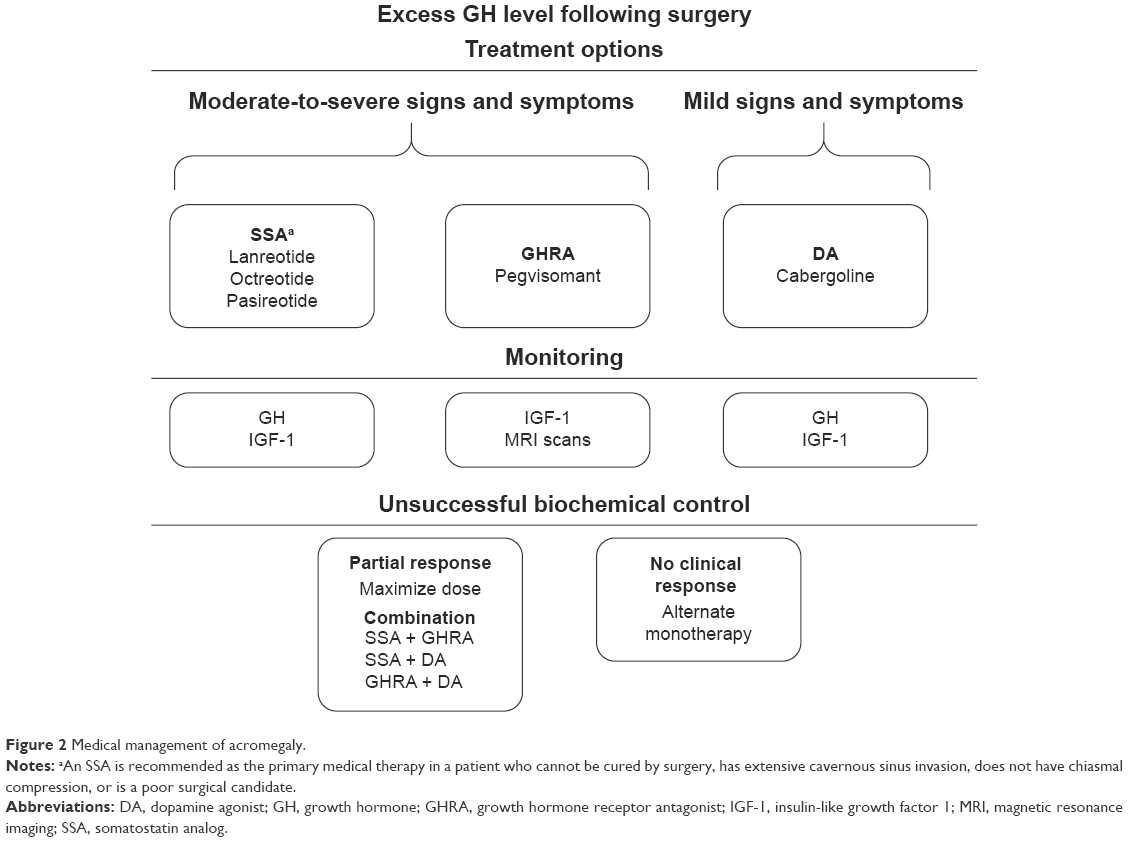 | Figure 2 Medical management of acromegaly. |
Combination therapy
Alternate strategies should be considered if biochemical control is not achieved with monotherapy. Combining different therapies, especially therapies that target different mechanisms of GH and IGF-1 secretion, may be an effective strategy to gain further biochemical control in patients with acromegaly. A number of combination therapies have been studied that yielded efficacious results in those who have not responded adequately to medical monotherapy. Recent clinical guidelines recommend the addition of pegvisomant or cabergoline to SSAs for patients with inadequate responses to SSAs.3 In a clinical study, combining pegvisomant with an SSA was shown to normalize IGF-1 levels in 95% of patients.41 In addition, combination therapy with cabergoline and an SSA provided normalization of IGF-1 levels in up to 50% of patients who had developed partial treatment resistance to prior SSA therapy. Combination of cabergoline with pegvisomant has also been considered.42 A prospective trial of cabergoline with low-dose pegvisomant provided normal IGF-1 levels to 68% of patients with acromegaly. These studies highlight that combination therapy could improve efficacy in cases where SSA monotherapy fails to provide adequate biochemical control in patients with acromegaly.
Surgical debulking
In patients who are not candidates for surgery, such as those with parasellar disease, surgical debulking has been shown to improve medical therapy.43–45 A retrospective study found that, prior to surgery, GH and IGF-1 levels were normalized with SSA therapy in 29% and 46% of patients, respectively.43 Following surgical debulking, normal GH levels were seen in 54% of patients and IGF-1 control was observed in 78% of patients with subsequent SSA retreatment. Another study documented a significant improvement in SSA success rate from 10% to 55% of patients obtaining normal GH and IGF-1 levels (P<0.0001) with surgical debulking.45
Importance of biochemical control
Acromegaly is an insidious and slowly progressing disease. Because of the probable lag in disease presentation and diagnosis, exposure to elevated levels of GH and IGF-1 in patients with acromegaly is likely to persist for years in the absence of timely diagnosis and proper treatment. A quick and effective restoration of GH and IGF-1 levels to normal levels is critical for improving patient outcomes. Biochemical control becomes more imperative in juveniles because excess GH exacerbates active linear growth at open epiphyseal growth plates, which leads to gigantism.46 Thus, timely biochemical control is an important treatment goal in the setting of excess GH levels. As previously discussed, different classes of medical therapy could be used as monotherapy or in combination to reduce GH and normalize IGF-1 levels in patients with persistent acromegaly who have failed to achieve controlled disease with surgery. This leads to improved patient outcomes and clinical burden of disease.
Achieving control of GH and IGF-1 levels in patients with acromegaly is important for health-care providers, as this translates to normalization of mortality rates and improvement in some comorbidities. In a retrospective study, an age-adjusted univariate analysis showed that patients with fasting GH levels >10 ng/mL at diagnosis and with heart disease or other malignancies had significantly increased mortality risk.47 A meta-analysis of mortality studies in acromegaly performed by Holdaway et al10 demonstrated that a serum GH level <2.5 μg/L or normalization of IGF-1 level was associated with improvements in mortality risk that were comparable to the mortality risk in the normal population. Along with decreasing mortality, achieving biochemical control has also been shown to be associated with improvements in a number of comorbidities, including respiratory disorders (eg, sleep apnea), skeletal complications (eg, carpal tunnel syndrome), and cardiovascular comorbidities (eg, hypertension).9 Moreover, it has been reported that patients treated with SSAs who achieve only suboptimal or partial control of disease demonstrated significant improvements in various cardiovascular risk markers (eg, reductions in systolic and diastolic blood pressure, total and low-density lipoprotein cholesterol levels, and triglyceride levels).48 However, it is important to note that not all cardiorespiratory comorbidities are reversible upon achieving safe GH and IGF-1 levels.49 Thus, it is imperative that patients with acromegaly are diagnosed in a timely manner if optimal management and treatment of comorbidities associated with disease are to be implemented. Patients should be comprehensively screened at diagnosis and periodically thereafter for any comorbidities that may arise. According to ENDO clinical guidelines, it is recommended that comorbidities also be longitudinally monitored and rigorously managed.3
Beyond GH and IGF-1 target goals, improvements in signs and symptoms and QoL are important parameters of disease control in patients with acromegaly. The chronic and debilitating nature of acromegaly diminishes the patient’s QoL50–53 and psychosocial function.28,29,54 In fact, it has been reported that patients with acromegaly who achieved long-term biochemical control scored worse on assessments of apathy, irritability, anxiety, and depression than did matched controls.55 However, in Phase III studies of patients with acromegaly who were treated with SSAs, patients demonstrated marked improvements in perspiration, fatigue, osteoarthralgia, paresthesia, and headaches, as well as in scores on the acromegaly QoL questionnaire.28,29 In another Phase III study, patients with acromegaly treated with lanreotide ATG demonstrated clinical benefits of treatment, including early and sustained reductions in tumor volume and improvements in biochemical control and QoL.56 Notably, a study of patients with IGF-1 levels within the age-adjusted normal range at baseline who were treated with SSA monotherapy and pegvisomant showed improved QoL in the absence of significant changes in IGF-1 levels, which suggests that IGF-1 levels may not be sufficient for assessing improvement in QoL.57 In contrast, a study demonstrated that scores on the acromegaly QoL questionnaire did not differ between patients with controlled disease and those with uncontrolled disease, although those with controlled disease had significantly better psychological subscale scores for appearance.53 Overall, these observations suggest that chronic exposure to elevated levels of GH and IGF-1 prior to diagnosis and proper treatment of disease could play an important role in the persistence of psychosocial symptoms, profoundly affecting QoL in patients with acromegaly. Although long-term biochemical control remains a targeted goal, implementation of long-term care in these patients may be needed to improve overall patient outcomes.
Despite significant clinical improvements in GH and IGF-1 levels, signs and symptoms of disease, comorbidities, and QoL, which are provided by medical therapy in patients with acromegaly, there remains a need for new agents to improve outcomes in patients with persistent disease. A number of pipeline therapies are currently being investigated to improve biochemical control in patients with acromegaly.
Investigational medical therapies
Somatoprim
Somatoprim is a novel SSA that binds preferentially to sst2, sst4, and sst5 and is currently under investigation for the treatment of acromegaly.58,59 It has been reported to be more potent than octreotide in reducing GH levels in vitro and in suppressing GH in somatotroph adenomas not responding to octreotide.58 Importantly, somatoprim demonstrated low insulin-suppressing activity, which is a possible unwanted side effect of octreotide and lanreotide. Given its unique receptor-binding profile and minimal effects on insulin release, preliminary data suggest that somatoprim could become a new medical therapy to treat patients with uncontrolled acromegaly.
Octreotide SC depot
A challenge with octreotide LAR is that it requires reconstitution and is administered intramuscularly.60 Octreotide SC depot is currently being developed as a long-acting octreotide for treatment of acromegaly and could address practical issues associated with the LAR formulation. Octreotide SC depot is administered subcutaneously as a low-volume, thin-needle injection and uses a proprietary ready-to-use FluidCrystal® Injection depot (Camurus AB, Lund, Sweden) that allows for controlled release of octreotide over extended periods.60,61 In a Phase I study, it has been reported that octreotide SC depot significantly reduced IGF-1 levels after 1–2 weeks of treatment in patients with acromegaly and was well tolerated.61 Octreotide SC depot also provided greater bioavailability and quicker initial reduction in IGF-1 levels as compared with octreotide LAR. Meanwhile, adverse events were reported at greater rates in patients receiving octreotide SC depot compared with those receiving the LAR formulation. Adverse events were generally well tolerated and were characterized primarily by mild-to-moderate gastrointestinal events. The potential convenience of administration and greater suppression of IGF-1 could allow octreotide SC depot to be a viable therapeutic option with certain practical advantages and could provide greater biochemical control to patients with acromegaly.
Oral octreotide
First-generation SSAs (eg, octreotide and lanreotide) require either intramuscular or deep SC injections that could present challenges associated with discomforting injections.62 Oral octreotide capsules (OOCs) are currently being studied as a switch therapy for patients with acromegaly who achieved biochemical control with injectable SSAs. The capsules are composed of a novel transient permeability enhancer formulation that facilitates intestinal absorption of octreotide. In a healthy volunteers study, a single dose of OOC 20 mg decreased basal and GH-releasing hormone-induced GH levels. In a recently completed Phase III study, 13 months of treatment with twice-daily OOC led to decreased GH and IGF-1 levels in 62% of patients with acromegaly compared with 89% of those receiving injectable SSAs at baseline. Most adverse events were mild to moderate in intensity and included gastrointestinal, neurological, and musculoskeletal events, which was consistent with the known safety profile of octreotide. Therefore, OOC may offer advantages over other SSA therapies, specifically injectable agents, including convenience associated with ease of administration, no pain or reactions associated with injections, and reduced number of monthly visits and dependence on health-care workers and/or family members for injections.
Clomiphene citrate
Clomiphene citrate (CC) is an oral selective estrogen receptor modulator that increases secretion of luteinizing hormone and follicle-stimulating hormone, which improves hypogonadism and fertility outcomes.63 Estrogens play a role in reducing IGF-1 in GH-deficient patients under GH replacement. In addition, it has been demonstrated that female patients with acromegaly who have mild elevations in IGF-1 levels can benefit from estrogens, either alone or in combination with SSAs. In a prospective, single-center study, CC was evaluated in male patients with acromegaly (n=16), with particular focus on those with low testosterone levels.63 After 3 months of treatment, CC decreased IGF-1 levels by 41% and provided normal IGF-1 levels to 44% of patients. Additionally, mean total testosterone levels increased by 209% in ten patients treated with CC, and normal testosterone levels were achieved in 67% of patients considered to be hypogonadal. No major side effects were reported. CC could be considered as a therapeutic option in patients with acromegaly who are not controlled by the currently available options.
Conclusion
Long-term biochemical control remains a goal in addressing acromegaly in the hopes of being able to reverse or stall the progression of disease (Figure 1). Decreased GH and IGF-1 levels can be accomplished through a number of different methods and mechanisms and have been associated with improved clinical outcomes. Identification of new therapeutic options will supplement the current treatment arsenal to facilitate multifaceted control of abnormalities associated with biochemical dysregulation in acromegaly. While long-term cure remains a current focus with clinicians, increased long-term care of patients will be needed to ensure that comorbidities, QoL, signs and symptoms, and psychosocial functions are properly managed.
Acknowledgments
Financial support for development of this paper was provided by Novartis Pharmaceuticals Corporation. Assistance with medical writing and editing was provided under the direction of the author by Andrea Eckhart and Meredith MacPherson (MedThink SciCom, Inc), with support from Novartis Pharmaceuticals Corporation.
Disclosure
EAC has received research funding from Pfizer. The author reports no other conflicts of interest in this work.
References
Ben-Shlomo A, Melmed S. Acromegaly. Endocrinol Metab Clin North Am. 2008;37(1):101–122, viii. | ||
Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest. 2009;119(11):3189–3202. | ||
Katznelson L, Laws ER Jr, Melmed S, et al; Endocrine Society. Acromegaly: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(11):3933–3951. | ||
Holdaway IM, Rajasoorya C. Epidemiology of acromegaly. Pituitary. 1999;2(1):29–41. | ||
Burton T, Le Nestour E, Neary M, Ludlam WH. Incidence and prevalence of acromegaly in a large US health plan database. Pituitary. Epub 2016 Jan 20. | ||
Rosario PW. Frequency of acromegaly in adults with diabetes or glucose intolerance and estimated prevalence in the general population. Pituitary. 2011;14(3):217–221. | ||
Chanson P, Salenave S. Acromegaly. Orphanet J Rare Dis. 2008;3:17. | ||
Anagnostis P, Efstathiadou ZA, Polyzos SA, et al. Acromegaly: presentation, morbidity and treatment outcomes at a single centre. Int J Clin Pract. 2011;65(8):896–902. | ||
Burton T, Le Nestour E, Bancroft T, Neary M. Real-world comorbidities and treatment patterns of patients with acromegaly in two large US health plan databases. Pituitary. 2013;16(3):354–362. | ||
Holdaway IM, Bolland MJ, Gamble GD. A meta-analysis of the effect of lowering serum levels of GH and IGF-I on mortality in acromegaly. Eur J Endocrinol. 2008;159(2):89–95. | ||
Sughrue ME, Chang EF, Gabriel RA, Aghi MK, Blevins LS. Excess mortality for patients with residual disease following resection of pituitary adenomas. Pituitary. 2011;14(3):276–283. | ||
Holdaway IM, Rajasoorya RC, Gamble GD. Factors influencing mortality in acromegaly. J Clin Endocrinol Metab. 2004;89(2):667–674. | ||
Carmichael JD, Bonert VS, Nuño M, Ly D, Melmed S. Acromegaly clinical trial methodology impact on reported biochemical efficacy rates of somatostatin receptor ligand treatments: a meta-analysis. J Clin Endocrinol Metab. 2014;99(5):1825–1833. | ||
Kreutzer J, Vance ML, Lopes MB, Laws ER Jr. Surgical management of GH-secreting pituitary adenomas: an outcome study using modern remission criteria. J Clin Endocrinol Metab. 2001;86(9):4072–4077. | ||
Starke RM, Raper DM, Payne SC, Vance ML, Oldfield EH, Jane JA Jr. Endoscopic vs microsurgical transsphenoidal surgery for acromegaly: outcomes in a concurrent series of patients using modern criteria for remission. J Clin Endocrinol Metab. 2013;98(8):3190–3198. | ||
Marquez Y, Tuchman A, Zada G. Surgery and radiosurgery for acromegaly: a review of indications, operative techniques, outcomes, and complications. Int J Endocrinol. 2012;2012:386401. | ||
Carlsen SM, Lund-Johansen M, Schreiner T, et al; Preoperative Octreotide Treatment of Acromegaly study group. Preoperative octreotide treatment in newly diagnosed acromegalic patients with macroadenomas increases cure short-term postoperative rates: a prospective, randomized trial. J Clin Endocrinol Metab. 2008;93(8):2984–2990. | ||
Shen M, Shou X, Wang Y, et al. Effect of presurgical long-acting octreotide treatment in acromegaly patients with invasive pituitary macroadenomas: a prospective randomized study. Endocr J. 2010;57(12):1035–1044. | ||
Mao ZG, Zhu YH, Tang HL, et al. Preoperative lanreotide treatment in acromegalic patients with macroadenomas increases short-term postoperative cure rates: a prospective, randomised trial. Eur J Endocrinol. 2010;162(4):661–666. | ||
Murray RD, Kim K, Ren SG, Chelly M, Umehara Y, Melmed S. Central and peripheral actions of somatostatin on the growth hormone-IGF-I axis. J Clin Invest. 2004;114(3):349–356. | ||
Marina D, Burman P, Klose M, et al. Truncated somatostatin receptor 5 may modulate therapy response to somatostatin analogues-observations in two patients with acromegaly and severe headache. Growth Horm IGF Res. 2015;25(5):262–267. | ||
Colao A, Auriemma RS, Pivonello R. The effects of somatostatin analogue therapy on pituitary tumor volume in patients with acromegaly. Pituitary. 2016;19(2):210–221. | ||
Freda PU, Katznelson L, van der Lely AJ, Reyes CM, Zhao S, Rabinowitz D. Long-acting somatostatin analog therapy of acromegaly: a meta-analysis. J Clin Endocrinol Metab. 2005;90(8):4465–4473. | ||
Bevan JS. Clinical review: the antitumoral effects of somatostatin analog therapy in acromegaly. J Clin Endocrinol Metab. 2005;90(3):1856–1863. | ||
Giustina A, Chanson P, Kleinberg D, et al; Acromegaly Consensus Group. Expert consensus document: a consensus on the medical treatment of acromegaly. Nat Rev Endocrinol. 2014;10(4):243–248. | ||
Losa M, Mortini P, Urbaz L, Ribotto P, Castrignanó T, Giovanelli M. Presurgical treatment with somatostatin analogs in patients with acromegaly: effects on the remission and complication rates. J Neurosurg. 2006;104(6):899–906. | ||
Bruns C, Lewis I, Briner U, Meno-Tetang G, Weckbecker G. SOM230: a novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur J Endocrinol. 2002;146(5):707–716. | ||
Colao A, Bronstein MD, Freda P, et al; Pasireotide C2305 Study Group. Pasireotide versus octreotide in acromegaly: a head-to-head superiority study. J Clin Endocrinol Metab. 2014;99(3):791–799. | ||
Gadelha MR, Bronstein MD, Brue T, et al; Pasireotide C2402 Study Group. Pasireotide versus continued treatment with octreotide or lanreotide in patients with inadequately controlled acromegaly (PAOLA): a randomised, phase 3 trial. Lancet Diabetes Endocrinol. 2014;2(11):875–884. | ||
Trainer PJ, Drake WM, Katznelson L, et al. Treatment of acromegaly with the growth hormone-receptor antagonist pegvisomant. N Engl J Med. 2000;342(16):1171–1177. | ||
Somavert [package insert]. New York, NY: Pfizer; 2016. | ||
van der Lely AJ, Biller BM, Brue T, et al. Long-term safety of pegvisomant in patients with acromegaly: comprehensive review of 1288 subjects in ACROSTUDY. J Clin Endocrinol Metab. 2012;97(5):1589–1597. | ||
Hodish I, Barkan A. Long-term effects of pegvisomant in patients with acromegaly. Nat Clin Pract Endocrinol Metab. 2008;4(6):324–332. | ||
Hofland LJ, Feelders RA, de Herder WW, Lamberts SW. Pituitary tumours: the sst/D2 receptors as molecular targets. Mol Cell Endocrinol. 2010;326(1–2):89–98. | ||
Abs R, Verhelst J, Maiter D, et al. Cabergoline in the treatment of acromegaly: a study in 64 patients. J Clin Endocrinol Metab. 1998;83(2):374–378. | ||
Gadelha MR, Kasuki L, Korbonits M. Novel pathway for somatostatin analogs in patients with acromegaly. Trends Endocrinol Metab. 2013;24(5):238–246. | ||
Duran-Prado M, Saveanu A, Luque RM, et al. A potential inhibitory role for the new truncated variant of somatostatin receptor 5, sst5TMD4, in pituitary adenomas poorly responsive to somatostatin analogs. J Clin Endocrinol Metab. 2010;95(5):2497–2502. | ||
Oriola J, Lucas T, Halperin I, et al. Germline mutations of AIP gene in somatotropinomas resistant to somatostatin analogues. Eur J Endocrinol. 2012;168(1):9–13. | ||
Jallad RS, Trarbach EB, Duarte FH, Jorge AA, Bronstein MD. Influence of growth hormone receptor (GHR) exon 3 and -202A/C IGFBP-3 genetic polymorphisms on clinical and biochemical features and therapeutic outcome of patients with acromegaly. Pituitary. 2015;18(5):666–673. | ||
Freda PU. Monitoring of acromegaly: what should be performed when GH and IGF-1 levels are discrepant? Clin Endocrinol (Oxf). 2009;71(2):166–170. | ||
Feenstra J, de Herder WW, ten Have SM, et al. Combined therapy with somatostatin analogues and weekly pegvisomant in active acromegaly. Lancet. 2005;365(9471):1644–1646. | ||
Higham CE, Atkinson AB, Aylwin S, et al. Effective combination treatment with cabergoline and low-dose pegvisomant in active acromegaly: a prospective clinical trial. J Clin Endocrinol Metab. 2012;97(4):1187–1193. | ||
Petrossians P, Borges-Martins L, Espinoza C, et al. Gross total resection or debulking of pituitary adenomas improves hormonal control of acromegaly by somatostatin analogs. Eur J Endocrinol. 2005;152(1):61–66. | ||
Karavitaki N, Turner HE, Adams CB, et al. Surgical debulking of pituitary macroadenomas causing acromegaly improves control by lanreotide. Clin Endocrinol (Oxf). 2008;68(6):970–975. | ||
Colao A, Attanasio R, Pivonello R, et al. Partial surgical removal of growth hormone-secreting pituitary tumors enhances the response to somatostatin analogs in acromegaly. J Clin Endocrinol Metab. 2006;91(1):85–92. | ||
Eugster EA, Pescovitz OH. Gigantism. J Clin Endocrinol Metab. 1999;84(12):4379–4384. | ||
Mercado M, Gonzalez B, Vargas G, et al. Successful mortality reduction and control of comorbidities in patients with acromegaly followed at a highly specialized multidisciplinary clinic. J Clin Endocrinol Metab. 2014;99(12):4438–4446. | ||
Delaroudis SP, Efstathiadou ZA, Koukoulis GN, et al. Amelioration of cardiovascular risk factors with partial biochemical control of acromegaly. Clin Endocrinol (Oxf). 2008;69(2):279–284. | ||
Gurnell M, Powlson AS. Cardiovascular disease and sleep disordered breathing in acromegaly. Neuroendocrinology. 2016;103(1):75–85. | ||
Kepicoglu H, Hatipoglu E, Bulut I, Darici E, Hizli N, Kadioglu P. Impact of treatment satisfaction on quality of life of patients with acromegaly. Pituitary. 2014;17(6):557–563. | ||
Mangupli R, Camperos P, Webb SM. Biochemical and quality of life responses to octreotide-LAR in acromegaly. Pituitary. 2014;17(6):495–499. | ||
Paisley AN, Rowles SV, Roberts ME, et al. Treatment of acromegaly improves quality of life, measured by AcroQol. Clin Endocrinol (Oxf). 2007;67(3):358–362. | ||
Matta MP, Couture E, Cazals L, Vezzosi D, Bennet A, Caron P. Impaired quality of life of patients with acromegaly: control of GH/IGF-I excess improves psychological subscale appearance. Eur J Endocrinol. 2008;158(3):305–310. | ||
Leon-Carrion J, Martin-Rodriguez JF, Madrazo-Atutxa A, et al. Evidence of cognitive and neurophysiological impairment in patients with untreated naive acromegaly. J Clin Endocrinol Metab. 2010;95(9):4367–4379. | ||
Tiemensma J, Biermasz NR, van der Mast RC, et al. Increased psychopathology and maladaptive personality traits, but normal cognitive functioning, in patients after long-term cure of acromegaly. J Clin Endocrinol Metab. 2010;95(12):E392–E402. | ||
Caron PJ, Bevan JS, Petersenn S, et al; PRIMARYS Investigators. Tumor shrinkage with lanreotide autogel 120 mg as primary therapy in acromegaly: results of a prospective multicenter clinical trial. J Clin Endocrinol Metab. 2014;99(4):1282–1290. | ||
Neggers SJ, van Aken MO, de Herder WW, et al. Quality of life in acromegalic patients during long-term somatostatin analog treatment with and without pegvisomant. J Clin Endocrinol Metab. 2008;93(10):3853–3859. | ||
Plöckinger U. Medical therapy of acromegaly. Int J Endocrinol. 2012;2012:268957. | ||
Fleseriu M. Advances in the pharmacotherapy of patients with acromegaly. Discov Med. 2014;17(96):329–338. | ||
Roberts J, Linden M, Cervin C, Tiberg F. Octreotide fluid crystal provides sustained octreotide bioavailability and similar IGF1 suppression to that of octreotide LAR (Sandostatin LAR): randomized, open-label, phase 1, repeat-dose study in healthy volunteers [abstract P914]. Endocr Abstr. 2014;35:329. | ||
Tiberg F, Roberts J, Cervin C, et al. Octreotide s.c. depot provides sustained octreotide bioavailability and similar IGF-1 suppression to octreotide LAR in healthy volunteers. Br J Clin Pharmacol. 2015;80(3):460–472. | ||
Melmed S, Popovic V, Bidlingmaier M, et al. Safety and efficacy of oral octreotide in acromegaly: results of a multicenter phase III trial. J Clin Endocrinol Metab. 2015;100(4):1699–1708. | ||
Duarte FH, Jallad RS, Bronstein MD. Clomiphene citrate for treatment of acromegaly not controlled by conventional therapies. J Clin Endocrinol Metab. 2015;100(5):1863–1869. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.