")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
Clinical feature and outcome of late-onset cobalamin C disease patients with neuropsychiatric presentations: a Chinese case series
Authors Wang S, Yan C, Wen B, Zhao Y
Received 4 December 2018
Accepted for publication 23 January 2019
Published 21 February 2019 Volume 2019:15 Pages 549—555
DOI https://doi.org/10.2147/NDT.S196924
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yuping Ning
Sheng-jun Wang, Chuan-zhu Yan, Bing Wen, Yu-ying Zhao
Department of Neurology, Qilu Hospital, Shandong University, Jinan, China
Objective: The Cobalamin C (cblC) disease is an inborn error of cobalamin metabolism. Late-onset cblC disease was diagnosed in patients having overt symptoms after 4 years of age. The late-onset cblC disease patients were rare and easily misdiagnosed. This study analyzed the clinical presentations, gene mutations, and treatments of Chinese patients with late-onset cblC disease.
Methods: The clinical data of 26 Han Chinese patients diagnosed with late-onset cblC disease were retrospectively analyzed. All patients underwent serum homocysteine level exam, urine concentrations of organic acids measurement, neuroimaging scans, gene analysis, and treatments evaluations.
Results: The mean age at disease onset and diagnosis was 17.8±7.0 years. The most frequent neuropsychiatric disturbances were lower limb weakness (50%), psychiatric disturbances (46.2%), and gait instability (42.3%). The mean methylmalonic acid level in urine was 107.4±56.6 µmol/L, and mean serum total homocysteine was 105.4±41.0 µmol/L. The most common abnormal radioimaging changes were observed in the spinal cord (88%) and brain (32%). Scoliosis was detected in 85.7% of patients. The methylmalonic aciduria and homocystinuria type C protein gene analysis showed that c.482G>A (57.7%) and c.609G>A (34.6%) mutations were the most frequent genotypes. After treatments with hydroxycobalamin, betaine, folic acid, L-carnitine, and compound vitamin B, the clinical features and biochemical parameters of patients with late-onset cblC disease were found to be alleviated.
Conclusion: In our late-onset cblC disease cases, lower limb weakness, psychiatric disturbances, and gait instability were the most frequent manifestations. Patients responded well to the drug treatments with hydrocobalamin and betaine. When juvenile or adult patients with hyperhomocysteinemia present with neurological symptoms, cblC disease needs to be considered.
Keywords: methylmalonic aciduria, homocysteine, late-onset, neuropsychiatric, cobalamin
Introduction
The Cobalamin C (cblC) disease is an inborn error of cobalamin metabolism caused by mutations in the methylmalonic aciduria and homocystinuria type C protein (MMACHC) gene.1,2 The defective MMACHC protein causes decreased levels of the adenosylcobalamin (AdoCbl) and methylcobalamin (MeCbl), cofactors for the enzymes methylmalonyl-CoA mutase and methionine synthase.1,2 The deficient activity of these enzymes causes elevation of methylmalonic acid (MMA) and homocysteine, and decreases production of methionine.1,2 The main biochemical markers for the disease are elevated total plasma homocysteine (Hcy) and MMA in plasma and urine. The onset age of most cblC disease patients is in the infancy period, while for few patients, it is in the adolescent or adult period, named late-onset cblC disease.1 The late-onset cblC disease commonly presenting with atypical clinical symptoms without family history is often misdiagnosed. The diagnosis of cblC disease is challenging because the clinical manifestations and age of presentation are highly variable. In the following, we retrospectively analyzed a large Chinese case series of late-onset cblC disease patients with neuropsychiatric presentations to raise awareness for this rare but treatable disease.
Methods
This study included 26 Han Chinese late-onset cblC disease patients with neuropsychiatric manifestations. This study was in compliance with the ethical principles of the Declaration of Helsinki, and was approved by the ethics committee of Qilu Hospital of Shandong University. All patients or their parents (for patients under the age of 18 years) had signed informed consent forms prior to inclusion in the study.
The plasma amino acid levels, including propionylcarnitine (C3)/cetylcarnitine (C2), were detected by tandem mass spectrometry, and urine concentrations of organic acids were measured by gas chromatography mass spectrometry in all patients’ samples (MILS International, Japan). All patients underwent urinalysis and blood tests, including electrolytes, glucose, liver and kidney function, blood coagulation, and total homocysteine concentrations. Most patients underwent brain and spinal cord MRI scans with a 1.5T MR system (GE Healthcare, Pittsburgh, PA, USA). The mutations in the MMACHC gene using PCR and DNA sequencing were detected in all patients’ blood samples (Kingmed Diagnostics, Guangzhou, China).
All patients received drug treatments of cyanocobalamin (CNCbl) (intramuscular injection; 0.5–1 mg/day) and/or hydrocobalamin (OHCbl) (intramuscular injection; 0.5–1 mg/day), and betaine (oral; 1–3 g/day). Some patients also received MeCbl (intravenous/intramuscular injection; 0.5–1 mg/day), folic acid (oral; 10–30 mg/day), L-carnitine (intravenous infusion; 1–3 g/day), and compound vitamin B (20–60 mg/day). When the disease was stable (4–8 weeks after treatments), all patients still received OHCbl (intramuscular injection; 0.5–1 mg/week) and betaine (oral administration; 1–3 g/day). Some patients also took oral administration of folic acid (10–30 mg/day) and compound vitamin B (20–60 mg/day). All patients were followed up for 0.5–5 years after treatments.
Results
There were 14 female and 12 male patients. The mean age at disease onset and diagnosis was 17.8±7.0 years (range =4–38 years) and 20.5±7.0 years (range =5–39 years), respectively. The mean delayed time from initial symptoms to diagnosis was 32.1±39.9 months (ranged from 0.5 months to 11 years). Lower extremity weakness (13/26) was the most frequent neuropsychiatric manifestation, followed by psychiatric disturbances (12/26), gait instability (spastic gait or ataxia gait) (11/26), cognitive impairment (7/26), and seizures (7/26). Moreover, two patients had nephropathy, and one patient had pulmonary embolism (Table 1).
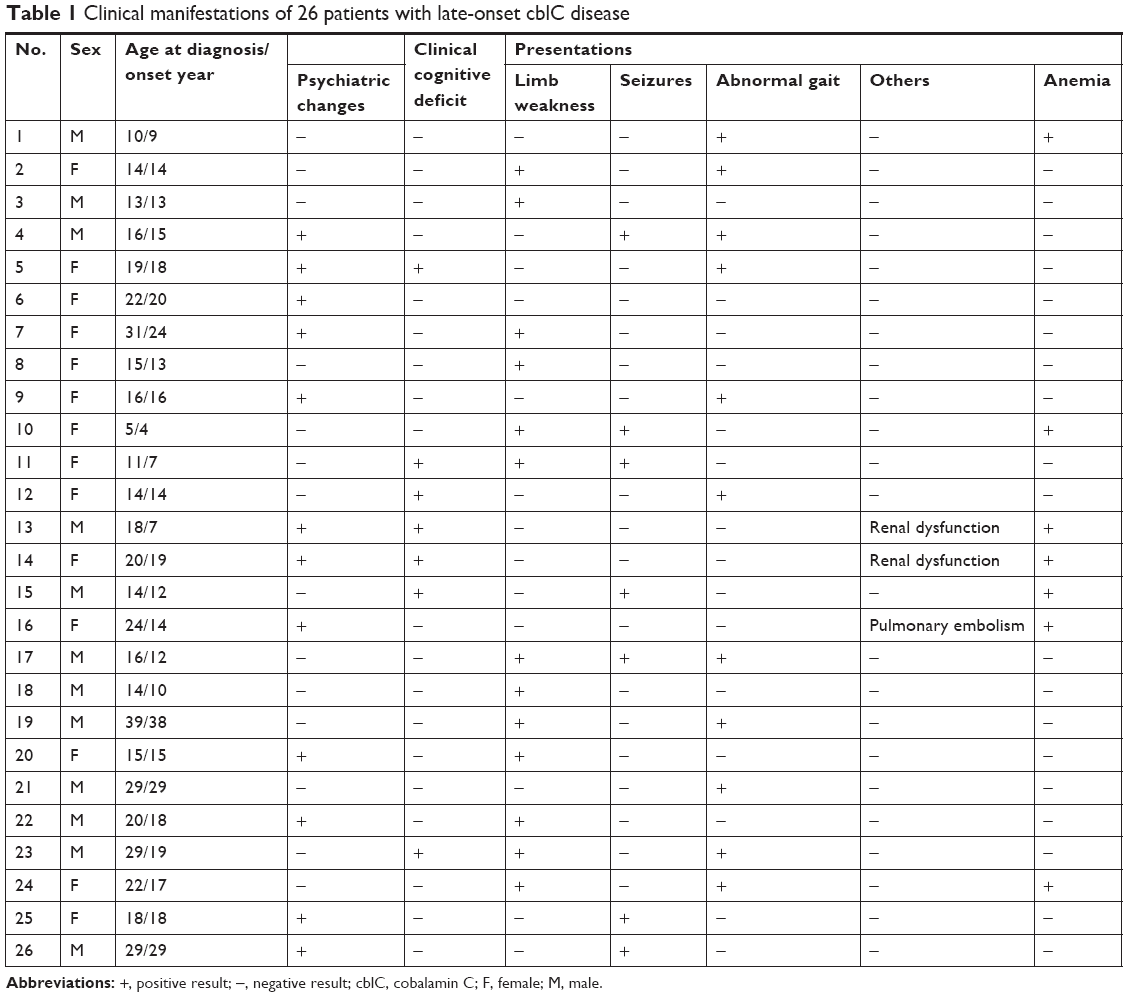 | Table 1 Clinical manifestations of 26 patients with late-onset cblC disease |
Anemia was detected in seven patients (7/26). The mean methylmalonic acid level in urine was 107.4±56.6 μmol/L (range =31.0–236.5 μmol/L; reference range =0.2–5.6 μmol/L) and mean serum total homocysteine was 105.4±41.0 μmol/L (range =61.4–237.3 μmol/L; reference range =0–15 μmol/L). The mean propionylcarnitine (C3)/acetylcarnitine (C2) was 0.61±0.18 (range =0.34–1.03; reference<0.30). The folate and vitamin B12 serum levels of all patients were normal. Eighteen patients had electromyographic (EMG) exams by which peripheral nerve damage, including decreased conduction velocity, was found in 15 patients (15/18) (Table 2). Twenty-five patients had brain MRI scans and 17 patients had spinal cord MRI scans. The neuroimaging exams showed spinal cord atrophy/myelin lesions (11/17), longitudinally extensive transverse myelitis (4/17), cerebral cortical atrophy/white matter lesions (8/25), cerebellum atrophy/lesions (3/25), basal ganglia lesions (1/25), reversible posterior leukoencephalopathy syndrome (RPLS) (1/25), and hydrocephalus (1/25) (Table 3). Twenty-one patients had X-ray spine exam, and 18 patients showed scoliosis (18/21) (Table 2).
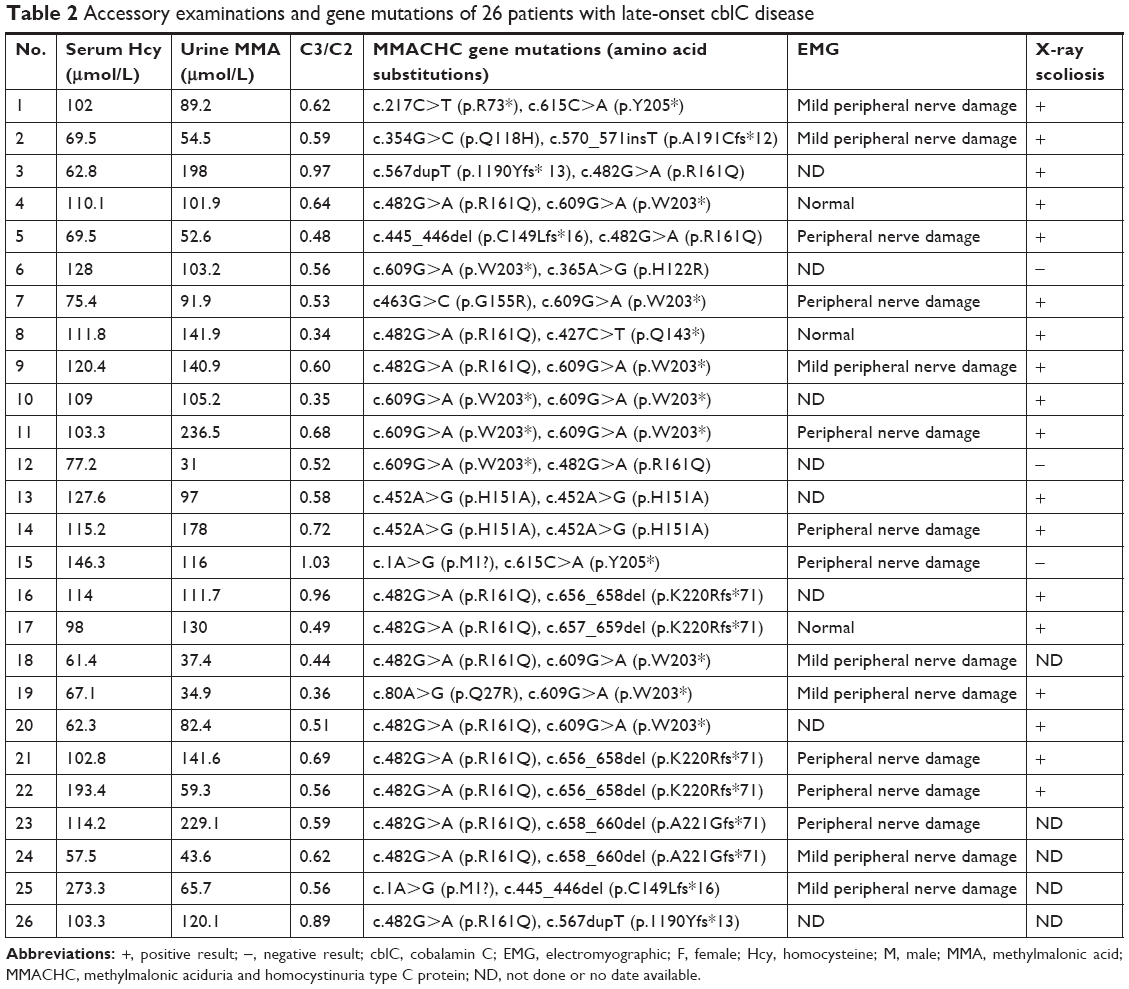 | Table 2 Accessory examinations and gene mutations of 26 patients with late-onset cblC disease |
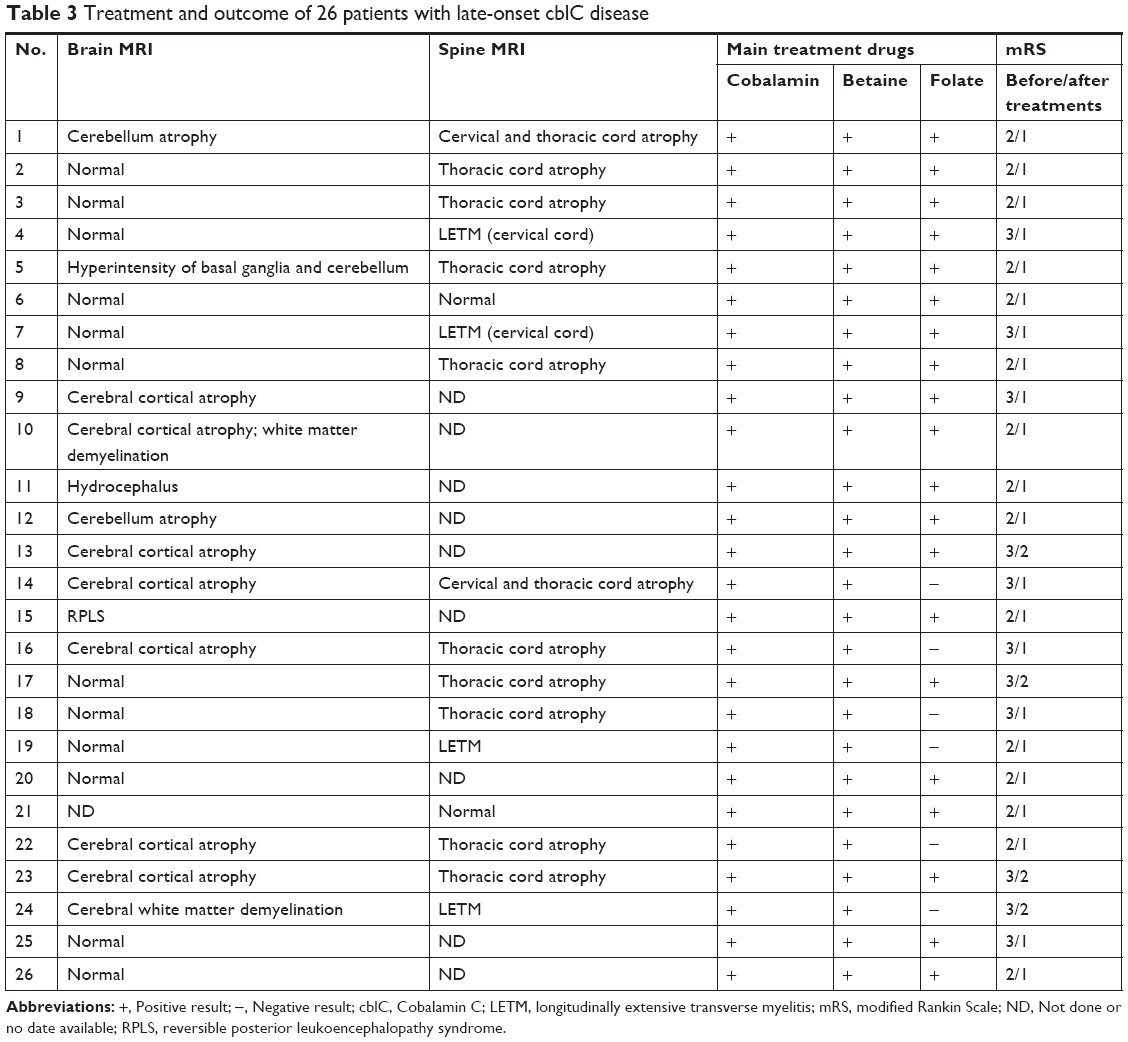 | Table 3 Treatment and outcome of 26 patients with late-onset cblC disease |
All 26 patients had MMACHC gene analysis. It showed homozygous mutations in two patients with c.609G>A (4/52) and two patients with c.452A>G (4/52). The other 22 patients showed the following heterozygous mutations: c.482G>A (15/52), c.609G>A (7/52), c.656_658del (5/52), c.658_660del (5/52), c.445_446delTG (2/52), c.567dupT (2/52), c.1A>G (2/52), c.463G>C (1/52), c.217C>T (1/52), c.365A>G (1/52), c.615C>A (1/52) c.80A>G (1/52), c.354G>C (1/52), c.657_659del (1/52), c.570_571insT (1/52), and c.427C>T (1/52) (Table 2).
After 2–4 weeks treatments, all patients showed alleviation of neurological manifestations, including psychiatric disturbances, lower extremity weakness, and gait instability. The patients’ neurological function evaluated with modified Rankin Scale (mRS) showed a median score of 2.42±0.50 (range =2–3) before treatments and 1.15±0.36 (range =1–2) after treatments. During the follow-up examinations, the neuropsychiatric presentations of all the patients were found to be alleviated with sustained drug therapy (Table 3).
Discussion
Five decades ago, Mudd et al3 first described the disorder involving vitamin metabolism abnormality due to defective synthesis of the active cobalamin. The combined MMA and homocysteinemia are associated with genetic defects of intracellular cobalamin metabolism (cblC, cblD, cblF, cblJ, and cblX).4 cblC disease is the most common type of cobalamin metabolic defect with the gene encoding for the MMACHC located at 1p34.1.2 The protein encoded by the MMACHC gene was identified as the cause of cblC disease in 2006.2 The MMACHC protein acts as a “trafficking chaperone” for cobalamin to accept its derivatives for passage into the cytoplasm.5 The estimated incidence of MMA ranges from 1:48,000 to 1:250,000 worldwide.6 The incidence of cblC was approximately 1:100,000 in New York State, USA.7 The incidence of MMA disease was recently reported to be approximately 1:3,920 live births in Jinan and 1:3,220 in Jining, Shandong province, China.8,9
cblC disease is a multisystem disease, including metabolic instability, pancreatitis, renal failure, intellectual impairment, optic nerve atrophy, spinal cord, and basal ganglia injury.1 Depending on the age of disease onset, cblC disease patients are divided into early-onset type and late-onset type. In early-onset patients, the disease develops in infancy during the first year after birth. Approximately 90% of reported patients with cblC disease present with the severe, infantile, early-onset form of the disease.1 Patients with late-onset cblC disease, first manifesting in adolescence or adulthood, are less common. “Late-onset” cblC disease was first reported in the 1980s.10 The term has been historically applied for patients that have overt symptoms after 1 year or 4 years of age.1,3,4,11 The age at disease onset of late-onset cblC disease reported in the literature ranged from 1.3 to 44 years.11 In this study, the disease onset age ranged from 4 to 38 years. To date, no more than 100 cases of late-onset cblC have been reported, which shows a significant variation of clinical symptoms.11,12 It is supposed that the wide clinical heterogeneity of cblC disease should depend on the nature of different MMACHC mutations and polymorphisms of other genes associated with cobalamin metabolism.1 The late-onset cblC disease may present with acute or insidious onset and show sudden deterioration as well as long stable phases.1,11 The clinical manifestations of late-onset patients are very heterogeneous and may be misdiagnosed. In this study, the mean time delay from initial symptoms to diagnosis was 32.1 months. Late-onset cblC patients often present with neurological regression, neuropsychiatric symptoms, subacute combined degeneration of the spinal cord, hematological manifestations, and thromboembolic complications. In this study, lower extremity weakness, gait instability, and psychiatric disturbances were the main clinical presentations of Chinese late-onset cblC patients. In the literature, cognitive impairment was the most frequent symptom of late-onset cblC disease, followed by myelopathy, ataxia, and seizures.11 It is suggested that, in older children/adolescents, psychiatric symptoms, ataxia and cognitive decline were most frequent; while, in adults, cognitive decline, ataxia, thromboembolic events, and neuropathy/myelopathy were usually seen.11 In our study, nephropathy was detected in two patients, and pulmonary embolism was also observed in one patient. We found about 80% patients suffered peripheral nerve damage detected by EMG exams. The radiologic features of cblC disease were usually nonspecific. The cerebral white matter involvement and basal ganglia lesions, especially infarcts of globus pallidus, were the most common findings in early-onset patients.13 On the other hand, diffuse cerebrum/spinal cord atrophy and bilateral hyperintensity in the deep white matter/cerebellum on the MRI imaging were frequently reported in the late-onset patients.1,11 In this study, more than half of the patients with cblC disease showed spinal cord atrophy or lesions. Moreover, scoliosis was also a common radioimaging abnormality.
MMACHC protein may act as an intracellular cobalamin-trafficking chaperone which catalyzes the reductive decyanation of cyanocobalamin to generate cobalamin.1 Since methylcobalamin is the essential cofactor for the enzyme methionine synthase, the cblC disease causes an impairment of the remethylation of Hcy to methionine. Adenosylcobalamin is a cofactor for the mitochondrial enzyme methylmalonyl-CoA mutase, a critical enzyme in the degradation of MMA.1 Thus, the elevated total Hcy concentrations in plasma and MMA in plasma and urine with low plasma Met concentration are the biochemical markers for the cblC disease. In this study, total plasma Hcy concentrations in all patients were markedly increasing, which is a useful indicator for the prompt diagnosis of cblC disease. Total Hcy is also the preferred parameter to monitor patients with cblC disease. Elevations of propionylcarnitine (C3) by newborn screening can detect cblC disease. Sequencing of the MMACHC gene is suggested for confirming the diagnosis of cblC disease.1 Although vitamin B12 concentrations were normal in our patient cohort, it should be measured to rule out nutritional deficiency disorders of the absorption of vitamin B12.
The wide clinical heterogeneity of cblC disease likely depends on the nature of the different MMACHC mutations. Until now, >80 different mutations of MMACHC have been reported.1,2 Patients with compound heterozygotes for missense alleles usually appear to have a milder phenotype.1 In the European cblC disease patients with early-onset, severe disease showed high frequency with c.271dupA or the c.331C>T mutations, while c.394C>T was related to late-onset disease.2,14 It seems that the c.394C>T mutation might induce a truncated protein with residual function, thus explaining its association with late-onset rather than early-onset disease.1 The c.609G>A nonsense mutation or c.482G>A missense mutation is encountered frequently in the Chinese population.11,12,15 In this study, we found the c.482G>A mutation was the most frequent genotype in our patients with a ratio of 57.7% (15/26). The second most frequent mutation was c.609G>A nonsense mutation. Seven patients were heterozygous, and two patients were homozygous for this mutation. In our previous study about four pairs of Chinese late-onset sibling cblC patients, c.482G>A mutation was also the most frequently identified.16
The management strategies of cblC disease have remained largely unchanged since its discovery. Treatment with high-calorie and low-protein diet is a mainstay to treat patients.1,17 Since low-protein diet therapy does not show a beneficial effect on the metabolic control of the patients, protein restriction is not recommended for the patients anymore.18 The therapy medicines for patients with cblC disease include cobalamin, L-carnitine, betaine, folic acid, and B group vitamins.1,17 The immediate treatment with parenteral OHCbl and betaine was strongly recommended, which seems to be effective in decreasing the infantile mortality rate.4 It also seems that most late-onset cblC patients respond well to the OHCbl treatment with significant improvements not only of biochemical parameters, but also of clinical neurological manifestations.11 It has been reported that high doses of cobalamin administration could have a greater benefit in cblC disease patients with severe neurological phenotypes.19 It was shown that mutant MMACHC proteins have increased binding affinity to OHCbl compared to CNCbl.20 Moreover, mutant MMACHC proteins are stable in the presence of high concentrations of intracellular cobalamin.21 It is suggested that long-term management of late-onset patients with OHCbl supplementation and metabolic monitoring is necessary.1
Conclusion
Above all, late-onset cblC disease is relatively rare and difficult to recognize due to a wide diversity of symptoms. When juvenile/adult patients with hyperhomocysteine in plasma present with neurological symptoms that are not consistent with common neurological diseases, cblC disease needs to be considered. The organic acid screening and genetic analysis for MMACHC gene mutations should be implemented to allow for early diagnosis and to improve prognosis.
Acknowledgments
This work was supported by grants from the Natural Science Foundation of Shandong Province, China (No. ZR2017MH082), the Innovative Research Project of Resident Standardization Training of Qilu Hospital, Shandong University (No. ZPZX2017B10), and the Taishan Scholars Program of Shandong Province.
Disclosure
The authors report no conflicts of interest in this work.
References
Carrillo-Carrasco N, Chandler RJ, Venditti CP. Combined methylmalonic acidemia and homocystinuria, cblC type. I. clinical presentations, diagnosis and management. J Inherit Metab Dis. 2012;35(1):91–102. | ||
Lerner-Ellis JP, Tirone JC, Pawelek PD, et al. Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nat Genet. 2006;38(1):93–100. | ||
Mudd SH, Levy HL, Abeles RH, Jennedy JP. A derangement in B 12 metabolism leading to homocystinemia, cystathioninemia and methylmalonic aciduria. Biochem Biophys Res Commun. 1969;35(1):121–126. | ||
Huemer M, Diodato D, Schwahn B, et al. Guidelines for diagnosis and management of the cobalamin-related remethylation disorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR deficiency. J Inherit Metab Dis. 2017;40(1):21–48. | ||
Kim J, Gherasim C, Banerjee R. Decyanation of vitamin B12 by a trafficking chaperone. Proc Natl Acad Sci U S A. 2008;105(38):14551–14554. | ||
Wang F, Han L, Yang Y, et al. Clinical, biochemical, and molecular analysis of combined methylmalonic acidemia and hyperhomocysteinemia (cblC type) in China. J Inherit Metab Dis. 2010;33(S3):435–442. | ||
Weisfeld-Adams JD, Morrissey MA, Kirmse BM, et al. Newborn screening and early biochemical follow-up in combined methylmalonic aciduria and homocystinuria, cblC type, and utility of methionine as a secondary screening analyte. Mol Genet Metab. 2010;99(2):116–123. | ||
Han B, Cao Z, Tian L, et al. Clinical presentation, gene analysis and outcomes in young patients with early-treated combined methylmalonic acidemia and homocysteinemia (cblC type) in Shandong Province, China. Brain Dev. 2016;38(5):491–497. | ||
Guo K, Zhou X, Chen X, Wu Y, Liu C, Kong Q. Expanded newborn screening for inborn errors of metabolism and genetic characteristics in a Chinese population. Front Genet. 2018;9:122. | ||
Shinnar S, Singer HS. Cobalamin C mutation (methylmalonic aciduria and homocystinuria) in adolescence. A treatable cause of dementia and myelopathy. N Engl J Med. 1984;311(7):451–454. | ||
Huemer M, Scholl-Bürgi S, Hadaya K, et al. Three new cases of late-onset cblC defect and review of the literature illustrating when to consider inborn errors of metabolism beyond infancy. Orphanet J Rare Dis. 2014;9:161. | ||
Liu YR, Ji YF, Wang YL, et al. Clinical analysis of late-onset methylmalonic acidaemia and homocystinuria, cblC type with a neuropsychiatric presentation. J Neurol Neurosurg Psychiatry. 2015;86(4):472–475. | ||
Deodato F, Boenzi S, Rizzo C, Dionisi-Vici C. The clinical picture of early-onset cobalamin C defect (methylmalonic aciduria and homocystinuria). Paediatr Child Health. 2008;18(1):S57–S60. | ||
Nogueira C, Aiello C, Cerone R, et al. Spectrum of MMACHC mutations in Italian and Portuguese patients with combined methylmalonic aciduria and homocystinuria, cblC type. Mol Genet Metab. 2008;93(4):475–480. | ||
Wang X, Sun W, Yang Y, Jia J, Li C, Li C. A clinical and gene analysis of late-onset combined methylmalonic aciduria and homocystinuria, cblC type, in China. J Neurol Sci. 2012;318(1–2):155–159. | ||
Wang SJ, Yan CZ, Liu YM, Zhao YY. Late-onset cobalamin C deficiency Chinese sibling patients with neuropsychiatric presentations. Metab Brain Dis 2018;33(3):829–835. | ||
Hauser NS, Manoli I, Graf JC, Sloan J, Venditti CP. Variable dietary management of methylmalonic acidemia: metabolic and energetic correlations. Am J Clin Nutr. 2011;93(1):47–56. | ||
Martinelli D, Deodato F, Dionisi-Vici C. Cobalamin C defect: natural history, pathophysiology, and treatment. J Inherit Metab Dis. 2011;34(1):127–135. | ||
Matos IV, Castejón E, Meavilla S, et al. Clinical and biochemical outcome after hydroxocobalamin dose escalation in a series of patients with cobalamin C deficiency. Mol Genet Metab. 2013;109(4):360–365. | ||
Froese DS, Zhang J, Healy S, Gravel RA. Mechanism of vitamin B12-responsiveness in cblC methylmalonic aciduria with homocystinuria. Mol Genet Metab. 2009;98(4):338–343. | ||
Froese DS, Healy S, McDonald M, et al. Thermolability of mutant MMACHC protein in the vitamin B12-responsive cblC disorder. Mol Genet Metab. 2010;100(1):29–36. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.