")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Clinical Evidence and Safety Profile of Emicizumab for the Management of Children with Hemophilia A
Authors Le Quellec S
Received 17 November 2019
Accepted for publication 22 January 2020
Published 3 February 2020 Volume 2020:14 Pages 469—481
DOI https://doi.org/10.2147/DDDT.S167731
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Georgios Panos
Sandra Le Quellec1,2
1Unité d’hémostase Clinique – Hôpital Cardiologique Louis Pradel – Hospices Civils de Lyon, Lyon, France; 2Service d’hématologie biologique, Groupement Hospitalier Est – Hospices Civils de Lyon, Lyon, France
Correspondence: Sandra Le Quellec
Unité d’Hémostase Clinique, Hôpital Cardiologique Louis Pradel, 59, boulevard Pinel, Bron 69677, France
Tel +33 4 72 11 88 10
Fax +33 4 72 11 88 17
Email [email protected]
Abstract: Emicizumab is a bispecific, humanized, monoclonal antibody mimicking the factor (F) VIII cofactor activity in mediating the generation of FXa by FIXa in patients with hemophilia A (HA). This subcutaneous non-factor agent has been recently extensively approved for the prophylaxis of patients of HA patients with and without FVIII-inhibitors of all ages, although few data are currently available in children. In Phase 3 clinical trials and case series, emicizumab prophylaxis significantly reduced bleeding rates compared to previous treatment in HA adolescents and children with or without FVIII-inhibitors and was generally well tolerated. In addition, subcutaneous administration of emicizumab provided beneficial effects on health-related quality of life, and lessened the burden of the disease in HA patients as well as in their caregivers. However, additional prospective studies are required to evaluate the long-term safety of emicizumab prophylaxis in very young patients, including previously untreated patients. The aim of this paper was to review the limited data available on the use of emicizumab prophylaxis in children and to highlight the need for further studies to address remaining concerns.
Keywords: emicizumab, hemophilia A, inhibitors, factor VIII, prophylaxis, subcutaneous
Introduction to Current Management of People with Hemophilia A with or Without Factor VIII Inhibitors
Hemophilia A (HA) is a rare congenital bleeding disorder characterized by coagulation factor VIII (FVIII) deficiency. In severe HA, defined as plasma FVIII clotting activity <1% of normal (FVIII:C < 1 IU/dl), bleeding may frequently occur spontaneously, notably in joints, leading to painful hemophilic arthropathy and loss of joint function. The standard of care for patients with severe HA, in developed countries, consists of regular intravenous infusions of FVIII concentrates to prevent bleeding episodes (prophylactic treatment).1,2 Owing to the short half-life of standard FVIII concentrates, of about 12 h, no less than three intravenous infusions per week may be required for maintaining FVIII levels at >1 IU/dl, which is effective at reducing incidence of life-threatening bleeds and chronic manifestations of recurrent bleeding episodes. However, FVIII replacement therapy is extremely expensive, not widely available, and invasive. The use of novel recombinant FVIII concentrates with prolonged half-life has increased the interval between treatments but still require lifelong intravenous infusions, which considerably alter patients’ quality of life. Frequent intravenous infusions are burdensome for pediatric patients and their caregivers, and may necessitate the use of central venous access with the risk of infection-related complications.3 In addition, the risk of developing inhibitory antibodies to infused FVIII, commonly called “FVIII-inhibitors”, is the major complication of replacement therapy that occurs in approximately 20–30% of patients with severe HA.4–6 In these patients, FVIII substitution becomes inefficient and bleeding occurrences are treated or prevented using bypassing agents (BPA). BPA include activated prothrombin complex concentrates (aPCC) and recombinant factor VIIa (rFVIIa).7,8 Although they are effective in restoring hemostasis, they are not able to completely normalize thrombin generation in HA patients with FVIII-inhibitors, in contrast of FVIII replacement therapy in these without FVIII-inhibitors.9 In addition, they have sometimes been associated with thrombotic adverse events.10,11 Thus, patients with persistent FVIII-inhibitors suffer from more morbidity and mortality than patients without FVIII-inhibitors.12–14 They should be offered immune tolerance induction (ITI), which consists of the daily infusion of large doses of FVIII concentrates, given until the FVIII-inhibitors disappear and the FVIII pharmacokinetic parameters normalize.15 However, ITI is not always successful in making FVIII-inhibitors disappear and is really inconvenient for patients and their caregivers.16 Recently, several strategies of non-replacement therapy have been developed. Non-replacement therapies aim to restore the hemostatic equilibrium and offer the opportunity to treat patients with HA with or without FVIII-inhibitors. Among them, emicizumab (HEMLIBRA®, Roche, Bazel, Switzerland) has been approved in Europe, the US, and Japan, for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in HA adult and pediatric patients of all ages with and without FVIII-inhibitors. The purpose of this review is to discuss the efficacy and the safety of prophylaxis emicizumab in children with HA.
Pharmacology, Mode of Action, and Pharmacokinetics of Emicizumab
Emicizumab is a recombinant, humanized, bispecific monoclonal antibody that partially mimics FVIII functions, by bridging factor IXa and factor X together, which is mandatory for effective hemostasis.17 Owing to a complete absence of structure homology compared to FVIII, emicizumab is not suspected to induce FVIII-inhibitors and can play its role irrespective of the presence of FVIII-inhibitors, no matter the FVIII-inhibitor titer.18 Indeed, emicizumab induced a dose-dependent shortening of activated partial prothrombin time and increase of thrombin generation in ex-vivo FVIII-neutralized plasma from healthy adult volunteers.19
Additionally, the convenient route of administration of emicizumab and its pharmacokinetic profile has rapidly made it particularly attractive for prophylactic use in patients with HA with and without FVIII-inhibitors. A single subcutaneous injection of emicizumab provided a linear pharmacokinetic profile with a half-life of approximately 4–5 weeks in healthy adult subjects,19 thus allowing for infrequent dosing regimen. Initially, once weekly (QW) administration of emicizumab at 0.3, 1, and 3 mg/kg bodyweight dose-dependently increased plasma emicizumab concentrations, and reached steady-state ~12 weeks after first injection, in HA adult patients with and without FVIII-inhibitors.20,21 Pharmacologic modeling determined a novel regimen, which would allow shortening the time to reach steady-state.22 This regimen was first studied in the phase 3 HAVEN 1 trial (ClinicalTrials.gov number, NCT02622321), which included 109 HA adult patients with FVIII-inhibitors.23 Mean steady-state plasma emicizumab concentrations were reached after 4 weeks of weekly loading dose of 3 mg/kg bodyweight followed by QW maintenance dose of 1.5mg/kg bodyweight. The resulting plasma emicizumab concentration was sustained throughout the trial with a mean level >50µg/mL. Such observation was particularly interesting, and supported the use of emicizumab for prophylaxis in HA patients with and without FVIII-inhibitors, avoiding peak and trough levels as obtained after BPA or FVIII replacement therapy, respectively. Trough levels of plasma emicizumab concentrations and the resulting hemostatic effect were expected to correspond to at least 10–15 IU/dl of equivalent FVIII activity (FVIII:C), which represents a level of FVIII:C associated with a low risk of joint bleeding.24,25 However, it is worth mentioning that extrapolation of emicizumab hemostatic activity to “real” equivalent FVIII:C has not been possible to date. Equivalent FVIII:C corresponding to therapeutic dose of emicizumab have varied widely across studies, depending on the coagulation assays that were used, whether they were global hemostatic assays or FVIII one-stage assays.25,26 Interestingly, similar pharmacokinetic profiles and through levels of plasma emicizumab concentrations were observed in the 152 HA adult patients without FVIII-inhibitors who were enrolled in the phase 3 HAVEN 3 study (ClinicalTrials.gov number, NCT02847637), whether they were included in the QW prophylaxis regimen cohort or in the maintenance dose of 3.0 mg/kg bodyweight emicizumab every 2 weeks cohort (Q2W). Other emicizumab prophylaxis regimens were studied in the HAVEN 4 trial (ClinicalTrials.gov number, NCT03020160), in which HA adult and adolescent patients with and without FVIII-inhibitors received emicizumab subcutaneous injection every 4 weeks (Q4W). In the expansion cohort, patients received the usual 3 mg/kg loading dose per week during 4 weeks followed by a maintenance dose of 6 mg/kg bodyweight Q4W. Mean trough plasma concentrations reached 50 μg/mL after loading dose and were maintained slightly above 40 μg/mL throughout the trial.27 However, unlike QW and Q2W regimens, the Q4W dosing regimen resulted in peaks and valleys of emicizumab concentration, which could lessen the efficiency of emicizumab prophylaxis over bleeding protection.27
In children, previous studies have shown that FVIII half-life were shorter than those reported in adults receiving the same FVIII product.28–30 However, with the exception of bridging FIXa and FX, emicizumab and FVIII have little in common, and emicizumab pharmacokinetic properties are less likely to be correlated with binding-capacity to VWF or ABO groups, as described recently for FVIII.31 As a result, same QW, Q2W, and Q4W emicizumab dosing regimens as those used for adults were studied in children with HA. Pharmacokinetic profile of emicizumab prophylaxis in HA pediatric patient was first studied in the HAVEN 2 trial (ClinicalTrials.gov number, NCT02795767). HAVEN 2 is a non-randomized, open-label, multicenter, phase 3 clinical trial designed to evaluate the efficacy, safety, and pharmacokinetics of emicizumab prophylaxis in children <12 years of age (12 to 17 years, if bodyweight < 40 kg) with HA and high-titer FVIII-inhibitors (≥5 BU/mL), previously treated with BPAs.32 First cohort of patients (n=68) received subcutaneous injections of loading dose emicizumab 3 mg/kg QW for 4 weeks, followed by maintenance dose 1.5 mg/kg QW thereafter. Two additional non-randomized cohorts were subsequently opened to enroll patients to receive similar loading dose regimen, followed by maintenance dose 3 mg/kg Q2W (n=10 patients), or 6 mg/kg Q4W (n=10 patients). Results were concordant with those observed in adult patients, revealing that through plasma emicizumab concentrations were maintained at approximately 50 µg/mL, 45 µg/mL, and 38 µg/mL with QW, Q2W, and Q4W dosing, respectively (Figure 1).32 Recently, the HOHOEMI study, a Japanese multicenter non-randomized trial, reported pharmacokinetic data of emicizumab administered subcutaneously at a maintenance dose of 3 mg/kg Q2W (n=6 patients) or 6 mg/kg Q4W (n=7 patients) in HA pediatric patients without FVIII-inhibitors,33 including only one previously untreated patient (PUP), after receiving a loading dose of 3 mg/kg QW subcutaneously for the first 4 weeks of treatment. The mean trough plasma concentrations of emicizumab were approximately 48 μg/mL at the completion of the loading dose in both cohorts, which were comparable with those in adult and adolescent patients receiving the same Q2W or Q4W regimen in HAVEN 2, HAVEN 3 and HAVEN 4 studies.23,27,34 However, mean steady-state trough plasma emicizumab concentrations during the subsequent maintenance period, were slightly lower for the Q2W and Q4W cohorts than those for the Q2W dosing patients in HAVEN 3 and for the Q4W dosing patients in HAVEN 4, at approximately 35 and 30 μg/mL, respectively. Noteworthy, mean trough plasma emicizumab concentrations varied widely between individuals during the maintenance period, ranging from 20.9 to 50.5 μg/mL for the Q2W cohort and from 13.4 to 55.2 μg/mL for the Q4W cohort, from 12 weeks after treatment initiation onwards.
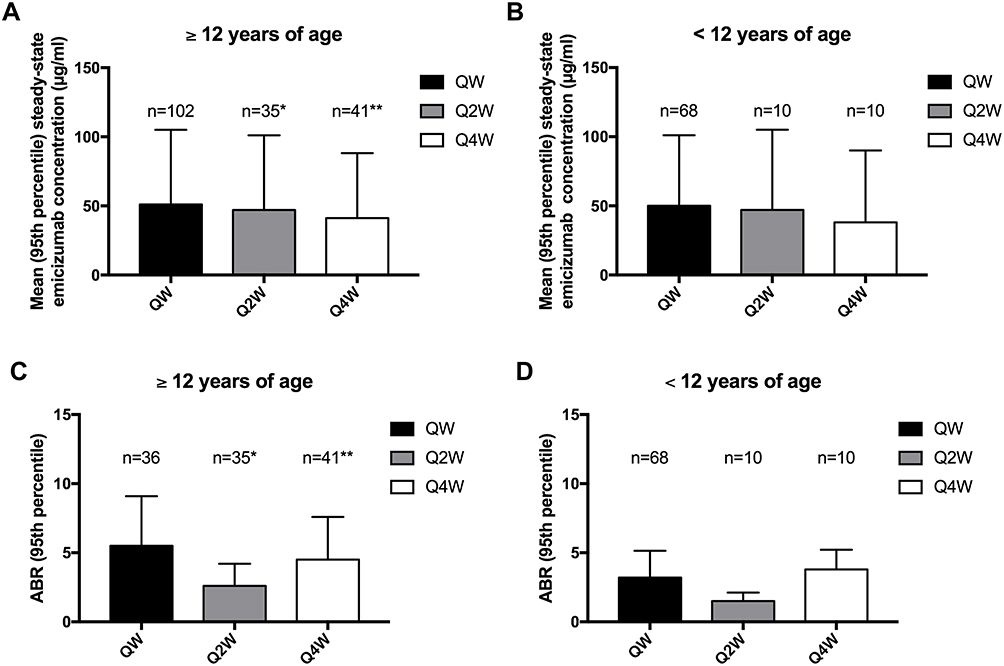 |
Figure 1 Comparison of pharmacokinetic and efficacy data for each dosing regimen of emicizumab prophylaxis, obtained across the various HAVEN studies. (A) Pharmacokinetic data in adult adolescents patients; (B) Pharmacokinetic data in children; (C) Efficacy data (all ABR) in adult and adolescents patients; (D) Efficacy data (all ABR) in children. QW: 3 mg/kg/week loading dose and 1.5 mg/kg/week maintenance dose; Q2W: 3 mg/kg/week loading dose and 3 mg/kg every 2 weeks; Q4W: 3 mg/kg/week loading dose and 6 mg/kg every 4 weeks; *adult and adolescent patients with severe HA without FVIII-inhibitors; **adult and adolescent patients with severe HA with and without FVIII-inhibitors. |
Altogether, these studies have shown that emicizumab prophylaxis exhibited similar pharmacokinetic profile in HA patients with or without FVIII-inhibitors. Recent quantitative analyses indicated that the relationship between plasma emicizumab concentrations and annualized bleeding rate (ABR) reached a plateau at above approximately 30 µg/mL,35 thus supporting the use of emicizumab prophylaxis with infrequent dosing regimen in children with HA. In contrast, another model-based study suggested that bleeding frequency may plateau more at a plasma emicizumab concentration ≥45 µg/mL.22 Mean trough emicizumab concentrations with Q4W emicizumab prophylaxis were slightly lower than those obtained with QW or Q2W emicizumab in HA adults/adolescents and children.27,32,33 Therefore, we can speculate that choosing the QW emicizumab regimen would be more appropriate in children in order to provide better long-term protection from bleeding, especially in these patients who are more prone to be highly active and to participate in high-impact activities and sports.
Efficacy of Emicizumab in Children with Severe Hemophilia A
Efficacy in Children with Active FVIII-Inhibitors
Considering pharmacokinetic profiles are equivalent between HA adults and children patients, we could expect that the hemostatic efficiency of emicizumab prophylaxis would also be similar. As a result, emicizumab prophylaxis should obviously be considered for HA pediatric patients with high-titer FVIII-inhibitors who will not receive ITI or for those who failed ITI attempts. Indeed, QW emicizumab prophylaxis significantly reduced the bleeding rate in HA adult patients with FVIII-inhibitors,20,21,23 as compared to prior treatment strategy. Once-weekly administration of emicizumab resulted in 87% (p<0.001) reduction of the ABR compared to the group with no prophylaxis, receiving on-demand treatment with BPA;23 and in 79% (p<0.0001) intra-individual reduction of ABR among patients who were previously receiving prophylaxis with BPAs.23 Such efficacy was recently confirmed in pediatric patients (<12 years of age), who are the most prone to develop FVIII-inhibitors and to receive ITI.36 Data about emicizumab prophylaxis clinical efficiency in HA children with FVIII-inhibitors are arising from single centers case series and the phase 3 HAVEN 2 trial (ClinicalTrials.gov number NCT02795767; Table 1). In both case series, no spontaneous bleeds were reported for up to 52 weeks after initiation of QW emicizumab prophylaxis in a total of 15 male HA children with high-titer FVIII-inhibitors.37,38 In the phase 3 HAVEN 2 trial, enrolling 88 HA children (2–12 years of age) with high-titer FVIII-inhibitors, 68 patients received the QW dosing regimen for a median efficacy period of 57.6 weeks (range 17.9–92.6). Among them, 94.7% had zero treated bleeds, and 72.3% experienced zero bleeds, whether they were treated or not. Once-weekly administration of emicizumab resulted in an ABR for treated bleeds of 0.3 (95% CI, 0.17; 0.5), with a median ABR of 0 for all treated bleed endpoints. An intra-individual comparison was performed in a total of 18 patients <12 years of age who had previously participated in the non-interventional study (ClinicalTrials.gov identifier NCT02476942) prior to enrolling in HAVEN 2, and received treatment with QW emicizumab prophylaxis. Intra-individual comparison analysis showed that emicizumab prophylaxis resulted in a 99% reduction in treated bleeds compared to previous episodic/prophylaxic treatment with BPA.39 Among 15 patients who received prior prophylactic BPA treatment, QW emicizumab prophylaxis at a median duration of 89.1 (range, 56–92.6) weeks demonstrated a substantially lower bleeding rate. In these HA pediatric patients with FVIII-inhibitors, ABR was 0.3 (95% CI, 0.12; 0.56) for QW emicizumab prophylaxis whereas it was 21.1 (95% CI, 15.99; 27.82) with prior BPA prophylaxis. Most treated bleeds observed in these studies were traumatic bleeds, with half of them being joint bleeds, which resolved after few injections of BPA.32,37,38 When comparing to other dosing regimens used in the HAVEN 2 study, we can notice that the Q4W dosing regimen performed less well than both QW and Q2W dosing regimens in terms of pharmacokinetic profile, which resulted in lower efficacy over bleeding prevention (Table 1; Figure 1).32 Interestingly, studies revealed that emicizumab prophylaxis considerably reduced target joints. Target joints were defined as a major joint, into which ≥3 bleeds occur over a 24-week period. In all studies available to date, no target joint developed in HA pediatric patients with FVIII-inhibitors who had no target joint at baseline, including 12 patients <2 years of age.32,37,38 In addition, among 38.6% of patients who met criteria for target joint at baseline in the HAVEN 2 study, all 45 target joints resolved after 52 weeks of emicizumab prophylaxis.32 The markedly better efficacy of emicizumab prophylaxis over BPA treatment for bleeding prevention and the reduction of target joints supports the use of this drug in patients who have developed FVIII-inhibitors and who failed ITI.
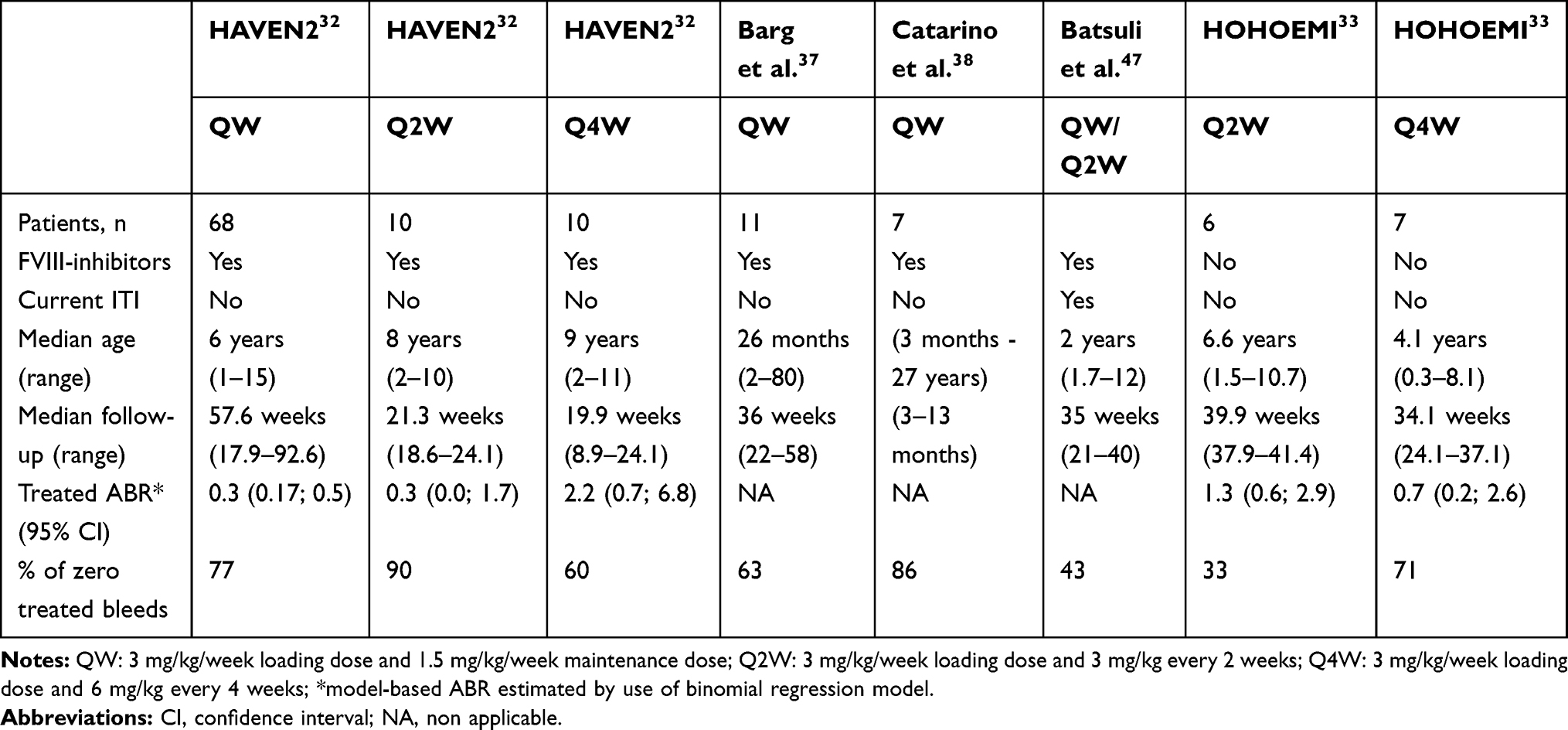 |
Table 1 Principal Studies and Case Series Evaluating Emicizumab Prophylaxis in HA Children < 12 Years of Age |
Efficacy in Children with Recently Developed FVIII-Inhibitors
For patients who recently developed FVIII-inhibitors, most experts recommend attempting ITI at least once.40,41 Although we are entering in the era of novel non-factor therapies including emicizumab, FVIII-inhibitors eradication should remain the ultimate goal in managing HA patients with FVIII-inhibitors for many valuable reasons: i) emicizumab prophylaxis does not completely prevent for all bleeds, ii) FVIII infusions for the on-demand treatment of breakthrough bleeds during emicizumab prophylaxis are more efficient and less likely to induce safety issues such as thrombotic events compared to BPA infusions.23,34,42 Yet questions remain with regard to the choice of ITI regimen and the potential use of concomitant emicizumab prophylaxis.40,43,44 It has been suggested that emicizumab prophylaxis could be given while waiting for FVIII-inhibitor titers to fall below 10 BU/mL before for starting ITI.41,43 During this period, which can last up to 6 months,45 providing emicizumab prophylaxis would be of particular interest as it would avoid FVIII antigen exposure, prevent bleeding occurrence more efficiently than rFVIIa, and lessen the burden of regular intravenous infusions owing to rFVIIa short half-life.15 However, most hemophilia comprehensive care centers now start ITI as soon as the diagnosis of FVIII-inhibitors is made, irrespective of FVIII-inhibitors titers.40 Using emicizumab prophylaxis during ITI would aim to reduce spontaneous joint bleeds in this period of time. This would eventually allow for lowering the dose and/or frequency of FVIII infusions, and for reducing the time to successfully achieve FVIII tolerance.43 In theory, emicizumab and FVIII as part of ITI could be used together without providing synergistic hemostatic effects as they compete for FIXa and FX.17,46 Such use of emicizumab and ITI are only described in case reports to date. Batsuli et al recently reported results from the “Atlanta Protocol” (Table 1).47 Seven patients received QW or Q2W emicizumab prophylaxis while on ITI regimen with either plasma-derived or recombinant FVIII at a dose of 50–100 IU/kg 3 times per week starting ≥4 weeks after initiation of emicizumab. While the success rate of the ITI was impossible to define in this study, 3 (43%) patients had a negative FVIII-inhibitor titer, and 2 achieved a normal FVIII recovery at the end of the follow-up. More importantly, 3 patients (43%) had no bleeds, and 9 bleeding events (8 were trauma-induced) occurred in the remaining 4 patients.47 These results confirmed that the bleeding rate in HA pediatric patients with FVIII-inhibitors undergoing ITI with emicizumab prophylaxis is expected to be low. However, there is still a special need for gaining insight in the management of FVIII-inhibitors and the treatment of breakthrough bleeds with the advent of emicizumab. Although the rapid implementation of low dose ITI combined with emicizumab may be recommended by some authors,40,41 efficacy, and safety of treating HA pediatric patients with FVIII-inhibitors with ITI in combination with emicizumab prophylaxis should be assessed in specifically designed clinical trials. Prospective studies are required to compare treatment outcomes of ITI and emicizumab to other ITI regimens, and to evaluate whether emicizumab prophylaxis modifies the immunologic response to FVIII exposition. The observational MOTIVATE study (ClinicalTrial.gov number, NCT04023019) and the prospective Emicizumab PUP and Nuwiq ITI study (ClinicalTrial.gov number, NCT03344003), will attempt to address these concerns. However, other issues are not resolved yet for HA patients with FVIII-inhibitors receiving emicizumab, such as: i) how to monitor and define ITI success?, ii) how to adapt ITI regimen?, iii) when to stop ITI?, iv) what to do after ITI completion?, etc. In case ITI was successful, should emicizumab be continued or should it be replaced by a return to FVIII prophylaxis? (Table 2).
 |
Table 2 Remaining Questions Requiring Additional Clinical Studies |
Efficacy in Children Without FVIII-Inhibitors
Whether emicizumab prophylaxis should be offered in HA patients without FVIII-inhibitors at baseline or after successful ITI might be of debate. It is currently not known what amount of regular FVIII exposure might be needed to achieve or maintain FVIII tolerance. In addition, FVIII prophylaxis remains the gold-standard treatment strategy in HA patients without FVIII-inhibitors in developed countries, which has been proven efficient in reducing bleeding occurrence for decades.48 However, recent results from clinical trials evaluating the efficacy of emicizumab prophylaxis may change this paradigm. Indeed, QW administration of emicizumab has clearly demonstrated a clinically meaningful efficiency for bleeding prevention in HA adult and adolescent patients without FVIII-inhibitors included in the HAVEN 3 study, with a median ABR (95% CI) at 1.5 (0.9–2.5). The intra-individual comparison performed in 48 patients showed that QW emicizumab prophylaxis resulted in a 68% lower ABR than with previous FVIII prophylaxis.34 Additionally, the HOHOEMI study published recently brings additional arguments in favor of the use of emicizumab in HA children without FVIII-inhibitors, despite the absence of QW dosing regimen evaluation. Six and 7 patients with a median age (range) at baseline at 6.6 years (1.5–10.7) and 4.1 years (0.3–8.1) were included in the Q2W and Q4W cohorts, respectively. During the on-treatment period, which lasted up to 40 weeks after first injection of emicizumab, two out of six patients in the Q2W cohort and five out of seven patients in the Q4W cohort, including a PUP aged 4 months, had no treated bleeds. A total of nine treated bleeds were reported in this study: six had occurred in the Q2W cohort, and three in the Q4W. All but one were traumatic bleeds (three joint bleeds and five no joint bleeds), which were resolved after a single administration of FVIII replacement therapy ranging from 32.5 to 64.7 IU/kg. One spontaneous joint bleed occurred in the QW2 cohort and was managed with FVIII replacement therapy given once daily for 5 days at approximately 30 IU/kg. Model-based ABRs for treated bleeding events were 1.3 (95% confidence interval [CI], 0.6–2.9) for the Q2W cohort and 0.7 (95% CI, 0.2–2.6) for the Q4W cohort, which is lower than for most FVIII prophylaxis using from 2 to 3 infusions per week.29,49 These results should be interpreted with caution and considered on longer term, because HA patients < 12 years of age exhibit an age-dependent increased bleeding risk owing to the association of their normal growth and development to increased physical activity. Attention should also be paid when discussing the initial treatment of previously untreated patients (PUPs). Optimal bleeding protection of a newly diagnosed severe HA children would ideally consist in initiating treatment as soon as the diagnosis is made. Such strategy would especially aim to avoid the occurrence of intracranial hemorrhage (ICH), which account for up to 4% of children with HA.50 While the current standard of care using FVIII prophylaxis is not amenable immediately given the need for frequent venous access, prophylaxis with subcutaneous injections of emicizumab may be of interest for early therapeutic strategy, as it would lessen the burden of prophylactic treatment for children patients and caregivers. However, owing to its pharmacokinetic profile, subcutaneous administration emicizumab is not expected to provide an immediate hemostatic effect after first injections at an early age after birth. So far, there is no data on the efficacy and safety of emicizumab prophylaxis in newborns, and children <2 years of age including newborns are rarely included in licensure studies. To date, clinical experience on the use of emicizumab for the treatment of PUPs with HA is only described in a single PUP who was enrolled at 4 months of age in the Q4W cohort of the HOHOEMI clinical trial.33 This PUP experienced no bleeds during a 24-week period follow-up. Although this result seems promising, prospective studies using emicizumab prophylaxis in PUPs may be required to better assess the outcome of such novel therapeutic strategy in pediatric patients with severe HA (Table 2).
Altogether, emicizumab represents an interesting therapeutic approach in HA pediatric patients with or without FVIII-inhibitors because it has been shown that the lower the bleeding rate, the lower the long-term complications of hemophilia such as hemophilic arthropathy.1,2 We can expect that if hemophilic arthropathy develops lately (or does not), the number of major orthopedic surgeries will decrease, thus partially reducing the risk of major bleeding in a perioperative setting. However, HA children are still at risk for minor surgeries to be performed. Interestingly, most studies have shown that emicizumab prophylaxis may be efficient to prevent bleeding complication during minor surgeries in severe HA adult and pediatric patients with or without FVIII-inhibitors.23,34 In children, minor surgeries described to date were mostly managed with or without additional episodic treatment with BPA in patients with FVIII-inhibitors, or with FVIII in patients without FVIII-inhibitors (Table 3). Further clinical data are required to help standardizing the management of minor and major surgeries in HA pediatric patients with or without FVIII-inhibitors receiving emicizumab prophylaxis. The use of emicizumab together with high dose and/or long-term exposure to BPA or to FVIII, which may be required to efficiently prevent bleeding complication in a perioperative setting, may impact patients’ safety.
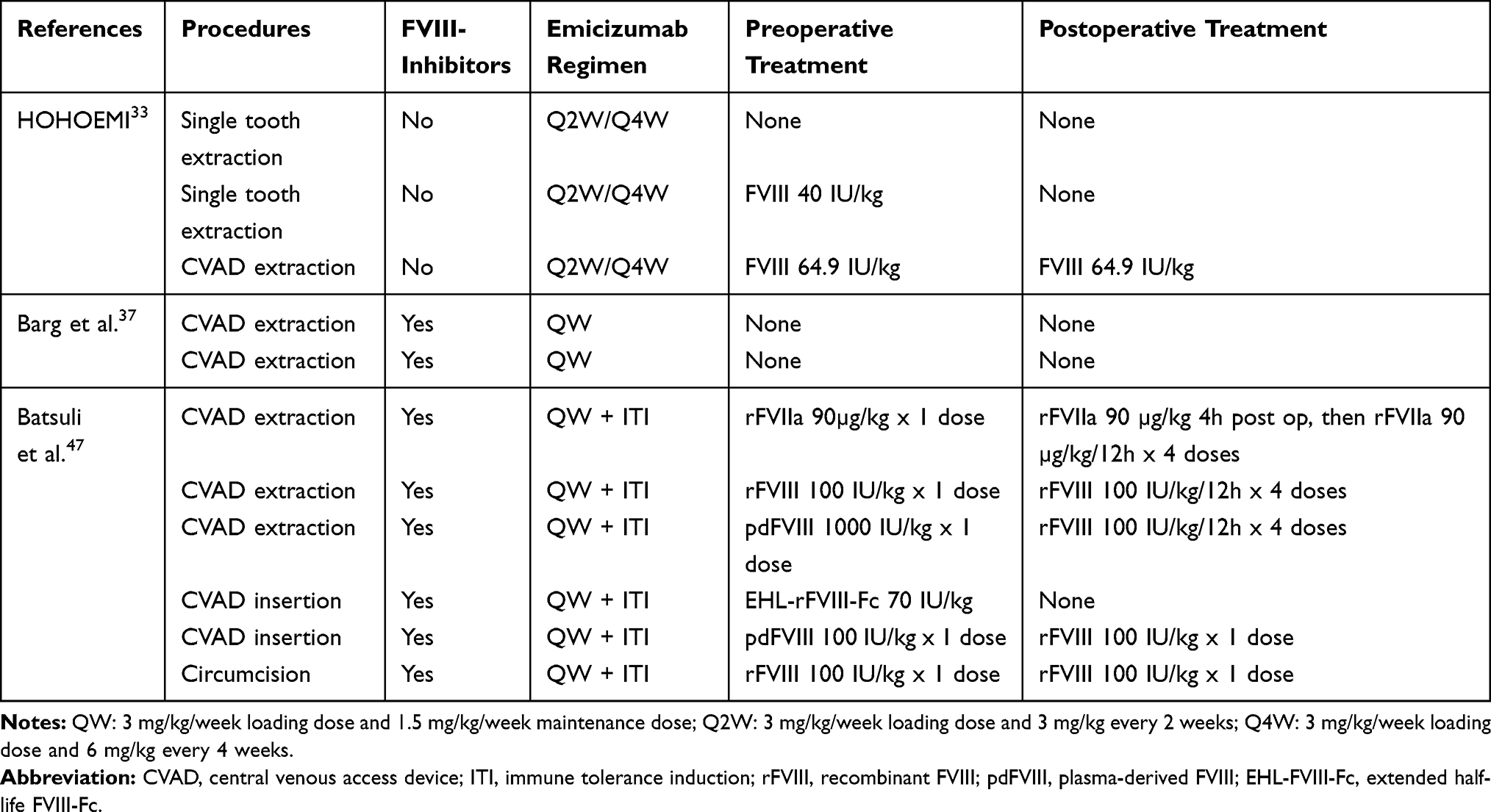 |
Table 3 Management of Surgeries in HA Children < 12 Years of Age Receiving Emicizumab Prophylaxis |
Safety and Tolerability of Emicizumab in Children with Severe Hemophilia A
Safety assessment is an essential component of studies in all phases of development of new drugs. In case of emicizumab, adverse events (AEs) are reported in the various clinical trial publications.27,32–34,51 No emicizumab-related serious AEs were reported during the Phase 1 studies enrolling adult/adolescent patients.19–21 However, serious safety concerns were raised from phase 3 studies, as soon as the HAVEN 1 study was published. Among 109 HA adult/adolescents patient with FVIII-inhibitors receiving QW emicizumab prophylaxis, 2 had thromboembolic events (TEs), and 3 experienced a thrombotic microangiopathy (TMA) among which one died from uncontrollable rectal hemorrhage.51 Reports demonstrated that all these adult patients had been treated with concomitant use of high cumulative doses of aPCC, at >100U/kg/day for more than 1 day, for the management of breakthrough bleeds.51 No such observations were made in patients receiving lower dose and/or shorter course of aPCC. Further, in vitro studies confirmed the synergistic hemostatic effect between emicizumab and aPCC. Such synergistic effect was explained by the presence of FIXa in aPCC,52 which is a substrate for emicizumab. Owing to the absence of on/off regulation of emicizumab, an excess of FIXa, may lead to uncontrolled FXa generation and subsequent thrombotic complications.17 These findings were also supported by the fact that none of the patients who have received emicizumab prophylaxis alone, with concomitant use of rFVIIa, or with FVIII, did develop TE or TMA.27,34,51 Subsequently, study protocols were amended and the manufacturer has provided recommendations for the use and dosing of BPA during emicizumab prophylaxis: aPCC should be avoided as far as possible or be used at the lowest dose possible, and rFVIIa should be preferably used in case of breakthrough bleeds or surgeries. Since then, no new potentially emicizumab-related serious TE and TMA have been recorded in clinical trials.53 However, four additional TEs had been reported in the postmarketing setting, among which three were venous TE, and one was arterial TE.54 Although few data are available with regards to these recent TE, it seems that emicizumab was used alone. Patients had received (off-license) emicizumab prophylaxis for treating acquired hemophilia. However, it is known that acquired haemophilia may be at risk of thrombosis because of older age, autoimmunity, and cancer;55 and it is unlikely to occur in children.55 In addition, non-hemophilia children are globally less at risk for thrombosis compared to adults, even in the presence of inherited thrombophilia.56 Interestingly, neither TMA nor thromboembolic events have been reported to date in HA pediatric patients <12 years of age with or without FVIII-inhibitors,27,33,37,38,47 thus suggesting that emicizumab prophylaxis is relatively safer in this age group.
However, it is worth mentioning that more than 90% of emicizumab-treated HA children presented at least one adverse events (AEs) across studies.32,33,37,38,47 In these patients, the most common emicizumab-related AEs were mild or moderate, consistent with previous studies in adults and adolescents,23,27,34 and resolved spontaneously. The most frequent (>10% incidence) were injection site reactions, nasopharyngitis, contusion, and headache.32,33,37,38,47 In the HAVEN 2 study, the majority of AEs occurred in the QW cohort, with a total of 615 AEs reported in 63 patients out of 68 (96,2%) patients. This was due to the higher number of patients enrolled in this cohort, and the longer period of follow-up, because the relative incidence of most AEs was similar between the three dosing cohorts. However, local injection site reactions were significantly more frequent in the Q4W cohort, occurring in 60% of children, than in QW and Q2W cohorts (approximately 20% each).32 We can speculate that the higher incidence of local injection site reactions in cohort Q4W may be due to the higher volume injected. Of note, local injection site reactions generally include swelling, erythema, and tenderness. In young children, injection site reactions may also include pain during subcutaneous injection (personal observation). Therefore, it might be preferred to use lower dose at higher frequency in young children, which would be in favor of choosing the QW or Q2W dosing regimen for emicizumab prophylaxis rather than the Q4W dosing regimen. Additionally, injecting less frequent but higher doses of emicizumab might eventually induce the development of anti-drug antibodies (ADA) directed against emicizumab. In literature, more than 18 patients were reported to have developed emicizumab ADA across studies, which represent an incidence of approximately 4–5%.27,32,57 All four emicizumab-treated children <12 years of age who developed ADA had been receiving Q4W emicizumab prophylaxis. Therefore, we can speculate that Q4W dosing regimen might induce a higher immunogenicity in children. However, it is worth mentioning that three out of four ADA who developed in children had no effect on the safety, efficacy, pharmacokinetics, or pharmacodynamics of emicizumab. The remaining one had a neutralizing potential inducing a loss of efficacy of emicizumab, which was discontinued.32
In contrast, none of the HA pediatric patients had recurrent or de novo FVIII-inhibitors.32,33 For patients without FVIII-inhibitors at baseline, and particularly for PUPs, using emicizumab instead of FVIII products may change the typical history of FVIII-inhibitor development. Using emicizumab alone as prophylaxis could actually result in a higher FVIII-inhibitor incidence by delaying the first and cumulative exposures to exogenous FVIII, which will be given in case of bleeding episodes, surgeries, or trauma. Other questions remain with regard to the long-term safety of emicizumab prophylaxis alone. Among these, we can suppose that emicizumab would not be able to compensate for potential extravascular role of FVIII. Indeed, a recent review reported that FVIII plays important roles beyond hemostasis, including in the cardiovascular system, in angiogenesis and in bone health.58 Not compensating the absence of FVIII might eventually result in increased bone resorption and an excess of osteoporosis in HA patients as it has been described recently.59 Therefore, additional prospective studies are required to evaluate the long-term safety and the impact of the use of emicizumab on joint status and FVIII-inhibitor development.
Quality of Life and Adherence to Treatment with Emicizumab Prophylaxis
Hemophilia A is a chronic disease that currently requires frequent intravenous infusions of FVIII or BPA, in HA patients without FVIII-inhibitors or with FVIII inhibitors, respectively, for the prevention and/or the treatment of frequent and prolonged bleeding episodes. Studies have shown that current treatment with regular IV infusions increases the burden for HA patients, notably in children and adolescents, and their caregivers. It induces financial, technical, and educational challenges that considerably impact their quality of life.60,61 Subsequently, it may affect adherence to prescribed treatments,62,63 which may lead to an increased risk of bleeding and joint damage.62 In addition, it has also been shown that frequent bleedings, especially in joints, and their long-term complications, alter the HA patients’ and caregivers’ health-related quality of life (HRQOL), especially on emotional, social, and physical aspects.64–67 This partially explains why the impairment of HRQOL is greater for HA patients with FVIII-inhibitors, who are more prone to recurrent bleedings, compared to HA patients without FVIII-inhibitors.68 Bleeding rates remained high in HA patients with FVIII-inhibitors > 12 years of age, but only 60% of bleeds were effectively treated. In addition, approximately 40% of patients exhibited low compliance to their BPA prophylactic dosing frequency.68 The presence of FVIII-inhibitors may also impact on caregivers as recently shown by the BBC study, which used the HEMOCAB questionnaire in 144 caregivers across 7 European countries. This study demonstrated that the burden of the disease was higher in caregivers caring for HA children with FVIII-inhibitors than in caregivers caring for HA children without FVIII-inhibitors, owing to the combination of poor bleed control and treatment burden.61,65 From a qualitative aspect, a recent Canadian study used one-to-one interviews of HA patients without FVIII-inhibitors or caregivers, for the assessment of HRQOL. This study helped identifying desired improvements over current treatment from patients’ perspectives. Alternate mode of administration and longer-lasting treatment effects were identified as being required to overcome the challenges represented by administration of IV infusion, and coordination of treatment schedules.63 Therefore, no one can deny that the arrival of non-factor therapies such as emicizumab is revolutionizing the landscape of hemophilia.
It seems now obvious that emicizumab has the potential to improve health outcomes by substantially reducing bleeding occurrence, especially in HA children with FVIII-inhibitors.32 In addition, once a week to once a month subcutaneous administration of emicizumab offers improvements on mode of administration. The favorable safety profile of emicizumab is another factor that may contribute to the improvement of HRQOL in HA patients with or without FVIII-inhibitors. To date, few studies about HRQOL in HA patients receiving emicizumab prophylaxis are available. They have mostly been conducted as part of interventional clinical trials.32,33 In HA children with FVIII-inhibitors enrolled in the HAVEN 2 study, QW emicizumab prophylaxis have resulted in marked improvements in self- and caregiver-reported HRQOL.32 The validated Haemophilia-specific Quality of Life questionnaire for children Short-Form (Haemo-QoL-SF)69 was used to assess nine specific domains (“Physical Health”, “Feelings”, “View of yourself”, “Family”, “Friends”, “Other People”, “Sports and School”, “Dealing with hemophilia”, and “Treatment”) in HA children aged 8–11, from baseline to week 25 after emicizumab prophylaxis initiation. Of note, the higher the Haemo-QoL SF scores, the greater the impairment of HRQOL. Significant improvements were observed between emicizumab prophylaxis and prior BPA treatment, with a mean Haemo-QoL SF “Total” score reduction of −9.8. Greatest improvements were seen in the following domains: “physical health”, “feelings”, and “treatment”, with a mean score reduction of −11.3, −14.3, and −14.3, respectively. Marked improvements of HRQOL were also seen in caregivers of HA children included in the QW cohort. The Adapted inhib-QoL total score, which reflects caregivers' perception of child’s health and caregivers' burden, showed substantial mean reduction of −21.8, from baseline to week 25 after emicizumab prophylaxis initiation. Domains with greatest improvements were “physical health” (mean reduction −31.7), “dealing with inhibitors” (mean reduction −26.8), and “family life” (mean reduction −25.8). In addition, emicizumab prophylaxis has allowed a rapid and increased school attendance in HA children with FVIII-inhibitors. The mean proportion of daycare/school days missed was 0.41 at baseline, whereas it was 0.25 at week 13 after emicizumab initiation. Similar observations were reported by caregivers of HA children without FVIII-inhibitors. In the HOHOEMI study, 23.1% of caregivers who completed the emicizumab patient preference survey (EmiPref) after the first 16 weeks of treatment with emicizumab, reported that emicizumab treatment had a lower effect on activities such as work, school, sports, and socials interactions. The easier route of administration and the lower frequency were other reasons why all caregivers have preferred emicizumab prophylaxis over the patient’s previous treatment.33 In total, it seems that emicizumab prophylaxis reduces worries about bleeding and other physical complications. In addition, lower frequency of treatment should provide a more relaxing schedule of daily life, which may allow more activities and social interactions for HA children patients and their caregivers.
Conclusions, Place in Therapy
Clinical data available to date have clearly shown that prophylaxis using subcutaneous injection of emicizumab is safe and effective for preventing bleeding in severe HA pediatric patients with and without FVIII-inhibitors. Considering that reduction of bleeding frequency provides better long-term clinical outcomes and improved quality of life, we believe that emicizumab should be prescribed early in HA pediatric patients, especially in those who have failed ITI. Looking upon pharmacokinetic, efficacy, and safety data, it seems that using QW or Q2W dosing regimen in severe HA children with or without FVIII-inhibitors would be preferred over the Q4W dosing regimen. However, not all pediatric patients may receive QW regimen. How to set optimal dosing regimen may be influenced by several factors including patient and/or caregiver preferences with regard to the frequency of administration, patient age and activity, patient adherence to prescribed treatment, vial size that best fit to therapeutic dose, etc. In addition, further argued-discussions are required about how to manage practical issues of emicizumab administration. For clinical practice, it is recommended to follow the approved prescribing information, which corresponds to a strict weight-based dosing. However, this is not always feasible owing to vial size and concentration of emicizumab it contains, bodyweight variations, etc. Whether to inject multiple doses, to change dose, or to round up or down to vial size, should be considered individually for each emicizumab-treated patient based on his age, weight, activity, own preference, but also vial availability, financial considerations, etc. These questions may be even more important to consider in children, because dosing adaptation will be required along the various stages of age in childhood (newborn, infant, pre-school age, school age, young adolescent). In addition, studies have shown that emicizumab does not provide sufficient hemostatic protection for serious trauma or major surgeries. Therefore, we are entering an era where different agents will be used for baseline prophylaxis and on-demand treatments. Conventional BPA and FVIII products will remain the main therapeutic options for on-demand treatment or prevention of bleeding during major surgery for HA patients with and without FVIII-inhibitors, respectively. This is of particular concerns because no validated assays exist for the accurate monitoring of the hemostatic effect of emicizumab used in combination with current therapeutic agents, which can put the patients at risk of thrombotic events. However, FVIII:C can be accurately measured using chromogenic FVIII assay based on bovine reagent, which are unaffected by the presence of emicizumab.26 All of these real-life situations highlight that the clinical management of HA patients receiving emicizumab may not be an easy task, despite the convenience of subcutaneous administration and infrequent dosing regimen. Therefore, we believe that the use of emicizumab prophylaxis should be overseen by experienced hemophilia comprehensive care centers. Hemophilia comprehensive care centers should be involved in additional clinical trials that are required to evaluate long-term safety of emicizumab prophylaxis in specific population such as HA children who will be offered ITI, as well as in PUPs. Long-term safety will be one of the key factors to take into account while choosing between lifelong emicizumab and other approaches for the treatment of severe HA, such as gene therapy. As pediatric patients get older, these with persistent FVIII-inhibitors may be at risk of exclusion from future gene therapy, whereas these without FVIII-inhibitors could eventually be offered gene therapy with a potential for a cure.
Abbreviations
ABR, annualized bleeding rate; ADA(s), anti-drug antibod(ies); AE(s), adverse event(s); aPCC, activated prothrombin complex concentrate; BPA(s), bypassing agent(s); BU, Bethesda units; CVAD, central venous access device; FVIII, factor VIII; HA, hemophilia A; HRQOL, health-related quality of life; ICH, intracranial hemorrhage; IQR, interquartile range; ITI, immune tolerance induction; NIS, Non-interventional Study; QW, once a week; Q2W, once every 2 weeks; Q4W, once every 4 weeks; rFVIIa, recombinant activated factor VII; rFVIII, recombinant factor VIII; pdFVIII, plasma-derived FVIII; TMA, thrombotic microangiopathy.
Acknowledgments
Acknowledgements go to Dr A. Veleno and Dr A. Gaude and their team from Roche/Chugai for providing useful references (scientific data about HAVEN 2 study) that helped writing this manuscript.
Disclosure
Sandra Le Quellec has received grants and/or honoraria for lectures and/or participation in advisory boards from CSL Behring, LFB, Shire, SOBI, Octapharma, Bayer. The author reports no other conflicts of interest in this work.
References
1. Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535–544. doi:10.1056/NEJMoa067659
2. Manco-Johnson MJ, Soucie JM, Gill JC. Joint outcomes committee of the universal data collection USHTCN. Prophylaxis usage, bleeding rates, and joint outcomes of hemophilia, 1999 to 2010: a surveillance project. Blood. 2017;129(17):2368–2374. doi:10.1182/blood-2016-02-683169
3. Rodriguez V, Mancuso ME, Warad D, et al. Central venous access device (CVAD) complications in haemophilia with inhibitors undergoing immune tolerance induction: lessons from the international immune tolerance study. Haemophilia. 2015;21(5):e369–e74. doi:10.1111/hae.12740
4. Gouw SC, van der Bom JG, Ljung R, et al. Factor VIII products and inhibitor development in severe hemophilia A. N Engl J Med. 2013;368(3):231–239. doi:10.1056/NEJMoa1208024
5. Peyvandi F, Mannucci PM, Garagiola I, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med. 2016;374(21):2054–2064. doi:10.1056/NEJMoa1516437
6. Calvez T, Chambost H, Claeyssens-Donadel S, et al. Recombinant factor VIII products and inhibitor development in previously untreated boys with severe hemophilia A. Blood. 2014;124(23):3398–3408. doi:10.1182/blood-2014-07-586347
7. Ettingshausen CE, Kreuz W. Early long-term FEIBA prophylaxis in haemophilia A patients with inhibitor after failing immune tolerance induction: a prospective clinical case series. Haemophilia. 2010;16(1):90–100. doi:10.1111/hae.2009.16.issue-1
8. Morfini M, Auerswald G, Kobelt RA, et al. Prophylactic treatment of haemophilia patients with inhibitors: clinical experience with recombinant factor VIIa in European haemophilia centres. Haemophilia. 2007;13(5):502–507. doi:10.1111/hae.2007.13.issue-5
9. Negrier C, Dargaud Y, Bordet JC. Basic aspects of bypassing agents. Haemophilia. 2006;12(Suppl 6):
10. Rosenfeld SB, Watkinson KK, Thompson BH, Macfarlane DE, Lentz SR. Pulmonary embolism after sequential use of recombinant factor VIIa and activated prothrombin complex concentrate in a factor VIII inhibitor patient. Thromb Haemost. 2002;87(5):925–926. doi:10.1055/s-0037-1613110
11. Aledort LM. Comparative thrombotic event incidence after infusion of recombinant factor VIIa versus factor VIII inhibitor bypass activity. J Thromb Haemost. 2004;2(10):1700–1708. doi:10.1111/jth.2004.2.issue-10
12. Walsh CE, Soucie JM, Miller CH. United States hemophilia treatment center N. Impact of inhibitors on hemophilia A mortality in the United States. Am J Hematol. 2015;90(5):400–405. doi:10.1002/ajh.v90.5
13. Monahan PE, Baker JR, Riske B, Soucie JM. Physical functioning in boys with hemophilia in the U.S. Am J Prev Med. 2011;41(6 Suppl 4):S360–S368. doi:10.1016/j.amepre.2011.09.017
14. Morfini M, Haya S, Tagariello G, et al. European study on orthopaedic status of haemophilia patients with inhibitors. Haemophilia. 2007;13(5):606–612. doi:10.1111/hae.2007.13.issue-5
15. DiMichele DM, Hoots WK, Pipe SW, Rivard GE, Santagostino E. International workshop on immune tolerance induction: consensus recommendations. Haemophilia. 2007;13(Suppl 1):1–22. doi:10.1111/j.1365-2516.2007.01497.x
16. Wight J, Paisley S, Knight C. Immune tolerance induction in patients with haemophilia A with inhibitors: a systematic review. Haemophilia. 2003;9(4):436–463. doi:10.1046/j.1365-2516.2003.00781.x
17. Lenting PJ, Denis CV, Christophe OD. Emicizumab, a bispecific antibody recognizing coagulation factors IX and X: how does it actually compare to factor VIII? Blood. 2017;130(23):2463–2468. doi:10.1182/blood-2017-08-801662
18. Kitazawa T, Igawa T, Sampei Z, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med. 2012;18(10):1570–1574. doi:10.1038/nm.2942
19. Uchida N, Sambe T, Yoneyama K, et al. A first-in-human phase 1 study of ACE910, a novel factor VIII-mimetic bispecific antibody, in healthy subjects. Blood. 2016;127(13):1633–1641. doi:10.1182/blood-2015-06-650226
20. Shima M, Hanabusa H, Taki M, et al. Factor VIII-mimetic function of humanized bispecific antibody in hemophilia A. N Engl J Med. 2016;374(21):2044–2053. doi:10.1056/NEJMoa1511769
21. Shima M, Hanabusa H, Taki M, et al. Long-term safety and efficacy of emicizumab in a phase 1/2 study in patients with hemophilia A with or without inhibitors. Blood Adv. 2017;1(22):1891–1899. doi:10.1182/bloodadvances.2017006684
22. Yoneyama K, Schmitt C, Kotani N, et al. A pharmacometric approach to substitute for a conventional dose-finding study in rare diseases: example of phase III dose selection for emicizumab in hemophilia A. Clin Pharmacokinet. 2017.
23. Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. 2017;377(9):809–818. doi:10.1056/NEJMoa1703068
24. den Uijl IE, Fischer K, Van Der Bom JG, Grobbee DE, Rosendaal FR, Plug I. Analysis of low frequency bleeding data: the association of joint bleeds according to baseline FVIII activity levels. Haemophilia. 2011;17(1):41–44. doi:10.1111/hae.2010.17.issue-1
25. Soeda T, Kitazawa T, Muto A, et al. In vitro characterization of ACE910, a humanized bispecific antibody to factors IXa and X. Haemophilia. 2014;20(S3):77.
26. Muller J, Pekrul I, Potzsch B, Berning B, Oldenburg J, Spannagl M. Laboratory monitoring in emicizumab-treated persons with hemophilia A. Thromb Haemost. 2019;119(9):1384–1393. doi:10.1055/s-0039-1692427
27. Pipe SW, Shima M, Lehle M, et al. Efficacy, safety, and pharmacokinetics of emicizumab prophylaxis given every 4 weeks in people with haemophilia A (HAVEN 4): a multicentre, open-label, non-randomised phase 3 study. Lancet Haematol. 2019;6(6):e295–e305. doi:10.1016/S2352-3026(19)30054-7
28. Chowdary P, Carcao M, Holme PA, et al. Fixed doses of N8-GP prophylaxis maintain moderate-to-mild factor VIII levels in the majority of patients with severe hemophilia A. Res Pract Thromb Haemost. 2019;3(3):542–554. doi:10.1002/rth2.12220
29. Lissitchkov T, Klukowska A, Pasi J, et al. Efficacy and safety of simoctocog alfa (Nuwiq(R)) in patients with severe hemophilia A: a review of clinical trial data from the GENA program. Ther Adv Hematol. 2019;10:2040620719858471. doi:10.1177/2040620719858471
30. Shah A, Coyle T, Lalezari S, et al. BAY 94-9027, a PEGylated recombinant factor VIII, exhibits a prolonged half-life and higher area under the curve in patients with severe haemophilia A: comprehensive pharmacokinetic assessment from clinical studies. Haemophilia. 2018;24(5):733–740. doi:10.1111/hae.13561
31. Swystun LL, Ogiwara K, Rawley O, et al. Genetic determinants of VWF clearance and FVIII binding modify FVIII pharmacokinetics in pediatric hemophilia A patients. Blood. 2019;134(11):880–891. doi:10.1182/blood.2019000190
32. Young G, Liesner R, Chang TY, et al. A multicenter, open-label, phase 3 study of emicizumab prophylaxis in children with hemophilia A with inhibitors. Blood. 2019;134(24):2127–2138.
33. Shima M, Nogami K, Nagami S, et al. A multicentre, open-label study of emicizumab given every 2 or 4 weeks in children with severe haemophilia A without inhibitors. Haemophilia. 2019. doi:10.1111/hae.13848
34. Mahlangu J, Oldenburg J, Paz-Priel I, et al. Emicizumab prophylaxis in patients who have hemophilia A without inhibitors. N Engl J Med. 2018;379(9):811–822. doi:10.1056/NEJMoa1803550
35. Jonsson F, Schmitt C, Petry C, Mercier F, Frey N, Retout S. Exposure-response modeling of emicizumab for the prophylaxis of bleeding in hemophilia A patients with and without inhibitors against factor VIII. Res Pract Thromb Haemost. 2019;3(Suppl 1):315.
36. Hay CR, Palmer B, Chalmers E, et al. Incidence of factor VIII inhibitors throughout life in severe hemophilia A in the United Kingdom. Blood. 2011;117(23):6367–6370. doi:10.1182/blood-2010-09-308668
37. Barg AA, Avishai E, Budnik I, et al. Emicizumab prophylaxis among infants and toddlers with severe hemophilia A and inhibitors-a single-center cohort. Pediatr Blood Cancer. 2019;66(11):e27886. doi:10.1002/pbc.27886
38. Catarino C, Campanico S, Rodrigues F, et al. Prophylaxis with emicizumab in children and adults with hemophilia A and inhibitors: preliminary data on 7 patients of a portuguese hemophilia center. Res Pract Thromb Haemost. 2019;3(Suppl 1):299.
39. Young G, Sidonio RF, Liesner R, et al. HAVEN 2 updated analysis: multicenter, open-label, phase 3 study to evaluate efficacy, safety and pharmacokinetics of subcutaneous administration of emicizumab prophylaxis in pediatric patients with hemophilia A with inhibitors. Blood. 2017;130(Suppl1):85.
40. Carcao M, Escuriola-Ettingshausen C, Santagostino E, et al. The changing face of immune tolerance induction in haemophilia A with the advent of emicizumab. Haemophilia. 2019;25(4):676–684. doi:10.1111/hae.13762
41. Young G. How I treat children with haemophilia and inhibitors. Br J Haematol. 2019;186(3):400–408. doi:10.1111/bjh.15942
42. Astermark J, Donfield SM, DiMichele DM, et al. A randomized comparison of bypassing agents in hemophilia complicated by an inhibitor: the FEIBA NovoSeven Comparative (FENOC) study. Blood. 2007;109(2):546–551. doi:10.1182/blood-2006-04-017988
43. Le Quellec S, Negrier C. Emicizumab should be prescribed independent of immune tolerance induction. Blood Adv. 2018;2(20):2783–2786. doi:10.1182/bloodadvances.2018015859
44. Young G. Implementing emicizumab in hemophilia inhibitor management: emicizumab should be prescribed after tolerance. Blood Adv. 2018;2(20):2780–2782. doi:10.1182/bloodadvances.2018015842
45. Dimichele DM, Hay CR. The international immune tolerance study: a multicenter prospective randomized trial in progress. J Thromb Haemost. 2006;4(10):2271–2273. doi:10.1111/jth.2006.4.issue-10
46. Nogami K, Matsumoto T, Tabuchi Y, et al. Modified clot waveform analysis to measure plasma coagulation potential in the presence of the anti-factor IXa/factor X bispecific antibody emicizumab. J Thromb Haemost. 2018;16(6):1078–1088. doi:10.1111/jth.2018.16.issue-6
47. Batsuli G, Zimowski KL, Tickle K, Meeks SL, Sidonio RF
48. Nilsson IM, Berntorp E, Lofqvist T, Pettersson H. Twenty-five years’ experience of prophylactic treatment in severe haemophilia A and B. J Intern Med. 1992;232(1):25–32. doi:10.1111/joim.1992.232.issue-1
49. Kruse-Jarres R, Oldenburg J, Santagostino E, et al. Bleeding and safety outcomes in persons with haemophilia A without inhibitors: results from a prospective non-interventional study in a real-world setting. Haemophilia. 2019;25(2):213–220. doi:10.1111/hae.13655
50. Klinge J, Auberger K, Auerswald G, Brackmann HH, Mauz-Korholz C, Kreuz W. Prevalence and outcome of intracranial haemorrhage in haemophiliacs–a survey of the paediatric group of the German Society of Thrombosis and Haemostasis (GTH). Eur J Pediatr. 1999;158(Suppl 3):S162–S165. doi:10.1007/PL00014346
51. Oldenburg J, Levy GG. Emicizumab prophylaxis in hemophilia A with Inhibitors. N Engl J Med. 2017;377(22):2194–2195. doi:10.1056/NEJMoa1703068
52. Hartmann R, Feenstra T, Valentino L, Dockal M, Scheiflinger F. In vitro studies show synergistic effects of a procoagulant bispecific antibody and bypassing agents. J Thromb Haemost. 2018. doi:10.1111/jth.14203
53. Mancuso ME, Callaghan MU, Kruse-Jarres R, et al. Emicizumab prophylaxis in adolescent/adult patients with hemophilia A previously receiving episodic or prophylactic bypassing agent treatment: updated analyses from the HAVEN 1 study. Blood. 2017;130(Suppl 1):1071.
54. Makris M, Iorio A, Lenting PJ. Emicizumab and thrombosis: the story so far. J Thromb Haemost. 2019;17(8):1269–1272. doi:10.1111/jth.v17.8
55. Knoebl P, Marco P, Baudo F, et al. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost. 2012;10(4):622–631. doi:10.1111/j.1538-7836.2012.04654.x
56. Chalmers EA. Heritable thrombophilia and childhood thrombosis. Blood Rev. 2001;15(4):181–189. doi:10.1054/blre.2001.0166
57. Yada K, Nogami K. Spotlight on emicizumab in the management of hemophilia A: patient selection and special considerations. J Blood Med. 2019;10:171–181. doi:10.2147/JBM.S175952
58. Samuelson Bannow B, Recht M, Negrier C, et al. Factor VIII: long-established role in haemophilia A and emerging evidence beyond haemostasis. Blood Rev. 2019;35:43–50. doi:10.1016/j.blre.2019.03.002
59. Rodriguez-Merchan EC, Valentino LA. Increased bone resorption in hemophilia. Blood Rev. 2019;33:6–10. doi:10.1016/j.blre.2018.05.002
60. Poon JL, Zhou ZY, Doctor JN, et al. Quality of life in haemophilia A: hemophilia Utilization Group Study Va (HUGS-Va). Haemophilia. 2012;18(5):699–707. doi:10.1111/j.1365-2516.2012.02791.x
61. von Mackensen S, Myrin Westesson L, Kavakli K, et al. The impact of psychosocial determinants on caregivers’ burden of children with haemophilia (results of the BBC study). Haemophilia. 2019;25(3):424–432. doi:10.1111/hae.2019.25.issue-3
62. De Moerloose P, Urbancik W, Van Den Berg HM, Richards M. A survey of adherence to haemophilia therapy in six European countries: results and recommendations. Haemophilia. 2008;14(5):931–938. doi:10.1111/hae.2008.14.issue-5
63. Wiley RE, Khoury CP, Snihur AWK, et al. From the voices of people with haemophilia A and their caregivers: challenges with current treatment, their impact on quality of life and desired improvements in future therapies. Haemophilia. 2019;25(3):433–440. doi:10.1111/hae.13754
64. Ferreira AA, Leite IC, Bustamante-Teixeira MT, et al. Health-related quality of life in hemophilia: results of the hemophilia-specific quality of life index (Haem-a-Qol) at a Brazilian blood center. Rev Bras Hematol Hemoter. 2013;35(5):314–318. doi:10.5581/1516-8484.20130108
65. Khair K, Klukowska A, Myrin Westesson L, et al. The burden of bleeds and other clinical determinants on caregivers of children with haemophilia (the BBC study). Haemophilia. 2019;25(3):416–423. doi:10.1111/hae.2019.25.issue-3
66. Poon JL, Doctor JN, Nichol MB. Longitudinal changes in health-related quality of life for chronic diseases: an example in hemophilia A. J Gen Intern Med. 2014;29(Suppl 3):S760–S766. doi:10.1007/s11606-014-2893-y
67. Witkop M, Neff A, Buckner TW, et al. Self-reported prevalence, description and management of pain in adults with haemophilia: methods, demographics and results from the Pain, Functional Impairment, and Quality of life (P-FiQ) study. Haemophilia. 2017;23(4):556–565. doi:10.1111/hae.2017.23.issue-4
68. Mahlangu J, Oldenburg J, Callaghan MU, et al. Health-related quality of life and health status in persons with haemophilia A with inhibitors: a prospective, multicentre, non-interventional study (NIS). Haemophilia. 2019;25(3):382–391. doi:10.1111/hae.2019.25.issue-3
69. Pollak E, Muhlan H, Von Mackensen S, Bullinger M, Haemo-Qol G. The Haemo-QoL Index: developing a short measure for health-related quality of life assessment in children and adolescents with haemophilia. Haemophilia. 2006;12(4):384–392. doi:10.1111/j.1365-2516.2006.01292.x
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.