")
Back to Journals » Journal of Blood Medicine » Volume 13
Clinical Epidemiology, Treatment Outcome and Mortality Rate of Newly Diagnosed Immune Thrombocytopenia in Adult Multicentre Study in Malaysia
Authors Hamzah R , Yusof N, Tumian NR , Abdul Aziz S , Mohammad Basri NS , Leong TS , Ho KW , Selvaratnam V, Tan SM, Muhamad Jamil SA
Received 24 January 2022
Accepted for publication 22 April 2022
Published 21 June 2022 Volume 2022:13 Pages 337—349
DOI https://doi.org/10.2147/JBM.S358993
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Martin H Bluth
Roszymah Hamzah,1,2 Nurasyikin Yusof,1 Nor Rafeah Tumian,3 Suria Abdul Aziz,1 Nur Syahida Mohammad Basri,1 Tze Shin Leong,2 Kim Wah Ho,2 Veena Selvaratnam,2 Sen Mui Tan,2 Siti Afiqah Muhamad Jamil4
1Department of Pathology, Faculty of Medicine, Universiti Kebangsaan Malaysia Medical Center, Kuala Lumpur, Malaysia; 2Department of Haematology, Ampang Hospital, Ampang, Selangor, Malaysia; 3Haematology Unit, Department of Medicine, Universiti Kebangsaan Malaysia Medical Center, Kuala Lumpur, Malaysia; 4Faculty of Computer and Mathematical Sciences, Universiti Teknologi Mara, Shah Alam, Selangor, Malaysia
Correspondence: Nurasyikin Yusof, Department of Pathology, Faculty of Medicine, Universiti Kebangsaan Malaysia Medical Center, Jalan Yaacob Latif, Bandar Tun Razak, Kuala Lumpur, 56000, Malaysia, Tel +60 3 91455373, Fax +60 3 91459485, Email [email protected]
Background: Immune thrombocytopenia (ITP) is well characterized in Western, European and other Asia-Pacific countries. Nevertheless, the clinical epidemiology, treatment pattern and disease outcome of ITP in Malaysia are still limited and not well known.
Objective: This study aimed to describe the clinical epidemiology, treatment outcome and mortality of ITP patients in haematology tertiary multicentre in Malaysia.
Methods: Clinical and laboratory data of newly diagnosed adults with ITP by a platelet count < 100 × 109/L from January 2010 to December 2020 were identified and analyzed.
Results: Out of 500 incident ITP, 71.8% were females with a striking age preponderance of both genders among those aged 18– 29 years. The median age was 36 years. The median platelet count was 17.5 × 109/L, 23.0% had a secondary ITP, 34.6% had a Charlson’s score ≥ 1, 53.0% had bleeding symptoms including 2.2% intracranial bleedings (ICB). Helicobacter pylori screening was performed in < 5% of cases. Persistency and chronicity rates were 13.6% and 41.8%, respectively. Most (80.6%) were treated at diagnosis onset and 31.2% needed second-line treatment. Throughout the course of ITP, 11.0% of patients died; 3.0% and 8.0% with bleeding and non-bleeding related ITP.
Conclusion: This study confirms the epidemiology of ITP is comparable with worldwide studies. Our incidence is high in the female, Malay ethnicity, primary ITP and events of cutaneous bleeding at ITP onset with 18– 29 years predominance age group for both genders. The frequency of persistent and chronic ITP is inconsistent with published literature. Corticosteroids and immunotherapies are the most prescribed first-line and second-line pharmacological treatments. Thrombopoietin receptor agonist medications (TPO-RAs) usage is restricted and splenectomy is uncommon. Our mortality rate is similar but ITP related bleeding death is fourth-fold lower than previous studies. Mortality risks of our ITP patients include age ≥ 60 years, male, severe bleeding at presentation, CCI≥ 1 and secondary ITP.
Keywords: epidemiology, immune thrombocytopenia, mortality, treatment outcome
Introduction
Immune thrombocytopenia (ITP) formerly described as idiopathic or immune thrombocytopenic purpura1 is an acquired autoimmune bleeding disorder secondary to premature platelet destruction, decelerated platelet production or a combination of both leading to thrombocytopenia and life-threatening bleeding in some patients.2–5 In the last several years, the basic understanding of the pathophysiology of ITP has significantly improved but the epidemiology and clinical course of ITP have not been well investigated in the general population, particularly in the Asian population. The current literature includes a limited number of published studies, each of which describes a relatively small number of ITP patients.6 ITP is an old disease characterized by bleeding disorder comprises heterogeneous clinical characteristics and response to therapy. Isolated thrombocytopenia (defined as platelet count <100 × 109/L) without an evident predisposing etiology is considered primary ITP as opposed to secondary ITP that is associated with coexisting systemic disease.7,8 ITP remains the exclusion of other causes of thrombocytopenia as no gold standard test to confirm ITP diagnosis. In adult ITP patients, bleeding is the most significant cause of morbidity, mortality and decision to start treatment.9 Treatment is indicated if the platelet counts less than 20 × 109/L to 30 × 109/L, even in the absence of bleeding manifestation. However, the risk factors that influence bleeding such as older age (eg >65 years), type of ITP, previous bleeding history, exposure to anticoagulants or antiplatelet drugs, the presence of comorbidities such as renal impairment and the risk of trauma from daily activities should be assessed before a decision to start treatment.10–13
First-line therapy for ITP is based on oral corticosteroids, plus administration of intravenous immunoglobulin (IVIg) in case of severe bleeding.11,14 Majority of patients will respond to initial corticosteroid treatment but relapse after the dose is reduced. Only about one-third of treated patients can expect a long-term response.15 Although chronic refractory patients are a minority, they appear to be at the highest risk of severe bleeding and mortality.16–18 ITP becomes persistent (lasting >3 months) or chronic (lasting >12 months) in about 70% of adult cases.19 Various corticosteroid-sparing agents are used in these cases such as splenectomy, rituximab or thrombopoietin receptor agonists (TPO-RAs) such as eltrombopag or romiplostim.11,14,20 Splenectomy is now frequently delayed until late in the disease course as it is an effective treatment for steroid-refractory or steroid-dependent ITP.21,22
Although ITP disease has been studied for more than a century, its robust clinical epidemiological data is not well known in Malaysia. This retrospective study was conducted to analyze the clinical epidemiology, treatment outcome and mortality of newly diagnosed adult ITP patients in the tertiary hospitals in Malaysia. To the best of our knowledge, this is the first article involving a multicentre study on the clinical epidemiology, treatment outcome and mortality of adult ITP in Malaysia at present.
Methods
Study Design
All consecutive adult patients with newly diagnosed ITP from January 2010 to December 2020 were identified through the discharge codes (International Classification of Diseases, 7th–10th). Codes D69.3 (immune thrombocytopenia) from the 10th revision of the International Classification of Diseases (ICD-10) was used in this retrospective, observational multicentre study. All data were retrieved from the database of the International Centre for Casemix and Clinical Coding for Hospital Canselor Tuanku Mukhriz, Universiti Kebangsaan Malaysia Medical Centre (HCTM, UKMMC), Kuala Lumpur, Malaysia and from the database of new cases in Haematology Department, Ampang Hospital, Selangor, Malaysia. Diagnosis of ITP was validated using medical records and Integrated Laboratory Information System (ILMS). There was no duplicated data between the two registries of patients included in this study.
Study Population
An adult was defined as a patient by an age of 18 years and above. Platelet count <100 × 109/L without a secondary cause was defined as ITP in our study as outlined by Rodeghiero et al.19 We obtained information on patients’ demographic and clinical data which includes age, gender, ethnicity, hospital visits, date of ITP diagnosis, platelet count and bleeding types at presentation, laboratory values, types and initiation date of pharmacological therapy and duration of ITP-treatment from the registered data and medical records.
Definitions
Phases of ITP disease were defined according to the International Working Group (IWG); 1. Newly diagnosed ITP: from the time of diagnosis up to 3 months; 2. Persistent: between 3 and 12 months from diagnosis; 3. Chronic phase: disease duration of more than 12 months.23,24 We defined treatment outcome to ITP treatments following American Society of Haematology (ASH) terminology: Complete response (CR): platelet count ≥100 × 109/L and absence of bleeding; Response (R): platelet count ≥30 × 109/L and at least 2-fold increase the baseline count and absence of bleeding; No response (NR): platelet count <30 × 109/L or less than 2-fold increase of baseline platelet count or bleeding; Loss of CR or R: platelet count below 100 × 109/L or bleeding (from CR) or below 30 × 109/L or less than 2-fold increase of baseline platelet count or bleeding (from R).19
Mortality
The outcomes of the patients including the date of death were recorded. For patients who passed away during the observation period, the cause of death was investigated by manually reviewing their medical records to identify diagnoses listed at the time of death. These were grouped into ITP related to bleeding and ITP with no specific bleeding.
Statistical Analysis
Patients’ clinical characteristics were analyzed using simple descriptive statistics, mean ± standard deviation or median and range for quantitative variables and percentages, n [%] for qualitative or categorical variables. All potential risk associations were assessed by Pearson chi-square test for categorical data. Data were considered statistically significant at p-values <0.05. Mortality relative risk was computed by odds ratio (OR) and 95% confidence interval (CI) for observed number of deaths. Statistical Package for the Social Sciences (IBM-SPSS) version 27 was used to perform all data analysis.
Ethics Approval
This study was conducted in accordance with the Declaration of Helsinki. As this is a retrospective study, informed consent is not required. The study was approved by the Universiti Kebangsaan Malaysia Medical Research and Ethics Committee (UKM MREC FF-2021-080) and National Medical Research Register (NMRR-20-2969-57701), regional ethical board in Malaysia. Permission to obtain data was approved by the respective unit and was kept anonymized and confidentiality was maintained using coding.
Results
Patient Selection
In a database of 740604 of total admission over the study period, we identified 832 patients with a diagnosis encoded D69.3 ICD-10 at any age. Following exclusion criteria, 330 patients were excluded including 12 with a platelet count at ITP diagnosis of ≥100 × 109/L but <150 × 109/L. Consequently, only 500 patients were included in the study.
Clinical Epidemiology at ITP Diagnosis
In total, 500 adult patients with newly diagnosed ITP were included in the cohort. Demographic characteristics of ITP patients are expressed in Table 1. The median age at ITP diagnosis of the entire population was 36 years, range 18–84 years. The incidence was more common in females, 359 (71.8%) with a median age of 35 years (18–84 years). The male-to-female ratio was 1:2.5. Male patients were slightly older with a median age of 41 years (range 18–82 years). The most frequent age at diagnosis was those aged 18–29 years in both genders (Figure 1).
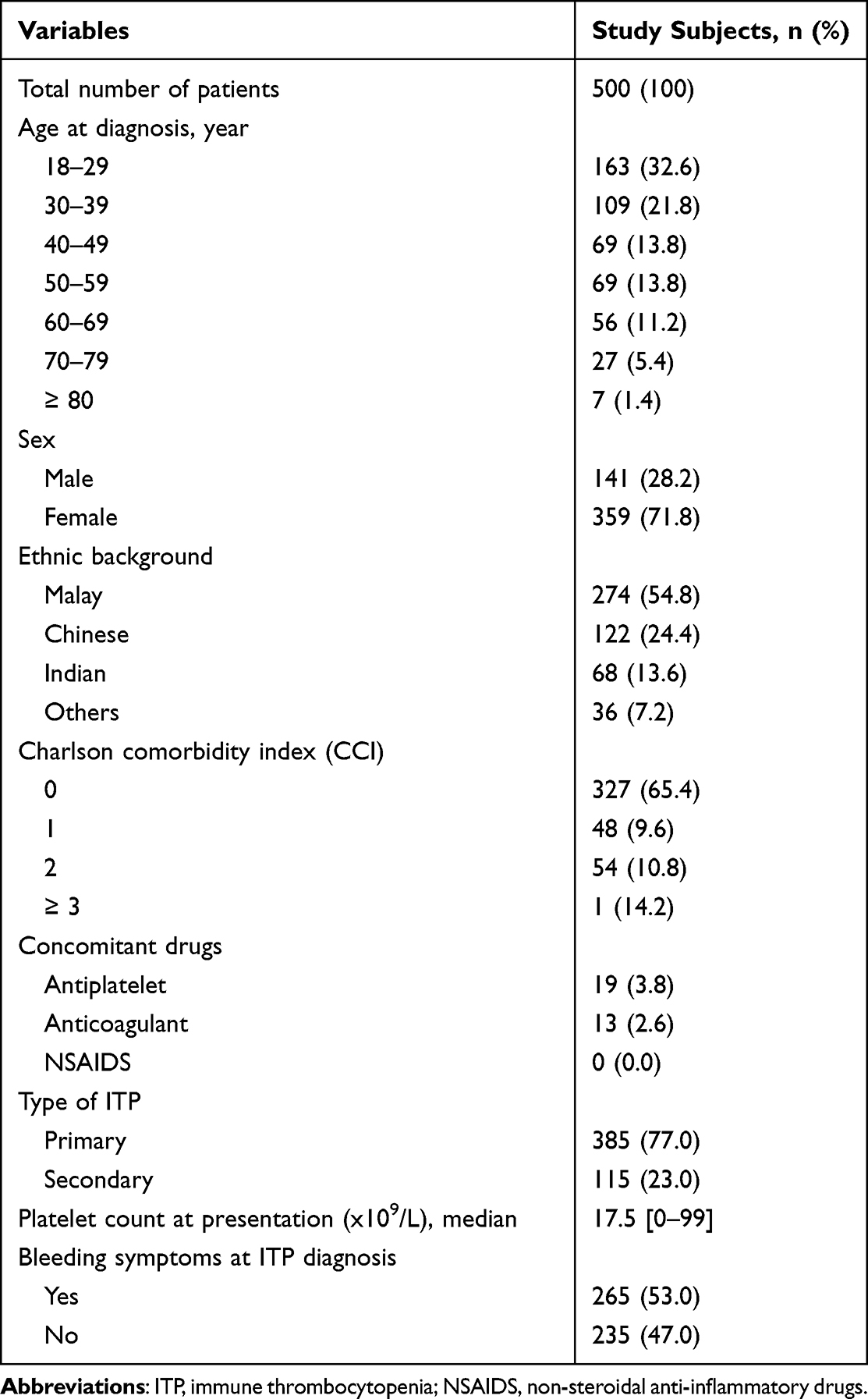 |
Table 1 Demographic Characteristics of ITP Patients |
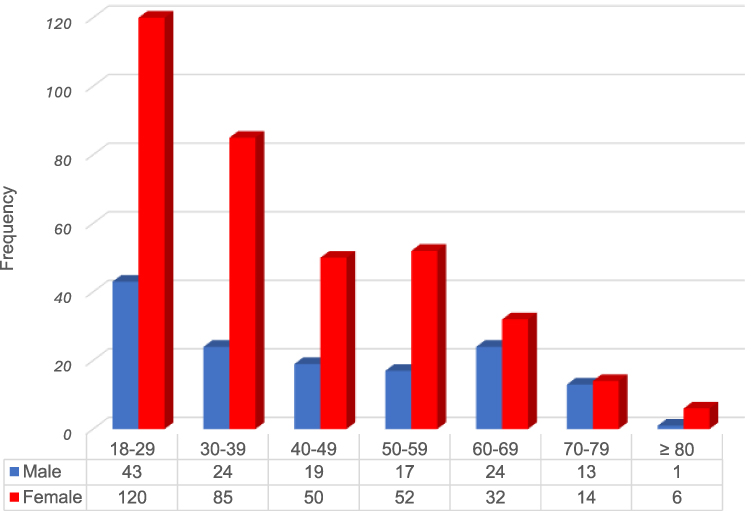 |
Figure 1 Distribution of age and gender of ITP patients. |
Ethnic background is presented in Table 1. The percentage of patients was higher in Malays and smaller in the other groups. Other ethnicities include Indonesian, Siamese, Myanmar, Australian and African. Two hundred and five patients (41%) cases had one or more comorbidities including hypertension, diabetes mellitus, autoimmune disease, renal and heart disease. We observed 173 patients had Charlson comorbidity index (CCI) of ≥1 (14.2% had a CCI of ≥3).
Out of all cohort patients, only 19 patients were on antiplatelet drugs and 13 patients were exposed to the anticoagulant, but none had exposure to non-steroidal anti-inflammatory drugs (NSAIDS). A total of 385 patients with primary ITP and 115 with secondary ITP were diagnosed on both participating sites. The secondary causes were related to other conditions including autoimmune disease (n = 79, 15.8%), viral infections (n = 22, 4.4%), malignancy (n = 6, 1.2%) and drug-induced (n = 8, 1.6%). Eight patients were identified with Evans syndromes.
Clinical symptoms of bleeding at diagnosis were assessed by location; no bleeding, skin, oral cavity, nose, gynaecological, gastrointestinal tract (GIT), genitourinary, intracranial, pulmonary, and ocular (Figure 2). Bleeding manifestations (any site) were observed more frequently in females (194 patients, 73.2%) than in males (71 patients, 26.8%) (p = 0.458) but no difference in age between males and females (peak at age 18–28 years). Multiple sites (≥1 site) of bleeding have been observed in 134 patients of the bleeding subgroup with cutaneous bleeding as the most common presentation.
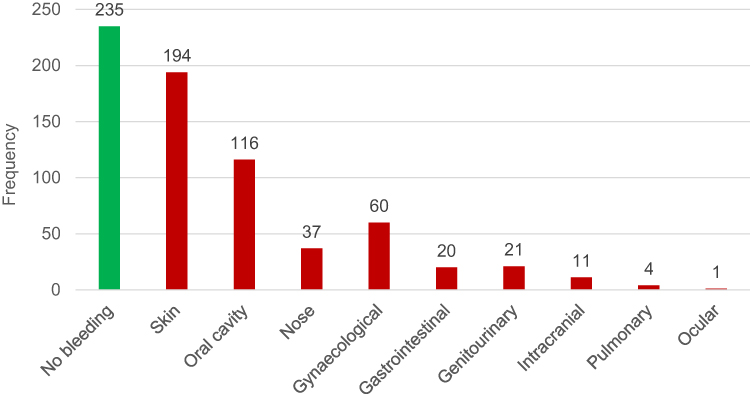 |
Figure 2 Bleeding symptoms at presentation by anatomical site. |
The median platelet count at diagnosis was 17.5 × 109/L (range 0–99 × 109/L) and 257 (51.4%) patients showed value <20 × 109/L at presentation. The distribution of the platelet count at initial diagnosis is shown in Figure 3. If data are analyzed in patients with a presenting platelet of <20 × 109/L, bleeding occurred in 190 (71.7%) of cases (p <0.001). ICB occurred in 11 of cases (2.2%) (p = 0.002), by which 2 patients with the platelet of ≥20 × 109/L. The mean age of patients with intracranial hemorrhage was 40 years. The median platelet at diagnosis was 7.0 × 109/L (range 1–23 × 109/L). Among them, only one patient died due to massive ICB after 5 days of admission. No association was found between risk of ICB and any of the following; age, gender, ethnicity, comorbidity, type of ITP or drug exposure except for platelet counts (p = 0.041).
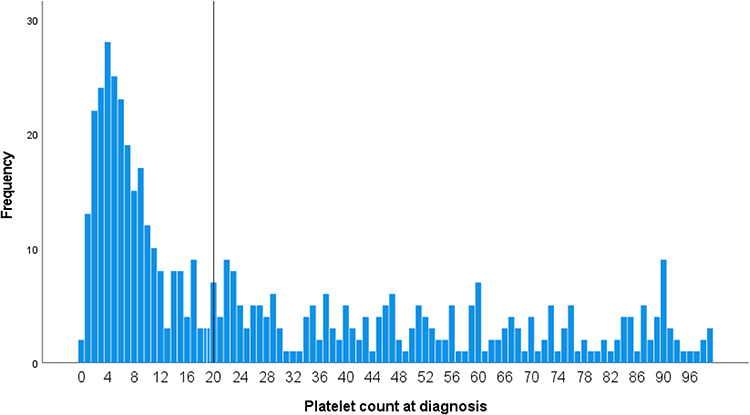 |
Figure 3 Platelet count at the time of ITP diagnosis. The vertical line indicates a platelet count of 20 × 109/L. |
Bone marrow examinations were performed in 134 (26.8%) cases, more often in patients who needed corticosteroids in contrast to other treatments. An antiphospholipid antibody, rheumatoid factor and Helicobacter pylori (H.pylori) testing were performed in less than 50% of patients. Antinuclear antibody (ANA) was the frequently positive laboratory test, while testing for H. pylori was not routinely done in ITP patients (Table 2).
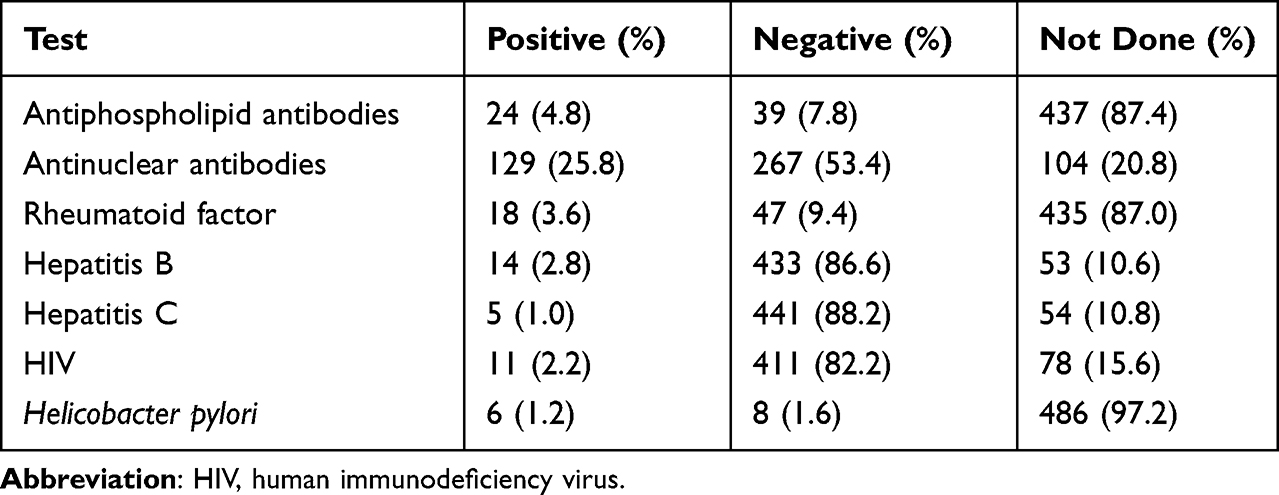 |
Table 2 Laboratory Test Results |
Treatment Exposure at Diagnosis
A total of 403 (80.6%) patients received the first ITP treatment during the study period. Among this subgroup, 246 (61.0%) were presented with bleeding and 157 (39.0%) without bleeding. All patients have recorded platelets count at the clinic before the start of treatment. Among the patients who did not need ITP treatment, the mean platelet count was 60.3 × 109/L. The median platelet count at treatment start was 11.0 × 109/L (range 0–99 × 109/L). Among them, 298 (59.6%) and 234 (46.8%) had platelet counts checked at 3 months and 12 months, respectively, after treatment started. In the subgroup of patients requiring treatment, 77 (19.1%) had severe bleedings and were mostly in the age group 18–29 years. Seventeen (4.2%) patients were aged 60 and above.
Corticosteroids were the most used first-line treatment. Corticosteroids alone were given in 398 patients (98.8%) in which all of them received high-dose prednisolone 1mg/kg at the initial treatment. Intravenous (IV) methylprednisolone was pulsed in 258 (51.6%) patients at starting of therapy while concurrent intravenous hydrocortisone and dexamethasone were given in 0.7% and 0.5% patients, respectively. Out of 104 (20.8%) who received platelet transfusion, 83 (79.8%) was due to platelet <20 × 109/L, 84 (80.8%) presented with any bleeding and 47 (45.2%) with severe bleeding. Among them, 4 patients with platelet >20 × 109/L were given platelet transfusion prior to surgical procedures. Second-line therapy is illustrated in Table 3.
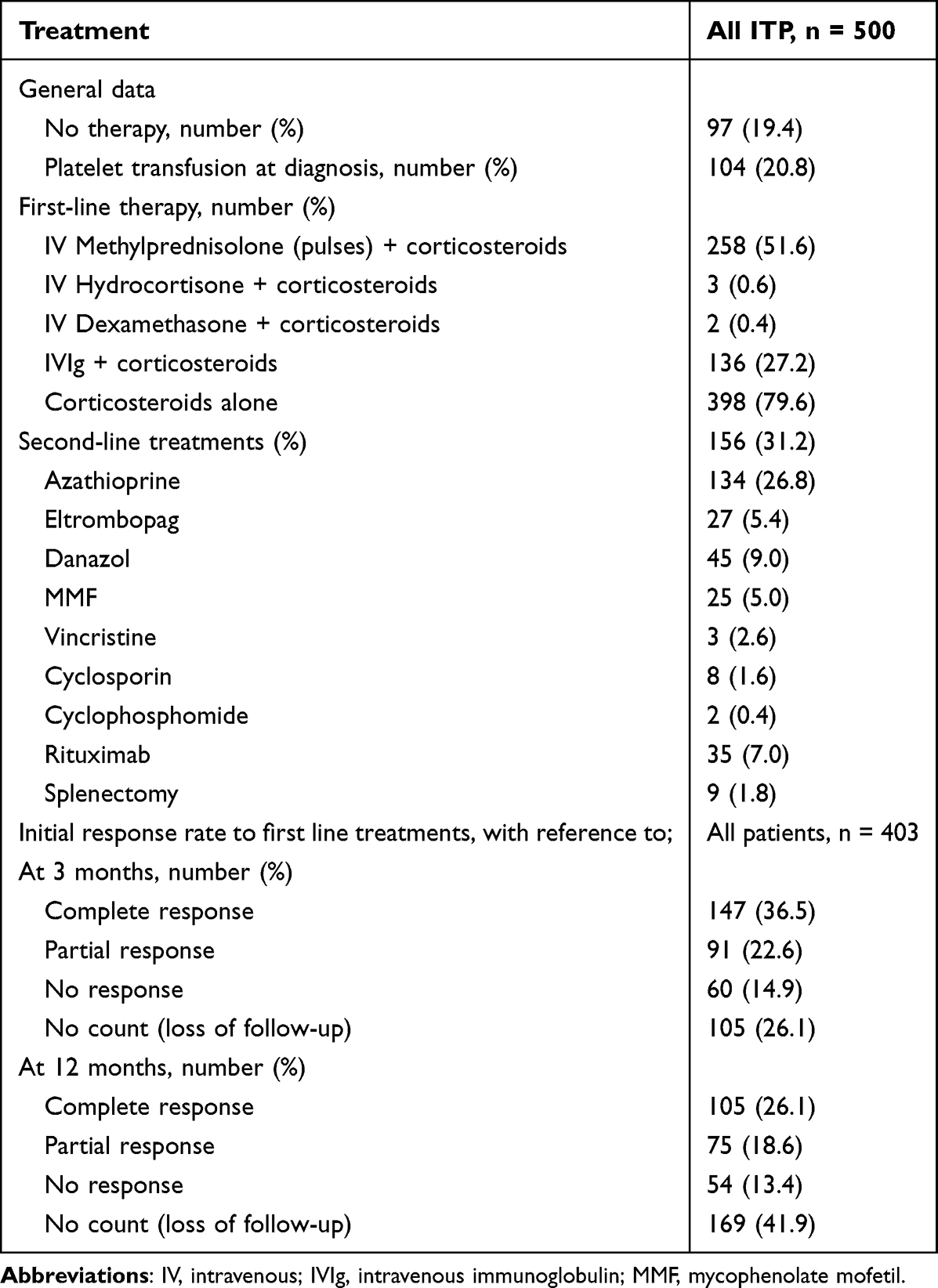 |
Table 3 Treatment Exposure at Diagnosis |
Treatment Outcomes and Mortality
The mean follow-up was 69 months after the first diagnosis. ITP course at 3 and 12 months is observed in the whole cohort by platelet counts. Out of the remaining 97 patients who did not require initial treatment, 15 (15.5%) had normal platelet count >100 × 109/L at 3 months and 23 (23.7%) at 12 months without any active treatment. Among those who did not require initial treatment, only 2 (2.1%) patients had platelet count <30 × 109/L at 3 months and none at 12 months monitoring. The initial response to first-line treatment is shown in Table 3. Disease outcome was assessed at the time of analysis according to their platelet count response at least of 3 to 12 months consequence follow-up. Acute ITP was observed in 185 (37.0%) patients, 68 (13.6%) became persistent ITP and 209 (41.8%) progress to chronic ITP (cITP). Another 38 (7.6%) cases were lost to follow-up. Among those who progress to persistent and cITP, 23 (33.8%) and 85 (40.7%) required second-line therapy, respectively.
At the time of analysis, 445 patients were alive (89.0%) whereby 55 patients had died (11%). Table 4 shows the mortality risk observed in our cohort. The death was two-fold in male patients with a mortality risk of 2.0 (95% CI, 1.2–3.2). When patients were reclassified as young age (<60 years) and old age (≥60 years), the old age had a higher mortality risk 2.8 (95% CI, 1.5–5.1). ITP patients presenting with severe bleeding symptoms suffered a 2.6-fold (95% CI, 1.4–4.9) increased mortality risk. CCI ≥1 and secondary ITP accelerated the mortality risk by 3.9 (95% CI, 2.2–7.0) and 2.1 (95% CI, 1.2–3.8), respectively.
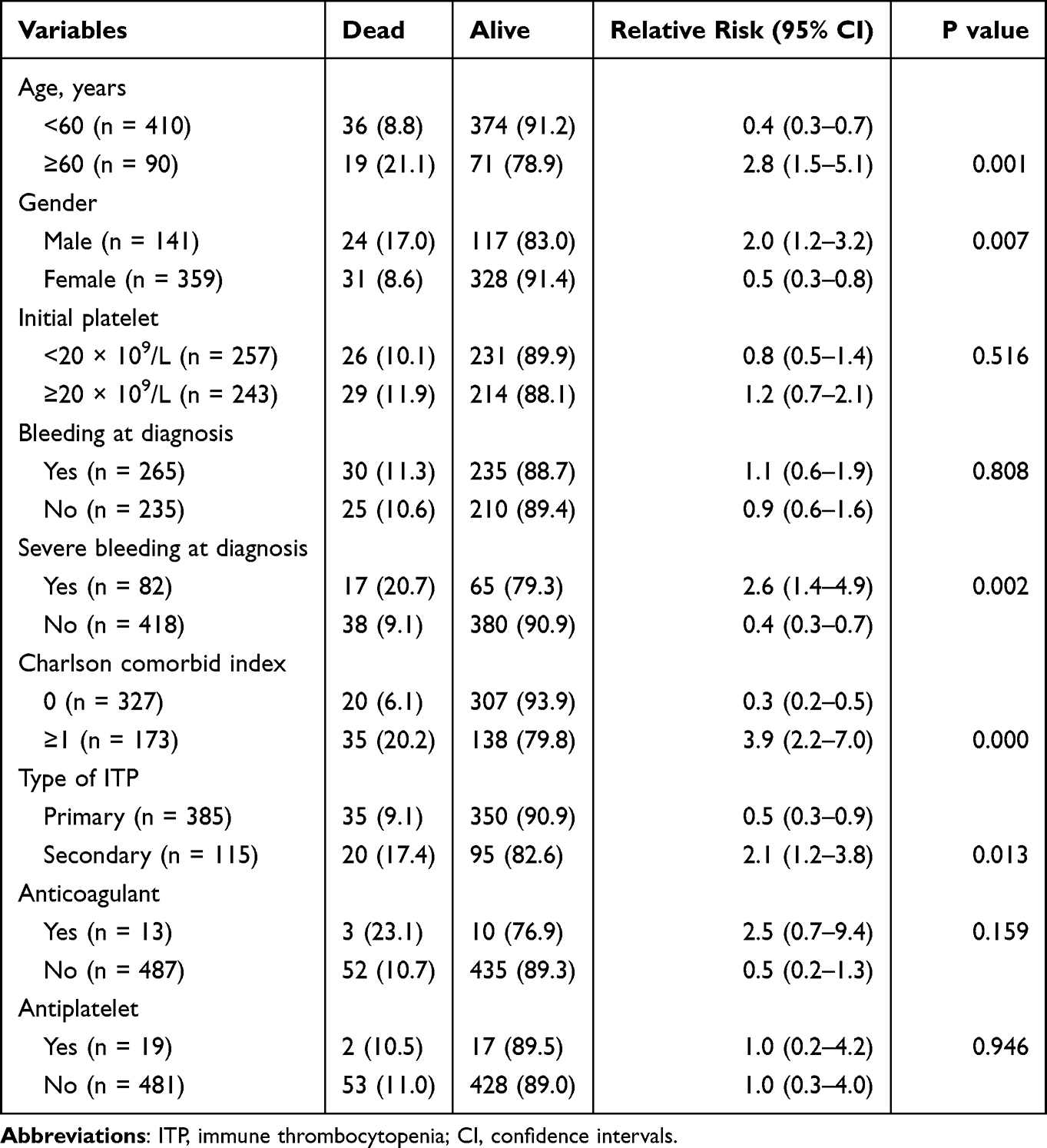 |
Table 4 Mortality Risk of ITP Patients |
Causes of death were classified into ITP related and non-ITP related deaths. A total of 15 (3.0%) patients with newly diagnosed ITP died due to bleeding ITP-related deaths during the entire period of follow-up. Among this subgroup, 7 were ICB, 7 with upper gastrointestinal bleeding (UGIB) and one case with epistaxis and massive bleeding. Causes of death considered unrelated to ITP or bleeding ITP consisted of malignancy (6), cardiovascular disease (7), severe infection (15), multiorgan failure (3), multiple underlying comorbid (3), both sepsis and underlying malignancy (1), trauma (1), old age (3) and one (1) undetermined cause of death.
Discussion
Clinical epidemiology, laboratory tests and mortality of newly diagnosed ITP in adults have not been systematically described in previous studies including in Asia and Malaysia. Published research suggests that there are differences in the clinical manifestations of ITP with variable clinical presentations, clinical consequences and treatment responses in different ages.11,25 Understanding the similarities and differences could affect the clinical practice and may provide a starting point for future study of the underlying etiologies and pathophysiological causes in each age group. The establishment of a regional ITP registry plays an important role to gather all the data, as a valuable resource for studies of rare diseases at the population level, clinical trials, improve treatment, disease burden and outcomes for all ITP patients.26 In addition, the local population study also will be able to guide future planning of social, health policies and services.27 Although there are few tertiary centres for referral for haematology cases in Malaysia, the national ITP registry has not yet been established. Currently, Japan is the only country in the Asia-Pacific region that has a national ITP registry.19,28
The incidence of ITP is reported in 1.5 to 5 per 100000 persons in the general population for both adults and children.6,29–33 The incidence of adult ITP patients is illustrated in Table 5. A multimodal incidence with 1 peak seen in childhood whereby the second and third peaks observed in young adults and the elderly.34 Our study confirms the higher incidence of ITP in females in younger adults as reported by previous population-based literature but not in the elderly (>65 years). The prevalence of ITP in men and women in the elderly is almost even in previous studies.4,6,18 We identify only one predominance peak of age at 18–29 years old for both genders. We did not see any multimodal peak as described in European4 and Japanese studies.28 It has previously been reported that most cases of adult ITP occur between 20 and 40 years old in female patients and that it is less common after 50 years old.35–37 These epidemiological data were similar in our study, instead not only in female but also in male patients. The incidence of ITP was found to be higher in Malay (54.9%) ethnicity. This is most likely contributed by the Malay being the highest ethnicity group in Malaysia.
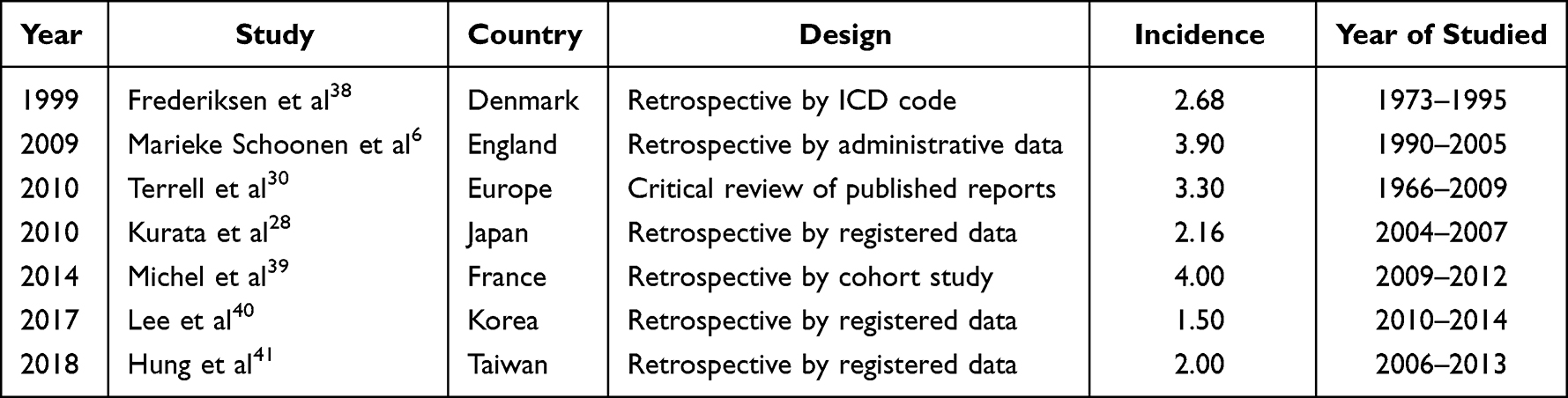 |
Table 5 Incidence of Adult ITP Patients in General Population |
This study demonstrated that infective screening (hepatitis B, hepatitis C and HIV) is frequently done (>80%) in our practice compared to autoimmune antibodies screening (<10% except ANA test). The antiphospholipid antibody, ANA and rheumatoid factor were not performed in all patients, restricting the analysis to exclude secondary causes and may underestimate the incidence. Furthermore, ANA provides information about the baseline risk of thrombosis which is associated with systemic lupus erythematosus (SLE)42 or antiphospholipid syndrome (APS).43 ITP patients concomitant with APS have an increased risk of thrombotic complications which may be higher if given TPO-RAs medications. For ITP patients at risk of thrombotic complications, there is limited evidence for the use of anticoagulation but stopping anticoagulation can result in more harmful outcomes than continuing anticoagulation.44 Positive H. pylori testing is common with increasing age and the prevalence of pathogenic strains can depend upon the country of birth. The overall prevalence rate of H. pylori infection was 35.9% in Malaysia and differs in the different regions within Malaysia. The Malay populations have the lowest prevalence of H. Pylori infection (11–29%) compared to Indians and Chinese, 49–52.3% and 26.7–57.5%, respectively.45,46 Despite the fact that the prevalence of H. pylori infection is high in Malaysia, the screening test by H. pylori serology and confirmation of active infection with a C14 urea breath test is not routinely done in our study (<5.0%). Moreover, eradication of H. pylori may rise a platelet count up to 50% of ITP patients with H. pylori–positive based on studies that were mostly conducted in Japan.47,48 Our frequency of secondary ITP (23%) is almost equal to the estimated incidence (~20%) by the expert opinion.41,49–51 In contrast, the prevalence of secondary ITP was substantially higher (64.8%) as compared to primary ITP (35.2%) in Pakistan.52 Our data also support that the female patients have a higher association with autoimmune disorders (n = 67, 18.6%) for secondary ITP, which may be an explanation for the greater number of women with ITP.
The incidence of bleeding events (53.0%) is in following previous French and the United Kingdom (UK) cohorts.12,53,54 Figure 2 presents the incidence of bleeding according to the site of bleeding at presentation. Estimated incidence, the weighted proportion of ICB and severe non-ICB bleeding have been reported in 1.4% (95% CI, 0.9–2.1%) and 9.6% (95% CI, 4.1–17.1%), respectively, in adults with ITP.55 The event of bleeding and rarity of serious bleeding at ITP onset in our cohort study is comparable with previous studies. Nonetheless, the incidence of ICB is higher but fewer GIT bleeding cases (2.2% and 4.0% respectively) compared to <1% for ICB and ~6.0% for GIT in the past.54,56,57 There was no association of ICB with age, gender, ethnicity, comorbidity, type of ITP or drugs exposure except for platelet counts (p = 0.041). The incidence of persistent and chronic disease in our study is smaller (<50%), inconsistent with France and Swiss populations.58–60 Interestingly, the persistent disease (13.6%) is much lower but higher rate of cITP (41.8%) in our population compared to Taiwanese population (24% persistent and 12.3% cITP). Our study demonstrated that 23 (18.0%) and 8 (6.3%) of partial response and no response ITP patients, respectively, at 3 months monitoring has switched into remission status at 12 months follow-up (p < 0.001). Out of 23 partial response who became remission, only 6 (26.1%) patients needed second-line treatment while 2 patients did not require further therapy among no response cases.
The rate of patients treated at the onset of ITP diagnosis (80.6%) was in accordance with the recent study by Grimaldi-Bensouda et al (87.4%).60 Corticosteroids remain the most frequently used first-line treatment followed by IVIg. A scoring system such as Khellaf’s bleeding score >8 has been recommended by the French group to aid clinicians in adding IVIg to corticosteroids particularly in patients with life-threatening bleeding when a rapid increase in platelet counts is required.61,62 This practice can also be implemented among Malaysian physicians. The use of anti-D Ig in the Asia-Pacific region is uncommon due to its ability to cause hemolysis and lack of availability in many countries in Asia.63,64 None of the patients was given anti-D Ig in our study. Second-line therapy (31.2%) is less frequently used in our population as compared to French nationwide population-based study (47.1%).20,51 Recent ASH ITP guidelines recommend TPO-RAs rather than rituximab;65 however, our rate of TPO-RAs usage is not as high as reported in ITP World Impact Survey (iWISh study).66 TPO-RAs such as eltrombopag or romiplostim are an option in patients after failure of steroids and splenectomy, prior to the use of rituximab and also can reduce the use of concomitant ITP medication.67–70 Nevertheless, long-term treatment should be used with caution as the most frequent adverse effects were headache, thrombocytosis and the risk of fibrosis in the bone marrow.69,71 Splenectomy is hardly practiced nowadays and was performed in only 9 (1.8%) of our refractory patients, as compared to 6–34% in the other countries.6 Splenectomy is not advisable in secondary ITP associated with an autoimmune or connective tissue disease since a subsequent flare of the connective tissue disease may necessitate immunosuppression. Asplenic patients have a higher risk of infection and the use of immunosuppressants is associated with poor survival in these patients.72
The mortality rate (11.0%) in our cohort is relatively consistent with the UK study (12.1%) but slightly lower compared to Netherland’s study (15.8%). Mortality risks of our ITP patients include age ≥60 years, male, severe bleeding at presentation, CCI ≥1 and secondary ITP. Surprisingly, platelet counts at ITP onset and medications (anticoagulant and antiplatelet) were not related to mortality risk in our study population. Of the 55 ITP patients for whom a cause of death was recorded, 3% died due to bleeding, which is a very small proportion than reported previously (~13.0%).53,73 This is probably due to established guidelines from the ITP-IWG and ASH on ITP management, with the inclusion of eltrombopag, romiplostim and rituximab in the treatment of cITP. Newer agents such as sutimlimab, fostamatinib and decitabine were shown promising results in recent a ITP study.74
The strengths of our study include the ability to include all cases of adult ITP during the study period. Thus, there is no selection bias of a certain group of ITP patients. The records from the computerized database were able to provide a relatively large number of cases. The study findings are comparable with other studies worldwide because we used IWG definitions and standardized terms and definitions. Nevertheless, this study has some limitations. As this study was done retrospectively in the tertiary hospital of haematology centres, the data were restricted which may contribute to analysis discrepancies. The local national ITP registry is not available in Malaysia, thus the completeness of the data registry cannot be exactly assessed. Consequently, the results cannot be extended to other regions or countries, particularly regarding laboratory results and treatment exposure.
Conclusions
In conclusion, this study confirms the epidemiology of newly diagnosed adults ITP is comparable with nationwide studies. Our study population demonstrated the high incidence in the female, Malay ethnicity, primary ITP and events of cutaneous bleeding with a predominance 18–29 years age group for both genders. Corticosteroids and immunotherapies are the most prescribed first-line and second-line pharmacological treatments. Splenectomy and TPO-RAs prescriptions are infrequent. Our mortality rate is even, but ITP-related bleeding death is fourth-fold lower than reported before. Mortality risks of our ITP patients include age ≥60 years, male, severe bleeding at presentation, CCI ≥1 and secondary ITP.
Future Directions
This study has paved the way for the development of a local national ITP registry or a population-based surveillance system. This registry will enable us to obtain data which is useful for standardization of local clinical practice, explore new research and novelty findings in ITP patients in Malaysia including study for possible platelet disorders in refractory and cITP group. It is also our hope that with this data/registry, we are able to pave easier access to TPO-RAs that will benefit all individuals diagnosed with ITP.
Data Sharing Statement
Study data are available on request from the corresponding author.
Acknowledgments
The authors are grateful to Assoc. Prof. Dr. Ahmad Sabry Mohamad, to all clinical and laboratory staff at the Haematology Department, Ampang Hospital and HCTM, UKMMC for their participation, dedication and valuable assistance in this study. We thank the Malaysian government and National Registration Department for assisting in data collection and the medical record library for providing medical record provision. Part of this paper was supported by a grant from the Malaysian Society of Haematology.
Disclosure
The authors report no conflict of interests.
References
1. McCrae K. Immune thrombocytopenia: no longer ‘idiopathic’. Cleve Clin J Med. 2011;78(6):358–373. doi:10.3949/ccjm.78gr.10005
2. Cines DB, McMillan R. Pathogenesis of chronic immune thrombocytopenic purpura. Curr Opin Hematol. 2007;14(5):511–514. doi:10.1097/MOH.0b013e3282ba5552
3. Imbach P, Lazarus A, Kühne T. Intravenous immunoglobulins induce potentially synergistic immunomodulations in autoimmune disorders. Vox Sang. 2010;98(3 p2):385–394. doi:10.1111/j.1423-0410.2009.01264.x
4. Moulis G, Palmaro A, Montastruc J-L, Godeau B, Lapeyre-Mestre M, Sailler L. Epidemiology of incident immune thrombocytopenia: a nationwide population-based study in France. Blood. 2014;124(22):3308–3315. doi:10.1182/blood-2014-05-578336
5. Iraqi M, Perdomo J, Yan F, Choi PY, Chong BH. Immune thrombocytopenia: antiplatelet autoantibodies inhibit proplatelet formation by megakaryocytes and impair platelet production in vitro. Haematologica. 2015;100(5):623. doi:10.3324/haematol.2014.115634
6. Marieke Schoonen W, Kucera G, Coalson J, et al. Epidemiology of immune thrombocytopenic purpura in the general practice research database. Br J Haematol. 2009;145(2):235–244. doi:10.1111/j.1365-2141.2009.07615.x
7. Douglas B, Cines HL, Stasi R. Pathobiology of secondary immune thrombocytopenia. Semin Hematol. 2009;46:S2–S14. doi:10.1053/j.seminhematol.2008.12.005
8. Cines DB, Bussel JB, Liebman HA, Luning Prak ET. The ITP syndrome: pathogenic and clinical diversity. Blood. 2009;113(26):6511–6521.
9. Frederiksen H, Maegbaek ML, Nørgaard M. Twenty‐year mortality of adult patients with primary immune thrombocytopenia: a Danish population‐based cohort study. Br J Haematol. 2014;166(2):260–267. doi:10.1111/bjh.12869
10. Cooper N, Ghanima W. Immune thrombocytopenia. N Engl J Med. 2019;381(10):945–955. doi:10.1056/NEJMcp1810479
11. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115(2):168–186. doi:10.1182/blood-2009-06-225565
12. Piel-Julian ML, Mahevas M, Germain J, et al. Risk factors for bleeding, including platelet count threshold, in newly diagnosed immune thrombocytopenia adults. J Thromb Haemost. 2018;16(9):1830–1842. doi:10.1111/jth.14227
13. Cohen YC, Djulbegovic B, Shamai-Lubovitz O, Mozes B. The bleeding risk and natural history of idiopathic thrombocytopenic purpura in patients with persistent low platelet counts. ArchInternMed. 2000;160(11):1630–1638.
14. Neunert C, Lim W, Crowther M, Cohen A, Solberg JL, Crowther MA. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011;117(16):4190–4207. doi:10.1182/blood-2010-08-302984
15. ITP A, British Committee for Standards in Haematology General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol. 2003;120(4):574–596. doi:10.1046/j.1365-2141.2003.04131.x
16. Stasi R, Provan D. Management of immune thrombocytopenic purpura in adults. Mayo Clin Proc. 2004;79(4):504–522. doi:10.4065/79.4.504
17. Lambert MP, Gernsheimer TB. Clinical updates in adult immune thrombocytopenia. Blood. 2017;129(21):2829–2835. doi:10.1182/blood-2017-03-754119
18. Fogarty PF. Chronic immune thrombocytopenia in adults: epidemiology and clinical presentation. Hematol Oncol Clin North Am. 2009;23(6):1213–1221. doi:10.1016/j.hoc.2009.08.004
19. Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113(11):2386–2393. doi:10.1182/blood-2008-07-162503
20. Moulis G, Lapeyre-Mestre M, Montastruc J-L, Sailler L. Exposure to non-corticosteroid treatments in adult primary immune thrombocytopenia before the chronic phase in the era of thrombopoietin receptor agonists in France. A nationwide population-based study. Autoimmun Rev. 2015;14(2):168–173. doi:10.1016/j.autrev.2014.10.017
21. Palandri F, Polverelli N, Sollazzo D, et al. Have splenectomy rate and main outcomes of ITP changed after the introduction of new treatments? A monocentric study in the outpatient setting during 35 years. Am J Hematol. 2016;91(4):E267–E272. doi:10.1002/ajh.24310
22. Chaturvedi S, Arnold DM, McCrae KR. Splenectomy for immune thrombocytopenia: down but not out. Blood. 2018;131(11):1172–1182. doi:10.1182/blood-2017-09-742353
23. Provan D, Arnold DM, Bussel JB, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019;3(22):3780–3817. doi:10.1182/bloodadvances.2019000812
24. Michel M. Immune thrombocytopenia nomenclature, consensus reports, and guidelines: what are the consequences for daily practice and clinical research? Semin Hematol. 2013;50(Suppl 1):S50–S54. doi:10.1053/j.seminhematol.2013.03.008
25. George JN, Woolf SH, Raskob GE, et al. Idiopathic thrombocytopenic purpura: a practice guideline developed by explicit methods for the American Society of Hematology. Blood. 1996;88(1):3–40. doi:10.1182/blood.V88.1.3.3
26. Christiansen CF, Bahmanyar S, Ghanima W, et al. Chronic immune thrombocytopenia in Denmark, Sweden and Norway: the Nordic country patient registry for romiplostim. EClinicalMedicine. 2019;14:80–87. doi:10.1016/j.eclinm.2019.07.015
27. Lamnisos D, Giannakou K, Jakovljevic M. Demographic forecasting of population aging in Greece and Cyprus: one big challenge for the Mediterranean health and social system long-term sustainability. Health Research Policy and Systems. 2021;19(1):21. doi:10.1186/s12961-020-00666-x
28. Kurata Y, Fujimura K, Kuwana M, Tomiyama Y, Murata M. Epidemiology of primary immune thrombocytopenia in children and adults in Japan: a population-based study and literature review. Int J Hematol. 2011;93(3):329–335. doi:10.1007/s12185-011-0791-1
29. Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytopenic purpura among adults: a population-based study and literature review. Eur J Haematol. 2009;83(2):83–89. doi:10.1111/j.1600-0609.2009.01247.x
30. Terrell DR, Beebe LA, Vesely SK, Neas BR, Segal JB, George JN. The incidence of immune thrombocytopenic purpura in children and adults: a critical review of published reports. Am J Hematol. 2010;85(3):174–180. doi:10.1002/ajh.21616
31. Segal JB, Powe N. Prevalence of immune thrombocytopenia: analyses of administrative data. J Thromb Haemost. 2006;4(11):2377–2383. doi:10.1111/j.1538-7836.2006.02147.x
32. Yong M, Schoonen WM, Li L, et al. Epidemiology of paediatric immune thrombocytopenia in the general practice research database. Br J Haematol. 2010;149(6):855–864. doi:10.1111/j.1365-2141.2010.08176.x
33. Neunert C, Terrell DR, Arnold DM, et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019;3(23):3829–3866. doi:10.1182/bloodadvances.2019000966
34. Schulze H, Gaedicke G. Immune thrombocytopenia in children and adults: what’s the same, what’s different? Haematologica. 2011;96(12):1739–1741. doi:10.3324/haematol.2011.055830
35. Wong GC, Lee LH. A study of idiopathic thrombocytopenic purpura (ITP) patients over a ten-year period. Ann Acad Med Singap. 1998;27(6):789–793.
36. Doan CA, Bouroncle BA, Wiseman BK. Idiopathic and secondary thrombocytopenic purpura: clinical study and evaluation of 381 cases over a period of 28 years. Ann Inter Med. 1960;53(5):861–876.
37. George J. Thrombocytopenia due to enhanced platelet destruction by immunologic mechanisms. In: Williams Hematology. McGraw-Hill; 1995:1315–1355.
38. Frederiksen H, Schmidt K. The incidence of idiopathic thrombocytopenic purpura in adults increases with age. Blood. 1999;94(3):909–913. doi:10.1182/blood.V94.3.909.415k02_909_913
39. Michel M, Suzan F, Adoue D, et al. Management of immune thrombocytopenia in adults: a population‐based analysis of the French hospital discharge database from 2009 to 2012. Br J Haematol. 2015;170(2):218–222. doi:10.1111/bjh.13415
40. Lee JY, Lee J-H, Lee H, et al. Epidemiology and management of primary immune thrombocytopenia: a nationwide population-based study in Korea. Thromb Res. 2017;155:86–91. doi:10.1016/j.thromres.2017.05.010
41. Hung GY, Lee CY, Yen HJ, Lin LY, Horng JL. Incidence of immune thrombocytopenia in Taiwan: a nationwide population-based study. Transfusion. 2018;58(11):2712–2719. doi:10.1111/trf.14915
42. Kavanaugh A, Tomar R, Reveille J, Solomon DH, Homburger HA. Guidelines for clinical use of the antinuclear antibody test and tests for specific autoantibodies to nuclear antigens. Arch Pathol. 2000;124(1):71–81.
43. Miyakis S, Lockshin M, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4(2):295–306. doi:10.1111/j.1538-7836.2006.01753.x
44. Kelton JG, Vrbensky JR, Arnold DM. How do we diagnose immune thrombocytopenia in 2018? Hematology Am Soc Hematol Educ Program. 2018;2018(1):561–567. doi:10.1182/asheducation-2018.1.561
45. Goh K-L, Parasakthi N. The racial cohort phenomenon: seroepidemiology of Helicobacter pylori infection in a multiracial South-East Asian country. Eur J Gastroenterol Hepatol. 2001;13(2):177–183. doi:10.1097/00042737-200102000-00014
46. Goh KL. Epidemiology of Helicobacter pylori infection in Malaysia–observations in a multiracial Asian population. Med J Malaysia. 2009;64(3):187–192.
47. Arnold DM, Bernotas A, Nazi I, et al. Platelet count response to H. pylori treatment in patients with immune thrombocytopenic purpura with and without H. pylori infection: a systematic review. Haematologica. 2009;94(6):850. doi:10.3324/haematol.2008.005348
48. Stasi R, Sarpatwari A, Segal JB, et al. Effects of eradication of Helicobacter pylori infection in patients with immune thrombocytopenic purpura: a systematic review. Blood. 2009;113(6):1231–1240.
49. Arnold DM, Nazy I, Clare R, et al. Misdiagnosis of primary immune thrombocytopenia and frequency of bleeding: lessons from the McMaster ITP registry. Blood Adv. 2017;1(25):2414–2420. doi:10.1182/bloodadvances.2017010942
50. Ko B-S, Wu GH-M, Wang Y-C, et al. Demographics and long-term outcome of incident immune thrombocytopenic purpura: a twelve-years nationwide population-based study in Taiwan. Blood. 2015;126(23):3259. doi:10.1182/blood.V126.23.3259.3259
51. Moulis G, Germain J, Comont T, et al. Newly diagnosed immune thrombocytopenia adults: clinical epidemiology, exposure to treatments, and evolution. results of the CARMEN multicenter prospective cohort. Am J Hematol. 2017;92(6):493–500. doi:10.1002/ajh.24702
52. Sadia Sultan F, Ahmed SI, Murad S, Irfan SM. Primary versus secondary immune thrombocytopenia in adults; a comparative analysis of clinical and laboratory attributes in newly diagnosed patients in Southern Pakistan. Med J Malaysia. 2016;71(5):269.
53. Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR, Group NRH. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population‐based cohort of 245 patients. Br J Haematol. 2003;122(6):966–974. doi:10.1046/j.1365-2141.2003.04547.x
54. Doobaree U, Nandigam R, Newland A, Provan D. The United Kingdom immune thrombocytopenia (UK-ITP) registry: preliminary findings on bleeding events experienced by its participants. Haematologica. 2015;100(S1):303.
55. Cuker A, Cines DB. Immune thrombocytopenia. Hematology Am Soc Hematol Educ Program. 2010;2010(1):377–384. doi:10.1182/asheducation.V2010.1.377.3643150
56. Neunert C, Noroozi N, Norman G, et al. Severe bleeding events in adults and children with primary immune thrombocytopenia: a systematic review. J Thromb Haemost. 2015;13(3):457–464. doi:10.1111/jth.12813
57. Kühne T, Berchtold W, Michaels LA, et al. Newly diagnosed immune thrombocytopenia in children and adults: a comparative prospective observational registry of the Intercontinental Cooperative Immune Thrombocytopenia Study Group. Haematologica. 2011;96(12):1831. doi:10.3324/haematol.2011.050799
58. Stasi R, Stipa E, Masi M, et al. Long-term observation of 208 adults with chronic idiopathic thrombocytopenic purpura. Am J Med. 1995;98(5):436–442. doi:10.1016/S0002-9343(99)80342-8
59. Vianelli N, Valdrè L, Fiacchini M, et al. Long-term follow-up of idiopathic thrombocytopenic purpura in 310 patients. Haematologica. 2001;86(5):504–509.
60. Grimaldi-Bensouda L, Nordon C, Michel M, et al. Immune thrombocytopenia in adults: a prospective cohort study of clinical features and predictors of outcome. Haematologica. 2016;101(9):1039. doi:10.3324/haematol.2016.146373
61. Khellaf M, Michel M, Schaeffer A, Bierling P, Godeau B. Assessment of a therapeutic strategy for adults with severe autoimmune thrombocytopenic purpura based on a bleeding score rather than platelet count. Haematologica. 2005;90(6):829–832.
62. Matzdorff A, Meyer O, Ostermann H, et al. Immune thrombocytopenia - current diagnostics and therapy: recommendations of a joint working group of DGHO, ÖGHO, SGH, GPOH, and DGTI. Oncol Res Treat. 2018;41(suppl 5):1–30. doi:10.1159/000492187
63. Heng LL, Caguioa P, Chin NS, et al. Chronic adult primary immune thrombocytopenia (ITP) in the Asia-Pacific region. Int J Hematol. 2011;94(2):142–149. doi:10.1007/s12185-011-0894-8
64. Neunert CE. Management of newly diagnosed immune thrombocytopenia: can we change outcomes? Blood Adv. 2017;1(24):2295–2301. doi:10.1182/bloodadvances.2017009860
65. Song F, Al-Samkari H. Management of adult patients with Immune Thrombocytopenia (ITP): a review on current guidance and experience from clinical practice. J Blood Med. 2021;12:653–664. doi:10.2147/JBM.S259101
66. Cooper N, Kruse A, Kruse C, et al. Immune thrombocytopenia (ITP) World Impact Survey (iWISh): patient and physician perceptions of diagnosis, signs and symptoms, and treatment. Am J Hematol. 2021;96(2):188–198. doi:10.1002/ajh.26045
67. Kuter DJ, Macahilig C, Grotzinger KM, et al. Treatment patterns and clinical outcomes in patients with chronic immune thrombocytopenia (ITP) switched to eltrombopag or romiplostim. Int J Hematol. 2015;101(3):255–263. doi:10.1007/s12185-014-1731-7
68. Bussel JB, Cheng G, Saleh MN, et al. Eltrombopag for the treatment of chronic idiopathic thrombocytopenic purpura. N Engl J Med. 2007;357(22):2237–2247. doi:10.1056/NEJMoa073275
69. Nurden AT, Viallard J-F, Nurden P. New-generation drugs that stimulate platelet production in chronic immune thrombocytopenic purpura. Lancet. 2009;373(9674):1562–1569. doi:10.1016/S0140-6736(09)60255-5
70. Shirasugi Y, Ando K, Hashino S, et al. A Phase II, open-label, sequential-cohort, dose-escalation study of romiplostim in Japanese patients with chronic immune thrombocytopenic purpura. Int J Hematol. 2009;90(2):157–165. doi:10.1007/s12185-009-0361-y
71. Giordano P, Lassandro G, Barone A, et al. Use of eltrombopag in children with chronic Immune Thrombocytopenia (ITP): a real life retrospective multicenter experience of the Italian Association of Pediatric Hematology and Oncology (AIEOP). Front Med. 2020;7:66.
72. Kueh Y. Adult idiopathic thrombocytopenic purpura. Singapore Med J. 1995;36(4):367–370.
73. Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood. 2001;97(9):2549–2554. doi:10.1182/blood.V97.9.2549
74. Khadka S, Kasireddy V, Dhakal PK, Dadiboyina C. Evolving treatment modalities for immune thrombocytopenia in adults. J Community Hosp Intern Med Perspect. 2021;11(1):115–119. doi:10.1080/20009666.2020.1843237
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.