")
Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 5
Clinical diagnosis and management in early Huntington's disease: a review
Authors Schiefer J, Werner CJ, Reetz K
Received 1 November 2014
Accepted for publication 20 January 2015
Published 25 March 2015 Volume 2015:5 Pages 37—50
DOI https://doi.org/10.2147/DNND.S49135
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Thomas Müller
Johannes Schiefer,1,* Cornelius J Werner,1,* Kathrin Reetz1,2
1Euregional Huntington Center, 2Jülich Aachen Research Alliance (JARA) – Translational Brain Medicine, Department of Neurology, RWTH Aachen University, Aachen, Germany
*These authors contributed equally to this work
Abstract: This review focuses on clinical diagnosis and both pharmacological and nonpharmacological therapeutic options in early stages of the autosomal dominant inherited neurodegenerative Huntington's disease (HD). The available literature has been reviewed for motor, cognitive, and psychiatric alterations, which are the three major symptom domains of this devastating progressive disease. From a clinical point of view, one has to be aware that the HD phenotype can vary highly across individuals and during the course of the disease. Also, symptoms in juvenile HD can differ substantially from those with adult-onset of HD. Although there is no cure of HD and management is limited, motor and psychiatric symptoms often respond to pharmacotherapy, and nonpharmacological approaches as well as supportive care are essential. International treatment recommendations based on study results, critical statements, and expert opinions have been included. This review is restricted to symptomatic and supportive approaches since all attempts to establish a cure for the disease or modifying therapies have failed so far.
Keywords: Neurodegeneration, clinical picture, early symptoms, therapy, treatment
Introduction
Huntington’s disease (HD) is an inherited, progressive neurodegenerative disease following an autosomal dominant pattern. It was first described by Huntington in 1872.1 The underlying gene defect was found in 1993.2 An unstable, expanded CAG triplet repeat located in the IT15 gene on chromosome 4p results in an enlarged polyglutamine stretch in the huntingtin protein (HTT). In HD, a repeat of more than 36 CAG triplets leads to clinical manifestation. Age of onset is related to the number of CAG repeats in the affected allele.3 Whereas the penetrance is reduced when carrying 35–39 repeats, only subtle clinical symptoms, if any, have been described in people showing 27–35 repeats in the affected allele.4,5 However, in these cases there is a risk for clinically relevant disease in offspring due to anticipation by intergenerational transmission, especially if transmission of the affected allele is paternal.6 Although the correlation between CAG repeat length and onset of symptoms has been proven to be robust, there are many exceptions that have been described, so the individual destiny must be waited for and cannot be predicted precisely. With regard to disease duration, the CAG repeat length seems to have some predictive value,7,8 implicating that progression of the pathological process is determined by additional factors such as environmental conditions and other genetic prerequisites once the disease has started.
Although our knowledge about the different pathophysiological processes that cause neuronal dysfunction and eventually lead to the specific pattern of neurodegeneration in HD has increased substantially over the last years, all attempts to establish a disease-modifying therapy have failed to show any beneficial effect. Thus, therapy still is symptomatic and restricted to individualized supportive treatment approaches. However, it has to be emphasized that symptomatic treatment in HD is much more sophisticated than 20 years ago. Nevertheless, psychological and social support for patients and their families needs to be extended substantially. This review focuses on the main clinical features in early HD and juvenile HD (JHD) and their pharmacological and nonpharmacological treatment.
Literature search
Electronic searches for English and German language journal articles were run within the PubMed and Medline from 1993 to January 2015. The following keywords were used for search: “(Huntington OR Huntington’s) AND (treatment OR medication OR therapy OR drugs OR non-pharmacological).” We considered only human studies that focused in particular on early HD. To identify additional studies that were not retrieved in the initial database searches, we further searched the reference lists of included studies as well as published reviews focusing on similar aspects. Case reports were included only when they were the sole source of evidence for a particular type of treatment.
Clinical symptoms of Huntington’s disease
There are three major pathognomonic domains of symptoms that characterize the clinical presentation of most HD patients: motor symptoms, cognitive deficits, and psychiatric/behavioral alterations. Which domain is affected first and how the different symptoms develop during the course of disease differ widely between individual subjects. One has to be aware that the clinical picture in JHD can differ a lot from what we expect by our knowledge about the typical course of the disease.9 It sometimes can be almost impossible even for specialists to distinguish between age-related temporary alterations and specific changes related to disease onset if psychiatric, cognitive, or behavioral disturbances appear first, especially in young premanifest people. The clinical picture can be complemented by less frequent symptoms such as circadian and sleep disturbances,10,11 dysfunctions of the autonomic nervous system, epileptic seizures, and myoclonus and general problems such as unintentional weight loss. Regardless of which symptoms appear first and how they develop, sooner or later they will progressively interfere with activities of everyday life, profession and career, and the subject’s social contacts, resulting in inevitable dependency. Thus taking care of HD patients and persons at risk in particular is a special responsibility, which requires recognition of disease-related symptoms at the earliest and, if needed, installation of an adequate therapy.
Motor symptoms
Chorea
In adults, chorea is the most prevalent motor dysfunction in the beginning of symptom manifestation in HD. Involuntary movements of the facial muscles and the distal extremities – fingers and toes in particular – occur predominantly. These movements are nonrepetitive, arrhythmic, and jerky. In the beginning, they are small and get integrated into voluntary movements unknowingly. Thus they may be misinterpreted as agitation or nervousness. Since the choreatic movements cannot be suppressed voluntarily and are associated with motor impersistence, chorea best can be observed if the subject is asked to absolutely rest or highly concentrated on a given (eg, cognitive) task. Surprisingly, unawareness of chorea by the patients themselves can last for quite a long time and still is present in almost 50%, even if the symptoms cannot be ignored by outsiders any longer.12 Motor impersistence also leads to the phenomenon that sustained tongue protrusion out of the wide-open mouth cannot be performed adequately. With disease progression, chorea often worsens. Proximal muscle groups and the axial trunk muscles are affected consecutively, while both the intensity and amplitude of the uncontrolled movements increase. Secondary problems result, which can lead to severe problems in everyday life, such as, for example, progressing imbalance, unstable walking, and frequent risk of falling. Involvement of the diaphragm and muscles located in larynx and pharynx leads to breathing disturbances, dysphagia, dysarthria, and involuntary vocalization interrupting the speech melody. In advanced stages of HD, chorea may plateau or decline and is replaced by rigidity and parkinsonian-like features eventually.
Dystonia
Dystonia can be also one of the early symptoms in JHD and is seen in advanced stages of HD almost regularly. Dystonia is accompanied by increased muscle tone of defined muscle groups, thus leading to abnormal posture of single limbs or the trunk and slowing of voluntary and involuntary movements. Dystonia can be completely absent while sitting and talking, but suddenly springs into attention if the subject is asked to walk. Thus, a slight abnormal finger posture of one hand or a sustained slightly supinated posture of one foot while walking can be subtle clinical signs of ongoing dystonia. With disease progression, dystonia will get more and more predominant not only by visible abnormal postures of trunk, head, and limbs but also by the increased muscle tone one will feel impressively when flexing/bending the extremities.
Bradykinesia/akinesia
Bradykinesia, ie, uncontrollable slowing of voluntary and involuntary movements, typically is combined with dystonia and increased muscle tone. Whereas both dystonia and bradykinesia contribute to the ever-increasing risk of falling, the latter causes a particular accident hazard due to the retarded ability to react in dangerous situations. The inability to start a certain movement is called akinesia. This situation is even worsened since motor dysfunction usually is accompanied by additional symptoms such as deficits in postural control, generalized slowing of cognitive functions, and defined neuropsychiatric deficits.
Eye movements
Pathological findings of eye movements can be the first symptoms in early stages of HD and also have been described in premanifest HD gene carriers. Therefore, special attention should be paid to the examination of eye movements during the neurological investigation in premanifest and young people in particular. The clinical examination can be complemented by vestibulooculography, which can objectify subtle findings and depict subclinical alterations. One of the first findings in early stages of HD can be an incomplete suppression of the optokinetic nystagmus. Slowing of saccade velocity and a delay in initiating volitional saccades is found additionally.13,14 In contrast, smooth pursuit is not altered in early stages. With disease progression, all mentioned functions are altered including refixation.
Clinical diagnosis
The total motor score (TMS) of the Unified Huntingon’s Disease Rating Scale (UHDRS)15 allows us to systematically check for motor symptoms that can occur in the course of HD. Due to its accuracy, this test therefore serves as the “gold standard” in both therapeutic and observational studies so far. Subtle chorea, dystonia, and slight disturbances of eye movements, as well as minimal changes in gait and balance, have been described to possibly appear in premanifest HD gene carriers quite long ago.7,8,16,17 The TMS, however, seems not to be the appropriate assessment to depict these subtle changes, especially when large groups of premanifest HD gene carriers are analyzed in total.18,19 This was revealed, for example, in the large observational studies PREDICT-HD and TRACK-HD.20,21 This insensitivity of the TMS is mainly seen as the result of both intra- and interindividual variability as well as inter- and intrarater variability.22,23
In order to detect even subtle motor symptoms in premanifest HD gene carriers at the earliest by objective clinical longitudinal monitoring and to define more accurate endpoints for ongoing clinical trials, several new motor assessments have been developed and tested in clinical settings to different degrees. These tests aim at best possible and objective accuracy based on quantifiable computerized tools (the so-called quantitative (Q)-Motor tests). Chorea can be recorded objectively and precisely by choreomotography.24–26 Other quantitative assessments measure variability of grasping force and fingertip coordination during isometric precision grip.25,27 Furthermore, sensitive Q-Motor tests have been established by analyzing variability of tongue protrusion force at different levels and both speeded and metronome-related finger tapping.28 Q-Motor measures have been used successfully even in large observational trials. The knowledge about the occurrence of subtle motor dysfunction in premanifest HD gene carriers could be confirmed by choosing these tests as endpoints.21,29,30 In addition, posturography may serve as an objective tool for recording and monitoring postural deficits.31
Pharmacological approaches
The time point at which symptomatic medication is started has no direct influence on the neurodegenerative process, and thus should be considered very critically based on the patients’ needs. The aim should be to guarantee the best quality of life. Intended benefit and side effects have to be assessed individually and questioned regularly. In particular, in early stages, drug therapy should be initiated according to the patient’s own wishes and should not be driven by the demand of relatives, who, for example, might feel embarrassed by the involuntary movements or cannot stand doing nothing while the patient is suffering according to their perception. Nevertheless, an ethical dilemma can result at the latest when the affected subject gets disabled in everyday life or his/her business, but does not feel like taking any medication since he/she is not aware of the choreatic movements – a typical feature in HD. In respect of this problem, we recommend the establishment of a mutual trust by seeing the patient and the relatives regularly and offering standardized screening visits routinely to objectify motor symptoms and to discuss medication options based on comparing these results with the HD subject’s own perception. Although it might cause misgivings, it sometimes may be prudent to wait to see how symptoms and the patient’s needs develop further and not to persuade him/her to start with medication.
A few substances have been shown in clinical trials to have beneficial effects on HD-related motor symptoms and chorea in particular. Based on these trials, first evidence-based treatment guidelines are available, for example published by the American Academy of Neurology.32 These guidelines, however, are discussed controversially by experts who have collected decades of experience in treating HD patients.33
Many agents have been tested to reduce choreatic movements including, for example, neuroleptics, monoamine-depleting agents, benzodiazepines, antiepileptics, acetylcholinesterase inhibitors, and glutamate antagonists. Nevertheless, there is not enough evidence to propose long-term guidelines for the symptomatic treatment of HD-related motor symptoms, and double-blinded controlled treatment trials are needed urgently.
Tetrabenazine
The monoamine depletor tetrabenazine (TBZ) has been shown to effectively reduce chorea in HD (Table 1) and is therefore recommended by American and European guidelines.32,34–36 In contrast to typical or atypical neuroleptics, TBZ carries a less severe risk of developing tardive dyskinesia.18,37 In general, TBZ is well tolerated and serves as a good candidate to start medical treatment of choreatic movements even in early stages.37 Possible side effects include depression, parkinsonism, insomnia, akathisia, and sedation. Starting with 12.5 mg per day, the dosage of TBZ can be increased weekly in 12.5-mg steps distributed three times per day. The maximum dose is discussed controversially, but usually limited to 100 mg per day. If comedication includes strong cytochrome P450 2D6 (CYP2D6) inhibitors eg, selective serotonin reuptake inhibitors (SSRIs) such as paroxetine, fluoxetine, or other antidepressants such as, eg, bupropion, or the subject turns out to be a CYP2D6 slow metabolizer by genotyping, the maximum dose should be restricted to half, although no distinguishing features have been found in patients exhibiting various CYP2D6 activities.38 Notably, despite undoubtable beneficial effects on chorea, certain cognitive functions can worsen under medication with TBZ.
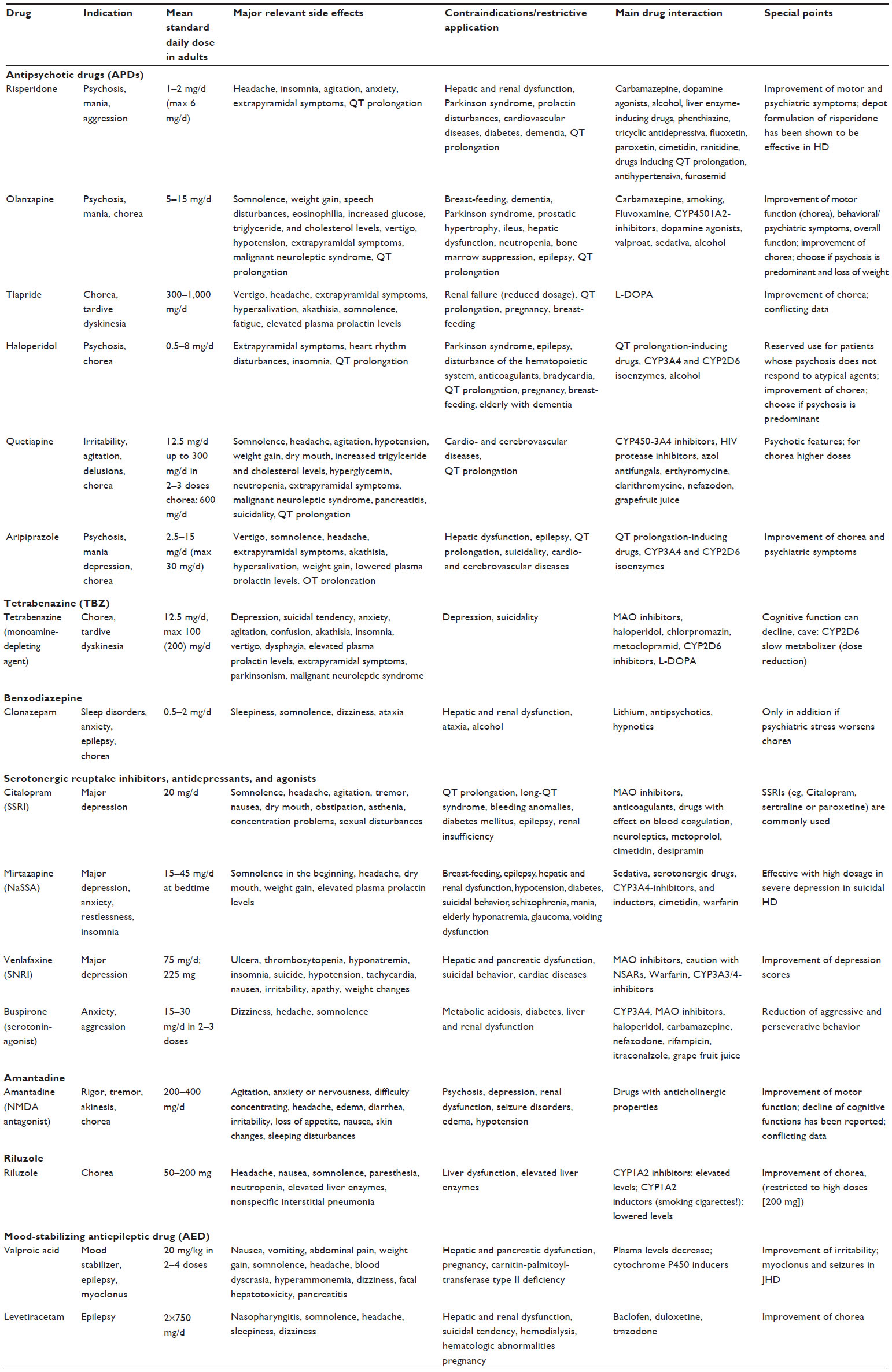 | Table 1 Drugs used in Huntington’s disease |
Neuroleptics
If both chorea and psychiatric symptoms like agitation and psychosis are predominant features, neuroleptics, which act by blocking the dopamine transmission, could be chosen preferentially (Table 1). Although atypical neuroleptics should be favored with respect to their better profile of adverse effects, typical neuroleptics such as haloperidole, fluphenazine, or chlorpromazine will have to be used if chorea is very severe or psychosis is accompanied by aggressiveness. Particular attention has to be paid if HD patients show parkinsonian-like symptoms. Medication with most neuroleptics can worsen parkinsonism. Several atypical neuroleptics have been chosen to treat chorea in HD patients. Olanzapine, risperidone, and aripiprazole have been reported to improve chorea in some small open-label studies or case series.39 Weight gain is a common side effect of neuroleptics but can even be of advantage for HD patients since unintended weight loss is in many cases a progressing burden in HD. Another relevant side effect is sedation. Aripiprazole is well tolerated and may also improve psychiatric symptoms. In European countries, tiapride (starting 2×50 mg, recommended maximum dose: 1000 mg/day) is widely used as a first-line medication.34 Much hope was placed in the dopamine-stabilizing agent pridopidine. Although no improvements in the modified motor-ranking scale after 6 months could be objectified, the UHDRS total motor score seemed to improve at a dose of 90 mg/day.40–42 At the moment, pridopidine is not recommended as a first-line treatment in HD-related chorea.43
Benzodiazepines
Chorea is known to worsen in psychologically demanding situations or under stress. Therefore, low dosages of benzodiazepines can be added to the medication transitionally to cushion these emotional effects. The risk of drug abuse and dependency, however, has to be respected.
Amantadine
The usefulness of amantadine (recommended dose: 300–400 mg/day) in the treatment of chorea is discussed controversially, since results of the present trials are not concordant.44,45 Accordingly, this N-methyl-D-aspartate (NMDA) receptor antagonist could be an option in individual patients to improve chorea. In our experience, it is not recommended as a first-line medication in early symptomatic patients although its use is recommended right after tetrabenazine by the American Academy of Neurology guidelines.
Riluzole
The initially high expectations in using riluzole for the treatment of chorea in HD have diminished since results of randomized controlled trials did not show the expected improvements. Besides, notable side effects were reported, but restricted to high doses of 200 mg/day. In conclusion, a time- and dose-dependent positive effect of riluzole in the treatment of chorea is discussed, but no generalized recommendation can be given for the use of this drug.18 Thus, in our experience riluzole does not serve as first-line medication in the treatment of chorea in early symptomatic HD patients, although it is recommended in third position for treating chorea in HD by the American Academy of Neurology guidelines.
Nonpharmacological approaches
Any nonpharmacological approach in treating motor symptoms in early HD faces a two-fold difficulty. Firstly, although we know that subtle motor deficits do occur, it is not known whether these deficits confer a relevant impairment in activities of daily living and, thus, should be treated. Even moderate chorea does not necessarily impair everyday functioning. Instability of gait and falls are much more predictive of relevant disability46 but whether these are actually motor-mediated (eg, chorea) or due to reduced cognition and more impulsive and aggressive behavior leading to reckless walking is not always clear. The second problem stems from the fact that, from a pathophysiological point of view, it could be expected that procedural motor learning, on which standard physiotherapy (PT) and occupational therapy (OT) typically are based, is selectively impaired in this disorder.47–49 However, from a more pragmatic point of view, there is evidence that even in basal ganglia disorders nonpharmacological treatment approaches can be a promising option, as has been shown repeatedly, eg, Tai-Chi Chuan in Parkinson’s disease.50
In HD, the most recent study focuses on gait and stability in mid-stage HD and shows small benefits by an intervention of comparatively low intensity (PT twice weekly).51 A randomized, controlled trial involving both group sessions and instructed home exercises over the course of 9 months showed medium to large effect sizes on chorea and gait52 while also improving some cognitive measures (see section‚ Cognitive dysfunction - nonpharmacological approaches).
Another randomized, controlled Phase II trial in HD showed good tolerability and safety in a program employing once-weekly physical exercise in a studio complemented by home exercises.53 While there was a positive effect on cognition, motor scores failed to reach significance due to the small number of participants. An uncontrolled interventional study revealed that serial courses of multidisciplinary in-patient rehabilitation (3 weeks up to three times a year) each had a significant effect on balance and physical fitness.54 While this gain usually was lost until the next admission, it could be recovered by the following intervention. A randomized, but not blinded study involving a DVD-based home exercise program showed rather large effect sizes (exceeding minimal detectable changes) in a multitude of gait- and balance-related outcomes while boasting a good adherence rate.55
Overall, it can be shown that, even in early stages of HD, nonpharmacological interventions of varying degrees of intensity can lead to substantial gains in motor function, particularly gait and balance, which need to be maintained by repeated application, however. Safety and tolerability usually are excellent and exceed that of pharmacotherapy. For a practical approach, the Huntington’s Disease Physiotherapy Working Group (PWG) has put together a multilayered, treatment-based classification proposing different interventions for the different stages of HD.56
Cognitive symptoms
Clinical diagnosis
Cognitive impairment is one of the triad of symptom domains in HD, and can be seen in persons with the HD gene expansion with no overt motor signs or symptoms, indicating that cognitive onset of HD precedes motor manifestation.57
For diagnosing cognitive impairment in clinical practice, brief cognitive screening tests can be used in a first step. The most common one is the Mini-Mental-State Examination (MMSE).58 However, recent studies show that the Montreal Cognitive Assessment Test (MoCA) is a useful instrument for assessing cognitive performance over a broad level of functioning in HD,59 showing even higher sensitivity without sacrificing specificity in many domains relative to the MMSE.60 Nevertheless, most HD centers use the more comprehensive neuropsychological test battery of the UHDRS,15 which includes the Symbol Digit Modality Test, the Stroop Color Word Test, and a Verbal Fluency test as part of a comprehensive examination and is suited to diagnose cognitive dysfunction in HD accurately.
Although the amount of cognitive decline varies highly and often is not recognized by the patient, it generally increases while the disease progresses. The term “mild cognitive impairment” (MCI) is used to describe a transitional stage between normal cognition and dementia.61 In the framework of the observational study PREDICT-HD, it has been shown that nearly 40% of those who are genetically at risk for HD meet criteria for MCI, using accepted cutoff scores (1.5 standard deviations below the mean of a comparison group), with higher rates in individuals closer to (predicted) HD diagnosis.62
The neuropsychological profile in premanifest and manifest HD was described in a large review, comprising 110 HD studies.63 They reported that premanifest gene carriers presented typically no to little deficiencies in memory, language, or global cognitive functions, but deficits in tasks assessing the functions of psychomotor speed, negative emotion recognition, and to some degree in executive functioning. In manifest HD, adequate functioning appeared to remain intact for the longest periods for language and global functional domains; however, as a result of the progressing cognitive decline resulting in dementia, during end-stage HD, these functions also showed marked deterioration. During the progression of the disease, impairments can be expected in memory (especially visuospatial), psychomotor speed, negative emotion recognition, and executive functioning. Thus, potential candidate biomarkers should be most expected from such domains as working memory, psychomotor speed, recognition of negative emotions, and attentional and visuospatial executive functions, which need to be investigated in longitudinal studies.
Pharmacological approaches
There is currently no cure or treatment that can halt, slow, or reverse the progression of the disease. There are very few controlled trials targeting the cognitive features of HD. The majority, however, have been focused on motor outcome measures. The current German guidelines for HD34 state that there are no evidence-based treatment recommendations for dementia in HD (Table 1). In the past, clinical trials failed to provide effective pharmacotherapy for neuropsychological deficits and dementia. Thus, they still remain untreatable. Several clinical trials have revealed that cholinesterase inhibitors are not effective (for review, see Refs 64–66). The nonstimulant norepinephrine reuptake inhibitor atomoxetine demonstrated no advantage over placebo for cognitive dysfunction in early HD in a randomized, double-blind crossover study.67 Also, the recent randomized, double-blind, placebo-controlled study HORIZON determining the effect of latrepirdine on cognition and global function in patients with mild to moderate HD did not improve these outcome measures.68 The anti-glutamate NMDA antagonist memantine has been investigated as symptomatic treatment for early HD in the MITIGATE-HD study. Preliminary analysis results presented at the HSG Huntington’s Disease Symposium in 2010 suggested that 20 mg dosing of memantine resulted in improvement in certain (not all) cognitive measures but worsening of some motor symptoms. However, as our understanding of the consequences of the HD mutation has highly improved in the last years, the range of tractable targets for therapeutic approaches has broadened and thus an exciting era with pathobiological targets of HD will hopefully enter clinical trials with a reasonable expectation of success soon.69
Nonpharmacological approaches
Given that cognitive impairment in HD is at least as predictive for the patients’ everyday capabilities and quality of life as motor function,70 every measure available should be taken to preserve or increase cognitive abilities in HD patients.
Early to mid-stage HD patients were admitted for 3 weeks of an inpatient rehabilitation program for up to three times a year. In the first study, marked motor improvements were accompanied by a preservation of cognitive abilities as measured by the admittedly rather unresponsive MMSE.54 This finding could be replicated, and the authors also observed significant increases in motor function while MMSE and UHDRS cognition scores were preserved.71 Both studies were observational and did not include a control group. Another study performed in Australia examined the effects of a continuous, interdisciplinary outpatient/home training program.52 Again, substantial improvements on motor function were shown against a control cohort, and this time cognitive measures increased. Taken together, these trials show the high potential of nonpharmacological interventions, particularly in the absence of pharmacological options. Yet, these interventions (interdisciplinary, focused rehabilitation programs) are not easily implemented or distributed evenly, as they require access to highly trained multidisciplinary teams. In those circumstances, however, where such a team can be established, acceptance rates and patient satisfaction are very high.72 In the absence of other similarly effective treatments, every effort should be taken to increase the availability of these interventions.
Psychiatric symptoms
Clinical diagnosis
Psychiatric disturbances constitute another part of the triad of symptom domains of HD and cover a broad variety of psychiatric symptoms such as depression, anxiety, impulsivity, and irritability, which can be accompanied by aggressiveness and obsessive-compulsive behavior. They may be categorized into 1) mental illnesses, including in particular major depression but also mania, obsessive-compulsive disorders, and various delusional and psychotic disorders; 2) organic personality disorders, frontal lobe syndromes, and executive dysfunctions; and, finally, 3) psychiatric issues such as delirium, agitation, or sexual disorders. They have a high prevalence in patients with HD, and often start before the onset of motor symptoms.73–75 The proportion of patients who first present with psychiatric symptoms ranges from 24% to 79%, and the prevalence of psychiatric disorders in HD ranges from 35% to 73%.76 Mood disorders have been reported to affect up to 41%,77 and the frequency of psychosis ranges from 3% to 12% in HD.76 Suicidal ideation is a frequent finding in patients with HD, especially in the early stages of the illness.78 In contrast, obsessive-compulsive behavior seems to be less frequent, but also occurs in premanifest stages in the disease.79
Most HD centers use the behavioral subscales of the UHDRS15 for diagnostic purposes.15
Pharmacological approaches
Although psychiatric features have a significant impact on a patient’s professional and personal life and thus often become the major focus of management, there is hardly any evidence-based support for a pharmacotherapeutic management of psychiatric disorders in HD (Table 1). Consequently, therapeutic decision making in HD is often based on clinical experience, expert opinions, and principles of psychiatric treatment in general.80 The choice is mainly governed by patient response, side-effect profile, and the need for secondary therapeutic effects on features such as anxiety, obsessive behavior, or sleep disorders.81
Depression
Depressive symptoms in HD seem to respond to conventional pharmacologic treatments for major depressive disorder. Venlafaxin was shown, in a small study evaluating depression scores of 26 patients with HD, to be effective in the treatment of major depression in HD, although it may produce unpleasant side effects such as nausea and irritability (one in five patients).82 However, this trial was not blinded, did not have a control group, and used an inadequate treatment duration compared with conventional antidepressant trials. Besides Venlafaxin, SSRIs are commonly used in HD. A randomized, double-blind, and placebo-controlled clinical trial with 20 mg citalopram for 20 weeks in 33 HD adults with no depression showed no evidence that short-term treatment improved executive function, but improved mood.83 A randomized, double-blind, placebo-controlled trial with fluoxetine in 30 nondepressed HD patients failed to exert substantial clinical benefits.84 The norepinephrine reuptake inhibitor atomoxetine demonstrated in a randomized controlled trial in 20 HD patients no advantages over placebo for attention as well as for psychiatric and motor symptoms.67 Evidence-based data are also lacking for the use of sulpiride in antidepressive treatment. Even though the small double-blind trial of sulpiride in eleven patients demonstrated a positive result for the median movement count and the median chorea severity, a corresponding functional improvement in HD patients was not observed.85 An alternative in patients suffering simultaneously from sleeping disorders might be mirtazapine. Moreover, the noradrenergic and specific serotonergic antidepressant mirtazepine was, based on a case report, shown to be effective without significant adverse effects and with high-dose mirtazapine in the treatment of severe depression in suicidal HD (for review, see Ref 65). Case reports also described beneficial lithium treatment in patients with HD and suicidal behavior.86 Notably, tricyclic antidepressants should be avoided or limited due to the fact that the anticholinergic profile may worsen hyperkinesias and cognition. Controlled trials on antidepressants in HD are urgently required.
Depression and psychosis
According to the Physician’s Guide to the Management of Huntington’s Disease by the Huntington’s Disease Society of America (HDSA), risperidone, olanzepine, quetiapine, ziprasidone, or aripiprazole may be prescribed for treatment of chorea, as these drugs may have a lower incidence of side effects and appear to be just as effective as the classic neuroleptic.87 In cases with severe psychosis, clozapine may be considered, as there are no extrapyramidal side effects, but the full blood count should be controlled on a regular basis. Benzodiazepines, particularly short-acting drugs such as lorazepam, may be another good choice for the short-term management of agitation. In any case, neuroleptics and benzodiazepines used for acute agitation should be re-evaluated as soon as the clinical picture allows. In an open-label, retrospective study, it was shown that HD patients taking risperidone demonstrated improved psychiatric functioning (sum of the product of frequency and severity for eleven psychiatric symptoms [eg, anxiety, hallucinations, depression] and motor stabilization).88
Depression and apathy
Apathy and depression often coexist. In this case, patients should be treated for their depression, which may cause the other symptoms to improve as well. HD patients with primary apathy may respond to psychostimulants such as methylphenidate or dextroamphetamine. Nevertheless, it has to be stressed that these medications are highly abusable and may exacerbate irritability and should be described in limited amounts and with much caution.
Anxiety, agitation, and sleeping disorders
Common agents used in treating anxiety include the SSRI class of antidepressants, benzodiazepines, and mirtazapine, and non-benzodiazepine anxiolytics such as buspirone. Again, it has to be considered that benzodiazepine and benzodiazepine receptor agonists (zolpidem, zopiclone) have a potential for dependency. This might or might not be an issue, depending on the patient and the stage of the disease.
Irritability and aggression
In case reports, improvement was observed using quetiapine, risperidone, olanzapine, valproat, benzodiazepines, beta blockers, SSRI, and buspirone (for review, see Ref 66). A small open-pilot, uncontrolled study of olanzapine in eleven HD patients showed a significant short-term (6 months) improvement regarding depression, anxiety, irritability, and obsessions.89 For obsessive-compulsive symptoms, SSRIs are recommended; atypical antipsychotics are reserved for severe or refractory symptoms.87
Again, insufficient evidence is available to guide the choice of psychiatric treatment. Evidence-based pharmacological empirical evidence for the treatment of psychiatric disorders is highly warranted.
Nonpharmacological approaches
In early disease or even in the premanifest phase, the period of genetic testing imposes significant stress on patients and persons at risk, even for those that ultimately refuse to undergo testing.90 Coping with the results of the test can also be difficult in both positive and negative results (eg, “survivor guilt”). Clinically relevant depressive symptoms up to major depression after positive predictive testing is prevalent in up to 20% of positively and in 12.6% of negatively tested persons91 and should be looked for. Psychological counseling should be offered in all instances.
With regard to specific psychiatric symptoms, nonpharmacological treatment options such as psychotherapy have not been studied systematically so far. Case reports indicate some benefit with behavioral therapy strategies.92,93 The intensive rehabilitation program mentioned earlier showed significant beneficial effects on scales of depression and anxiety.71
From clinical experience, education of the patient’s spouse and family cannot be overestimated. Creating an insight into the psychiatric and psychological alterations typically related to HD can reduce the frequency and severity of domestic conflicts. Establishing a stable and predictable daily routine and reducing environmental complexity is a good way of reducing symptoms of irritability and anxiety. Constant, but good-natured exhortations might address symptoms of apathy, although this might take quite an enduring caregiver. However, the compensatory abilities of the patient’s family and spouse are often overestimated.94 Again, from clinical experience, this is particularly true in patients with reduced insight into their deficits, something that is quite common, unfortunately.95 Social counseling should be provided wherever possible in order to provide the family with the necessary knowledge of financial, legal, and occupational consequences of the diagnosis as well as with actual assistance.
In any case, psychiatric symptoms should give rise to a critical re-evaluation of any existing pharmacotherapy because depression, apathy, but also aggression and delirium can be side effects of frequently used drugs in HD.
Juvenile Huntington’s disease
The mean age of symptom onset in HD is in mid-life at approximately 40 years with a wide range between childhood and senility.96 Since the clinical feature and the sequence in which the distinct symptoms appear differ statistically between young people and patients with adult HD onset, two variants have been established: JHD, the so-called Westphal variant, and adult onset of HD (AHD). Although the line is blurred, there is general arrangement that people younger than 20 years of age at onset of first symptoms belong to the JHD group. If the CAG repeat length does reach 60 or more, it is very likely, but not mandatory, that JHD will develop.97,98 On the other hand, more than 50% of JHD patients show CAG triplet repeats less than 60.19 When looking for early clinical symptoms of HD, one has to be aware that in JHD dystonia, bradykinesia, and parkinsonian-like features as well as myoclonus and epileptic seizures are much more predominant than chorea, which is the major motor symptom in adult patients.99–101 Cognitive and behavioral alterations may be the leading symptoms especially in young HD patients, something that taxes families in particular.9,102 Hence, one has to be aware that findings like lack of concentration and unexpected decline in school performance as well as personality or behavioral changes may be the first symptoms of HD in young people.103,104 The distinction of early symptoms in JHD from normal age-related changes, eg, due to puberty, can be difficult in some cases.
The treatment of symptoms in JHD should not be restricted to pharmaceutical approaches but has to involve a multidisciplinary team respecting all needs of the affected individual and his/her surrounding to guarantee best supportive therapy. The teachers at school, for example, have to be informed about the illness so they can react professionally to certain symptoms such as aggression, inattention, or cognitive decline. Various pharmaceutics may be helpful if specific symptoms lead to loss of quality of life. Since polypharmacology will be needed in most cases, special attention has to be paid to possible side effects and interactions. A summary of substances prescribed for treating symptoms in JHD is given by Robertson et al.105 Symptoms and medication have to be assessed regularly and considered critically. Dopamine agonists, muscle relaxants, and antiepileptic drugs may be used more often as in AHD because parkinsonism, dystonia, myoclonus, and seizures occur predominately.
Future perspectives
Recent biomarker studies such as TRACK-HD21,29,30 and PREDICT-HD106–108 already have advanced our knowledge of the neurobiology underlying HD enormously. Well-characterized and distinct biomarkers are needed as valuable tools to recognize subtle changes in preclinical HD gene carriers and to identify individuals who could benefit from early installation of potentially neuroprotective treatment. These biomarkers will serve as objective surrogate parameters in clinical trials, testing new disease-modifying approaches. We can expect that several new designed drugs and treatment approaches will be proposed for testing in clinical trials in the near future. In addition, other nonpharmacological strategies such as deep brain stimulation, genetic approaches, and training strategies have to be taken in to account as well. The outcome of HD patients who underwent intrastriatal stem cell transplantation will have to be awaited.
Conclusion
Although major advances have been made in the clinical, genetic, and neuropathological understanding after the discovery of the mutation that causes HD, therapeutic options are still limited to symptomatic medication and supportive approaches. Symptoms of HD are multifaceted, including physical alterations as well as cognitive and behavioral problems, and in addition do concern the entire family and the social environment. Apart from symptom-oriented pharmacotherapy, which has to be assessed regularly and considered critically, special emphasis needs to be put in installing a broad multidisciplinary therapeutic setting addressing all individual needs of the patient and his/her family.
The identification of easily obtainable, reliable, and robust biomarkers of even subtle symptoms in HD progression is of upmost importance for the development and evaluation of disease-modifying treatments. Nevertheless, as there are several promising therapeutic agents progressing through current development pipelines, hopes are still high for a better treatment or even preventive medicine in HD.
Acknowledgment
KR was partly funded by the German Federal Ministry of Education and Research (BMBF 01GQ1402).
Disclosure
The authors report no conflicts of interest in this work.
References
Huntington G. On Chorea. Philadelphia: Medical and Surgical Reporter; 1987:317–321. | |
Group HS. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72: 971–983. | |
Langbehn DR, Hayden MR, Paulsen JS; PREDICT-HD Investigators of the Huntington Study Group. CAG-repeat length and the age of onset in Huntington disease (HD): a review and validation study of statistical approaches. Am J Med Genet B Neuropsychiatr Genet. 2010;153B: 397–408. | |
Ha AD, Jankovic J. Exploring the correlates of intermediate CAG repeats in Huntington disease. Postgrad Med. 2011;123:116–121. | |
Semaka A, Creighton S, Warby S, Hayden MR. Predictive testing for Huntington disease: interpretation and significance of intermediate alleles. Clin Genet. 2006;70:283–294. | |
Telenius H, Kremer HP, Theilmann J, et al. Molecular analysis of juvenile Huntington disease: the major influence on (CAG)n repeat length is the sex of the affected parent. Hum Mol Genet. 1993;2:1535–1540. | |
Gusella J, MacDonald M. No post-genetics era in human disease research. Nat Rev Genet. 2002;3:72–79. | |
Rosenblatt A, Liang KY, Zhou H, et al. The association of CAG repeat length with clinical progression in Huntington disease. Neurology. 2006;66:1016–1020. | |
Siesling S, Vegter-van der Vlis M, Roos RA. Juvenile Huntington disease in the Netherlands. Pediatr Neurol. 1997;17:37–43. | |
Morton AJ. Circadian and sleep disorder in Huntington’s disease. Exp Neurol. 2013;243:34–44. | |
Morton AJ, Rudiger SR, Wood NI, et al. Early and progressive circadian abnormalities in Huntington’s disease sheep are unmasked by social environment. Hum Mol Genet. 2014;23:3375–3383. | |
McCusker EA, Gunn DG, Epping EA, et al; PREDICT-HD Investigators of the Huntington Study Group. Unawareness of motor phenoconversion in Huntington disease. Neurology. 2013;81:1141–1147. | |
Blekher T, Johnson SA, Marshall J, et al. Saccades in presymptomatic and early stages of Huntington disease. Neurology. 2006;67:394–399. | |
Blekher TM, Yee RD, Kirkwood SC, et al. Oculomotor control in asymptomatic and recently diagnosed individuals with the genetic marker for Huntington’s disease. Vision Res. 2004;44:2729–2736. | |
Group HS. Unified Huntington’s disease rating scale: reliability and consistency. Mov Disord. 1996;11:136–142. | |
Kirkwood SC, Siemers E, Hodes ME, Conneally PM, Christian JC, Foroud T. Subtle changes among presymptomatic carriers of the Huntington’s disease gene. J Neurol Neurosurg Psychiatry. 2000;69: 773–779. | |
McCusker E, Richards F, Sillence D, Wilson M, Trent RJ. Huntington’s disease: neurological assessment of potential gene carriers presenting for predictive DNA testing. J Clin Neurosci. 2000;7:38–41. | |
Jankovic J, Roos RA. Chorea associated with Huntington’s disease: to treat or not to treat? Mov Disord. 2014;29:1414–1418. | |
Quarrell OW, Nance MA, Nopoulos P, Paulsen JS, Smith JA, Squitieri F. Managing juvenile Huntington’s disease. Neurodegener Dis Manag. 2013;3:1–16. | |
Biglan KM, Ross CA, Langbehn DR, et al; PREDICT-HD Investigators of the Huntington Study Group. Motor abnormalities in premanifest persons with Huntington’s disease: the PREDICT-HD study. Mov Disord. 2009;24:1763–1772. | |
Tabrizi SJ, Scahill RI, Owen G, et al; TRACK-HD Investigators. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol. 2013;12:637–649. | |
Paulsen JS, Wang C, Duff K, et al; PREDICT-HD Investigators of the Huntington Study Group. Challenges assessing clinical endpoints in early Huntington disease. Mov Disord. 2010;25:2595–2603. | |
Tang C, Feigin A. Monitoring Huntington’s disease progression through preclinical and early stages. Neurodegener Dis Manag. 2012;2: 421–435. | |
Reilmann R, Bohlen S, Kirsten F, Ringelstein EB, Lange HW. Assessment of involuntary choreatic movements in Huntington’s disease – toward objective and quantitative measures. Mov Disord. 2011;26:2267–2273. | |
Reilmann R, Bohlen S, Klopstock T, et al. Grasping premanifest Huntington’s disease – shaping new endpoints for new trials. Mov Disord. 2010;25:2858–2862. | |
Reilmann R, Kirsten F, Quinn L, Henningsen H, Marder K, Gordon AM. Objective assessment of progression in Huntington’s disease: a 3-year follow-up study. Neurology. 2001;57:920–924. | |
Rao AK, Gordon AM, Marder KS. Coordination of fingertip forces during precision grip in premanifest Huntington’s disease. Mov Disord. 2011;26:862–869. | |
Bechtel N, Scahill RI, Rosas HD, et al. Tapping linked to function and structure in premanifest and symptomatic Huntington disease. Neurology. 2010;75:2150–2160. | |
Tabrizi SJ, Langbehn DR, Leavitt BR, et al; TRACK-HD Investigators. Biological and clinical manifestations of Huntington’s disease in the longitudinal TRACK-HD study: cross-sectional analysis of baseline data. Lancet Neurol. 2009;8:791–801. | |
Tabrizi SJ, Scahill RI, Durr A, et al; TRACK-HD Investigators. Biological and clinical changes in premanifest and early stage Huntington’s disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet Neurol. 2011;10:31–42. | |
Reilmann R, Rumpf S, Beckmann H, Koch R, Ringelstein EB, Lange HW. Huntington’s disease: objective assessment of posture – a link between motor and functional deficits. Mov Disord. 2012;27: 555–559. | |
Armstrong MJ, Miyasaki JM; American Academy of Neurology. Evidence-based guideline: pharmacologic treatment of chorea in Huntington disease: report of the guideline development subcommittee of the American Academy of Neurology. Neurology. 2012;79:597–603. | |
Reilmann R. Pharmacological treatment of chorea in Huntington’s disease-good clinical practice versus evidence-based guideline. Mov Disord. 2013;28:1030–1033. | |
(DGN) DGfN. Leitlinien für die Huntington-Krankheit. Berlin: Deutsche Gesellschaft für Neurologie (DGN); 2012. | |
Frank S, Ondo W, Fahn S, et al. A study of chorea after tetrabenazine withdrawal in patients with Huntington disease. Clin Neuropharmacol. 2008;31:127–133. | |
Group HS. Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology. 2006;66:366–372. | |
Chen JJ, Ondo WG, Dashtipour K, Swope DM. Tetrabenazine for the treatment of hyperkinetic movement disorders: a review of the literature. Clin Ther. 2012;34:1487–1504. | |
Mehanna R, Hunter C, Davidson A, Jimenez-Shahed J, Jankovic J. Analysis of CYP2D6 genotype and response to tetrabenazine. Mov Disord. 2013;28:210–215. | |
Brusa L, Orlacchio A, Moschella V, Iani C, Bernardi G, Mercuri NB. Treatment of the symptoms of Huntington’s disease: preliminary results comparing aripiprazole and tetrabenazine. Mov Disord. 2009;24: 126–129. | |
de Yebenes JG, Landwehrmeyer B, Squitieri F, et al; MermaiHD Study Investigators. Pridopidine for the treatment of motor function in patients with Huntington’s disease (MermaiHD): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2011;10:1049–1057. | |
Lundin A, Dietrichs E, Haghighi S, et al. Efficacy and safety of the dopaminergic stabilizer pridopidine (ACR16) in patients with Huntington’s disease. Clin Neuropharmacol. 2010;33:260–264. | |
Huntington Study Group HART Investigators. A randomized, double-blind, placebo-controlled trial of pridopidine in Huntington’s disease. Mov Disord. 2013;28:1407–1415. | |
Ha AD, Fung VS. Huntington’s disease. Curr Opin Neurol. 2012;25: 491–498. | |
Heckmann JM, Legg P, Sklar D, Fine J, Bryer A, Kies B. IV amantadine improves chorea in Huntington’s disease: an acute randomized, controlled study. Neurology. 2004;63:597–598. [author reply 597–598]. | |
O’Suilleabhain P, Dewey RB Jr. A randomized trial of amantadine in Huntington disease. Arch Neurol. 2003;60:996–998. | |
Grimbergen YA, Knol MJ, Bloem BR, Kremer BP, Roos RA, Munneke M. Falls and gait disturbances in Huntington’s disease. Mov Disord. 2008;23:970–976. | |
Gabrieli JD, Stebbins GT, Singh J, Willingham DB, Goetz CG. Intact mirror-tracing and impaired rotary-pursuit skill learning in patients with Huntington’s disease: evidence for dissociable memory systems in skill learning. Neuropsychology. 1997;11:272–281. | |
Heindel WC, Butters N, Salmon DP. Impaired learning of a motor skill in patients with Huntington’s disease. Behav Neurosci. 1988;102: 141–147. | |
Knopman D, Nissen MJ. Procedural learning is impaired in Huntington’s disease: evidence from the serial reaction time task. Neuropsychologia. 1991;29:245–254. | |
Yang Y, Li XY, Gong L, Zhu YL, Hao YL. Tai Chi for improvement of motor function, balance and gait in Parkinson’s disease: a systematic review and meta-analysis. PLoS One. 2014;9:e102942. | |
Quinn L, Debono K, Dawes H, et al; Members of the TRAIN-HD Project Group. Task-specific training in Huntington disease: a randomized controlled feasibility trial. Phys Ther. 2014;94:1555–1568. | |
Thompson JA, Cruickshank TM, Penailillo LE, et al. The effects of multidisciplinary rehabilitation in patients with early-to-middle-stage Huntington’s disease: a pilot study. Eur J Neurol. 2013;20:1325–1329. | |
Busse M, Quinn L, Debono K, et al; Members of the COMMET-HD Management Group. A randomized feasibility study of a 12-week community-based exercise program for people with Huntington’s disease. J Neurol Phys Ther. 2013;37:149–158. | |
Zinzi P, Salmaso D, De Grandis R, et al. Effects of an intensive rehabilitation programme on patients with Huntington’s disease: a pilot study. Clin Rehabil. 2007;21:603–613. | |
Khalil H, Quinn L, van Deursen R, et al. What effect does a structured home-based exercise programme have on people with Huntington’s disease? A randomized, controlled pilot study. Clin Rehabil. 2013;27: 646–658. | |
Quinn L, Busse M. Physiotherapy clinical guidelines for Huntington’s disease. Neurodegen Dis Manag. 2012;2:21–31. | |
Paulsen JS, Long JD. Onset of Huntington’s disease: can it be purely cognitive? Mov Disord. 2014;29:1342–1350. | |
Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. | |
Gluhm S, Goldstein J, Brown D, Van Liew C, Gilbert PE, Corey-Bloom J. Usefulness of the montreal cognitive assessment (MoCA) in Huntington’s disease. Mov Disord. 2013;28:1744–1747. | |
Mickes L, Jacobson M, Peavy G, et al. A comparison of two brief screening measures of cognitive impairment in Huntington’s disease. Mov Disord. 2010;25:2229–2233. | |
Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183–194. | |
Duff K, Paulsen J, Mills J, et al; PREDICT-HD Investigators and Coordinators of the Huntington Study Group. Mild cognitive impairment in prediagnosed Huntington disease. Neurology. 2010;75:500–507. | |
Dumas EM, van den Bogaard SJ, Middelkoop HA, Roos RA. A review of cognition in Huntington’s disease. Front Biosci (Schol Ed). 2013;5: 1–18. | |
Mestre T, Ferreira J, Coelho MM, Rosa M, Sampaio C. Therapeutic interventions for symptomatic treatment in Huntington’s disease. Cochrane Database Syst Rev. 2009;8:CD006456. | |
Bonelli RM, Hofmann P. A systematic review of the treatment studies in Huntington’s disease since 1990. Expert Opin Pharmacother. 2007;8: 141–153. | |
Bonelli RM, Wenning GK. Pharmacological management of Huntington’s disease: an evidence-based review. Curr Pharm Des. 2006;12:2701–2720. | |
Beglinger LJ, Adams WH, Paulson H, et al. Randomized controlled trial of atomoxetine for cognitive dysfunction in early Huntington disease. J Clin Psychopharmacol. 2009;29:484–487. | |
HORIZON Investigators of the Huntington Study Group and European Huntington’s Disease Network. A randomized, double-blind, placebo-controlled study of latrepirdine in patients with mild to moderate Huntington disease. JAMA Neurol. 2013;70:25–33. | |
Wild EJ, Tabrizi SJ. Targets for future clinical trials in Huntington’s disease: what’s in the pipeline? Mov Disord. 2014;29:1434–1445. | |
Van Liew C, Gluhm S, Goldstein J, Cronan TA, Corey-Bloom J. The functional implications of motor, cognitive, psychiatric, and social problem-solving states in Huntington’s disease. Psychiatry. 2013;76: 323–335. | |
Piira A, van Walsem MR, Mikalsen G, Nilsen KH, Knutsen S, Frich JC. Effects of a one year intensive multidisciplinary rehabilitation program for patients with Huntington’s disease: a prospective intervention study. PLoS Curr. 2013;5:1–18. | |
Veenhuizen RB, Kootstra B, Vink W, et al. Coordinated multidisciplinary care for ambulatory Huntington’s disease patients. Evaluation of 18 months of implementation. Orphanet J Rare Dis. 2011;6:77. | |
Duff K, Paulsen JS, Beglinger LJ, Langbehn DR, Stout JC. Predict HDIotHSG. Psychiatric symptoms in Huntington’s disease before diagnosis: the predict-HD study. Biol Psychiatry. 2007;62:1341–1346. | |
Julien CL, Thompson JC, Wild S, et al. Psychiatric disorders in preclinical Huntington’s disease. J Neurol Neurosurg Psychiatry. 2007;78: 939–943. | |
Rosenblatt A. Neuropsychiatry of Huntington’s disease. Dialogues Clin Neurosci. 2007;9:191–197. | |
Mendez MF. Huntington’s disease: update and review of neuropsychiatric aspects. Int J Psychiatry Med. 1994;24:189–208. | |
Folstein SE, Folstein MF. Psychiatric features of Huntington’s disease: recent approaches and findings. Psychiatr Dev. 1983;1:193–205. | |
Schoenfeld M, Myers RH, Cupples LA, Berkman B, Sax DS, Clark E. Increased rate of suicide among patients with Huntington’s disease. J Neurol Neurosurg Psychiatry. 1984;47:1283–1287. | |
Beglinger LJ, Paulsen JS, Watson DB, et al. Obsessive and compulsive symptoms in prediagnosed Huntington’s disease. J Clin Psychiatry. 2008;69:1758–1765. | |
Killoran A, Biglan KM. Current therapeutic options for Huntington’s disease: good clinical practice versus evidence-based approaches? Mov Disord. 2014;29:1404–1413. | |
Killoran A, Biglan KM. Therapeutics in Huntington’s disease. Curr Treat Options Neurol. 2012;14:137–149. | |
Holl AK, Wilkinson L, Painold A, Holl EM, Bonelli RM. Combating depression in Huntington’s disease: effective antidepressive treatment with venlafaxine XR. Int Clin Psychopharmacol. 2010;25:46–50. | |
Beglinger LJ, Adams WH, Langbehn D, et al. Results of the citalopram to enhance cognition in Huntington disease trial. Mov Disord. 2014;29: 401–405. | |
Como PG, Rubin AJ, O’Brien CF, et al. A controlled trial of fluoxetine in nondepressed patients with Huntington’s disease. Mov Disord. 1997;12:397–401. | |
Quinn N, Marsden CD. A double blind trial of sulpiride in Huntington’s disease and tardive dyskinesia. J Neurol Neurosurg Psychiatry. 1984;47:844–847. | |
Raja M, Soleti F, Bentivoglio AR. Lithium treatment in patients with Huntington disease and suicidal behavior. J Clin Psychopharmacol. 2013;33:819–821. | |
Nance M, Paulsen J, Rosenblatt A, Wheelock V. A Physician’s Guide to the Management of Huntington’s Disease. New York: Huntington’s Disease Society of America; 2012. | |
Duff K, Beglinger LJ, O’Rourke ME, Nopoulos P, Paulson HL, Paulsen JS. Risperidone and the treatment of psychiatric, motor, and cognitive symptoms in Huntington’s disease. Ann Clin Psychiatry. 2008;20:1–3. | |
Squitieri F, Cannella M, Porcellini A, Brusa L, Simonelli M, Ruggieri S. Short-term effects of olanzapine in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol. 2001;14:69–72. | |
van der Steenstraten IM, Tibben A, Roos RA, van de Kamp JJ, Niermeijer MF. Predictive testing for Huntington disease: nonparticipants compared with participants in the Dutch program. Am J Hum Genet. 1994;55:618–625. | |
Codori AM, Slavney PR, Rosenblatt A, Brandt J. Prevalence of major depression one year after predictive testing for Huntington’s disease. Genet Test. 2004;8:114–119. | |
Fecteau GW, Boyne J. Behavioural relaxation training with Huntington’s disease patients: a pilot study. Psychol Rep. 1987;61:151–157. | |
Silver A. Cognitive-behavioural therapy with a Huntington’s gene positive patient. Patient Educ Couns. 2003;49:133–138. | |
Leimkuhler AM. [“Self-help potential of the family” can overtax the abilities of the marital partner. Report on a family group of Huntington chorea patients]. “Selbsthilfepotential der Familie” als Überforderung des Ehepartners. Bericht über eine Angehörigengruppe von Chorea-Huntington-Patienten. Psychiatr Prax. 1987;14:78–83. German. | |
Hoth KF, Paulsen JS, Moser DJ, Tranel D, Clark LA, Bechara A. Patients with Huntington’s disease have impaired awareness of cognitive, emotional, and functional abilities. J Clin Exp Neuropsychol. 2007;29:365–376. | |
Roos RA. Huntington’s disease: a clinical review. Orphanet J Rare Dis. 2010;5:40. | |
Andresen JM, Gayán J, Djoussé L, et al; US-Venezuela Collaborative Research Group, HD MAPS Collaborative Research Group. The relationship between CAG repeat length and age of onset differs for Huntington’s disease patients with juvenile onset or adult onset. Ann Hum Genet. 2007;71:295–301. | |
Duyao M, Ambrose C, Myers R, et al. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat Genet. 1993;4: 387–392. | |
Chuo YP, Hou PH, Chan CH, Lin CC, Liao YC. Juvenile Huntington’s disease presenting as difficult-to-treat seizure and the first episode of psychosis. Gen Hosp Psychiatry. 2012;34(436):e411–e439. | |
Cloud LJ, Rosenblatt A, Margolis RL, et al. Seizures in juvenile Huntington’s disease: frequency and characterization in a multicenter cohort. Mov Disord. 2012;27:1797–1800. | |
Knupp K, Wirrell E. Progressive myoclonic epilepsies: it takes a village to make a diagnosis. Neurology. 2014;82:378–379. | |
Brewer HM, Eatough V, Smith JA, Stanley CA, Glendinning NW, Quarrell OW. The impact of juvenile Huntington’s disease on the family: the case of a rare childhood condition. J Health Psychol. 2008;13:5–16. | |
Ribaï P, Nguyen K, Hahn-Barma V, et al. Psychiatric and cognitive difficulties as indicators of juvenile huntington disease onset in 29 patients. Arch Neurol. 2007;64:813–819. | |
Yoon G, Kramer J, Zanko A, et al. Speech and language delay are early manifestations of juvenile-onset Huntington disease. Neurology. 2006;67:1265–1267. | |
Robertson L, Santini H, O’Donovan KL, et al. Current pharmacological management in juvenile Huntington’s disease. PLoS Curr. 2012;4:RRN1304. | |
Paulsen JS, Long JD, Johnson HJ, et al; PREDICT-HD Investigators and Coordinators of the Huntington Study Group. Clinical and biomarker changes in premanifest Huntington disease show trial feasibility: a decade of the PREDICT-HD study. Front Aging Neurosci. 2014;6:78. | |
Paulsen JS, Long JD, Ross CA, et al; PREDICT-HD Investigators and Coordinators of the Huntington Study Group. Prediction of manifest Huntington’s disease with clinical and imaging measures: a prospective observational study. Lancet Neurol. 2014;13:1193–1201. | |
Paulsen JS, Langbehn DR, Stout JC, et al; Predict-HD Investigators and Coordinators of the Huntington Study Group. Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry. 2008;79:874–880. | |
Rote Liste® Service GmbH [homepage]. ROTE LISTE®. Available from: https://www.google.co.nz/webhp?sourceid=chrome-instant&ion=1&espv=2&ie=UTF-8#q=google%20translate. Accessed November 1, 2014. [German]. | |
Suche [Search]. ROTE LISTE, Fachinfo – Service®. Available from: http://www.fachinfo.de/. Accessed November 1, 2014. [German]. | |
EUROPEAN MEDICINES AGENCY, SCIENCE MEDICNES HEALTH [homepage]. Available from: http://www.ema.europa.eu. Accessed November 1, 2014. | |
US Food and Drug Administration [homepage]. US Department of Health and Human Services. Available from: http://www.fda.gov. Accessed November 1, 2014. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.