")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
Clinical characteristics and magnetic resonance imaging findings in nine patients with nonalcoholic Wernicke’s encephalopathy: a retrospective study
Authors Liu YL , Xiao WM, Liang MQ, Wu ZQ, Wang YZ, Qu JF, Chen YK
Received 27 May 2019
Accepted for publication 3 August 2019
Published 26 August 2019 Volume 2019:15 Pages 2433—2441
DOI https://doi.org/10.2147/NDT.S217237
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jun Chen
Yong-Lin Liu,1 Wei-Min Xiao,1 Man-Qiu Liang,2 Zhi-Qiang Wu,1 Ya-Zhi Wang,1 Jian-Feng Qu,1 Yang-Kun Chen1
1Department of Neurology, Affiliated Dongguan People’s Hospital, Southern Medical University (Dongguan People’s Hospital), Dongguan, Guangdong Province, People’s Republic of China; 2Department of Radiology, Affiliated Dongguan People’s Hospital, Southern Medical University (Dongguan People’s Hospital), Dongguan, Guangdong Province, People’s Republic of China
Correspondence: Yang-Kun Chen
Department of Neurology, Affiliated Dongguan People’s Hospital, Southern Medical University (Dongguan People’s Hospital), 3 South Wandao Road, Wanjiang District, Dongguan City, Guangdong Province 523059, People’s Republic of China
Tel +86 7 692 863 6830
Fax +86 07 692 222 2353
Email [email protected]
Purpose: Wernicke’s encephalopathy (WE) is a severe neurological disorder caused by thiamine deficiency. The most common cause of WE is alcoholism. However, there is a significant paucity of information in the existing literature relating to nonalcoholic WE. In this study, we investigated the clinical characteristics and neuroimaging findings of nine patients with nonalcoholic WE.
Patients and methods: We retrospectively collated clinical data from nine patients who had been diagnosed with WE in accordance with established criteria including age, gender, risk factors and clinical manifestations. We also collated initial hematological and neuroimaging findings.
Results: The mean age of the nine patients was 54.0±17.1 years; four of these patients (44.4%) were male. All nine patients had a history of fasting (range, 5–47 days) prior to WE. Four of the nine patients (44.4%) exhibited the classical triad, and eight (88.9%) showed alterations in mental status. Magnetic resonance imaging (MRI) scans showed that all nine patients had symmetric lesions of the medial thalamus. MRI also revealed other WE-related lesions in mammillary bodies (22.2%), the periaqueductal region (55.6%), the tectal plate of the midbrain (77.8%), cranial nerve nuclei (77.8%) and in the symmetric subcortical white matter (11.1%).
Conclusion: Our analysis showed that fasting is a common cause of WE in nonalcoholic patients and that MRI is a useful tool for the diagnosis of WE. The most common MRI findings were symmetrical lesions of the medial thalamus lesions, followed by the tectal plate of the midbrain and cranial nerve nuclei.
Keywords: Wernicke’s encephalopathy, altered mental status, magnetic resonance imaging, fasting, thiamine, alcoholism
Introduction
Wernicke’s encephalopathy (WE) is an uncommon but severe neurological disorder which results from thiamine (vitamin B1) deficiency. The most common cause of WE is chronic alcohol abuse. However, the risk factors for nonalcoholic patients include cancer, gastrointestinal surgery and hyperemesis gravidarum.1–3 Previous postmortem studies have estimated that the prevalence of WE in the general population ranges from 0.4% to 2.8%.4,5 This condition is characterized by altered mental state or mild memory impairment, along with oculomotor abnormalities and cerebellar dysfunction collectively, which are referred to as the classic clinical triad of WE. Nevertheless, this classic triad is only present in approximately 16.5% of patients.6 Furthermore, approximately 19% of patients do not exhibit any of these classic symptoms at the presentation of WE.6,7 Magnetic resonance imaging (MRI) is a useful tool for confirming a diagnosis of WE, particularly when clinical presentation is nonspecific. This is because MRI can readily demonstrate typical (thalamus, mammillary bodies, tectal plate and periaqueductal region) and atypical (cerebellum, cranial nerve nuclei and cerebral cortex) alterations in signal intensity.8 The prognosis of WE is good if a correct diagnosis is made early. However, some cases of WE become irreversible if diagnosis is delayed or misdiagnosis occurs. Therefore, the timely and accurate diagnosis of WE are critical for improving the prognosis of patients affected by this disease. Most of the published literature relating to WE refer to alcoholic patients. However, there have been only a few published studies focusing on nonalcoholic WE,9–14 which is easily neglected by health care professionals. Thus, we aimed to investigate the clinical and neuroimaging characteristics of nonalcoholic patients with WE.
Materials and methods
This study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of Dongguan People’s Hospital (Reference Number: KYKT2018-007). As this study featured a retrospective design, the Ethics Committee judged it as minimal risk research and the participants could not be located. Thus, they approved a waiver of informed consent. All study data of the participants were kept confidentially by the investigators according to the guideline of the Ethics Committee.
Participants
This was a retrospective study of patients diagnosed with WE at Dongguan People’s Hospital between 1 January 2011 and 31 December 2017. Dongguan People’s Hospital, which is the largest public tertiary hospital in Dongguan City, is located in the south of China. The population in Dongguan City is almost 8 million. According to recent statistics, there were 105,055 inpatients admitted into our hospital in 2017. We included patients with a clinical diagnosis of WE and showing at least two criteria from the classification previously published by Caine et al.15 Patients were excluded if they were younger than 14 years of age and/or neuroimaging data were not available. Those who had a history of alcohol abuse were excluded as well.
Demographic and clinical variables
For each subject, we collected a range of demographic and clinical variables, including age, gender, risk factors (eg, acute pancreatitis, cancer and hyperemesis gravidarum), clinical manifestations (including ocular signs, cerebellar signs, altered mental status or mild memory impairment) and initial hematological findings. We used the mini-mental state examination (MMSE) to assess global cognitive impairment. Hematological examinations included hemoglobin, serum creatinine, blood glucose, sodium, potassium, mean corpuscular volume (MCV), gamma-glutamyl transpeptidase (GGT) and serum albumin levels.
Diagnosis of WE
WE was diagnosed in accordance with specific criteria previously published by Caine et al.15,16 According to Caine et al, two of the following four signs must be observed: 1) dietary deficiencies; 2) oculomotor abnormalities; 3) cerebellar dysfunction and 4) altered mental state or mild memory impairment.15 Dietary deficiencies were defined as a body mass index lower than 2 standard deviation below normal as evidence of undernutrition, a history of grossly impaired dietary intake or an abnormal thiamine status.15
MRI analysis
All nine patients underwent MRI scans with a 3.0T system (Skyra, Siemens Medical, Berlin, Germany) within 3 days of the onset of prominent WE-related symptoms and before the administration of thiamin. All patients were examined with T1-weighted imaging (T1WI) and T2-weighted imaging (T2WI), as well as fluid-attenuated inversion recovery (FLAIR) imaging. Seven patients were examined with diffusion-weighted imaging (DWI) and four with enhanced scanning by gadolinium contrast. We also acquired axial spin-echo (SE) T1 images (time of repetition [TR]/time of echo [TE]/excitation =1500/11/1; field of view [FOV] =220 mm; slice thickness/gap =4 mm/1.2 mm; matrix =320×320; time of acquisition =1 min 26 s) and turbo spin-echo (TSE) T2 images (TR/TE/excitation =4720/96/2; turbo factor: 15; FOV =220 mm; slice thickness/gap =4 mm/1.2 mm; matrix =512×512; time of acquisition =1 min 50 s). Coronal position FLAIR (TR/TE/excitation =9000/84/1; FOV =230 mm; slice thickness/gap =5 mm/1.5 mm; matrix =320×320; time of acquisition =1 min 50 s) and DWI spin-echo echo-planar imaging (EPI) (TR/TE/excitation =4640/67/1; matrix =192×192; FOV =230 mm; slice thickness/gap =4 mm/1.2 mm; EPI factor =91; acquisition time =1 min 44 s) were also carried out; these images utilized three orthogonally applied gradients, with b values of 0 and 1000.
Follow-up
Patients were followed up via telephone or by face-to-face interview. For each patient, we used a modified Rankin scale (mRS)17 to quantitate the level of disability at follow-up.
Statistical analysis
Statistical analyses were conducted using SPSS for Windows (version 20.0, SPSS Inc., Chicago, IL, USA). Variables are presented as a mean (with SD), frequency or median (IQR), as appropriate.
Results
Participant characteristics
A total of nine patients with nonalcoholic WE were recruited, none of whom had a history of alcohol abuse. Four of the nine patients (44.4%) were male. Mean patient age was 54.0±17.1 years (range, 29–81 years). The levels of serum albumin were all below the normal range, with a mean of 31.5 g/L (range, 24.7–36.6 g/L). Six of the nine patients had lower blood sodium levels than normal, with a mean of 136.1 mmol/L (range, 131.0–147.7 mmol/L). Five of the patients had lower levels of hemoglobin than normal, with a mean of 106.8 g/L (range, 89–128 g/L). The levels of GGT were all normal, with a mean of 28.6 U/L (range, 8.6–53.1 U/L). The mean of MCV was 86.6 fl (range, 79.0–90.9fl).The demographic characteristics and hematological findings of each participant at the onset of WE are shown in Table 1.
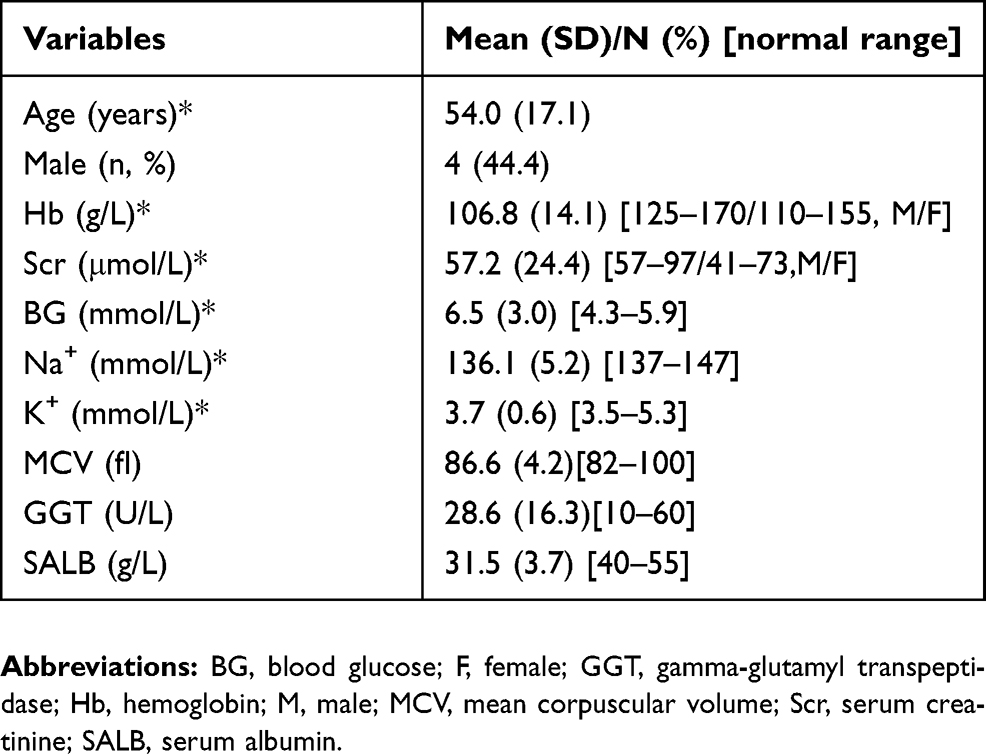 |
Table 1 Demographic characteristics and hematological findings of patients |
All nine patients had a history of fasting (ranging from 5 to 47 days) before WE as a result of acute pancreatitis (two cases), gastrointestinal tumors (four cases), intestinal obstruction (one case), pyloric obstruction (one case) and hyperemesis gravidarum (one case). During fasting, the patients received total parenteral nutrition without thiamine supplementation. The classic triad was evident in four patients (44.4%), while two patients (22.2%) exhibited two of the three classic manifestations; the other three patients (33.4%) exhibited only one of the classic manifestations. Eight of the nine patients (88.9%) exhibited changes in mental status; five patients presented with a reduced level of consciousness (drowsiness in four cases, coma in one case), while the other three patients presented with delirium. Three patients had psychiatric symptoms, including persecutory delusion (one case) and visual hallucination (two cases). Four participants (44.4%) showed memory impairment. Oculomotor abnormalities were present in seven participants (77.8%) as well as in four patients (44.4%) with nystagmus. Four patients (44.4%) showed cerebellar signs, which manifested as gait instability. All patients were treated with intramuscular (IM) thiamine (100 mg, 3 times per day). The median interval between the onset of WE-related symptoms and the first administration of IM thiamin was 7 days (range, 5–12 days). The median duration of IM thiamin was 13 days (range, 8–28 days). The clinical characteristics of the nine patients with WE, along with relevant frequency data, are shown in Tables 2 and 3.
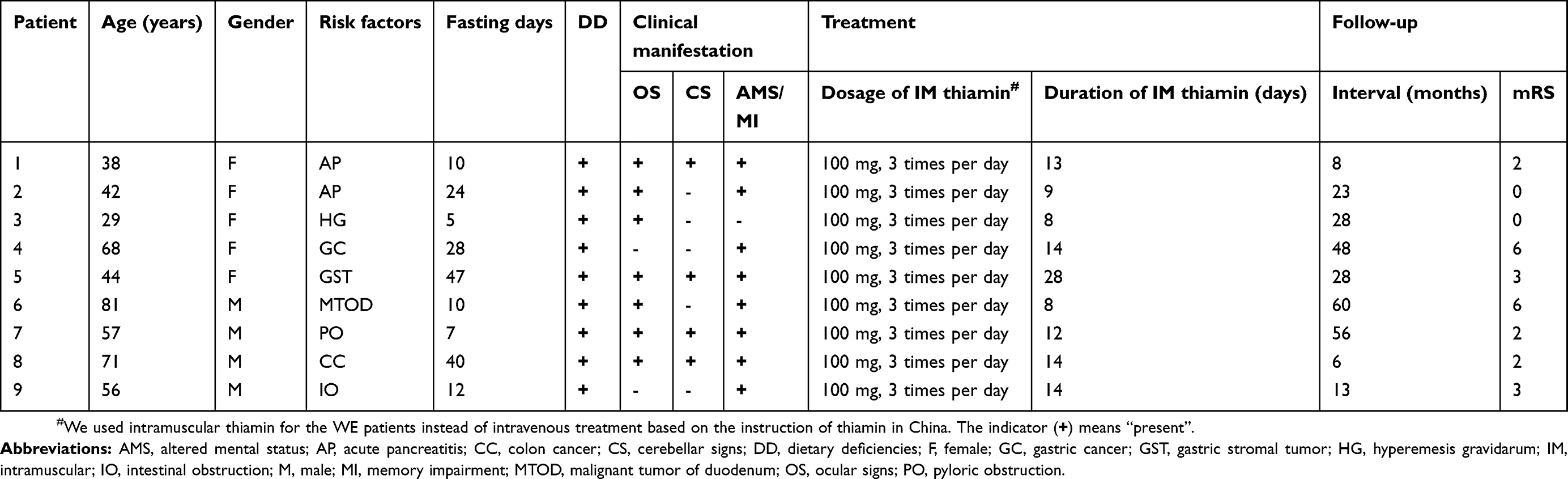 |
Table 2 Clinical characteristics and follow-up of 9 patients with Wernicke’s encephalopathy |
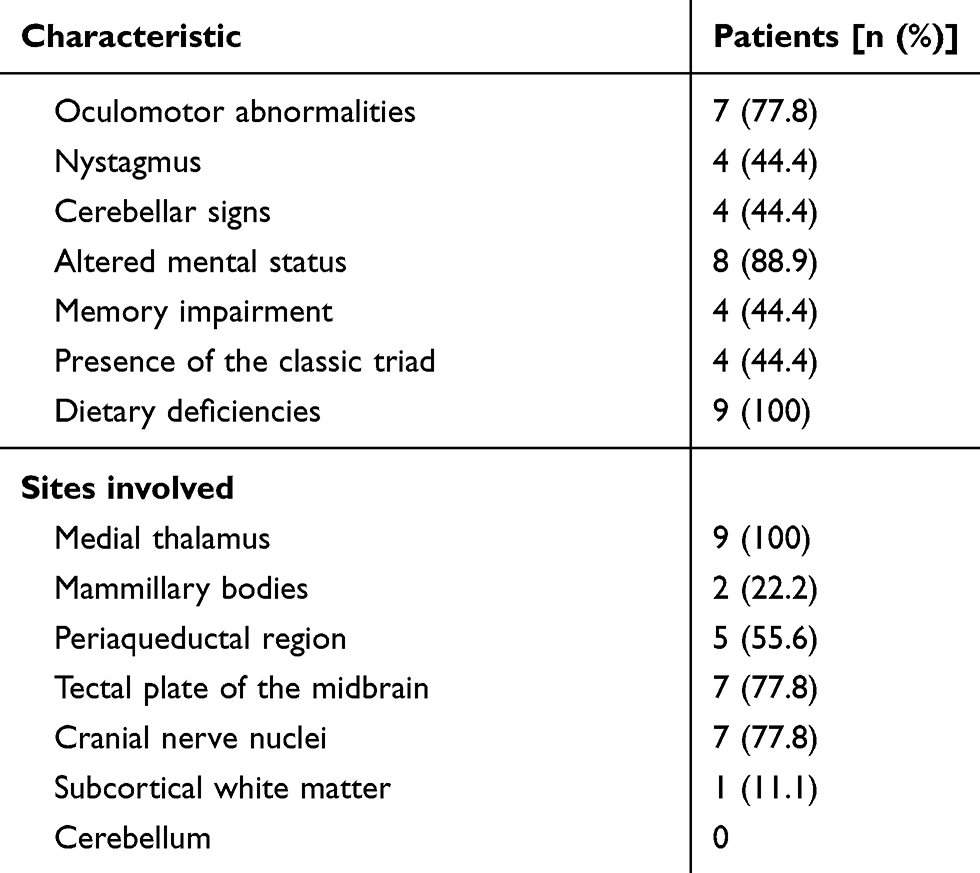 |
Table 3 Frequency of key clinical characteristics and topographic distribution of lesions in patients with Wernicke’s encephalopathy |
MRI findings
MRI showed symmetric hyperintensity on T2WI and FLAIR imaging and symmetrical hypointensity or no abnormalities on T1WI. Of the seven patients who underwent DWI scans, all showed symmetric hyperintensity lesions on DWI, while only three showed hypointensity on apparent diffusion coefficient (ADC). No enhanced lesions were observed in the four patients who underwent enhanced scanning.
All participants showed symmetrical increases of T2WI and FLAIR signals in the medial thalamus. T2WI and FLAIR imaging identified hyperintense lesions in the midbrain tectal plate in seven of the participants (77.8%). The periaqueductal region was involved in five patients (55.6%), and mammillary bodies in two patients (22.2%). Furthermore, atypical lesions were found in the cranial nerve nuclei (bilateral medial vestibular nuclei; seven patients, 77.8%) and subcortical white matter lesions (one patient, 11.1%) (Figure 1). We did not observe any lesion in the cerebellum. The topographic distribution of lesions in the nine patients with WE is shown in Table 3.
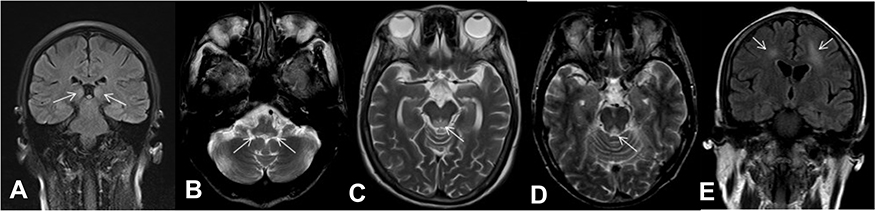 |
Figure 1 (A) A 38-year-old woman (Patient 1) had a history of acute pancreatitis and presented with the classic triad. FLAIR coronal imaging showed hyperintensity of the symmetric medial thalamus. The white arrows represent the symmetric medial thalamus. (B) A 57-year-old man (Patient 7) had pyloric obstruction and presented with the classic triad. T2WI axial imaging showed hyperintensity of the medial vestibular nuclei. (C) A 68-year-old woman (Patient 4) had a history of surgery and fasting for her gastric cancer and presented with delirium and persecutory delusion. T2WI axial imaging showed hyperintensity of the tectal plate. (D) A 71-year-old man (Patient 8) had a history of colon cancer and presented with the classic triad. T2WI axial imaging showed hyperintensity of the periaqueductal region. (E) A 42-year-old woman (Patient 2) had a history of acute pancreatitis and presented with drowsiness and oculomotor abnormalities. FLAIR coronal imaging showed hyperintensity of the symmetric subcortical white matter. The white arrows represent the symmetric subcortical white matter. Abbreviations: FLAIR, fluid-attenuated inversion recovery; T2WI, T2-weighted imaging. |
Patient outcomes
The mean follow-up period was 30.0±20.3 months after discharge. Two patients died within 6 months of discharge because of malignant tumors. Two patients had an mRS score of 0, three patients had an mRS score of 2 and two patients had an mRS score of 3. One patient (Patient 5) underwent repeated MRI scans 8 months after the onset of WE; the hyperintensity signal on DWI had disappeared, with an attenuated hyperintensity signal on T2WI (Figure 2).
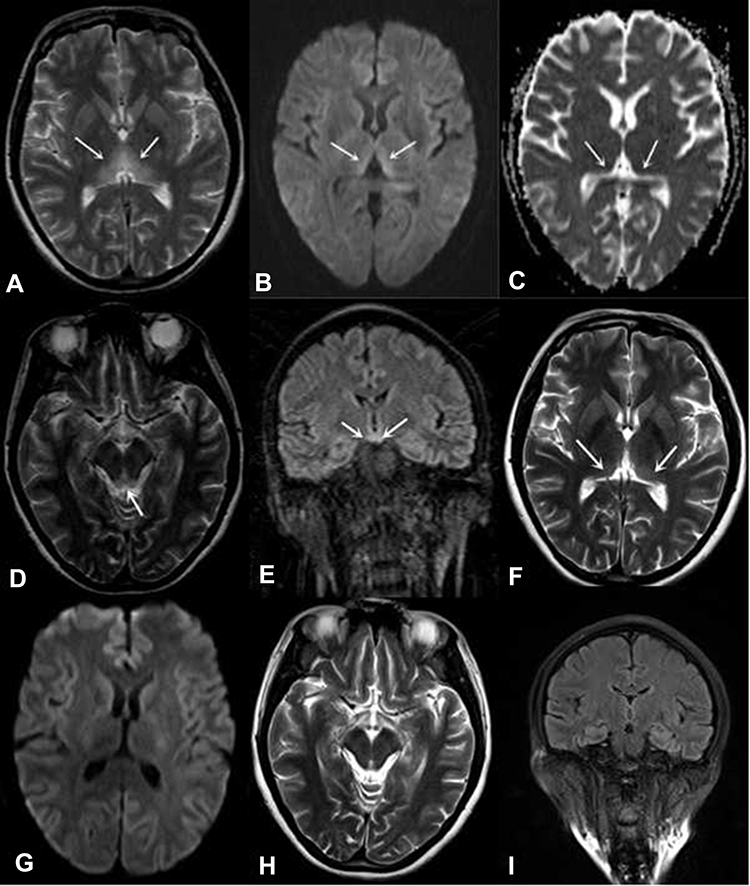 |
Figure 2 A 44-year-old woman (Patient 5) had a history of surgery and fasting for her gastric stromal tumor and presented with the classic triad. MRI showed hyperintensity of the symmetric medial thalamus (A), tectal plate (D) and mammillary bodies (E). DWI showed hyperintensity of the symmetric medial thalamus (B), while ADC showed hypointensity (C). Repeated MRI scans 8 months after presentation (F–I). The hyperintensity of the symmetric medial thalamus had decreased on T2WI (F), while intensity was normal on DWI (G). The intensity of the tectal plate observed by T2WI (H) and the mammillary bodies observed by FLAIR (I) were normal. Notes: The white arrows in panel A represent the hyperintensive symmetric medial thalamus on T2WI. The white arrows in panel B represent the hyperintensive symmetric medial thalamus on DWI. The white arrows in panel C represent the hypointensive symmetric medial thalamus on ADC. The white arrow in panel D represents the hyperintensive tectal plate of midbrain on T2WI. The white arrows in panel E represent the hyperintensive mammillary bodies on FLAIR. The white arrows in panel F represent the hyperintensive symmetric medial thalamus on T2WI 8 months after presentation.Abbreviations: ADC, apparent diffusion coefficient; DWI, diffusion-weighted imaging; FLAIR, fluid-attenuated inversion recovery; MRI, magnetic resonance imaging; T2WI, T2-weighted imaging. |
Discussion
WE is a metabolic disease caused by thiamine deficiency. Although chronic alcoholism is the leading cause of WE, it is also important to examine the causes of WE in nonalcoholic patients. It has previously been reported that nonalcoholic WE may occur after most gastrointestinal surgical procedures, including gastric bypass surgery,2,18,19 gastrojejunostomy20 and gastrectomy.21 In addition, acute pancreatitis,22,23 pyloric obstruction24 and hyperemesis gravidarum3,25 are also regarded as being predisposing factors for WE. In all of our current patients, WE was attributed to nonalcoholism-related causes, such as gastrointestinal surgery, hyperemesis gravidarum, acute pancreatitis and pyloric obstruction. Our nine nonalcoholic WE patients all had a history of fasting, ranging from 5 to 47 days, which may have resulted in thiamine deficiency. This indicated that even 5 days of fasting without thiamine supplement could result in WE. These findings suggest that fasting is a common cause of WE in nonalcoholic patients. Therefore, it is very important to provide thiamine supplementation to those who have dietary deficiencies or those who are at risk of WE.10,26
We found that 44.4% of our patients showed the classic triad of clinical manifestations; this was higher than that reported by previous studies,6,11,14 but lower than that in another study by Gascon-Bayarri et al.12 Changes in mental status and oculomotor abnormalities occurred in 88.9% and 77.8% of our patients, respectively; these findings were similar to those reported previously by Zuccoli et al.11 The present results were also in accordance with previous studies in that the most common manifestation of WE was changes in mental status.6,7,27 Collectively, these findings show that WE should be considered when a patient presents with changes to their mental status and dietary deficiency.
The MRI findings in this study revealed the presence of typical lesions, including those in the symmetric medial thalamus, mammillary bodies, periaqueductal region, and the tectal plate. Cranial nerve nuclei and subcortical white matter were also evident as atypical lesions; these findings were consistent with those reported by previous studies.8,28 In our study, lesions were most frequently found in the bilateral medial thalamus (9/9); these findings were similar to those published previously.10,11,14 Lesions were also frequently found in the tectal plate and the cranial nerve nuclei. These findings were in accordance with one previous study,11 but not others.10,14 No lesions in the cerebellum were observed in our study, which was similar to a previous study.12 We speculate that the involvement of the tectal plate and the cranial nerve nuclei may be more specific to nonalcoholic cases of WE. In Table 4, we present a summary of previously published studies that have investigated nonalcoholic cases of WE and compared these studies with data arising from our present study. According to previous literature, lesions may exhibit hyperintensity on DWI images with reduced, normal or even increased ADC values.29 In the present study, all seven patients who underwent DWI scans presented with hyperintensity. Three patients showed hypointensity on ADC, while the other four patients showed normal ADC values. DWI hyperintensity with reduced ADC is indicative of cytotoxic edema in the neurons and glial cells; this condition is characterized by restricted diffusion.30,31 However, according to Chu et al, hyperintensity on DWI, as well as reduced ADC, does not always indicate irreversible tissue damage, but rather, indicates the presence of tissues which are at risk of irreversible damage.32 In the present study, two patients (Patient 1 and Patient 7), who showed hyperintensity on DWI and reduced ADC, had a relatively good outcome (mRS =2) after sufficient thiamine therapy. Contrast enhancement in the mammillary bodies and thalamus is a typical finding for this disease in alcoholic patients.11 In the present study, no contrast enhancement was observed in the mammillary bodies of any of the four nonalcoholic patients who underwent enhanced scanning.
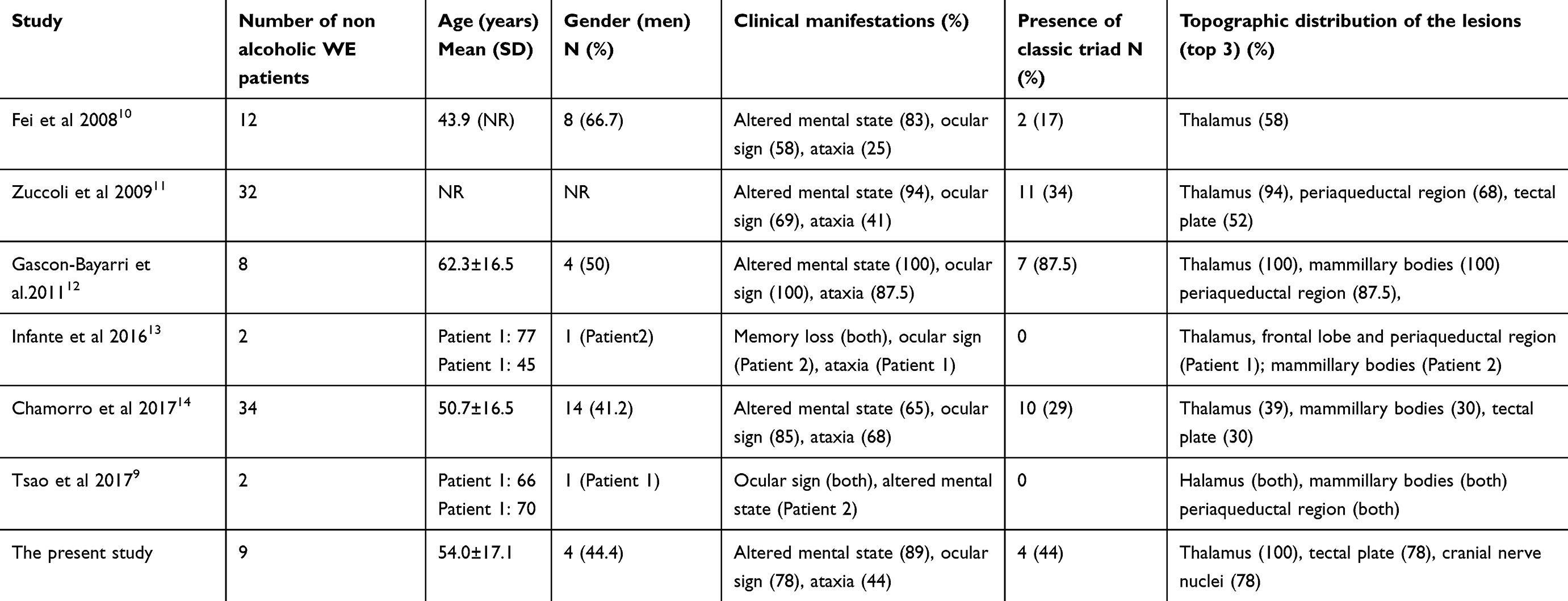 |
Table 4 Summary of studies investigating cases of non-alcoholic WE |
There were several advantages to our study. First, we recruited nine nonalcoholic cases of WE, each with relatively complete datasets, including neuroimaging data. In addition, our neuroimaging findings were in accord with a previous study by Zuccoli et al,11 which featured a large sample size and indicated that the tectal plate of the midbrain and cranial nerve nuclei might be prone to an involvement in nonalcoholic WE. However, there were several limitations to our study which also need to be considered. First, the sample size was relatively small. Second, we were unable to evaluate the basal thiamine levels of patients prior to its administration. Third, due to our retrospective experimental design, it was difficult to evaluate how the findings evolved on imaging.
Conclusion
This study suggests that fasting is a common cause of WE in nonalcoholic patients. MRI, which revealed differential patterns in the distribution of lesions between alcoholic patients and nonalcoholic patients, is a useful tool for the early diagnosis of WE. Involvement of the tectal plate and cranial nerve nuclei on MRI may be specific to nonalcoholic WE. These findings should now be confirmed by further studies which incorporate larger sample sizes.
Acknowledgment
The authors declare that there are no sources of funding to be acknowledged.
Author contributions
YL Liu and YK Chen participated in the conception and design of the study, the analysis of clinical data and critical revision of the manuscript for scientific validity. WM Xiao and MQ Liang analyzed the imaging data. ZQ Wu and JF Qu helped to acquire raw data. YZ Wang assisted in patient follow-ups. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Aasheim ET. Wernicke encephalopathy after bariatric surgery: a systematic review. Ann Surg. 2008;248(5):714–720. doi:10.1097/SLA.0b013e3181884308
2. Singh S, Kumar A. Wernicke encephalopathy after obesity surgery: a systematic review. Neurology. 2007;68(11):807–811. doi:10.1212/01.wnl.0000256812.29648.86
3. Chiossi G, Neri I, Cavazzuti M, Basso G, Facchinetti F. Hyperemesis gravidarum complicated by Wernicke encephalopathy: background, case report, and review of the literature. Obstet Gynecol Surv. 2006;61(4):255–268. doi:10.1097/01.ogx.0000206336.08794.65
4. Harper C, Fornes P, Duyckaerts C, Lecomte D, Hauw JJ. An international perspective on the prevalence of the Wernicke-Korsakoff syndrome. Metab Brain Dis. 1995;10(1):17–24.
5. Harper C. The incidence of Wernicke’s encephalopathy in Australia–a neuropathological study of 131 cases. J Neurol Neurosurg Psychiatry. 1983;46(7):593–598. doi:10.1136/jnnp.46.7.593
6. Harper CG, Giles M, Finlay-Jones R. Clinical signs in the Wernicke-Korsakoff complex: a retrospective analysis of 131 cases diagnosed at necropsy. J Neurol Neurosurg Psychiatry. 1986;49(4):341–345. doi:10.1136/jnnp.49.4.341
7. Torvik A, Lindboe CF, Rogde S. Brain lesions in alcoholics. A neuropathological study with clinical correlations. J Neurol Sci. 1982;56(2–3):233–248. doi:10.1016/0022-510x(82)90145-9
8. Zuccoli G, Pipitone N. Neuroimaging findings in acute Wernicke’s encephalopathy: review of the literature. AJR Am J Roentgenol. 2009;192(2):501–508. doi:10.2214/AJR.07.3959
9. Tsao WC, Ro LS, Chen CM, Chang HS, Kuo HC. Non-alcoholic Wernicke’s encephalopathy with cortical involvement and polyneuropathy following gastrectomy. Metab Brain Dis. 2017;32(5):1649–1657. doi:10.1007/s11011-017-0055-8
10. Fei GQ, Zhong C, Jin L, et al. Clinical characteristics and MR imaging features of nonalcoholic Wernicke encephalopathy. AJNR Am J Neuroradiol. 2008;29(1):164–169. doi:10.3174/ajnr.A0827
11. Zuccoli G, Santa CD, Bertolini M, et al. MR imaging findings in 56 patients with Wernicke encephalopathy: nonalcoholics may differ from alcoholics. AJNR Am J Neuroradiol. 2009;30(1):171–176. doi:10.3174/ajnr.A1280
12. Gascón-Bayarri J, Campdelacreu J, García-Carreira MC, et al. [Wernicke’s encephalopathy in non-alcoholic patients: a series of 8 cases]. Neurologia. 2011;26(9):540–547. doi:10.1016/j.nrl.2011.03.001
13. Infante MT, Fancellu R, Murialdo A, Barletta L, Castellan L, Serrati C. Challenges in diagnosis and treatment of Wernicke encephalopathy: report of 2 cases. Nutr Clin Pract. 2016;31(2):186–190. doi:10.1177/0884533615621753
14. Chamorro AJ, Roson-Hernandez B, Medina-Garcia JA, et al. Differences between alcoholic and nonalcoholic patients with Wernicke Encephalopathy: a multicenter observational study. Mayo Clin Proc. 2017;92(6):899–907. doi:10.1016/j.mayocp.2017.02.019
15. Caine D, Halliday GM, Kril JJ, Harper CG. Operational criteria for the classification of chronic alcoholics: identification of Wernicke’s encephalopathy. J Neurol Neurosurg Psychiatry. 1997;62(1):51–60. doi:10.1136/jnnp.62.1.51
16. Galvin R, Brathen G, Ivashynka A, Hillbom M, Tanasescu R, Leone MA. EFNS guidelines for diagnosis, therapy and prevention of Wernicke encephalopathy. Eur J Neurol. 2010;17(12):1408–1418. doi:10.1111/j.1468-1331.2010.03153.x
17. de Haan R, Limburg M, Bossuyt P, van der Meulen J, Aaronson N. The clinical meaning of Rankin ‘handicap’ grades after stroke. Stroke. 1995;26(11):2027–2030. doi:10.1161/01.str.26.11.2027
18. Nolli M, Barbieri A, Pinna C, Pasetto A, Nicosia F. Wernicke’s encephalopathy in a malnourished surgical patient: clinical features and magnetic resonance imaging. Acta Anaesthesiol Scand. 2005;49(10):1566–1570. doi:10.1111/j.1399-6576.2005.00879.x
19. Loh Y, Watson WD, Verma A, Chang ST, Stocker DJ, Labutta RJ. Acute Wernicke’s encephalopathy following bariatric surgery: clinical course and MRI correlation. Obes Surg. 2004;14(1):129–1232. doi:10.1381/096089204772787437
20. Worden RW, Allen HM. Wernicke’s encephalopathy after gastric bypass that masqueraded as acute psychosis: a case report. Curr Surg. 2006;63(2):114–116. doi:10.1016/j.cursur.2005.06.004
21. Hamilton LA, Darby SH, Hamilton AJ, Wilkerson MH, Morgan KA. Case report of Wernicke’s encephalopathy after sleeve gastrectomy. Nutr Clin Pract. 2018;33(4):510–514. doi:10.1177/0884533617722758
22. Krokos N, Karakatsanis A, Sarafianos P, Koukou S. Wernicke’s encephalopathy complicating acute necrotic pancreatitis. Am Surg. 2012;78(9):E390–E392.
23. Kayar Y, ElShobaky M, Danalioglu A, et al. Wernicke encephalopathy in a patient with severe acute pancreatitis. Acta Gastroenterol Belg. 2015;78(1):58–59.
24. Tanaka K, Aoki M, Hamada Y, et al. Wernicke’s encephalopathy caused by pyloric stenosis after endoscopic submucosal dissection. Gastrointest Endosc. 2009;69(6):1170–1171. doi:10.1016/j.gie.2008.11.023
25. Mathew NR, Menon SG, Mathew M. Ocular manifestations in a case of Wernicke’s encephalopathy due to hyperemesis gravidarum. Oman J Ophthalmol. 2018;11(1):85–87. doi:10.4103/ojo.OJO_137_2017
26. Sechi G, Serra A. Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007;6(5):442–455. doi:10.1016/S1474-4422(07)70104-7
27. Lindboe CF, Loberg EM. Wernicke’s encephalopathy in non-alcoholics. An autopsy study. J Neurol Sci. 1989;90(2):125–129. doi:10.1016/0022-510x(89)90095-6
28. Zuccoli G, Motti L. Atypical Wernicke’s encephalopathy showing lesions in the cranial nerve nuclei and cerebellum. J Neuroimaging. 2008;18(2):194–197. doi:10.1111/j.1552-6569.2007.00188.x
29. Lapergue B, Klein I, Olivot JM, Amarenco P. Diffusion weighted imaging of cerebellar lesions in Wernicke’s encephalopathy. J Neuroradiol. 2006;33(2):126–128.
30. Loh Y, Watson WD, Verma A, Krapiva P. Restricted diffusion of the splenium in acute Wernicke’s encephalopathy. J Neuroimaging. 2005;15(4):373–375. doi:10.1177/1051228405279037
31. Halavaara J, Brander A, Lyytinen J, Setala K, Kallela M. Wernicke’s encephalopathy: is diffusion-weighted MRI useful? Neuroradiology. 2003;45(8):519–523. doi:10.1007/s00234-003-1043-8
32. Chu K, Kang DW, Kim HJ, Lee YS, Park SH. Diffusion-weighted imaging abnormalities in wernicke encephalopathy: reversible cytotoxic edema? Arch Neurol. 2002;59(1):123–127. doi:10.1001/archneur.59.1.123
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.