")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 15
Clinical Characteristics and Gene Mutations of Two Families with MODY 3 in Inner Mongolia
Authors Ren XY, Xue MR, Yan ZL, Zhang SJ, Liu M, Li AZ
Received 16 June 2022
Accepted for publication 29 September 2022
Published 19 December 2022 Volume 2022:15 Pages 1019—1027
DOI https://doi.org/10.2147/PGPM.S371141
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Xiao-Yan Ren,1,* Meng-Ruo Xue,2,* Zhao-Li Yan,1 Shao-Jie Zhang,3 Min Liu,1 Ai-Zhen Li1
1Department of Endocrinology, Affiliated Hospital of Inner Mongolia Medical University, Hohhot, 010050, People’s Republic of China; 2Department of Interventional Radiology, The Affiliated Hospital of Inner Mongolia Medical University, Hohhot, 010050, People’s Republic of China; 3Department of Anatomy, Inner Mongolia Medical University, Hohhot, 010050, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhao-Li Yan, Department of Endocrinology, Affiliated Hospital of Inner Mongolia Medical University, No. 1, North Tongdao Street, Huiming District, Hohhot, 010050, People’s Republic of China, Tel +86 13848177245, Email [email protected]
Objective: This study aimed to analyze the clinical characteristics and gene mutations of two families with maturity-onset diabetes of the young 3 (MODY 3) in Inner Mongolia.
Methods: Fifty-three patients in Inner Mongolia suspected of having MODY 3 were enrolled in this study according to clinical manifestations. Blood samples were collected, and all exons of the HNF1α gene were analyzed; the second-generation DNA of the splicing regions of the gene was determined by direct sequencing.
Results: In Family 1, the proband, mother, and uncle all carried the missense heterozygous mutation on exon 2 of the HNF1α gene (c.512G>A, p.Arg171Gln), and both the proband and uncle had MODY 3. In Family 2, the proband, grandfather, father, uncle I, and uncle II all carried a missense mutation on exon 2 (c.391C>t, p.Arg131Trp), and all had MODY 3. The blood glucose control in these patients was stable while they were being treated with oral sulfonylurea hypoglycemic drugs alone or with insulin. Uncle II had serious macrovascular and microvascular complications.
Conclusion: Maturity-onset diabetes of the young 3 gene mutations (c.512G>A, p.Arg171Gln) and (c.391C>T, p.Arg131Trp) may be the main pathogenic genes of the two families with MODY 3. The two gene mutations found in this study have not been reported previously in China.
Keywords: gene, HNF1α, MODY, c.512G>A, (Arg171Gln), c.391C>T, (Arg131Trp)
Introduction
One of the most important contributions to diabetes is the identification of monogenic forms in recent years. Monogenic diabetes represents only a small portion of the diabetes cases. The most frequent monogenic diabetes type is the maturity onset diabetes of the young (MODY), seen in 1–2% of overall diabetes patients and including a number of single gene disorders which effect pancreatic beta-cell functions.1 It was first described by Tattersall and Fajans in 1973.Maturity-onset diabetes of the young (MODY) is an autosomal dominant type of diabetes; it is characterized by an early age of onset (usually < 25 years) and an islet β cell function defect.2 At present, MODY is divided into 13 types. Mutations in GCK, HNF1α, HNF4α, and HNF1β are the most frequently identified etiologies of MODY.3
Maturity-onset diabetes of the young 3 (MODY 3) is caused by a mutation of the HNF1α gene, which is located on chromosome 12. HNF1α is expressed in the liver, kidney, intestine, and islet β cells and is a key transcription factor regulating insulin secretion. To date, 450 species of HNF1α gene mutations have been found.4 The condition is characterized by strong clinical heterogeneity, early age of onset, autosomal dominant inheritance, and high penetrance. Because its clinical manifestations overlap with type 1 diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM), approximately 80% of MODY is misdiagnosed as T1DM or T2DM, resulting in its actual prevalence being underestimated.5
In this study, 53 cases of early onset diabetes were screened, and two families with MODY that have not been reported in China were discovered. Two Chinese families with MODY 3 with heterozygous HNF1α mutations c.512G>A (Arg171Gln) and c.391C>T (Arg131Trp) were identified. The HNF1α gene mutation sites of these two families have not been reported previously in China.
Subjects and Method
Subjects
1. Family 1: The proband was a 35-year-old female (height [HT]: 161cm, weight [WT]: 54 kg, and body mass index [BMI]: 20.83 kg/m2). Both birth weight and hearing were normal. One year ago, a physical examination found that her fasting plasma glucose (FPG) was 11.7 mmol/L, and her 2-hour postprandial blood glucose (2hPBG) was 15 mmol/L. Diabetes mellitus (DM) was diagnosed. After diet and exercise interventions, the patient’s 2hPBG was approximately 13.8 mmol/L (the glucose oxidase method was adopted for PBG determination), and saxagliptin (5 mg q.d. orally) was prescribed to reduce blood glucose levels. Blood glycated hemoglobin (HbA1c) was controlled at about 8.5% (HbA1c was determined by a high-pressure liquid-phase method). The family history is presented in Figure 1A. The patient’s medication was subsequently changed to glimepiride (0.5 mg q.d. orally). Her blood glucose control was stable, FBG was 5.7–6.7 mmol/L, and 2hPBG was 6.8–8.9 mmol/L. The patient’s HbA1c was controlled at about 6%, and her urine glucose was positive.
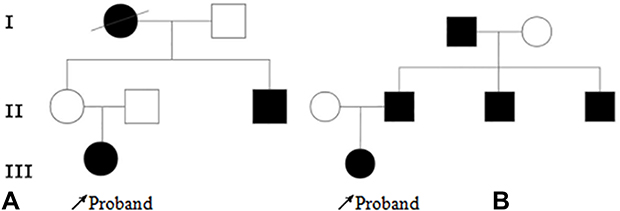 |
Figure 1 Pedigree diagram of family 1 (A) and family 2 (B). Arrow: Proband; Bias: Dead and undetected; Square: Male; Circle: Female; Blank: No clinical symptoms; Black: diabetes mellitus. |
The proband’s grandmother (I.1), who had diabetes for 4 years, was prescribed Glucobay® (50 mg tid orally) and has since died. The proband’s uncle (II.3) developed diabetes at the age of 35. He has had diabetes for 21 years, and diabetic retinopathy was diagnosed. Medications were adjusted to insulin and glimepiride (1 mg q.d. orally); the patient’s FBG was 7–8 mmol/L, his 2hPBG was 10 mmol/L, and his HbA1c was controlled at 7–8%. In the proband’s mother (II.1), FBG was 6.7 mmol/L, and she was not diagnosed with diabetes.
2. Family 2: The proband was a 17-year-old female (HT: 160 cm, WT: 50 kg, and BMI: 19.53 kg/m2). Both birth weight and hearing were normal. A physical examination 50 days ago found that her FBG was 8.5 mmol/L, and her 2hPBG was 20 mmol/L. The patient’s HbA1c was 10.2%. Glimepiride was prescribed (0.5 mg q.d. orally). The proband’s FBG was controlled at 6–7 mmol/L, her 2hPBG was controlled at 8–9 mmol/L, her HbA1c was 7.9%, and her urine glucose was positive.
The proband’s father (II.2) developed diabetes at the age of 37. He has had diabetes for 6 years. After insulin treatment, he was prescribed glimepiride (currently 1 mg q.d. orally). Uncle I (II.3) developed diabetes at the age of 40, and he has had diabetes for 1 year. At present, he takes metformin (0.25 g tid orally), and his blood glucose control is not stable. Uncle II (II.4) developed diabetes at the age of 24 and has had the disease for 9 years. He has been diagnosed with diabetic retinopathy and nephropathy. At present, he is prescribed insulin (Novavax 30) and glimepiride (1 mg q.d. orally). His HbA1c was 7.2%. The family history is presented in Figure 1B.
The BMI of the patients and relatives of the above two families were within the normal range, and their serum anti-glutamate decarboxylase antibody, islet cell antibody, and insulin autoantibody were all negative. Type 1 DM antibodies were detected by immunofluorescence. All participants provided written informed consent. This study was approved by the Ethics Committee of the Affiliated Hospital of the Inner Mongolia Medical University, China.
Research Method
Fifty-three patients suspected of having MODY 3, according to clinical manifestations, were enrolled as the study subjects. The diagnostic criteria for screening for MODY were as follows: (1) At least three generations of family members had DM, and their transmission was consistent with the autosomal dominant inheritance rule. (2) At least one patient with diabetes had a diagnostic age of 25 years or younger. (3) After diabetes was diagnosed, insulin was not needed to control blood sugar for at least 2 years. Because of the low incidence of MODY, our screening criteria were increased to the age of 40 years and younger. Two families suspected of having MODY 3 were treated, and a follow-up was conducted. The proband of Family 1 was prescribed glimepiride (0.5 mg q.d. orally). The proband of Family 2 was also prescribed glimepiride (0.5 mg q.d. orally), and blood glucose control and chronic complications in these two families were followed up after three months. Five members of Family 1 and six members of Family 2 underwent molecular screening for the HNF1α gene by blood sampling.
There are 10 exons in the HNF1α gene, according to the data of the human genome library. Ten pairs of primers were designed for the amplification of promoter regions using Primer 5.0 software. All primers (Table 1) were synthesized by Shanghai Sangon Biotech Service Co., Ltd., China. All exons of the HNF1α gene and the DNA of the splicing regions of the gene were analyzed by second-generation sequencing. The sequencing results were subject to online sequence alignment via the database of the University of California Santa Cruz (http://genome.ucsc.edu/cgi.bin/hgBlat) to determine the mutation sites and types. The mutation sites were compared with those in dbSNP, the 1000 Genomes Project database, and the ExAC database to analyze the conservation of mutant amino acids in different species (https://www.uniprot.org). The mutation function prediction software Mutation Taster (http://www.mutationtaster.org/) and PolyPhen2 (http://sift.bii.a-star.edu.sg) were used for pathogenicity predictions.
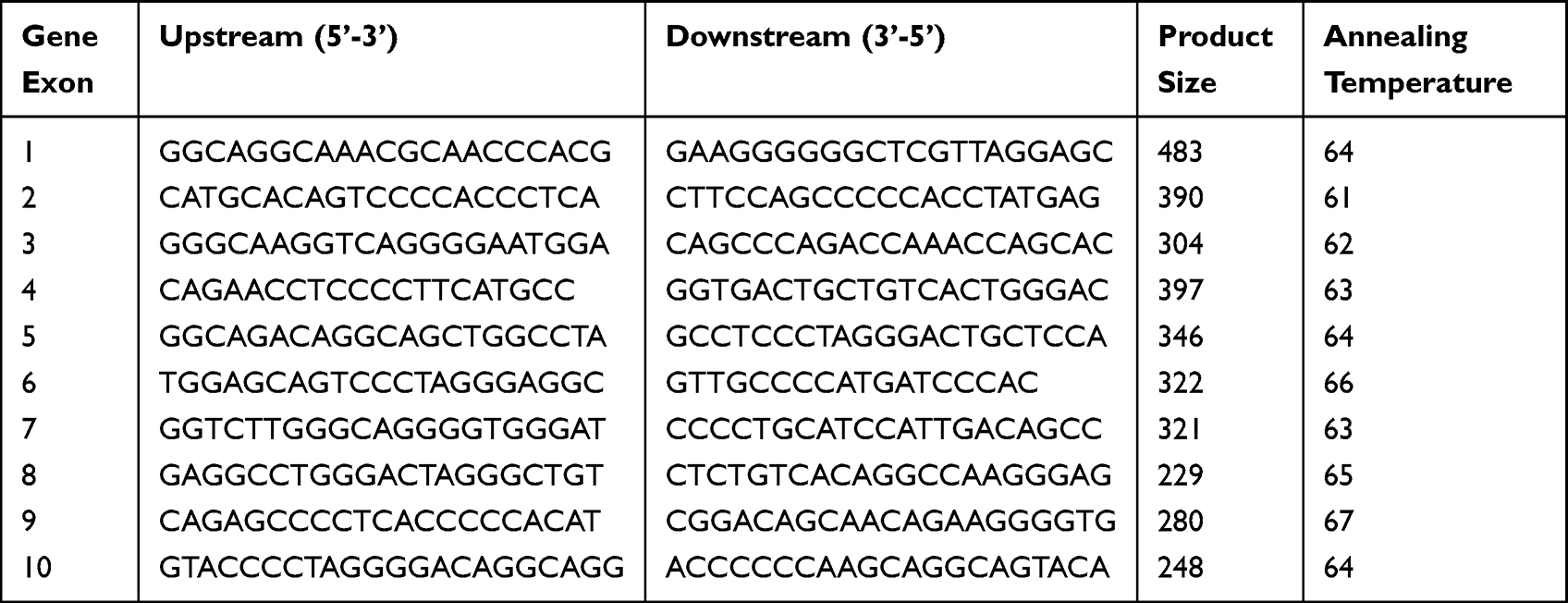 |
Table 1 HNF1α Gene Amplification and Primers Sequence |
Results
Among the five members of the three generations in Family 1, the analysis of the sequencing results of the proband (III.1), mother, and uncle (II.3) revealed that there was a missense heterozygous mutation on exon 2 of the HNF1α gene (c.512G>A, p.Arg171Gln). The sequencing results are presented in Figure 2. However, this mutation was not detected in the father or grandfather. The mutation makes a G base on exon 2 of the HNF1α gene, which becomes an A base, resulting in a change in the amino acid 17 that it encodes from arginine to glutamine. The proband met the characteristics of MODY 3. The mother was diagnosed with impaired glucose tolerance (IGT), and the uncle (II.3) was diagnosed with diabetes. Furthermore, five gene polymorphic loci were screened in Family 1 (Table 2). Codon 17 CTC → CTG of exon 1 was a synonymous mutation of leucine (Leu17Leu), Codon 27 ATC → CTC was a missense mutation that changes isoleucine to leucine (Ile27Leu), and Codon 459 CTG → TTG of exon 7 was a synonymous mutation of leucine (Leu459Leu). Codon 487 AGC → AAC was a missense mutation that changes serine to asparagine (Ser487Asn), and Codon 574 AGU → GGU of exon 9 was a missense mutation that changes serine to glycine (Ser574Gly). After retrieving from the ExAC human exome integration database, it suggested that the minimum allele frequency (MAF) of G/A at this site to be A = 0.000008 (rs765241951). A functional prediction of the mutation was conducted; the PolyPhen-2 score was 0.999, and the SIFT score was −2.436. The Mutation Taster software demonstrated that arginine at position 171 was a highly conserved amino acid. These results suggest that this mutation is pathogenic, thereby affecting protein function. According to the American College of Medical Genetics and Genomics guidelines, it is evidence of the moderate pathogenicity of PM5.6
 |
Table 2 Polymorphism of MODY3 Gene in Family 1 |
 |
Figure 2 Sequencing peak of probands of family 1. |
In Family 2, there was a missense mutation on exon 2 of the HNF1α gene (c.391C>T, p.Arg131Trp) in the proband (III.1), father (II.2), uncle 1 (II.3), uncle 2 (II.4), and grandfather (I.1) (Figure 3). This mutation makes a C base on exon 2 of the HNF1α gene, which becomes a T base, resulting in a change from arginine to tryptophan in the amino acid 131 it encodes. However, this mutation was not detected in the mother. Furthermore, four gene polymorphic loci were screened in Family 2 (Table 3). Except for Codon 27 of exon 1 (c.79A>C, p.Ile27Leu), the other polymorphic loci were the same as that of Family 1.
 |
Table 3 Polymorphism of MODY3 Gene in Family 2 |
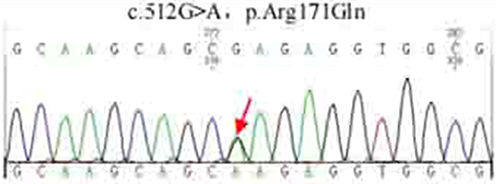 |
Figure 3 Sequencing peak of probands of family 2. |
The abovementioned mutation sites and polymorphic loci have been reported in the literature at home and abroad.7 After retrieving the ExAC human exome integration database, it suggested the MAF of G/A at this site to be T = 0.00000 (rs137853244). A functional prediction of the mutation was conducted; the PolyPhen-2 score was 0.999, and the SIFT score was −7.321. The Mutation Taster software demonstrated that arginine at position 131 was a highly conserved amino acid.
These results suggest that this mutation is pathogenic, thereby affecting protein function.No base variation at the two sites on exon 2 of gene HNF1α was detected in 53 cases of early onset T2DM and 50 normal controls. It can be considered that the base variation of these two sites is due to gene mutation.
The basic information of base mutation carriers in the two families is presented in Figures 2 and 3. Both were families with early onset diabetes (some members in both families developed diabetes before the age of 25), and the family members involved were found in three successive generations (DM/IGT); this met autosomal dominant inheritance. The proband and other affected persons had typical symptoms of “three high and one low”: The patients’ PBG was significantly higher than their FBG by >5 mmol/L, and their blood glucose control was poor after simple diet control and exercise. Drugs, including insulin treatment, were needed in the later stages of the disease. The family members had diabetic complications and progressively decreased pancreatic function, which required insulin and oral drug maintenance. After the medication was changed to low-dose sulfonylureas, blood glucose was significantly stabilized. In the follow-ups, probands 1 and 2, which were treated with low-dose sulfonylureas alone, had HbA1c values of 6% and 7.2%, respectively. The clinical indicators and characteristics of the probands of Family 1 and Family 2 at admission are shown in Tables 4 and 5.
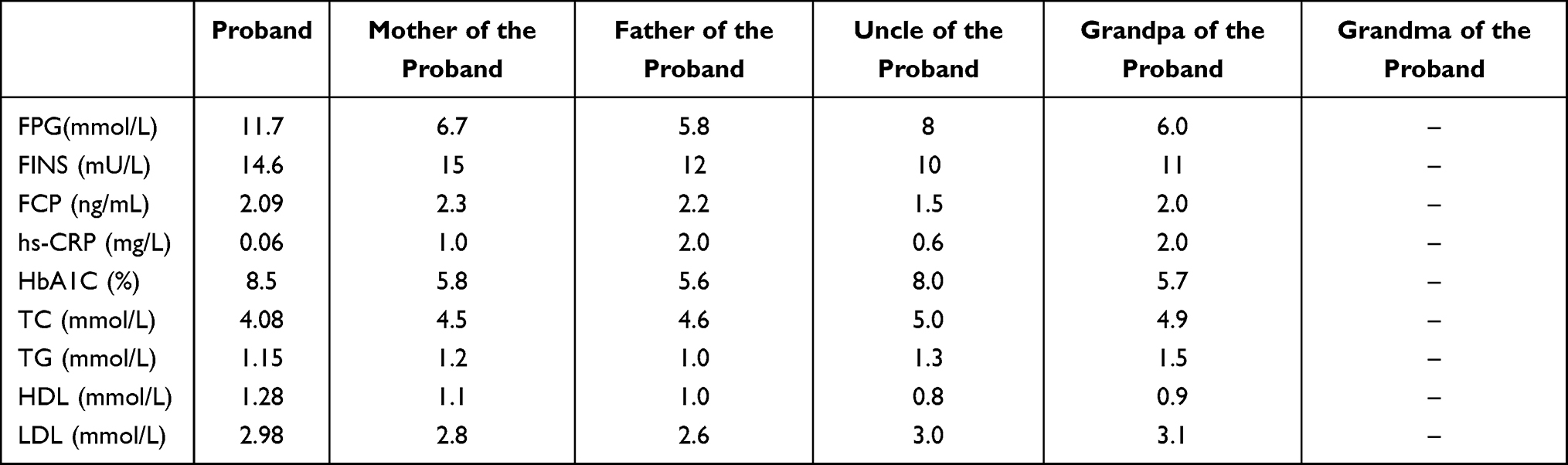 |
Table 4 Clinical Indicators of Proband of Family 1 |
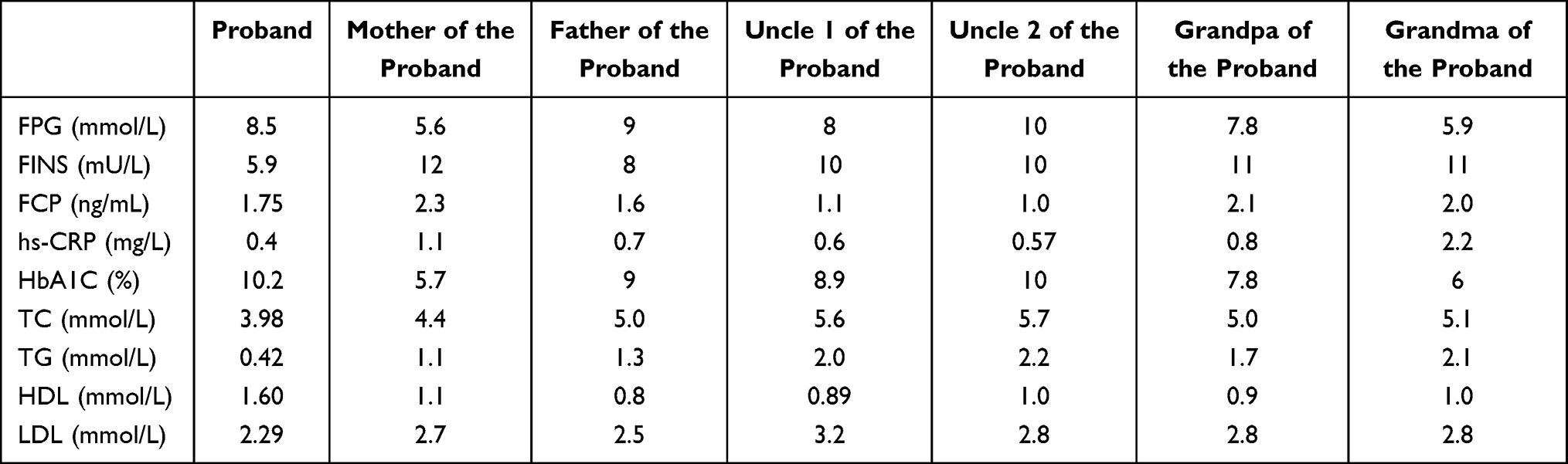 |
Table 5 Clinical Indicators of Proband of Family 2 |
Discussion
Maturity-onset diabetes of the young 3 is caused by a mutation located on the HNF1α gene of chromosome 12: 12q24.2. The HNF1α gene is composed of 10 exons with a size of about 23 kb. As a transcription factor, HNF1α participates in regulating metabolism related to glucose, fatty acids, and cholesterol, and its mutation may lead to islet β cell dysfunction.8 The onset age of MODY 3 in these families was less than 45 years, and at least one case was less than 25 years of age, suggesting a tendency for family aggregation. Of the two families, the age at the diagnosis of DM was <25 for the proband. Immediate family members in two generations had DM, and all were consistent with the characteristics of autosomal dominant inheritance, meeting the minimum diagnostic criteria of MODY.
In addition to family history and early onset, patients with MODY 3 have a decreased level of high-sensitivity C-reaction protein (hs-CRP).9 A previous study revealed that, the promoter region of the hs-CRP gene has HNF1α binding sites, resulting in the decrease of hs-CRP in patients with MODY 3.10 Compared with TIDM, MODY 3 had higher C-peptide levels (1.34 ± 1.51 vs 0.29 ± 0.22 ng/mL) and lower hs-CRP levels (0.18 ± 0.15 vs 1.22 ± 1.49 mg/L). In the probands of the two families, the hs-CRP levels were decreased below the normal range, although the levels of C-peptide were not. In addition, MODY 3 is sensitive to sulfonylureas.11
The initial sulfonylurea dose should be low to avoid hypoglycemia. Reports suggest that optimal glycemic control without problematic hypoglycemia at gliclazide doses of 20–40 mg.12 Another sulfonylurea, glimepiride, might offer a similar glucose-reducing effect with fewer episodes of hypoglycemia. It also exerts extrapancreatic effects such as decreased glucose output from the liver and enhanced sensitivity of peripheral tissues to insulin.
The proband of Family 1 was prescribed glimepiride (0.5 mg q.d. orally). When retested, their HbA1c level was 6%, and their urine glucose was positive in the same batch of urine, proving that the renal glucose threshold (RTG) decreases in patients with MODY 3. The uncle was prescribed glimepiride (1 mg q.d. orally), and his blood glucose level stabilized. In the proband of Family 2, the FPG was controlled within 6–7 mmol/L, and 2h-PBG was controlled within 8–9 mmol/L. When retested, HbA1c was 7.2%, and urine glucose was positive in the same batch of urine. The father (II.2) developed diabetes at 37 years of age and has had the disease for 6 years. He was treated with insulin and prescribed glimepiride (1 mg q.d. orally). Uncle 1 (II.3) developed diabetes at 40 years of age and has had the disease for 1 year. He was prescribed metformin, but his blood glucose remained unstable. Uncle 2 (II.4) developed diabetes at 24 years of age and has had the disease for 9 years. He was diagnosed with diabetic retinopathy and nephropathy and treated with insulin NovoRapid® 30. He received glimepiride (1 mg q.d. orally).
Compared with other subtypes of MODY, MODY 3 can lead to an insulin secretion defect and a decrease in RTG. Due to the mutation, insulin secretion is seriously damaged, and β cell function decreases progressively with age. The risk of complications is similar to that of T1DM and T2DM, and there is an elevated risk of microvascular complications related to DM. In Family 1, patient II.3 had fundus lesions 21 years after onset due to irregular treatment. In Family 2, patient II.4 developed diabetes at 24 years of age and had stage-II diabetic retinopathy and nephropathy 9 years after onset. Relative to other subtypes of MODY, MODY 3 progresses faster and is more prone to macrovascular and microvascular diseases.13
The mutation of the HNF1α gene may lead to islet β cell dysfunction. After an analysis of the Prabi prediction website, it can be speculated that base mutations cause the secondary structure of DNA to change from an α helix to an irregular curl and an extended chain. This change in secondary structure is likely to form a new active site, resulting in changes in protein function, an abnormal binding of transcription factors to target DNA, a decreased transcription level of insulin-related genes, and insufficient insulin secretion,14 leading to DM.
Vaxillaire et al15 studied 10 families with non-insulin-dependent DM. The results revealed that there was a co-separation between multiple gene mutation sites of the HNF1α gene and DM. There were no obvious associations of natural mutations (including frameshift, deletion, and gene location) with the clinical characteristics of DM. The cause of the HNF1α gene mutation leading to diabetes is unclear. It may involve the abnormal pancreatic development of the fetus, resulting in abnormal functioning at a later stage.
Similarly, the corresponding transcriptional genes play a key role in cell functions in normal islet β cells. Yamagata et al16 revealed that the MODY 3 transcription factor HNF1α was expressed in the liver, pancreas, and other tissues. Co-separation between genes and DM was found in a total of seven families with MODY. In Families 1 and 2, patients with diabetes carried mutation sites. In Family 1, the mother was diagnosed with IGT. Taking the patient’s long-term strict diet control and exercise regime into consideration, the age of onset was late. The two families met family co-separation.
In the present study, second-generation sequencing was used to perform the direct sequencing of all exon DNA of the HNF1α gene in 53 patients. MODY 3 was discovered in two patients, including c.512G>A (p.Arg171Gln) and c.391C>T (p.Arg131Trp) mutation sites and five polymorphic loci, but no new hot spot mutations or other pathogenic mutation sites were found. V. Radha17 stated that the c.512G>A (p.Arg171Gln) mutation has been reported in the Indian population. However, no functional analysis has been conducted for this coding region mutation. The mechanism of islet function impairment caused by this mutation is still unclear and needs further clinical research.
The c.391C>T (p.Arg131Trp) mutation was detected in a British family;7 the proband received treatment for diabetic ketoacidosis, and diagnosis and treatment were delayed for 11 years because the patient was treated as a T1DM. It is proven that the cell function of islet β cells deteriorates progressively, and the condition of this type of patient was more serious than in the other subtypes. In this study, the two probands had no deterioration of islet function due to the short course of their disease. The hot spot mutation site of the HNF1α gene is Pro291fs. This mutation has been found in more than 65 families with MODY worldwide.18 In this study, two groups of gene mutations were detected for the first time in Inner Mongolia. The mutation has not been reported in China.
The polymorphic loci found in this study were L17L, I27L, L459L, S487N, and S574G. A related report by Xu Cao et al19 also detected L17l, L459L, I27L, and S487N polymorphisms in the molecular screening of HNF1α gene defects of familial early onset families with MODY in the Chinese population. The CC genotype homozygous variants of I27L were all found in patients with T2DM; therefore, it is speculated that the CC genotype of this locus may be involved in the pathogenesis of T2DM.
A previous study revealed that Ser487Asn polymorphism was associated with an insulin secretion defect. Holmkvist et al20 reported that in the INS-1 and Hela cell lines, the HNF1α gene carrying S487N and I27L can reduce the transcriptional activity of albumin and glucose transcription 2 in the target transcription region. It is associated with decreased insulin secretion, especially in elderly and obese populations. Similarly, a study revealed that the HNF1α gene with I27L can increase high-density lipoprotein (HDL) levels, suggesting that the HNF1α gene has complex interactions with cardiovascular risk.21 The HDL level of the proband of Family 1 was 1.60 mmol/L, exceeding the upper limit of the normal HDL value by 1.55 mmol/L. Therefore, it may be speculated that the patient carries the I27L gene.
Of the five HNF1α polymorphisms found in the present study, L459L, S487N, and S574G were in the coding regions for transcription activation, and L17L and I27L variations were in the dimer coding regions. The three functional regions encoded by the HNF1α gene include a dimer region (codons 1–32), a DNA-binding region (codons 150–278), and a transcription activation region (codons 281–631).22 A study has revealed that a missense mutation in the transcriptional activation region of the HNF1α was associated with older age at diagnosis. In this study, L17L of the proband of Family 2 was in a dimer region, and its missense mutation in the DNA- binding region was associated with a younger age at diagnosis.23
When compared with other MODY subtypes, patients with MODY 3 have an earlier age of onset and an obvious insulin deficiency. Clinically, it is easy to be misdiagnosed as T1DM or T2DM. Early screening is important for providing a correct diagnosis and timely treatment. Because the sample size of the present study was small and limited to certain family members and their probands, it is impossible to determine whether gene mutation is the main pathogenic factor of MODY in Inner Mongolia. The mutations and polymorphic loci found in this research may be involved in the occurrence of diabetes in these two family members. However, the specific mechanism and molecular pathological significance need to be explored further. Finally, the number of families should be increased to improve the accuracy of MODY gene screening in Inner Mongolia.
Ethics Approval and Consent to Participate
This study was conducted in accordance with the declaration of Helsinki.This study was conducted with approval from the Ethics Committee of Affiliated Hospital of Inner Mongolia Medical University.A written informed consent was obtained from all participants.
Consent for Publication
Consent for publication was obtained from every individual whose data are included in this manuscript.
Funding
Youth Backbone project of Affiliated Hospital of Inner Mongolia Medical University(2022NYFY FG026, Natural Science Foundation of Inner Mongolia (Project no. 2016MS(LH)0803) and Inner Mongolia Health Scientific Research Project (Project No. 201701064).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Murphy RES, Ellard S, Hattersley,AT. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab. 2008;4(4):200–213. doi:10.1038/ncpendmet0778
2. Wang X, Wang T, Yu M, et al. Screening of HNF1A and HNF4A mutation and clinical phenotype analysis in a large cohort of Chinese patients with maturity-onset diabetes of the young. Acta Diabetol. 2019;56(3):281–288
3. Hattersley AT, Greeley SA, Polak M, et al. ISPAD clinical practice consensus guidelines 2018: the diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes. 2018;19(Suppl.27):47–63. doi:10.1111/pedi.12772
4. Valkovicova T, Skopkova M, Stanik J, et al. Novel insights into genetics and clinics of the HNF1A-MODY. Endocr Regul. 2019;53(2):110–134. doi:10.2478/enr-2019-0013
5. Shields BM, Hicks S, Shepherd MH, et al. Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia. 2010;53(12):2504–2508. doi:10.1007/s00125-010-1799-4
6. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
7. Glucksmann MA, Lehto M, Tayber O, et al. Novel mutations and a mutational hotspot in the MODY gene. Diabetes. 1997;46(6):1081–1086. doi:10.2337/diab.46.6.1081
8. Shih DQ, Screenan S, Munoz KN, et al. Loss of HNF1α function in mice leads to abnormal expression of genes involved in pancreatic islet development and metabolism. Diabetes. 2001;50(11):2472–2480. doi:10.2337/diabetes.50.11.2472
9. Ohki T, Utsu Y, Morita S, et al. Low serum level of high-sensitivity C-reactive protein in a Japanese patient with maturity onset diabetes of the young type 3 (MODY3). J Diabetes Investig. 2014;5(5):513–516. doi:10.1111/jdi.12237
10. Fu JL, Wang T, Liu JY. Using clinical indices to distinguish MODY2 (GCK Mutation) and MODY3 (HNF1A Mutation) from type 1 diabetes in a young Chinese population. Diabetes Ther. 2019;10(4):1381–1390. doi:10.1007/s13300-019-0647-x
11. Raile K, Schober E, Konrad K, et al. Treatment of young patients with HNF1A mutations (HNF1A-MODY). Diabet Med. 2015;32(4):526–530. doi:10.1111/dme.12662
12. Bishay RH, Greenfield JR. A review of maturity onset diabetes of the young (MODY) and challenges in the management of glucokinase-MODY. Med J Austr. 2016;205(10):480–485. doi:10.5694/mja16.00458
13. Thanabalasingham G, Owen KR. Diagnosis and management of maturity onset diabetes of the young(MODY). BMJ. 2011;343(7828):837–842. doi:10.1136/bmj.d6044
14. Kang XL, Ding WY, Chen X, et al. Clinical characteristics and HNF1α gene analysis of a family with early onset diabetes. J Pract Diabetes. 2017;13(3):36–39.
15. Vaxillaire M, Rouard M, Yamagata K. Identification of nine novel mutations in the hepatocyte nuclear factor 1 alpha gene associated with maturity-onset diabetes of the young (MODY3). Hum Mol Genet. 1997;6(4):583–586. doi:10.1093/hmg/6.4.583
16. Yamagata K, Oda N, Kaisaki PJ, et al. Mutations in the hepatocyte nuclear factor 1 alpha gene in maturity-onset diabetes of the young (MODY3). Nature. 1996;384(6608):455–458. doi:10.1038/384455a0
17. Radha V, Ek J, Anuradha S, et al. Identification of novel variants in the hepatocyte nuclear factor-1alpha gene in South Indian patients with maturity onset diabetes of young. J Clin Endocrinol Metab. 2009;94(6):1959–1965. doi:10.1210/jc.2008-2371
18. Ek J, Rose CS, Jensen DP, et al. The function Thr 130Ile and Val255Met polymorphisms of the hepatocyte nuclear factor-4alpha (HNF4a): gene associations with type 2 diabetes or altered beta-cell function among Danes. J Clin Endocrinol Metab. 2005;90(5):3054–3059. doi:10.1210/jc.2004-2159
19. Cao X, Tian MH. Molecular screening of HNF1α gene defects in Chinese familial early onset type 2 diabetes /MODY families. J New Knowl Med. 2009;19(4):133–134.
20. Holmkvist J, Cervin C, Lyssenko V, et al. Common variants in HNF1alpha and risk of type 2 diabetes. Diabetologia. 2006;49(12):2882–2891. doi:10.1007/s00125-006-0450-x
21. Babaya N, Ikegami H, Fujisawa T, et al. Association of I27L polymorphism of hepatocyte nuclear factor-1ɑ gene with high-density lipoprotein cholesterol level. J Clin Endocrinol Metab. 2003;88(6)::2548–2551. doi:10.1210/jc.2002-021891
22. Mohamati T, Yiming THT, Gongli R, et al. Gene study of hepatocyte nuclear factor-α in a Uygur patient with early onset diabetes. Chin J Diabetes. 2016;24(1):7–11.
23. Bellanne-Chantelot C, Carette C, Riveline JP, et al. The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity -onset diabetes of the young (MODY3). Diabetes. 2008;57(2):503–508. doi:10.2337/db07-0859
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.