")
Back to Journals » OncoTargets and Therapy » Volume 15
Clinical Applications of Aneuploidies in Evolution of NSCLC Patients: Current Status and Application Prospect
Received 25 June 2022
Accepted for publication 22 October 2022
Published 10 November 2022 Volume 2022:15 Pages 1355—1368
DOI https://doi.org/10.2147/OTT.S380016
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Arseniy Yuzhalin
Xing Yan,1 Shan Mei Liu,2 Changhong Liu1
1The Second Affiliated Hospital of Dalian Medical University, Dalian, 116000, People’s Republic of China; 2Inner Mongolia Medical University, Hohhot, 150110, People’s Republic of China
Correspondence: Changhong Liu, The Second Affiliated Hospital of Dalian Medical University, Dalian, 116000, People’s Republic of China, Tel +86 17709870870, Email [email protected]
Abstract: As one of the first characteristics of cancer cells, chromosomal aberrations during cell division have been well documented. Aneuploidy is a feature of most cancer cells accompanied by an elevated rate of mis-segregation of chromosomes, called chromosome instability (CIN). Aneuploidy causes ongoing karyotypic changes that contribute to tumor heterogeneity, drug resistance, and treatment failure, which are considered predictors of poor prognosis. Lung cancer (LC) is the leading cause of cancer-related deaths worldwide, and its genome map shows extensive aneuploid changes. Elucidating the role of aneuploidy in the pathogenesis of LC will reveal information about the key factors of tumor occurrence and development, help to predict the prognosis of cancer, clarify tumor evolution, metastasis, and drug response, and may promote the development of precision oncology. In this review, we describe many possible causes of aneuploidy and provide evidence of the role of aneuploidy in the evolution of LC, providing a basis for future biological and clinical research.
Keywords: aneuploidy, chromosome instability, mitosis, mutation, non-small cell lung cancer
Corrigendum for this paper has been published.
Introduction
As one of the most common malignancies globally, non-small cell lung cancer (NSCLC) has a higher morbidity and a poorer prognosis. At present, the most common causes of death for lung cancer patients are recurrence and metastasis.1 Therefore, it is important to explore the pathogenesis of NSCLC in order to find more effective treatment strategies. There are a number of efficient and specific biomarkers that can be used to diagnose, prognosis, and monitor the progression of NSCLC. In recent years, high-throughput sequencing of the whole genome of cancer patients correlate aneuploidy with cancer development and chromosomal instability.2 Cancers with an unequal number of chromosomes (increase or loss of copy number), aneuploidy, have been observed for more than 100 years. Its functional effect is usually thought to be achieved through inhibition of tumor suppressor genes and overexpression of oncogenes.3 In approximately 90% of cancer patients, the genomic sequence has undergone considerable changes which are closely related to clinical outcomes4 Duesberg et al5 put forward the “carcinogenicity theory of chromosome aneuploidy” 20 years ago, which indicated that aneuploidy was the key cause of genomic instability and tumorigenesis, and challenged the mainstream gene mutation theory at that time. However, whether aneuploidy causes tumorigenesis or is the consequence of it remains debated, aneuploidy has emerged as new regulators for cancer biology.
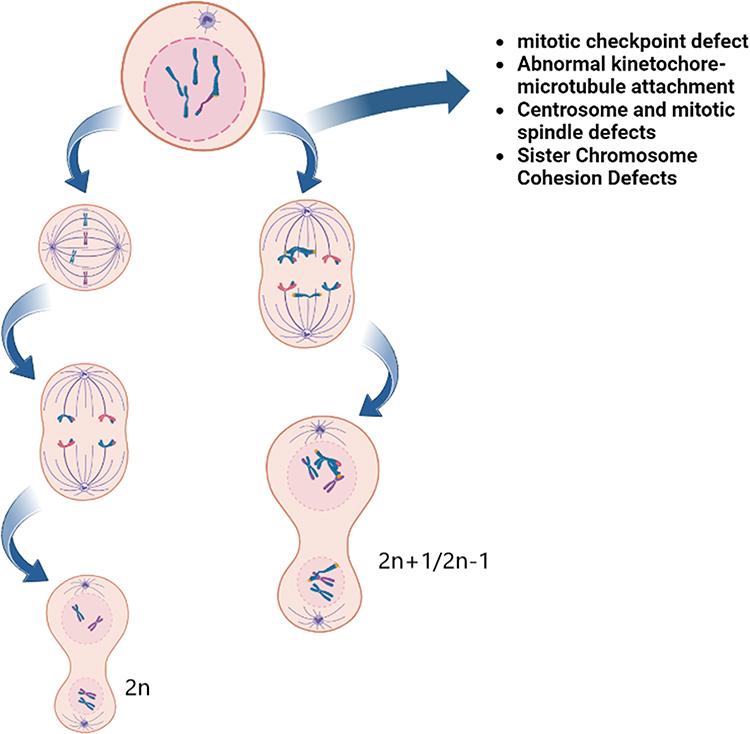 |
Figure 1 The figure illustrates defects in several processes involving chromosomes, spindle microtubules and centrosome targets, which, in addition to abnormal cytokinesis, may also lead to uneven distribution of chromosomes during mitosis, leading to aneuploidy. Because aneuploidy represents the digital imbalance of chromosomes, it can be reasonably expected that aneuploidy is caused by chromosome segregation errors in the process of cell division. |
Aneuploids include whole chromosomes, chromosome arms, and local chromosomes (this article focuses on the introduction of whole chromosomes), which are mainly caused by the misseparation of chromosomes in the cell cycle caused by mitotic checkpoint defects.6 Aneuploidy can also be found in tissue cells of healthy people, and that aneuploidy contributes to the development and selection of malignant clones and promotes the phenotypic development of tumor progression (Figure 1).7 About 60% of NSCLC patients are aneuploid, and aneuploid NSCLC is one of the most common tumors.8 Sundaresan et al9 observed the phenomenon of somatic chromosome deletions in lung squamous cell precancerous lesions as early as 1992. Subsequent studies found that the incidence of aneuploidy in small cell lung cancer was 77.8%, which was higher than that in any other histological type of lung cancer.10 Studying the involvement of aneuploidy in cancer may provide new ideas for future research on cancer treatment. Tumorigenesis and aneuploidy are known to be closely related, but their precise relationship remains unclear. In this paper, we review the results of a large number of clinical studies on NSCLC published since 2000, which discuss the significance of aneuploidy in tumor evolution and evaluate the use of aneuploid which may serve as prognostic biomarkers for NSCLC outcome. This paper describes the potential strategies of chromosome aneuploidy targeted therapy in patients with NSCLC.
The Cause of Chromosome Aneuploidy
Mitotic Checkpoint Defect
The mitotic checkpoint or spindle assembly checkpoint (SAC) monitors centromere attachment to the mitotic spindle. As long as the chromosome is not attached to the spindle microtubules, SAC blocks the mitotic process.11 SAC induces the formation of a mitotic checkpoint complex (MCC) consisting of MAD2, BubR1, Bub1, and Bub3.12 When MCC binds to the late promoter complex/ring (APC/C), the WD-40 domain of the agonist CDC20 binds to the APC/C substrate shifts and cannot recognize the substrate, thus inhibiting the initiation of anaphase and premature chromosome segregation.13 When the SAC mechanism fails, MCC is decomposed, and CDC20 binds to APC/C to target the ubiquitination of CyclinB and Securin, which are degraded by protease.14 The degradation of Securin and CyclinB leads to the separation of sister chromatids, which in turn promotes the end of mitosis.15 Cahill et al16 reported for the first time that Bub1 mutations produce a weakened checkpoint response in a subset of cancer cell lines of colon cancer patients, leading to chromosome aneuploidy. Lee et al17 found acquired mutations in the Bub1 and p53 genes and loss of SAC in BRCA2-deficient animal tumors. They concluded that inactivation mutations of these checkpoint genes lacked synergy with BRCA2, leading to hereditary breast cancer. An accurate SAC is important for cell survival. Once the checkpoint is damaged and the mechanism is defective, it will lead to an increase in the chromosome error separation rate, leading to an imbalance in chromosome number and the formation of aneuploid.18 However, it is controversial whether SAC defects represent common drivers of chromosome aneuploidy because SAC gene mutations are rarely found in solid tumors. According to the sequencing data of the next-generation cancer genome map (TCGA),19 only 9.6%, 7.4%, 6.2%, 6.4%, and 7.6% of patients with endometrial cancer (with MAD1L1, BUB1, BUB1B, CDC20, or BUB3 mutations) and colon adenocarcinomas (5.5% with MAD1L1 or BUB1 mutations), respectively, were detected with mutations of more than 5%. The effects of SAC mutations on lung cancer have also been reported. The relationship of MAD1L1 and MAD2L1 mutations and lung cancer was estimated by researchers between 1000 patients and 1000 healthy controls.20 It was also confirmed that aneuploidy caused by MAD1L1 and MAD2L1 gene mutations increased the susceptibility to lung cancer. This may be due to the decrease of SAC function caused by the decrease of MAD1L1 and/or MAD2L1 function. Mis localization of Mad2, causing SAC defects and chromosomal instability. However, this instability will not affect tumor regression caused by KRAS elimination21 Transient overexpression of MAD2 and chromosome instability may be important stimulating factors for tumor occurrence and development, and the tumor recurrence rate in these patients is significantly increased.22 Except for a few reported cases of decreased expression, SAC is generally considered to be overexpressed in lung cancer, and high expression levels are associated with a high proliferation index and metastatic potential. These results also suggest that abnormal gene expression caused by SAC deficiency is likely to lead to aneuploidy and promote lung cancer development.
Abnormal Kinetochore-Microtubule Attachment
Microtubule are dynamic polymers of protein heterodimers that rotate continuously at the end of the spindle, and the kinetochore is attached to the end of the microtubule.23 The Repeated binding and dissociation of single microtubules and kinetochores results in the dynamic attachment of microtubules to microtubules, which is necessary to promote microtubule stability and correct errors.24 Because of the randomness of microtubule-kinetochore attachment, the initial attachment of kinetochores and spindle microtubules is an error-prone process. In the process of somatic cell mitosis, the dynamic attachment of kinetochore microtubules slows down quickly and gradually stabilizes. Therefore, the early and middle stages of division are the key stages of abnormal attachment.25,26 Once abnormal attachment can lead to mitotic errors and produce chromosome nondiploid. Aurora B kinase, which exists in the centromere, participates in and regulates the kinetochore-microtubule dynamic attachment mechanism. Although the related mechanism is still unclear, the emerging view is that Aurora B-mediated phosphorylation of the key substrate Hec1 occurs in the early stage of mitosis, which destroys the stability of abnormal attachment and decomposes incorrectly attached kinetochores from microtubules. The physical tension generated by the correct attachment is sufficient to stabilize the attachment and drive the biologically oriented kinetoplast out of the Aurora B area of influence.27 The probability of chromosome mis segregation caused by Aurora B kinase inhibition was significantly increased. Kinetochore-microtubule attachment becomes too stable under the interference of these proteins, which in turn affects the efficiency of correcting abnormal attachment, which leads to CIN.28 The study by Takeshita et al29 also responded to this conclusion. They found that 87 of the 157 patients (57%) with non-small cell lung cancer were aneuploid, and 56 of the 83 patients (67%) with overexpression of Aurora B were aneuploid. Overexpression of Aurora B was significantly correlated with aneuploidy, researchers have also conducted clinical studies on Aurora A. Among 94 tumors overexpressing Aurora A, 60 were aneuploidy (63%), and the overexpression of Aurora A was significantly associated with aneuploidy. The overall survival rate of aneuploid tumor patients was significantly lower than that of diploid tumor patients. In addition, overexpression of the SAC protein MAD2,30 the depletion of adhesin subunit STAG2,31 and activation of the DNA damage response32 all lead to abnormal kinetochore-microtubule adhesion. In contrast, the overexpression of Kinesin-13 microtubule depolymerase MCAK and Kif2b reduced the stability of kinetochore-microtubule attachment and restored the non-diploid chromosome segregation of cancer cells that originally showed CIN.33 Therefore, the adhesion of microtubules and kinetoplasts in cancer cells is more stable than that in normal diploid cells, which provides strong evidence of the non-diploid nature of tumor cells. The effect of mutant genes on the dynamic attachment of kinetochore microtubules is rare. At present, only APC deletion has been found to increase the stability of motile microtubules in colon cancer, induce non-diploid chromosomes in other stable somatic cells, and finally induce tumorigenesis.34
Centrosome and Mitotic Spindle Defects
The centrosome consists of a pair of vertically arranged organelles that form the spindle poles. These two centromeres each contain nine groups of microtubules, which are surrounded by pericentriolar material (PCM) during mitosis and regulate spindle assembly in an orderly manner.35,36 More than a century ago, Theodore Boffrey first proposed that an additional centrosome would promote multipolar cell division, leading to genetic instability and malignant transformation, driving multipolar cell division aneuploidy, followed by tumorigenesis.37 A large number of studies have shown that centrosome abnormalities exist in most solid and hematological malignancies, especially in lung cancer cells.38 This abnormality, also known as centrosome amplification, is more frequently observed in cancer cells with a CIN phenotype or aneuploidy.39 It is not clear what mechanism somatic cells produce additional centrosome defects during mitosis. At present, it is believed that there are many mechanisms that may lead to excessive centrosomes, including excessive centrosome replication or additional centrosome assembly from scratch, mitotic sliding, intercellular fusion and cytokinesis failure.40–42 When too many extra centrosomes are produced, the following steps further make the cell susceptible to CIN: (1) random attachment to the chromosome in the early stage of mitosis, creating a transient multipolar spindle; and (2) decomposing this abnormal mitotic configuration into a bipolar spindle.43 This transition from multipolar spindles to bipolar spindles promotes the formation of incorrect kinetoplast-microtubule attachment, causing subsequent chromosome segregation errors in the late stage of division and forming aneuploid chromosomes during cytokinesis. The mechanism of tetraploid production also leads to the emergence of additional centrosomes, and has long been considered to be the pioneer of aneuploidy. Fujiwara et al44 showed that tetraploid cells caused by failed cytokinesis showed CIN and spontaneously produced tumors when injected into mice. This supports the argument that the long-term correlation between the extra centrosome and CIN in tumor cells drives tumorigenesis. Clinical studies have found that when the aggregation in vitro of the center is antagonized, cancer cells undergo multipolar division, leading to the death of daughter cells. This pro-apoptotic pathway, known as late disaster, occurs when centrosome protein CP110, cyclin-dependent protein 1 (CDK1), and CDK2 are deleted or inhibited, and aneuploid cancer cells are eliminated first. At the same time, CP110 is inhibited by the KRAS oncoprotein, which makes CDK1/2 inhibitors sensitive to lung cancer driven by the KRAS gene.45
Sister Chromosome Cohesion Defects
During the mitotic cycle of somatic cells, DNA replicates during the synthesis phase. Each chromosome produces two identical copies, which are packaged into sister chromatids. At the late stage of development, sister chromatids begin to be accurately separated and evenly distributed to the two daughter cells to ensure the uniform distribution of genetic information and maintain the stability of the genome.46,47 In fact, in order to prevent the symmetrical separation of sister chromatids between the formed daughter cells, the physical connection between sister chromatid cohesion is maintained from S phase to metaphase.48 This cohesion is opposite to the tension produced by the microtubules attached to the kinetochore, so that the chromosomes on the mitotic spindle are located in two directions.49 Therefore, if there is a sister chromatid cohesion defect, the daughter cells accept too many or too few chromosomes during mitosis, and the euploid chromosomes become aneuploidy.50 A protein complex called adhesin (SCC1 (RAD21), SCC3 (STAG1, STAG2), SMC1A, and SMC3) dynamically binds to DNA during the S phase of DNA replication and mediates and maintains sister chromatid cohesion. Its reduction and dysfunction can lead to chromosome dislocation and CIN.51 Recent studies have shown that menthol NIPBL,52 as an adhesin loading factor, interacts with the adhesin complex and loads adhesin onto chromosomes. The low expression and gene mutation of NIPBL may interfere with its interaction with adhesin complex, and lead to genomic instability by interfering with the separation and concentration of sister chromatids, resulting in aneuploidy in the process of carcinogenesis. Rare NIPBL mutations found in lung, breast, and colorectal tumors have been confirmed.53–55 In the process of sister chromatid separation, the failure of two steps of adhesin removal also induces aneuploidy and carcinogenic transformation: (1) at the end of metaphase, the adhesin on the chromosome arm is dissociated by the phosphorylation of the adhesin SA subunit, which is noteworthy, compared with other mitosis-related genes with few mutations. T mutations in the adhesin complex subunit STAG2 in tumor cell lines and tumor samples accounted for 39% of STAG2 mutations reported in the cancer genome map (TCGA). (2) In the later stage, protease suddenly starts to activate and cleaves the adhesin subunit SCC1 (RAD21) of the adhesin complex from sister chromatids, which immediately becomes an inhibitor of CDK1 and promotes the movement of sister chromatids to the poles of mitotic spindle.56,57 Premature and overexpression will destroy the normal regulation of the enzyme isolates. Zhang et al58 conditionally induced the overexpression of dissociating enzymes in the epithelial cells of diploid mice, resulting in premature chromatid separation and chromosome lag within 5 days, leading to aneuploidy and tumorigenesis. Correspondingly, Shugoshin1 (Sgo1) and PP2A protect adhesin on the centromere from cleavage by isolating enzymes. Interestingly, SGO1 on the centromere interacts with the chromosome passenger complex containing AURKB, and the mis localization of SGO1 hinders error-correction repair and may lead to CIN by stabilizing kinetochore-microtubule attachment defects.
The Contradictory Relationship Between Aneuploidy and Cancer
Cancer-Promoting Effect
According to the relevant tumor gene database, approximately 90% of solid tumors and 50% of blood tumors are aneuploid.59 Some views have been supported by a large number of experimental and clinical evidence that aneuploidy can not only induce tumorigenesis but also promote different stages of tumor progression, such as tumor initiation, recurrence, drug resistance and metastasis.2,60 This suggests that aneuploidy can promote tumorigenesis, even if it cannot completely induce tumorigenesis. Studies in yeast and mice have shown that aneuploidy cells are randomly produced due to accidental mitotic errors or induced by carcinogens, and the presence of additional chromosomes will lead to a disorder in which the transcription of coding genes is proportional to the protein level.61 Lead to a chain reaction, and eventually there will be cells with cancer-specific chromosome combinations and rearrangements. Therefore, it can be said that the occurrence of cancer is the result of abnormal mutations in large doses of aneuploid genes, which damage cell proliferation and metabolic changes. However, there is no clear answer to the specific mechanism of aneuploidy promoting cancer. Benezra et al62 reported that when some chromosomes in tetraploid mouse embryonic fibroblasts (MEF) were deliberately deleted, aneuploidy cells showed higher chromosomal instability and DNA damage, and formed huge invasive tumors in immunocompromised mice. This is a mutant phenotype related to tumorigenesis in vivo, indicating that the carcinogenic potential of chromosome loss is an early consequence of animal cell aneuploidy.
What are the advantages of aneuploidy? Although aneuploid cells have weak proliferative ability at first, they have high genomic instability and mutation rate. This may enable them to acquire the characteristics of cancer. Baker et al63 used the whole chromosome instability (W-CIN) mouse model to study the trend of division of each cell in the object to produce daughter cells with random karyotype abnormalities, proving that aneuploidy can promote tumorigenesis by losing the heterozygosity of tumor suppressor genes. SAC is one of the most critical components of mitosis. Genetic changes that affect the function or stability of sac proteins show an increase in spontaneous or carcinogenic tumorigenesis19,64 such as heterozygous mutations in BubR1, Mad1/2, Bub3, and other genes or subtypes. In addition, the overexpression of MAD2, Hec1, or Bub1 alleviates the regulation of SAC and promotes the occurrence of CIN and tumors, while genetic variation of MAD1L1 and MAD2L1 leads to decreased sac function and increased susceptibility to cancer. TP53 mutations are also significantly associated with aneuploid tumors. Galc í an et al65 analyzed TCGA data and showed that it was the only gene significantly associated with CIN (2013A). Mice expressing TP53 mutants did not induce apoptosis, but retained G1 phase blocking function to prevent tetraploid cell proliferation. Aneuploidy cells show extensive karyotype heterogeneity and chromosome rearrangement, and can cause tumor formation in mice.66 TP53 mutations are mainly clonal and significantly associated with genomic doubling tumors.67 In these cases, wild-type tumor suppressor genes were lost, whereas mutated alleles were obtained to compensate for gene dose imbalance. Specific mutations in oncogenes or tumor suppressor genes can increase cell proliferation and survival but may also affect chromosome integrity and segregation, leading to aneuploidy. Conversely, aneuploidy can actively increase genomic instability and mutation rate and accelerate tumor progression by increasing or decreasing the number of copies of oncogenes or tumor suppressor genes.68
Aneuploidy imbalances the expression of many proteins that may be involved in DNA synthesis and repair, as well as chromosome segregation.69 Subsequent aneuploid tolerance mutations lead to stronger proliferation of tumor cells. CENP-E is a centromere-binding motility protein attached to SAC microtubules, which guides the aggregation of initially dislocated chromosomes and controls the cell cycle. During mitosis, CENP-E accumulates in late G2 and degrades at the end of mitosis. Normally, even a single kinetoplast is sufficient to maintain the activation of mitotic checkpoints.70,71 However, after the decrease in CENP-E, the mitotic checkpoints are weakened, and the percentage of abnormal mitosis and the number of chromosomal aberrations per division increase, resulting in aneuploid tumors. Experiments on animals with CENP-E levels half the normal level showed that the incidence rate of splenic lymphoma and pulmonary adenoma increased, which was similar to the proportion of lung cancer among smokers 74, but the tumor developed late and was not completely exposed (10%). Other proposed aneuploid cancer-promoting mechanisms include the deletion of pro-apoptotic protein bcl9L and stress protein p38 α (also known as MAPK14), although there are relatively few mutations in the genes encoding these proteins in human tumors, suggesting that they may not be a common mechanism of aneuploidy tolerance. Uncontrolled expression of genes involved in oxidative phosphorylation and protection against oxidative stress also contributes to the effect of aneuploidy on tumours.73 Overall, these studies clearly show that the evolution of cancer seems to be a transition from random chromosomal protein changes to early selection of aneuploidy cloning to a large extent. The existence of additional chromosomes leads to the disorder of transcription of coding genes in proportion to protein levels, thereby increasing tumor invasiveness, which may be due to the effect of gene dose on proteins related to maintaining mitotic fidelity. While maintaining a highly aneuploid karyotype, the reasons for the strong growth of cancer need further study.
Inhibition of Tumor
All mammals have a certain proportion of aneuploidy cells, and adult animals have a higher proportion of aneuploidy cells, but they all have spontaneous tumors. Aneuploidy plays a role as a booster in promoting tumor development, but it is clear that this is not a one-way effect because aneuploid cells show a decrease in cell adaptability. It effectively participates in tumor growth inhibition.74 Weaver team75 found that the lack of the p19/ARF tumor suppressor gene caused an increase in the rate of single chromosome deletion in tumors, especially spontaneous liver cancer, and had a significant and unexpected impact on survival rate. The increase of aneuploidy increased the average tumor-free survival time of these animals by 93 days (P=0.0079) and the tumor incubation period of almost all animals was significantly prolonged. It was also confirmed that aneuploidy from whole chromosome loss (or acquisition) can strongly delay tumorigenesis in vivo after loss of the p19/ARF tumor suppressor gene. These results provide evidence of aneuploidy inhibition rather than the enhancement of tumorigenesis. Silk et al72 showed that the effect on tumors is determined by the chromosome mis selection rate rather than the accumulated aneuploid level. They found that the decrease of CENP-E resulted in an increase in the percentage of abnormal mitosis and the number of chromosomes misdivisions per division. When the increase in chromosome misselection rate exceeded a certain threshold, the misaggregation of a few chromosomes per division was not enough to increase the level of cell death. It can inhibit tumor by causing cell death. In other words, in the case of aneuploidy, a low proportion of chromosome missegregation can promote tumorigenesis, while a high proportion of chromosome missegregation leads to cell death and tumor inhibition. It has also been reported that a small amount of aneuploid MEF caused by Bubr1 mutations does not cause tumor growth defects,76 but Bubr1 deletions lead to missegregation of a large number of chromosomes at a time, which can accelerate cell death.77 These results suggest that the reason for cancer inhibition by aneuploidy is likely to be the increase in the number of misseparated chromosomes during mitosis, resulting in unscheduled cell death.
Amon et al successively conducted two groundbreaking studies to overturn Boveri’s hypothesis that aneuploidy causes cancer. In the first experiment,78 in the cultured mouse embryonic fibroblast (MEF) line, it was found that the MEF carrying the spindle checkpoint module BubR1 subtype mutation often carried one or two extra chromosomes. In addition to the unique characteristics of chromosomes, each MEF also contained different extra frontal chromosomes. It also exhibits the characteristics of delayed cell proliferation and metabolic changes. In the second,79 the cells with six different chromosomes (mChr1, mchr13, mchr16, mchr19, hchr3 and hchr5) were tested for carcinogenicity, and the results showed that the carcinogenicity of euploid cells was lower than that of gene-matched euploid cells. In addition, the activation of several carcinogenic pathways (HRAS, BRAF, PIK3CA, and MYC) and the functional removal of tumor suppressors p53 and Rb were not sufficient to overcome the adaptive punishment caused by aneuploidy, and no transformation of primary cells induced by aneuploidy or synergistic behavior with carcinogenic mutations was observed. In a related supplementary experiment,80 additional chromosomes were injected into human colorectal cancer cells with stable chromosomes, although the rates of proliferation of transduced trisomy and euploid cell lines were almost the same. However, the tumor formation in the three systems in vivo was smaller, indicating that aneuploidy characterized by chromosome acquisition can effectively inhibit tumor formation and growth. The above studies show that the aneuploidy of a single chromosome is not sufficient to induce the malignant transformation of euploid cells, but is antitumorigenic. Patients with Down syndrome characterized by extra chromosome 21 showed the same tumor pattern; that is, the incidence of solid tumors (including lung, stomach, breast, and prostate cancer) was lower than that of normal people.81 The study found that the apoptosis rate of trisomy 21 increased, which may indicate that cell death is a more common response to decreased adaptation of aneuploid cells and a protective event to prevent malignant transformation of these tissues, thereby inhibiting tumor formation. This reduces the risk of cancer.82 In addition, trisomy mouse cells designed for specific chromosomes showed delayed proliferation, possibly when aneuploid cells were introduced into normal diploid cancer cell lines, and aneuploid cells were defeated by diploid cells.83 Therefore, under normal circumstances, aneuploidy may act as a barrier to tumorigenesis by reducing the growth of precancerous cells.
The Role of Aneuploidy in NSCLC
Different Pathological Type of NSCLC Aneuploidy the Similarities and Differences
Lung cancer can be roughly divided into small cell lung cancer (SCLC), accounting for about 15% of all diagnoses, and non-small cell lung cancer (NSCLC), accounting for the majority, mainly including adenocarcinoma (AC), squamous cell carcinoma (SqCC) and large cell carcinoma (LCC). The chromosome ploidy was also different among the main tissue subsets of NSCLC. In the last century, Petersen et al84 used genomic hybridization to evaluate the chromosomal imbalance between 25 cases of lung adenocarcinoma and 25 cases of lung squamous cell carcinoma. A decrease in the number of common DNA copies of the two lung cancer subtypes was observed on chromosome 1p, 3p, 4qQ, 5Q, 6Q, 8p, 9p, 13q, 18Q and 21q. Similarly, the DNA gains of chromosomes 5p, 8q, 11q, 13, 16p, 17q and 19q were observed. Adenocarcinomas showed more frequent DNA overexpression of chromosome 1Q and loss of DNA on chromosomes 3Q, 9Q, 10p and 19Q, while squamous cell carcinoma was characterized by increased overexpression of chromosomes 3Q, 12p and deletion of 2Q. In particular, the overexpression of chromosome band 1q23 and the deletion of 9q22 were significantly associated with adenoid differentiation, while the loss of DNA of chromosome band 2q36-37 and the overexpression of 3q21-22 and 3q24-qter were statistically significant markers of squamous cell types. This study for the first time used statistical methods to strengthen the concept that different tumor subgroups of NSCLC have different chromosome change patterns. Jemal et al85 also demonstrated the overall relationship between different pathological subtypes of lung cancer (AC, n=1206, SqCC, n=467, LCC, n=37 and SCLC, n=88) and chromosome ploidy by DNA flow cytometry analysis. The study found that compared with other histological groups, SCLC cases generally showed more chromosome ploidy changes, while AC showed the least. In various histology, 4Q and 5Q loss (SCLC and SqCC), 10p loss (SCLC, LCC) and 7p increase (AC, LCC), these chromosome changes are relatively common. However, the increase of chromosome 18 and 19 (SCLC), the loss of 22q and 10q (SCLC) and the increase of 22q (SqCC) seem to be more frequent. Six gains on chromosome 3Q and seven losses on 3p, 4p, 4q and 5Q distinguish AC from SqCC. All of these regions have higher frequency of change in SqCC. In addition, the gain on chromosome 1p, 2p, 3Q and 22q, and the loss on chromosome 2p, 4p, 4Q, 5Q, 9p, 10Q, 13q and 22q were found to distinguish AC, sqCC and SCLC. Interestingly, AC and SqCC cases showed the highest percentage of interstitial ploidy values close to 2N (that is, diploids were the most common). In contrast, LCC cases showed a more mixed distribution of 2N and 3N, while SCLC cases showed the most common triploid (3N). At the same time, studies have shown that chromosome instability has less effect on AC than other histological groups, but on the contrary, mutation-driven tumorigenesis may be more important in AC, such as frequent EGFR, STK11, TP53 and KRAS mutations observed in this subtype.
Main Chromosome Changes
NSCLC is a heterogeneous disease, and its mutation rate is second only to that of melanoma, which indicates that lung cancer has inherent genomic instability. Studies have shown that NSCLC is characterized by widespread acquisition and loss of low and high levels of genetic material on different chromosomes.86 Approximately 70% of 80% of lung cancer patients carry aneuploid karyotypes caused by the mis separation of chromosomes during mitosis.87 Chromosome aneuploidy was detected in 46 lung cancer specimens according to Gao et al.8 All samples showed aneuploidy and showed the acquisition of chromosomes 7, 8, and 12 (67.4%, 60.9%, 28.3%) and the deletion of chromosome 9 (56.5%). Liu et al88 detected FISH probes on chromosomes 2, 3, 6, 7, 8, 9, 11, 12, and 17 in 74 cases of non-small cell lung cancer, and the aneuploidy rate was 62–93% in lung tumor tissues. Jemal et al85 also found that the most frequent chromosome arm changes were the increase of chromosomes 5p and 20 (excluding AC) and the loss of 3p, 13 Q (excluding SCLC) and 17p. These results provide strong evidence for the adverse relationship between aneuploidy and lung cancer. High levels of aneuploidy are related to several manifestations of NSCLC invasiveness, including poor prognosis and metastasis. Here, we explored the biological interaction between the aneuploid environment and LC to understand the role of aneuploidy in the progression of lung cancer.
Somatic epigenetic and genetic abnormalities caused by aneuploidy (including specific genome amplification or deletion) play an important role in the occurrence and development of lung cancer.89 The most common chromosomal changes in lung cancer include amplification of 1Q, 3Q, 5p, 8Q, 11q, 16p, and 17Q, and loss/deletion of 3p, 4Q, 5Q, 8p, 9p, 13Q, 17p, and 19p.90 Here, we focus on abnormalities in chromosomes 3, 7, and 8, which are most closely related to lung cancer. Studies91 have shown that the aneuploidy rate of chromosomes and chromosome 3 is the highest in NSCLC, which is 21.35%, and the aneuploidy rate of monomer and trisomy is more prominent (P < 0.05). From precancerous lesions to invasive cancer, the changes on chromosome 3 run through the entire development and metastatic stage of NSCLC, and the amplification of the 3Q distal part may be the main sign of tumor transformation. At the same time, the researchers also pointed out that chromosome 3 may contain many genes that may play a role in carcinogenesis. In Atasoy’s study,91 the aneuploidy rate of chromosome 7 was 9.06%, second only to that of chromosome 3. Overexpression of the epidermal growth factor receptor (EGFR) on chromosome 7 occurs frequently in patients with non-small cell lung cancer and is associated with disease progression and poor survival.92 The expression level of EGFR is not only related to its amplification. In NSCLC, the imbalance of chromosome 7 is a key genetic event that changes the expression of the EGFR protein.93 In some cases, it is independent of EGFR gene amplification. It has been reported that trisomy 7, the only abnormal malignant clone,94 is found in malignant cells of cultured non-small cell lung cancer. It is well known that the frequently amplified proto-oncogene c-myc is related to the promotion of lymphatic metastasis in NSCLC and is a significant prognostic factor in early lung adenocarcinoma. C-myc is located on chromosome.95 Kubokura et al96 performed FISH analysis on the amplification of the c-myc gene and the change in the copy number of chromosome 8 (aneuploid) in patients with non-small cell lung cancer. It was found that an abnormal number of chromosome 8 was significantly associated with poor prognosis. Yakut’s parallel correlation between c-myc amplification, trisomy 8 metastasis, and survival time was also supported by statistical analysis (P < 0.05). They observed that a patient with NSCLC with c-myc amplification relapsed 6 months after the operation, and trisomy 8 was detected in the tumor tissue.
Aneuploidy Changes the Initial Tumor Microenvironment of Lung Cancer
Aneuploidy alters the initial tumor microenvironment of NSCLC and accelerates the progression of precancerous lesions.97 At the end of the last century, Smith et al98 determined that aneuploidy is closely related to precancerous lesions of NSCLC, and that there is widespread aneuploidy in the respiratory epithelium (including bronchioles, bronchioles, and alveoli) in patients with NSCLC. The degree and incidence of cancer increase with the aggravation of histopathological changes and occasionally appear in hyperplasia, usually in the stage of dysplasia and carcinoma in situ, and then induce precancerous lesions to upgrade to invasive carcinoma in a variety of ways. Zojer et al94 also used centromere specific fluorescence in situ hybridization (FISH) to study the chromosome status of 7 cases of invasive non-small cell lung cancer and its precancerous lesions. The percentage of aneuploid cells in invasive precancerous lesions and invasive lung cancer tissues was 8.5% and 59%, respectively. Increasing evidence shows that aneuploidy is involved in the early pathological changes in NSCLC and contributes to the further development of precancerous lesions to invasive NSCLC. The initiation of lung cancer is inseparable from the construction of the microenvironment. In order to better understand and describe the role of aneuploidy in the initial tumor environment, Alikhanyan et al97 constructed a mouse model of NSCLC. In this model, the aneuploid environment of epithelial cells adjacent to the lung tumor increases the number of alveolar macrophages (AM), tumor-associated macrophages (TAM), neutrophils, and NK cells. However, the levels of cytotoxic CD8+T cells and IFN-γ in lung tissue decreased. Interestingly, although NK cells have increased, NK cells no longer have the normal function of natural immune cells, which indicates that higher levels of aneuploidy help to create an immunosuppressive microenvironment, thereby promoting the initiation and growth of tumors. SU et al,99 it was found that the unfolded protein response (UPR) directly driven by aneuploidy polarized macrophages and dendritic cells into tumor-promoting phenotypes, endowed them with immunosuppressive function, and had a negative effect on T cell activation, thus reshaping the tumor immune microenvironment to evade immune surveillance. Taken together, these findings suggest that aneuploidy plays a key role in shaping the immune pattern of lung tumors, resulting in an immunosuppressive environment that awakens dormant cancer cells and initiates tumors, leading to a higher tumor burden.
The Relationship Between Aneuploidy and Metastasis or Recurrence of Lung Cancer
In the case of chromosomal instability, high levels of aneuploidy significantly increase the recurrence rate of NSCLC, which is a sign of poor prognosis. Sotillo et al100 induced MAD2 overexpression to form tumors in mice with lung cancer, and a high level of aneuploidy was observed. After removing the induction, the recurrent tumor appeared in the same anatomic position as the primary tumor, similar to the primary tumor, and was also highly aneuploid (39.4%). This shows that a high level of aneuploidy in residual cancer cells can enable the survival of oncogenes after revocation, thus promoting the recurrence of NSCLC and significantly increasing its recurrence rate. At present, the prognosis of patients with lung cancer after surgical resection depends on two main prognostic variables: the stage of the disease and the state of manifestation (including imaging findings). Choi et al101 analyzed the surgical specimens of 63 patients with lung adenocarcinoma and found 32 cases of aneuploidy positivity (39.7%). The 5-year disease-free survival rates was 58.7% and 46.9% for aneuploid-positive patients and 71.0% (95% confidence interval [CI] 1.04–5.26, P= 0.04) for aneuploid-negative patients, respectively. In the multivariate analysis, after adjusting for pathological lymph node staging, tumor staging, sex, age, and smoking history, the overall survival rate of positive patients was still significantly lower than that of aneuploid-negative patients (95% CI, 1.66–43.42; p = 0.010). Choma et al102 observed aneuploidy in 64% of patients with non-small cell lung cancer through 4033 patients with lung cancer, and the proportion of aneuploid cells in cancer cells gradually increased with the progression of the tumor stage. The percentage of aneuploid tumors in stages I, II, and III was 58%, 57%, and 70%, respectively. Patients with aneuploid-positive non-small cell lung cancer had a higher risk of postoperative death, and their survival probability decreased by 11% within one year compared with other patients, accounting for 21% of the patients who worsened 5 years after operation. The purpose of this study was to evaluate the importance of aneuploidy in the prognosis of NSCLC and its application as an independent prognostic factor after NSCLC surgery. Currently, the real-time progression of lung cancer is mostly tracked by imaging, and the complex relationship between aneuploidy and lung cancer may provide information related to early cancer surveillance and prevention.
Effect of Smoking on Chromosome Ploidy in Patients with Lung Cancer
It is well known that toxins in tobacco smoke increase the risk of lung cancer; it accounts for almost 90% of lung cancer causes in men and 65% in women.103 Whether chromosome ploidy changes play an important role in the evolution of smoking-related lung cancer is still under discussion. Sanchez-Cespedes et al104 selected 18 lifelong non-smoking and 27 smoking lung adenocarcinoma patients and used microsatellite markers to test normal and tumor tissues to find chromosome changes in each tumor. The results showed that alleles in the chromosome arm were lost or increased by 3p (37 vs 6%), 6Q (46 vs 12%), 9p (65 vs 22%), 16p (28 vs 0%), 17p (45 vs 11%). The frequency of 19p (58 vs 16%) in adenocarcinoma of smokers was significantly higher than that of non-smokers. Chromosome arms showing allelic imbalance are rare in lung tumors in non-smokers, but are more common at 19q (22%), 12p (22%) and 9p (22%). Smokers have higher levels of FAL (partial allele loss or gain) (48%) than non-smokers (11%). In the study of Sy et al105 comparative genomic hybridization (CGH) technique was used to find that smoking can cause a significant increase in chromosome amplification in primary lung adenocarcinoma, including 13q, 17q, 19Q and 22q. At the same time, Yan et al106 found that there was a significant increase in chromosome amplification in patients with smoking lung SCC. The amplification rates of 3Q and 8q in smoking patients were significantly higher than those in non-smoking patients (suggesting that there may be targets for smoking carcinogens in these chromosome regions) smoking carcinogens may eventually lead to lung cancer by changing the amplification of these chromosomal regions (resulting in the activation of oncogenes). There are also obvious commonalities in chromosome amplification and deletion between smoking and non-smoking patients (such as higher 5p and 18p amplification rates and higher 3p, 4Q, 5Q deletion rates), indicating that there is consistency in the changes of chromosome ploidy in lung SCC caused by different causes. Other studies have shown that there is no significant difference in chromosome ploidy changes in lung cancer patients between smokers and non-smokers. For example, Farkas et al107 found that smoking had no effect on chromosome aneuploidy in 2145 Hungarian subjects. However, it is agreed that quitting smoking may promote the reversibility of genotoxicity, and because of the directness of tobacco, their cells may be more vulnerable to cytogenetic changes.
The Application Direction of Aneuploidy in Clinical Treatment of Lung Cancer
At present, the incidence of NSCLC is relatively high, and aneuploidy in tumors provides fuel for the evolution and drug resistance of NSCLC. The aneuploidy of chromosome aneuploidy (ANE) also has significance in predicting the efficacy of radiotherapy in patients with NSCLC. Jia et al108 evaluated the predictive value of ANE in radiotherapy. For patients who did not receive radiotherapy, the survival prognosis of different age groups was similar. After radiotherapy, compared with different ANE patients, the survival rate of patients in the lower ANE group was significantly improved (95% CI, 0.9202–3.2770; P=0.0450), which supported the ANE score as an adverse indicator of benefit from radiotherapy.
A aneuploidy is usually quantified by measuring DNA content or chromosome structure and number in cells. Therefore, an increasing number of techniques can be used to detect aneuploidy, including various molecular cytogenetic methods, FISH or ICM, single nucleotide polymorphism array (SNP array), and genome-wide DNA and RNA (ribonucleic acid) sequencing.109 However, an important question is whether aneuploidy can also provide treatment decisions for targeted treatment of NSCLC, thus predicting the benefit of patients from treatment. Epidermal growth factor receptor (EGFR) is the tumor-driving gene of lung cancer, which guides the occurrence and growth of tumors by activating the EGFR signaling pathway.110 It has been reported that111 EGFR amplification and overexpression are usually associated with aneuploidy and tumor cell proliferation to support the strong correlation between aneuploidy and patient efficacy. Wei et al112 demonstrated that patient with aneuploid NSCLC who received EGFR-TKIs did not gain more benefits in terms of progression-free survival (PFS), objective effective rate (ORR), and disease control rate (DCR) than euploidy. Analysis of aneuploidy may help determine which patients are most likely to respond to EGFR-TKI-based treatment.
As mentioned above, Aurora B is considered to be an oncogene that induces abnormal cell division and subsequent aneuploidy, and is associated with a significant decrease in overall and disease-free survival in patients with NSCLC. Yu et al113 found that aneuploidy caused by overexpression of Aurora B was observed in non-small cell lung cancer cells with reduced response to cisplatin and paclitaxel, which was related to the poor prognosis of patients with non-small cell lung cancer. Under normal circumstances, uncontrolled cell cycle processes trigger the activation of p53 and downstream repair and apoptosis pathways to clear cells in a state of abnormal proliferation.114 However, in the background of Aurora-B overexpression, most p53 is degraded by ubiquitin-mediated proteins, and the cell cycle is stagnated in the G1 and S phases and activates the apoptosis cascade reaction, which causes DNA-damaged and aneuploid cells to bypass the cell cycle checkpoint, evade damage repair, and ultimately weaken the efficacy of drugs targeting tumor cell DNA and cell cycle, leading to drug resistance.115 Recently, several inhibitors of Aurora B have been developed (such as small molecular inhibitors GSK650394 and LY3295668, etc.).116,117 These inhibitors can effectively inhibit proliferation and induce apoptosis of NSCLC cells by reducing the expression of Aurora B and inactivating Aurora B. For aneuploidy, specific aneuploid drivers or passengers have been shown to be useful in selectively killing aneuploid cells. The use of aneuploidy in treatment is likely to depend on the factors and mechanisms of aneuploidy formation in aneuploidy cells. As a process of promoting aneuploidy in vivo, deregulation of mitotic checkpoint genes is a new treatment for aneuploidy tumor inhibition. Taxane is a microtubule targeting agent, which can lead to wrong chromosome pairing, thus strengthening chromosome disaggregation and increasing aneuploid cells. However, when combined with different CDK2 inhibitors to treat lung cancer, it showed synergistic or additive anti-tumor effects.118
There is evidence that aneuploidy is a new driving force of NSCLC and can be used as a new biomarker with recognizable tumor-specific characteristics. As a predictor of drug response and potential therapeutic target, it may have clinical relevance. In view of the increasing sensitivity of aneuploid analysis methods, it is necessary to promote research designs aimed at more accurately tracking the actions of aneuploidy in tumors and the therapeutic benefits of aneuploid cells.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
All authors report no conflicts of interest in this work.
References
1. Jones GS, Baldwin DR. Recent advances in the management of lung cancer. Clin Med. 2018;18(Suppl 2):s41–s46. doi:10.7861/clinmedicine.18-2-s41
2. Ben-David U, Amon A. Context is everything: aneuploidy in cancer. Nat Rev Genet. 2020;21(1):44–62. doi:10.1038/s41576-019-0171-x
3. Chi YH, Jeang KT. Aneuploidy and cancer. J Cell Biochem. 2007;102(3):531–538. doi:10.1002/jcb.21484
4. Williams BR, Amon A. Aneuploidy: cancer’s fatal flaw? Cancer Res. 2009;69(13):5289–5291. doi:10.1158/0008-5472.CAN-09-0944
5. Duesberg P, Li R, Fabarius A, Hehlmann R. The chromosomal basis of cancer. Cell Oncol. 2005;27(5–6):293–318. doi:10.1155/2005/951598
6. Lens SMA, Medema RH. Cytokinesis defects and cancer. Nat Rev Cancer. 2019;19(1):32–45.
7. Duesberg P, Li R, Fabarius A, Hehlmann R. Aneuploidy and cancer: from correlation to causation. Contrib Microbiol. 2006;13:16–44.
8. Gao SG, Dong XY, Wang LJ, He J. The study on the chromosome aneuploidy in human lung cancer. Zhonghua Yi Xue Za Zhi. 2007;87(24):1701–1703.
9. Sundaresan V, Ganly P, Hasleton P, et al. p53 and chromosome 3 abnormalities, characteristic of malignant lung tumours, are detectable in preinvasive lesions of the bronchus. Oncogene. 1992;7(10):1989–1997.
10. Fodina V, Dudorova A, Alksere B, et al. The application of PGT-A for carriers of balanced structural chromosomal rearrangements. Gynecol Endocrinol. 2019;35(sup1):18–23. doi:10.1080/09513590.2019.1632091
11. Diogo V, Teixeira J, Silva PM, Bousbaa H. Spindle assembly checkpoint as a potential target in colorectal cancer: current status and future perspectives. Clin Colorectal Cancer. 2017;16(1):1–8. doi:10.1016/j.clcc.2016.06.006
12. Kops GJPL, Snel B, Tromer EC. Evolutionary dynamics of the spindle assembly checkpoint in eukaryotes. Curr Biol. 2020;30(10):R589–R602. doi:10.1016/j.cub.2020.02.021
13. Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Curr Biol. 2012;22(22):R966–R980. doi:10.1016/j.cub.2012.10.006
14. Chao WC, Kulkarni K, Zhang Z, Kong EH, Barford D. Structure of the mitotic checkpoint complex. Nature. 2012;484(7393):208–213. doi:10.1038/nature10896
15. Castro A, Bernis C, Vigneron S, Labbé JC, Lorca T. The anaphase-promoting complex: a key factor in the regulation of cell cycle. Oncogene. 2005;24(3):314–325. doi:10.1038/sj.onc.1207973
16. Cahill DP, Lengauer C, Yu J, et al. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392(6673):300–303. doi:10.1038/32688
17. Lee H, Trainer AH, Friedman LS, et al. Mitotic checkpoint inactivation fosters transformation in cells lacking the breast cancer susceptibility gene, Brca2. Mol Cell. 1999;4(1):1–10. doi:10.1016/S1097-2765(00)80182-3
18. Bokros M, Wang Y. Spindle assembly checkpoint silencing and beyond. Cell Cycle. 2016;15(13):1661–1662. doi:10.1080/15384101.2016.1176396
19. Simonetti G, Bruno S, Padella A, Tenti E, Martinelli G. Aneuploidy: cancer strength or vulnerability? Int J Cancer. 2019;144(1):8–25. doi:10.1002/ijc.31718
20. Guo Y, Zhang X, Yang M, et al. Functional evaluation of missense variations in the human MAD1L1 and MAD2L1 genes and their impact on susceptibility to lung cancer. J Med Genet. 2010;47(9):616–622. doi:10.1136/jmg.2009.074252
21. Kato T, Daigo Y, Aragaki M, et al. Overexpression of MAD2 predicts clinical outcome in primary lung cancer patients. Lung Cancer. 2011;74(1):124–131. doi:10.1016/j.lungcan.2011.01.025
22. Sotillo R, Schvartzman JM, Socci ND, Benezra R. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature. 2010;464(7287):436–440. doi:10.1038/nature08803
23. Tanaka K, Hirota T. Chromosomal instability: a common feature and a therapeutic target of cancer. Biochim Biophys Acta. 2016;1866(1):64–75. doi:10.1016/j.bbcan.2016.06.002
24. Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol. 2010;20(6):R285–R295. doi:10.1016/j.cub.2010.01.034
25. Nicklas RB, Ward SC. Elements of error correction in mitosis: microtubule capture, release, and tension. J Cell Biol. 1994;126(5):1241–1253. doi:10.1083/jcb.126.5.1241
26. Bakhoum SF, Thompson SL, Manning AL, Compton DA. Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat Cell Biol. 2009;11(1):27–35. doi:10.1038/ncb1809
27. Zhao G, Cheng Y, Gui P, et al. Dynamic acetylation of the kinetochore-associated protein HEC1 ensures accurate microtubule-kinetochore attachment. J Biol Chem. 2019;294(2):576–592. doi:10.1074/jbc.RA118.003844
28. Oh HJ, Kim MJ, Song SJ, et al. MST1 limits the kinase activity of Aurora B to promote stable kinetochore-microtubule attachment. Curr Biol. 2010;20(5):416–422. doi:10.1016/j.cub.2009.12.054
29. Takeshita M, Koga T, Takayama K, et al. Aurora-B overexpression is correlated with aneuploidy and poor prognosis in non-small cell lung cancer. Lung Cancer. 2013;80(1):85–90. doi:10.1016/j.lungcan.2012.12.018
30. Kabeche L, Compton DA. Checkpoint-independent stabilization of kinetochore-microtubule attachments by Mad2 in human cells. Curr Biol. 2012;22(7):638–644. doi:10.1016/j.cub.2012.02.030
31. Kleyman M, Kabeche L, Compton DA. STAG2 promotes error correction in mitosis by regulating kinetochore-microtubule attachments. J Cell Sci. 2014;127(Pt 19):4225–4233. doi:10.1242/jcs.151613
32. Bakhoum SF, Kabeche L, Murnane JP, Zaki BI, Compton DA. DNA-damage response during mitosis induces whole-chromosome missegregation. Cancer Discov. 2014;4(11):1281–1289. doi:10.1158/2159-8290.CD-14-0403
33. Manning AL, Ganem NJ, Bakhoum SF, Wagenbach M, Wordeman L, Compton DA. The kinesin-13 proteins Kif2a, Kif2b, and Kif2c/MCAK have distinct roles during mitosis in human cells. Mol Biol Cell. 2007;18(8):2970–2979. doi:10.1091/mbc.e07-02-0110
34. Caldwell CM, Kaplan KB. The role of APC in mitosis and in chromosome instability. Adv Exp Med Biol. 2009;656:51–64.
35. Regolini M. The centrosome as a geometry organizer. Results Probl Cell Differ. 2019;67:253–276.
36. Bornens M. Centrosome organization and functions. Curr Opin Struct Biol. 2021;66:199–206. doi:10.1016/j.sbi.2020.11.002
37. Hardy PA, Zacharias H. Reappraisal of the hansemann-boveri hypothesis on the origin of tumors. Cell Biol Int. 2005;29(12):983–992. doi:10.1016/j.cellbi.2005.10.001
38. Zhao JZ, Ye Q, Wang L, Lee SC. Centrosome amplification in cancer and cancer-associated human diseases. Biochim Biophys Acta Rev Cancer. 2021;1876(1):188566. doi:10.1016/j.bbcan.2021.188566
39. Rhys AD, Godinho SA. Dividing with extra centrosomes: a double edged sword for cancer cells. Adv Exp Med Biol. 2017;1002:47–67.
40. Brinkley BR. Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol. 2001;11(1):18–21. doi:10.1016/S0962-8924(00)01872-9
41. Godinho SA, Kwon M, Pellman D. Centrosomes and cancer: how cancer cells divide with too many centrosomes. Cancer Metastasis Rev. 2009;28(1–2):85–98. doi:10.1007/s10555-008-9163-6
42. Fukasawa K. Centrosome amplification, chromosome instability and cancer development. Cancer Lett. 2005;230(1):6–19. doi:10.1016/j.canlet.2004.12.028
43. Ryniawec JM, Rogers GC. Centrosome instability: when good centrosomes go bad. Cell Mol Life Sci. 2021;78(21––22):6775–6795. doi:10.1007/s00018-021-03928-1
44. Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437(7061):1043–1047. doi:10.1038/nature04217
45. Kawakami M, Mustachio LM, Liu X, Dmitrovsky E. Engaging anaphase catastrophe mechanisms to eradicate aneuploid cancers. Mol Cancer Ther. 2018;17(4):724–731. doi:10.1158/1535-7163.MCT-17-1108
46. Peters JM, Nishiyama T. Sister chromatid cohesion. Cold Spring Harb Perspect Biol. 2012;4(11):a011130. doi:10.1101/cshperspect.a011130
47. Tumini E, Aguilera A. The Sister-chromatid exchange assay in human cells. Methods Mol Biol. 2021;2153:383–393.
48. Biggins S, Murray AW. Sister chromatid cohesion in mitosis. Curr Opin Cell Biol. 1998;10(6):769–775. doi:10.1016/S0955-0674(98)80120-8
49. Zheng G, Yu H. Regulation of sister chromatid cohesion during the mitotic cell cycle. Sci China Life Sci. 2015;58(11):1089–1098. doi:10.1007/s11427-015-4956-7
50. Barbero JL. Sister chromatid cohesion control and aneuploidy. Cytogenet Genome Res. 2011;133(2–4):223–233. doi:10.1159/000323507
51. Cheng H, Zhang N, Pati D. Cohesin subunit RAD21: from biology to disease. Gene. 2020;758:144966. doi:10.1016/j.gene.2020.144966
52. Hill VK, Kim JS, Waldman T. Cohesin mutations in human cancer. Biochim Biophys Acta. 2016;1866(1):1–11.
53. Xu H, Tomaszewski JM, McKay MJ. Can corruption of chromosome cohesion create a conduit to cancer? Nat Rev Cancer. 2011;11(3):199–210.
54. Zhou H, Zheng L, Lu K, et al. Downregulation of cohesin loading factor nipped-B-like protein (NIPBL) induces cell cycle arrest, apoptosis, and autophagy of breast cancer cell lines. Med Sci Monit. 2017;23:4817–4825.
55. Barber TD, McManus K, Yuen KW, et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc Natl Acad Sci U S A. 2008;105(9):3443–3448.
56. Losada A. Cohesin in cancer: chromosome segregation and beyond. Nat Rev Cancer. 2014;14(6):389–393.
57. De Koninck M, Losada A. Cohesin mutations in cancer. Cold Spring Harb Perspect Med. 2016;6(12):a026476.
58. Zhang N, Pati D. Biology and insights into the role of cohesin protease separase in human malignancies. Biol Rev Camb Philos Soc. 2017;92(4):2070–2083.
59. Delhanty JD, Pellestor F. Aneuploidy. Preface. Cytogenet Genome Res. 2011;133(2–4):89–90.
60. Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet. 2012;13(3):189–203.
61. Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature. 2004;432(7015):338–341.
62. Thomas R, Marks DH, Chin Y, Benezra R. Whole chromosome loss and associated breakage-fusion-bridge cycles transform mouse tetraploid cells. EMBO J. 2018;37(2):201–218.
63. Baker DJ, Jin F, Jeganathan KB, van Deursen JM. Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell. 2009;16(6):475–486.
64. Marques S, Fonseca J, Silva PM, Bousbaa H. Targeting the spindle assembly checkpoint for breast cancer treatment. Curr Cancer Drug Targets. 2015;15(4):272–281.
65. López-García C, Sansregret L, Domingo E, et al. BCL9L dysfunction impairs caspase-2 expression permitting aneuploidy tolerance in colorectal cancer. Cancer Cell. 2017;31(1):79–93.
66. Narkar A, Johnson BA, Bharne P, et al. On the role of p53 in the cellular response to aneuploidy. Cell Rep. 2021;34(12):108892.
67. Duensing A, Duensing S. Guilt by association? p53 and the development of aneuploidy in cancer. Biochem Biophys Res Commun. 2005;331(3):694–700.
68. Fang X, Zhang P. Aneuploidy and tumorigenesis. Semin Cell Dev Biol. 2011;22(6):595–601.
69. Milán M, Clemente-Ruiz M, Dekanty A, Muzzopappa M. Aneuploidy and tumorigenesis in Drosophila. Semin Cell Dev Biol. 2014;28:110–115.
70. Kim Y, Heuser JE, Waterman CM, Cleveland DW. CENP-E combines a slow, processive motor and a flexible coiled coil to produce an essential motile kinetochore tether. J Cell Biol. 2008;181(3):411–419.
71. Wood KW, Sakowicz R, Goldstein LS, Cleveland DW. CENP-E is a plus end-directed kinetochore motor required for metaphase chromosome alignment. Cell. 1997;91(3):357–366.
72. Silk AD, Zasadil LM, Holland AJ, Vitre B, Cleveland DW, Weaver BA. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc Natl Acad Sci U S A. 2013;110(44):E4134–E4141.
73. Kaya A, Gerashchenko MV, Seim I, Labarre J, Toledano MB, Gladyshev VN. Adaptive aneuploidy protects against thiol peroxidase deficiency by increasing respiration via key mitochondrial proteins. Proc Natl Acad Sci U S A. 2015;112(34):10685–10690.
74. Hernando E. Cancer. Aneuploidy advantages? Science. 2008;322(5902):692–693.
75. Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11(1):25–36.
76. Baker DJ, Jeganathan KB, Cameron JD, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36(7):744–749.
77. Janssen A, Kops GJ, Medema RH. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc Natl Acad Sci U S A. 2009;106(45):19108–19113.
78. Williams BR, Prabhu VR, Hunter KE, et al. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322(5902):703–709.
79. Torres EM, Williams BR, Amon A. Aneuploidy: cells losing their balance. Genetics. 2008;179(2):737–746.
80. Sheltzer JM, Ko JH, Replogle JM, et al. Single-chromosome gains commonly function as tumor suppressors. Cancer Cell. 2017;31(2):240–255.
81. Osuna-Marco MP, López-Barahona M, López-Ibor B, Tejera ÁM. Ten reasons why people with down syndrome are protected from the development of most solid tumors -a review. Front Genet. 2021;12:749480.
82. Roizen NJ, Patterson D. Down’s syndrome. Lancet. 2003;361(9365):1281–1289.
83. Siegel JJ, Amon A. New insights into the troubles of aneuploidy. Annu Rev Cell Dev Biol. 2012;28:189–214.
84. Petersen I, Bujard M, Petersen S, et al. Patterns of chromosomal imbalances in adenocarcinoma and squamous cell carcinoma of the lung. Cancer Res. 1997;57(12):2331–2335.
85. Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90.
86. Nasim F, Sabath BF, Eapen GA. Lung Cancer. Med Clin North Am. 2019;103(3):463–473.
87. Sansregret L, Swanton C. The role of aneuploidy in cancer evolution. Cold Spring Harb Perspect Med. 2017;7(1):a028373.
88. Liu YZ, Wang Z, Fang LL, et al. A potential probe set of fluorescence in situ hybridization for detection of lung cancer in bronchial brushing specimens. J Cancer Res Clin Oncol. 2012;138(9):1541–1549.
89. Gao B, Yang F, Han M, et al. Genomic landscape and evolution of arm aneuploidy in lung adenocarcinoma. Neoplasia. 2021;23(9):870–878.
90. Qian J, Massion PP. Role of chromosome 3q amplification in lung cancer. J Thorac Oncol. 2008;3(3):212–215.
91. Atasoy S, Erturan SS, Yılmaz N, et al. Analysis of chromosome 3, 7 and 8 centromeric regions in bronchial lavage specimens by FISH. Turk Thorac J. 2016;17(4):141–147.
92. Wang J, Zhang Y, Wan H, et al. Global analyses of subtype- or stage-specific genes on chromosome 7 in patients with lung cancer. Cancer Metastasis Rev. 2015;34(2):333–345.
93. Dorđević G, Matušan Ilijaš K, Hadžisejdić I, Maričić A, Grahovac B, Jonjić N. EGFR protein overexpression correlates with chromosome 7 polysomy and poor prognostic parameters in clear cell renal cell carcinoma. J Biomed Sci. 2012;19(1):40.
94. Zojer N, Dekan G, Ackermann J, et al. Aneuploidy of chromosome 7 can be detected in invasive lung cancer and associated premalignant lesions of the lung by fluorescence in situ hybridisation. Lung Cancer. 2000;28(3):225–235.
95. Baykara O, Bakir B, Buyru N, Kaynak K, Dalay N. Amplification of chromosome 8 genes in lung cancer. J Cancer. 2015;6(3):270–275.
96. Kubokura H, Tenjin T, Akiyama H, et al. Relations of the c-myc gene and chromosome 8 in non-small cell lung cancer: analysis by fluorescence in situ hybridization. Ann Thorac Cardiovasc Surg. 2001;7(4):197–203.
97. Alikhanyan K, Chen Y, Somogyi K, Kraut S, Sotillo R. Mad2 induced aneuploidy contributes to Eml4-Alk driven lung cancer by generating an immunosuppressive environment. Cancers. 2021;13(23):6027. doi:10.3390/cancers13236027
98. Smith AL, Hung J, Walker L, et al. Extensive areas of aneuploidy are present in the respiratory epithelium of lung cancer patients. Br J Cancer. 1996;73(2):203–209. doi:10.1038/bjc.1996.36
99. Xian S, Dosset M, Almanza G, et al. The unfolded protein response links tumor aneuploidy to local immune dysregulation. EMBO Rep. 2021;22(12):e52509. doi:10.15252/embr.202152509
100. Sotillo R, Hernando E, Díaz-Rodríguez E, et al. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11(1):9–23. doi:10.1016/j.ccr.2006.10.019
101. Choi CM, Seo KW, Jang SJ, et al. Chromosomal instability is a risk factor for poor prognosis of adenocarcinoma of the lung: fluorescence in situ hybridization analysis of paraffin-embedded tissue from Korean patients. Lung Cancer. 2009;64(1):66–70. doi:10.1016/j.lungcan.2008.07.016
102. Choma D, Daurès JP, Quantin X, Pujol JL. Aneuploidy and prognosis of non-small-cell lung cancer: a meta-analysis of published data. Br J Cancer. 2001;85(1):14–22. doi:10.1054/bjoc.2001.1892
103. Nagah A, Amer A. Different mechanisms of cigarette smoking-induced lung cancer. Acta Biotheor. 2021;69:37–52. doi:10.1007/s10441-020-09394-9
104. Sanchez-Cespedes M, Ahrendt SA, Piantadosi S, et al. Chromosomal alterations in lung adenocarcinoma from smokers and nonsmokers. Cancer Res. 2001;61(4):1309–1313.
105. Sy SM, Wong N, Mok TS, et al. Genetic alterations of lung adenocarcinoma in relation to smoking and ethnicity. Lung Cancer. 2003;41(1):91–99. doi:10.1016/S0169-5002(03)00138-7
106. Yan WS, Song LY, Wei WD, et al. Chromosomal imbalance in primary lung squamous cell carcinoma and their relationship with smoking. Ai Zheng. 2005;24(1):47–52.
107. Farkas G, Jurányi Z, Székely G, Kocsis ZS, Gundy S. Relationship between spontaneous frequency of aneuploidy and cancer risk in 2145 healthy Hungarian subjects. Mutagenesis. 2016;31(5):583–588. doi:10.1093/mutage/gew024
108. Jia Q, Chu Q, Zhang A, et al. Mutational burden and chromosomal aneuploidy synergistically predict survival from radiotherapy in non-small cell lung cancer. Commun Biol. 2021;4(1):131. doi:10.1038/s42003-021-01657-6
109. Danielsen HE, Pradhan M, Novelli M. Revisiting tumour aneuploidy - The place of ploidy assessment in the molecular era. Nat Rev Clin Oncol. 2016;13(5):291–304. doi:10.1038/nrclinonc.2015.208
110. da Cunha Santos G, Shepherd FA, Tsao MS. EGFR mutations and lung cancer. Annu Rev Pathol. 2011;6:49–69. doi:10.1146/annurev-pathol-011110-130206
111. Khalil FK, Altiok S. Advances in EGFR as a predictive marker in lung adenocarcinoma. Cancer Control. 2015;22(2):193–199. doi:10.1177/107327481502200210
112. Wei B, Zhao C, Yang K, et al. Comprehensive analysis of aneuploidy status and its effect on the efficacy of EGFR-TKIs in lung cancer. J Thorac Dis. 2022;14(3):625–634. doi:10.21037/jtd-22-73
113. Yu J, Zhou J, Xu F, Bai W, Zhang W. High expression of Aurora-B is correlated with poor prognosis and drug resistance in non-small cell lung cancer. Int J Biol Markers. 2018;33(2):215–221. doi:10.1177/1724600817753098
114. Ryan KM, Phillips AC, Vousden KH. Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol. 2001;13(3):332–337. doi:10.1016/S0955-0674(00)00216-7
115. Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20(15):1803–1815. doi:10.1038/sj.onc.1204252
116. He Y, Fu W, Du L, et al. Discovery of a novel Aurora B inhibitor GSK650394 with potent anticancer and anti-aspergillus fumigatus dual efficacies in vitro. J Enzyme Inhib Med Chem. 2022;37(1):109–117. doi:10.1080/14756366.2021.1975693
117. Du J, Yan L, Torres R, et al. Aurora A-selective inhibitor LY3295668 leads to dominant mitotic arrest, apoptosis in cancer cells, and shows potent preclinical antitumor efficacy. Mol Cancer Ther. 2019;18(12):2207–2219. doi:10.1158/1535-7163.MCT-18-0529
118. Rodrigues-Ferreira S, Nahmias C. From tumorigenesis to cell death: the aneuploidy paradox. Mol Cell Oncol. 2020;7(2):1709390. doi:10.1080/23723556.2019.1709390
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.