")
Back to Journals » OncoTargets and Therapy » Volume 8
Clinical and pathological analysis of 116 cases of adult adrenal cortical adenoma and literature review
Authors Jia A, Du H, Fan M, Li Y, Xu J, Niu G, Bai J, Zhang G, Ren Y
Received 29 January 2015
Accepted for publication 13 February 2015
Published 28 May 2015 Volume 2015:8 Pages 1251—1257
DOI https://doi.org/10.2147/OTT.S81831
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Daniele Santini
Ai-Hua Jia,1 Hong-Quan Du,2 Min-Hua Fan,2 Yu-Hong Li,1 Jun-Long Xu,1 Gui-Fen Niu,2 Jie Bai,2 Guang-Zhen Zhang,2 Yu-Bo Ren1
1Department of Pathology, Liaocheng People’s Hospital, Liaocheng, People’s Republic of China; 2Department of Endocrinology, Liaocheng People’s Hospital, Liaocheng, People’s Republic of China
Background: The aim of this study is to investigate origin, gross features, microscopic features, immunohistochemical properties, and differential diagnosis of adrenal cortical adenoma (ACA) in patients ≥20 years old.
Methods: The clinicopathological features of 116 cases of ACA and the immunohistochemical features of 50 cases of ACA were evaluated, and the relevant literature was reviewed.
Results: In our cohort, 76.72% (89/116) of the cases were functional, and 27 cases had non-functional, benign adrenal adenomas. ACA presented as an island tumor with an envelope, and the mean tumor size was 3.6 cm (range 1–5 cm), with a mean tumor weight of 9.28 g (range 5–113 g). The shape of the tumor cells was consistent, and mitosis was rarely observed. Forty of the 46 patients with cortisol-secreting ACA had tumors containing granule cells. Primary aldosteronism was observed in 43 cases. Thirty-eight cases had endoscopically visible tumors, with clear cells and lipid-rich cytoplasm arranged in irregular patches or strips. Cortisol-producing ACAs were associated with atrophy of the non-tumorous cortex. Adrenocortical adenomas displayed positive immunohistochemical staining for MELAN-A, Syn (46 of 50 cases of ACA), NSE (44 of 50 cases of ACA), Vim (42 of 50 cases of ACA) and Ki-67 <5% (24 of 50 cases of ACA; the remaining 26 cases were negative for Ki-67).
Conclusion: Prediction of endocrine syndrome in functional ACA was possible based on its structure and morphologic features, which could prevent an unanticipated postoperative crisis. However, a clinical study is needed to validate these findings.
Keywords: adrenal cortical adenoma, pathological features, immunohistochemical staining, pathological analysis
Introduction
Primary adrenal cortical adenomas (ACAs) are common. Based on a summary of 25 autopsy studies, the prevalence of these tumors was approximately 6%.1,2 More than 85% of adrenal neoplasms, incidentally discovered by imaging techniques, ultimately clinically proved to be adenomas.3 However, adenomas are difficult to recognize in practice because many patients are asymptomatic, or they present with only non-specific symptoms. With recent advances in imaging techniques and new methods of hormone determination, the detection of ACA will continue to improve in the future.4
Only a few reports have been published regarding the pathology of ACA.5 The differences between malignant and benign adrenal disease,6,7 the characteristics of functioning and non-functioning adenomas,8,9 and the distinction between patients with bilateral and unilateral adrenal incidentalomas10 have been described, but little is known about the differentiation of adult and pediatric ACAs.
In this study, we reviewed a series of 116 adult ACAs treated at our institution over the last 6 years. General information, preoperative examinations, immunohistochemical properties, follow-up information, and symptoms and signs related to hormonal hypersecretion were analyzed. Furthermore, the origin, gross features, microscopic features, pathologic diagnosis, and differential diagnosis of ACA (patients ≥20 years old) were investigated.
Materials and methods
Patients
Patients with ACA who were admitted to Liaocheng Hospital and other hospitals between 2006 and 2012 were reviewed. The presence of ACA was confirmed in all 116 patients by pathology, and all of the patients provided informed consent to be included in this study. Eighty cases were obtained from our hospital, and 36 cases were received from the surrounding counties and cities.
Methods
Analysis was performed in all cases by a standardized Envision method, using 10% formalin-fixed paraffin-embedded tissue sections that were approximately 4 μm in thickness. Hematoxylin and eosin-stained slides from all of the cases were thoroughly reviewed by two investigators. A comprehensive list of macroscopic and histologic observations was noted and recorded as follows: tumor size (greatest dimension in centimeters); tumor weight (in grams); location of the tumor (right or left); and the presence of capsular invasion. For the 50 cases with suspicious diagnoses, immunohistochemical staining, using a hypersensitive two-step method, was recommended for further confirmation of ACA. Immunohistochemical analysis of melan-A, CK (AE1/AE3), Syn, Vim, CgA, NSE, S-100, P16, and Ki-67 was performed. In addition, we reviewed detailed patient histories, the patients’ demographics (sex and age), physical examination findings, and the symptoms and signs related to hormonal hypersecretion. We also recorded the macroscopic and microscopic pathologic features of the tumors. Follow-up data included current patient and disease status.
Results
Clinical data
One hundred and sixteen patients who were diagnosed with ACA and hospitalized in the Department of Urology of Liaocheng Hospital (Liaocheng, People’s Republic of China) and in other hospitals between January 2006 and December 2012 were included in this retrospective study (68 women, 48 men). Study protocols involving human materials were approved by the institutional ethics committee of Liaocheng Hospital. The median age of the 116 patients was 50.8 years (range 26–79 years; male:female, 1:1.417), and only one case was present in a patient younger than 30 years old. Unilateral incidentalomas were present on the left side in 56 patients and on the right side in 60 patients, all of which were island tumors with envelopes. The tumors ranged in size from 0.5 to 22.0 cm, and the size distribution of the present series peaked at 1.5–4.0 cm (94.8%, 110/116). Biochemical and baseline hormonal evaluations revealed that 76.7% (89/116) of the tumors were functional (hyperaldosteronism 37.1% [43/116], hypercortisolism 39.7% [46/116]).
The duration between disease onset and diagnosis ranged from 2 months to 6 years. Forty-six patients with cortisol-producing adenomas exhibited symptoms, including headache and/or dizziness (38/46), central obesity and sanguineous temperament (24/46), muscle weakness (22/46), purple skin striae or ecchymosis (14/46), edema (16/46), and dwarfism (12/46). Forty-three patients had symptoms and signs of primary aldosteronism, eg, headache and/or dizziness (35/43), myasthenia gravis or periodic paralysis (23/43), polydipsia/polyuria (12/43), tic (5/43), and blurred vision (5/43). In the non-functional adenoma group, 20 patients were asymptomatic, and tumors were discovered incidentally over the course of work-ups or during treatment for unrelated disorders. The remaining seven patients presented with other clinical features, such as abdominal pain or other non-specific complaints.
All of the cases received follow-up exams (from 5 months to 5 years). No recurrence or metastasis was found in any of the 116 pathologically confirmed benign adenomas.
Pathological examinations
Gross examination: solitary masses in the unilateral adrenal gland occurred in 116 cases of ACA. The tumors were oval, flat, and round; they contained an envelope and appeared aurantium or brown on the cut surface with a soft texture. The tumors ranged in size from 0.5 to 22.0 cm, with a mean tumor weight of 9.28 g (range 5–113 g). One hundred and twelve (96.55%) tumors were ≤50 g in weight.
Microscopic features: ACAs were covered with a fibrous capsule, and the adrenal cortex was compressed at the periphery in some cases. The tumors were composed of different proportions of clear cells and compact cells, and the tumor cells were slightly larger than normal cortical cells. The cell shape was consistent, and mitosis was rare. Scattered irregular nuclei, hyperchromatic cells and giant cells were observed in some adenomas. The tumor cells were arranged in nests, cords or trabecular shapes, with rare solid areas, rich blood sinuses and smaller numbers of interstitial cells.
Cortisol-producing ACAs were associated with atrophy of the non-tumorous cortex. In contrast, the non-tumorous adrenal cortex was not atrophic in glands harboring an aldosterone-producing ACA (Figure 1A), and they occasionally exhibited hyperplastic changes, especially in the zona glomerulosa layer (paradoxical hyperplasia). There was no obvious morphological difference between non-functional and functional ACAs, although the latter lesion can cause primary aldosteronism or hypercortisolism.11 Primary aldosteronism was observed in 43 cases, and there were 38 cases of endoscopically visible tumors containing clear cells, with increased amounts of lipofuscin in the cytoplasm arranged in irregular patches or strips (Figure 1B). Forty of the 46 patients with cortisol-secreting ACAs presented with granule cell tumors, and the cells were densely arranged in small mesh or fasciculate patterns, with cell cords exhibiting sinus gap shapes and blood sinus segmentation (Figures 1C and D).
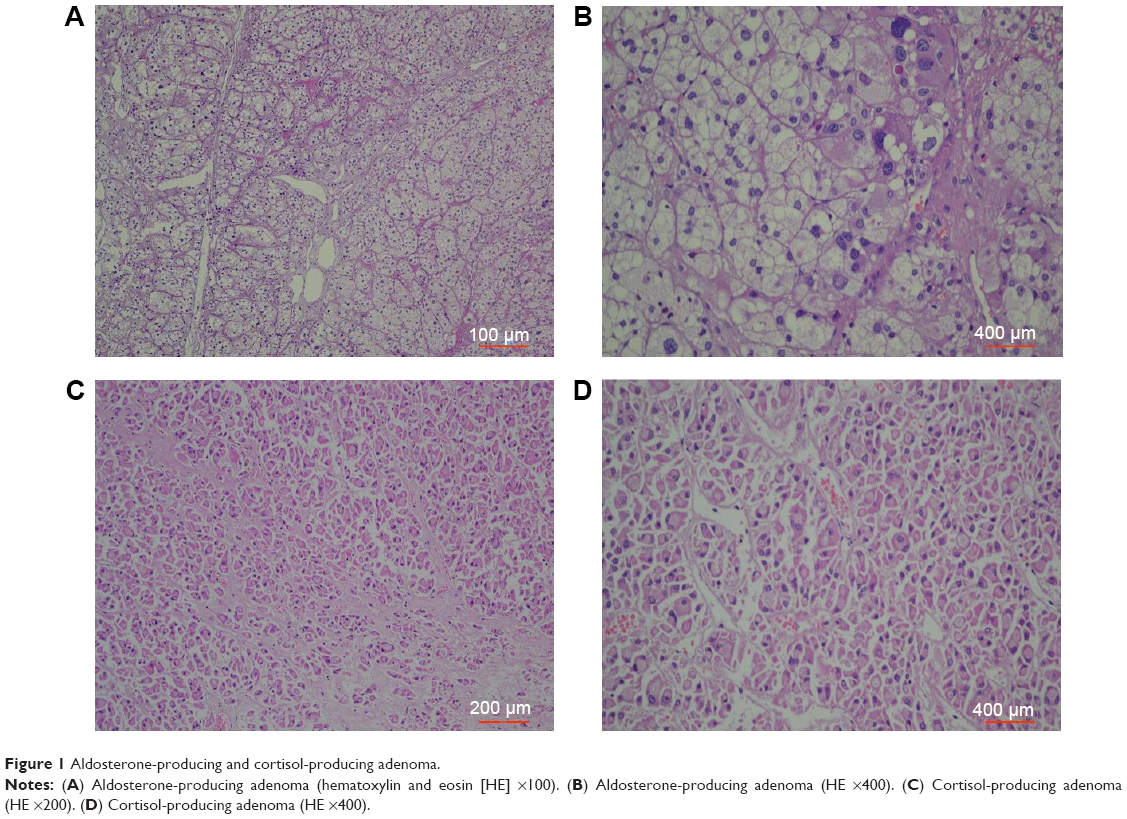 | Figure 1 Aldosterone-producing and cortisol-producing adenoma. |
Immunohistochemical results
Fifty cases had suitable material for immunohistochemical evaluation. None of the adenomas showed S-100 or CgA expression; however, Syn staining was positive in 92% (46/50) of cases tested, NSE (+) staining was observed in 88% (44/50) of cases, and Vim (+) staining was found in 84% (42/50) of cases. All samples were positive for melan-A, and 24 cases displayed Ki-67 staining <5% (+).
Discussion
The incidence of adult ACA is low. In our study, most of the patients were in the third to fifth decades of life, and they were predominantly female. Adrenal cortical neoplasms in the pediatric population are rare, and it is estimated that approximately 25 cases of pediatric adrenal cortical neoplasms occur in the United States per year. In an American study, 83 cases of pediatric adrenal cortical neoplasms were found, representing 0.13% of the 60,732 benign and malignant primary neoplasms diagnosed in pediatric patients, ranging in age from 4 months to 19 years old (mean 7.6 years; median 4 years).12 No biphasic age distribution was observed, with 53% of the cases occurring in children younger than 5 years old and 37% of the cases occurring in those older than 10 years old. Only 10% of the cases occurred in children between the ages of 5 and 10 years old.12 Although girls were affected more frequently than boys, sex did not prove to be a statistically significant predictor of an adverse patient outcome.12
ACA is a benign tumor of the adrenal cortical cells. The pathogenesis of adrenocortical adenomas is currently obscure. Some researchers believe that adenoma nodules are formed from adenomatoid hyperplasia and that the formation of hyperplastic nodules by ACTH gradually transforms these hyperplastic nodules into independent functional nodules. Depending upon the ability of these tumors to produce hormones, they were divided into non-functional and functional ACAs. Clinical symptoms were usually associated with tumor size, the position of the tumor, and the secreted hormone. The synthesis and secretion of hormones in the tumor often fluctuated, and the symptoms were not obvious when secretion levels were low. Non-functional adult adrenal cortical neoplasms accounted for 23.28% (27/116) of the cases in our cohort, and 19.3% (16/83) of the studied pediatric population displayed non-functional lesions.
Forty-six patients had symptoms and signs of Cushing’s syndrome, eg, central obesity, moon face, buffalo hump, headache and/or dizziness, sanguineous temperament, muscle weakness, purple skin striae or ecchymosis, edema, and dwarfism. The 43 patients with primary aldosteronism presented with headache and/or dizziness, myasthenia gravis or periodic paralysis, polydipsia/polyuria/polydipsia, tic, and blurred vision. Therefore, ACAs were mostly found due to the presentation of clinically related symptoms. The serendipitous discovery of an adrenal tumor (adrenal incidentaloma) is a byproduct of modern technology. The routine use of sophisticated radiological techniques, such as abdominal ultrasound, computed tomography, and magnetic resonance imaging, has revealed a problem that is increasingly recognized in current medical practice. Of 27 patients with non-functional adrenal adenomas, seven patients had abdominal pain or other non-specific complaints unrelated to the suspicion of adrenal disease, and the remaining 20 adenomas were discovered inadvertently in the course of other work-ups.
Identification of ACA and adrenal nodular hyperplasias: occasionally, it was difficult to distinguish ACA from adrenal nodular hyperplasias. Generally, the latter was composed of bilateral multiple nodules, there were no fibrous septa between the nodule and the surrounding tissue, and diffuse hyperplasia of the adrenal cortex adjacent to the nodule was observed. In contrast, ACAs usually presented as well-circumscribed and encapsulated solitary neoplasms, and the non-neoplastic adrenal glands demonstrated cortical atrophy to a different extent. The ACA cells were larger than normal cortical cells, and there were no differences between these cells regarding intracytoplasmic lipid content, nuclear morphology or the presence/absence of lipofuscin.
The distinction between aldosterone-producing and cortisol-producing adrenal cortical neoplasms: Peppelman et al13 showed that CD56 is expressed in adrenal cortical neoplasms and concluded that CD56 immunohistochemistry could not distinguish between aldosterone- and cortisol-producing ACAs.
While this finding might be true, the authors also stated that “the distinction between aldosterone-producing and cortisol-producing adrenal cortical neoplasms is hardly possible by conventional histology”. However, some scientists14–16 suggested that aldosterone-producing and cortisol-producing adenomas could be distinguished by assessing the status of the non-tumorous adrenal cortex on hematoxylin and eosin stained slides. Lack believed that cortisol-producing ACAs were associated with atrophy of the non-tumorous cortex due to the negative feedback–suppression effects of the hypothalamic–pituitary axis. In contrast, the non-tumorous adrenal cortex is not atrophic in glands harboring an aldosterone-producing ACA, and it occasionally exhibits hyperplastic changes, especially in the zona glomerulosa layer (paradoxical hyperplasia).
The real challenge lies in distinguishing aldosterone-producing adenomas from non-functioning ACAs that also display a normal adjacent cortex. Because the ultrastructural features of aldosterone-producing adenomas are unique, this distinction could be made reliably by electron microscopy. Aldosterone-producing cells contained mitochondria with lamellar-type or plate-like cristae, whereas glucocorticoid-producing and non-functioning adenoma cells contained mitochondria with tubulovesicular cristae. The endoscopic manifestation of the aldosterone-secreting ACA that we observed was consistent with the above results.
Importantly, assessment of the non-tumorous adrenal cortex is a critical function of the surgical pathologist, which could have acute clinical implications, yet it is often overlooked. Even the gross identification of atrophy of the non-tumorous adrenal cortex in a gland with a dominant cortical adenoma should prompt the surgical pathologist to determine whether the patient has been diagnosed with Cushing’s syndrome. If the surgical pathologist recognizes the atrophy, then timely contact with the relevant clinician could prevent the unanticipated postoperative crisis of acute Addison’s disease.17
Distinguishing ACAs from carcinomas can pose a difficult diagnostic problem, even if clinical imaging and cellular morphology are taken into account. The major differential diagnostic criteria for ACA and adenocarcinoma include capsular invasion, vascular envelope presence, and metastasis. Metastatic disease is the most reliable evidence of malignancy. In a few cases, especially when a smooth envelope was observed, a small number of irregular tumor nuclei was scattered throughout the sample, and hyperchromatic cells or tumor giant cells were observed in specimens. Due to improper treatment or slices of poor quality, it was difficult to determine whether invasion of the capsule and of the blood vessels occurred. Sometimes capsular invasion can be difficult to recognize because the expanding capsule might be a pre-existing adrenal capsule.
Invasion of adjacent soft tissue, the kidney or the liver is a definitive sign of malignancy. Criteria for the diagnosis of adrenal cortical carcinoma were recently proposed by Weiss’ group. Weiss’ criteria18 are useful because of their reliance on histologic features. These features include 1) high nuclear grade; 2) mitotic rates >5 per 50 high-power fields; 3) atypical mitotic figures; 4) eosinophilic tumor cell (≥75% of tumor); 5) diffuse architecture (≥33% of tumor); 6) confluent necrosis; 7) vena cava invasion (smooth muscle in wall); 8) sinusoidal invasion (no smooth muscle in wall); and 9) capsular invasion. Three or more of the nine criteria were indicative of an adrenal cortical carcinoma. The major criteria for adrenal cortical carcinomas included high mitotic rate, atypical mitoses, and vena cava invasion, whereas two or fewer would be more suggestive of the presence of an adenoma. Therefore, tumor weight ≥90 g, tumor size ≥6 cm, confluent necrosis, mitotic rate >5 per 50 high-power fields and invasive growth could be useful in the diagnosis of adrenal cortical carcinomas. Three or more of the five criteria would be indicative of an adrenal cortical carcinoma, one to two criteria indicate borderline tumors, and the absence of any criteria indicates a benign tumor. All 116 cases met these criteria, and 110 cases displayed tumor sizes of 1.5–4.0 cm in diameter. Our results are in agreement with those reported by Terzolo et al.19
It was reported18 that pediatric ACA patients, ranging in age from 4 months to 19 years old (mean 7.6 years; median 4 years), can have tumors that weigh up to 500 g; however, some tumors may weigh as much as 2,413 g. Most carcinomas in adults are >100 g, and adrenal cortical carcinomas weighing <50 g are extremely uncommon, whereas adenomas generally weigh 50 g or less. Most of the cortical adenomas in our series were small, with cortical adenomas <50 g accounting for 96.55% of the cases. Our findings are also in accordance with those of the aforementioned studies.
Adrenal cortical neoplasms in pediatric patients are more difficult to diagnose and to differentiate from adenomas and carcinomas. Wieneke et al12 undertook an outcome-based analysis study of 83 adrenal cortical neoplasms to determine whether adult clinical and histological features could be applied to pediatric patients. Only 31% of histologically malignant tumors behaved in a clinically malignant fashion. Features associated with an increased probability of malignant clinical behavior included tumor weight (>400 g), tumor size (>10.5 cm), vena cava invasion, capsular and/or vascular invasion, extension into periadrenal soft tissue, confluent necrosis, severe nuclear atypia, >15 mitotic figures/20 high-power fields and the presence of atypical mitotic figures. Vena cava invasion, necrosis, and increased mitotic activity (>15 mitotic figures/20 high-power fields) independently suggested malignant clinical behavior in multivariate analysis.
Functional adult adrenal cortical neoplasms accounted for 76.72% (89/116) of the lesions in our cohort, and 80.7% (67/83) of the pediatric population had functional lesions. In contrast to tumors in the adult population, tumors in children were more often hormonally functional, with clinical evidence of virilization, Cushing’s syndrome, mixed endocrine syndromes and, rarely, feminization or Conn’s syndrome.12 Furthermore, feminizing tumors in adults are fatal, which is a fate not shared by pediatric patients, although deaths from such tumors have been reported in this population. As mentioned earlier, virilizing tumors secreting adrenal androgens are most common in children, and the reported prevalence is 20%–30% of all adrenocortical tumors.20,21 Purely feminizing tumors are very rare. While a functional tumor can be indicative of poor outcomes in adult patients, the same conclusion cannot be extrapolated to the pediatric population.12
Application of immunohistochemistry in diagnosis and differential diagnosis
Immunohistochemical determinants that were useful in differentiating ACA from carcinoma included melan-A, inhibin A and calretinin. These tumors were usually positive for melan-A by immunostaining.22 Tartour et al23 noted that immunohistochemical studies have demonstrated positive immunostaining of CK (AE1/AE3), Vim, CgA, Syn, NSE, and S-100 in adrenal cortical tumors to different degrees. These adrenal cortical hyperplasia cells were usually positive for CK by immunostaining in a belt-like distribution; however, CK was usually negative in these adrenal cortical carcinomas, and a variable degree of expression in the intensity of CK immunostaining of ACA was observed, indicating prompt CK loss during the process of adrenocortical tumor carcinogenesis.
CgA only appeared in the cytoplasm of the adrenal medulla and pheochromocytoma. Syn and NSE were positive in adrenal tumor cells with neuroendocrine differentiation, but they were negative in normal adrenal cortex cells. CD56 immunohistochemistry is not an adequate marker for discriminating between adrenal aldosterone-producing and cortisol-producing adenomas.13 In addition, Ki-67 expression was not detected in the normal adrenal cortex adjacent to the neoplasm, and it was weakly positive in some of the adenomas. Wachenfeld et al24 reported that a Ki-67 index greater than 5% was a good marker for malignancy in adrenocortical neoplasms. Szajerka et al25 previously reported that the difference in Ki-67 expression by immunohistochemistry between adrenocortical carcinomas and adenomas was statistically significant (P<0.05), and it provided an objective criterion for differential diagnosis, with higher Ki-67 expression in patients with adrenal carcinoma suggesting an unfavorable prognosis. There were no differences in the expression of Ki-67 among the adult patients with tumors diagnosed as cortisol-secreting, aldosterone-secreting, and non-functional neoplasms, suggesting that these tumor cells might be consistent in their biological behavior, although differential endocrine functions exist.
Currently, specific immunohistochemical markers of adrenal cortical tumors have not been found. According to the melan-A, CK (AE1/AE3), Vim, CgA, Syn, NSE, S-100 and Ki-67 staining performed in this study, some of the biological characteristics of cortical tumors were identified, and although we did not make a specific diagnosis of adrenocortical tumor, these features could be used to differentiate whether a tumor is derived from the adrenal gland (the cortex or medulla) or from other parts of the body.
In summary, it is possible to predict the endocrine syndrome of functional ACA based on the structure and morphologic features of the tumor. In combination with clinical endocrine detection, the diagnosis of a given adenoma can be determined with greater accuracy. Immunohistochemical staining is an important parameter for the differential diagnosis of adenoma and adenocarcinoma of the adrenal cortex. Compared with the pediatric population (<20 years old), there were different pathologic criteria for adrenal cortical neoplasms in the adult population. For example, adult ACAs were lighter, generally weighing 50 g or less. Although the incidence of non-functioning adenomas tended to increase, no significant difference was observed, likely due to the small sample size; thus, our ability to draw definitive conclusions was limited.
Disclosure
The authors have no conflicts of interest to disclose.
References
Kanagarajah P, Ayyathurai R, Manoharan M, Narayanan G, Kava BR. Current concepts in the management of adrenal incidentalomas. Urol Ann. 2012;4(3):137–144. | ||
Glover AR, Ip JC, Zhao JT, Soon PS, Robinson BG, Sidhu SB. Current management options for recurrent adrenocortical carcinoma. Onco Targets Ther. 2013;6:635–643. | ||
Young WF Jr. Management approaches to adrenal incidentalomas. A view from Rochester, Minnesota. Endocrinol Metab Clin North Am. 2000;29(1):159–185. | ||
Chen G, Yao J, Mou L, et al. Clinical Analysis of 249 Cases of Adrenal Tumors in a Chinese Hospital. Urol Int. 2010;85(3):270–275. | ||
Aron D, Terzolo M, Cawood TJ. Adrenal incidentalomas. Best Pract Res Clin Endocrinol Metab. 2012;26(1):69–82. | ||
Boland GW, Dwamena BA, Jagtiani Sangwaiya M, et al. Characterization of adrenal masses by using FDG PET: a systematic review and meta-analysis of diagnostic test performance. Radiology. 2011;259(1): 117–126. | ||
Liu C, Zhao Z, Zhang Q, Wu Y, Jin F. Primary malignant fibrous histiocytoma of the breast: report of one case. Onco Targets Ther. 2013;6: 315–319. | ||
Rao SK, Caride VJ, Ponn R, Giakovis E, Lee SH. F-18 fluorodeoxyglucose positron emission tomography-positive benign adrenal cortical adenoma: imaging features and pathologic correlation. Clin Nucl Med. 2004;29(5):300–302. | ||
Treglia G, Oragano L, Fadda G, De Gaetano AM, Raffaelli M, Rufini V. Markedly Increased 18F-FDG Uptake in a Nonfunctioning Adrenal Adenoma Mimicking Malignancy. Clin Nucl Med. 2013;38(8):e333–e335. | ||
Morelli V, Palmieri S, Salcuni AS, et al. Bilateral and unilateral adrenal incidentalomas: biochemical and clinical characteristics. Eur J Endocrinol. 2013;168(2):235–241. | ||
Ng VW, Ma RC, So WY, et al. Evaluation of functional and malignant adrenal incidentalomas. Arch Intern Med. 2010;170(22):2017–2020. | ||
Wieneke JA, Thompson LD, Heffess CS. Adrenal cortical neoplasm in the pediatric population: a clinicopathologic and immunophenotypic analysis of 83 patients. Am J Surg Pathol. 2003;27(7):867–881. | ||
Peppelman M, Timmers HJ, Lenders JW. CD56 immunohistochemistry does not discriminate between cortisol-producing and aldosterone-producing adrenal cortical adenomas. Histopathology. 2011;58(9):994–996. | ||
Tanase C, Albulescu R, Codrici E, et al. Decreased expression of APAF-1 and increased expression of cathepsin B in invasive pituitary adenoma. Onco Targets Ther. 2014;8:81–90. | ||
Mete O, Asa SL. Aldosterone-producing adrenal cortical adenoma with oncocytic change and cytoplasmic eosinophilic globular inclusions. Endocr Pathol. 2009;20(3):182–185. | ||
Liu Z, Ren X, Yang Z, et al. Effects and mechanisms of indol-2,3-dione on atherosclerosis. Int J Clin Exp Med. 2014;7(8):2087–2091. | ||
Mete O, Asa SL. Morphological distinction of cortisol-producing and aldosterone-producing adrenal cortical adenomas: not only possible but a critical clinical responsibility. Histopathology. 2012;60(6): 1015–1016. | ||
Lloyd RV. Adrenal cortical tumors, pheochromocytomas and paragangliomas. Mod Pathol. 2011;24 Suppl 2:S58–S65. | ||
Terzolo M, Bovio S, Pia A, Reimondo G, Angeli A. Management of adrenal incidentaloma. Best Pract Res Clin Endocrinol Metab. 2009;23(2): 233–243. | ||
Young WF. Clinical practice. The incidentally discovered adrenal mass. N Engl J Med. 2007;356(6):601–610. | ||
Mansmann G, Lau J, Balk E, Rothberg M, Miyachi Y, Bornstein SR. The clinically inapparent adrenal mass: Update in diagnosis and management. Endocr Rev. 2004;25(2):309–340. | ||
Busam KJ, Iversen K, Coplan KA, et al. Immunoreactivity for A103, an antibody to Melan-A ( Mart-1), in adrenocortical and other steroid tumours. Am J Surg Pathol. 1998;22(1):57–63. | ||
Tartour E, Caillou B, Tenenbaum F. Immunohistochemical study of adrenocortical carcinoma. Predictive value of the D11 monoclonal antibody. Cancer. 1993;72(11):3296–3303. | ||
Wachenfeld C, Beuschlein F, Zwermann O, et al. Discerning malignancy in adrenocortical tumors: are molecular markers useful? Eur J Endocrinol. 2001;145(3):335–341. | ||
Szajerka A, Dziegiel P, Szajerka T, Zabel M, Winowski J, Grzebieniak Z. Immunohistochemical evaluation of metallothionein, Mcm-2 and Ki-67 antigen expression in tumors of the adrenal cortex. Anticancer Res. 2008;28(5B):2959–2965. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.