")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
Clinical and neuropsychological outcomes for children with phenylketonuria in Upper Egypt; a single-center study over 5 years
Authors Sadek AA , Hassan MH , Mohammed NA
Received 3 June 2018
Accepted for publication 18 July 2018
Published 5 October 2018 Volume 2018:14 Pages 2551—2561
DOI https://doi.org/10.2147/NDT.S176198
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Video abstract presented by Abdelrahim Sadek.
Views: 393
Abdelrahim A Sadek,1 Mohammed H Hassan,2 Nesreen A Mohammed3
1Department of Pediatrics, Pediatric Neurology and Psychiatry Unit, Faculty of Medicine, Sohag University, Sohag, Egypt; 2Medical Biochemistry Department, Faculty of Medicine, South Valley University, Qena, Egypt; 3Public Health and Community Medicine Department, Faculty of Medicine, Sohag University, Sohag, Egypt
Background: Phenylketonuria (PKU) is considered to be a rare inborn error of metabolism but one of the commonest causes of mental retardation if untreated.
Objectives: The present study was done to characterize the clinical patterns of PKU and analyze various neuropsychiatric outcomes in PKU children in Sohag Province, Egypt.
Patients and methods: A prospective cohort study was conducted on 113 PKU patients, diagnosed during the period from 2012 to 2017, at the Pediatric Neurology Clinic of Sohag University Hospital, Upper Egypt. One hundred and ten cases were diagnosed based on clinical suspicion combined with laboratory confirmation by measuring their plasma phenylalanine levels using amino acid analyzer, while 3 cases were detected during neonatal screening. With the exception of the 3 cases detected during neonatal screening, all patients were clinically diagnosed and treated late. Psychometric evaluations of PKU patients were done using intelligence quotient (IQ, Stanford–Binet V), Childhood Autism Rating Scale, and Children’s Attention and Adjustment Survey. Dietetic management was applied. The results of neuroimaging (computed tomography or magnetic resonance imaging of the brain) and electroencephalography were included when available.
Results: The overall results showed that 15.9% had hyperphenylalaninemia, 35.4% had mild to moderate PKU, and classic PKU was diagnosed in 48.7%. Global developmental delay (54.9%) and delayed language (29.2%) were the most frequent presentations. Moderately impaired or delayed overall IQ was present in 77%. While, 83.2% had moderately impaired or delayed verbal IQ, autism was diagnosed in 50.4%. Super average and average probability of attention-deficit hyperactivity disorder was diagnosed in 88.5%. Abnormal neuroimaging (white matter abnormalities and brain atrophy) was the most important significant predictor for poor language and motor developmental outcome (P<0.05).
Conclusion: PKU children had variable neuropsychological outcomes, mainly attention-deficit hyperactivity disorder and impaired verbal IQ, both of which were not related to the initial phenylalanine levels or to duration of dietary therapy, but were significantly related to early dietary intervention.
Keywords: phenylketonuria, IQ, autism, CARS, ADHD, Upper Egypt
Introduction
Phenylketonuria (PKU; OMIM212600) is considered to be a rare inborn error of metabolism, with an estimated prevalence of 1: 10,000. PKU is attributed to phenylalanine hydroxylase deficiency,1,2 with reduction of brain tyrosine and tryptophan levels, leading to deficiency of the neurotransmitters dopamine and serotonin.3,4
Untreated PKU patients suffer from irreversible intellectual disability. In addition, neurological affection in the form of microcephaly, motor deficits, autism, developmental affection, aggressive behavior, and hyperactivity were also reported.5,6 Seizures are commonly found to occur, affecting severely retarded PKU children.7 Hypsarrhythmic electroencephalographic (EEG) patterns, were recorded in PKU children, even in the absence of seizures.8 White matter abnormalities (WMA) could be detected in both early- and late-treated PKU patients, using magnetic resonance imaging (MRI).9–11 The extent and severity of WMA could be affected by patient age and dietary adherence.12–18
Autism spectrum disorder could be the only presentation of untreated PKU, and may be misdiagnosed among patients who have no other features suggesting PKU.19,20 The introduction of treatment leads to difficulties in determining the frequency of autistic symptoms in PKU.21–24
The early diagnosis and therapy immediately after birth in PKU patients could help produce a better outcome regarding developmental performance and educational achievement. However, neuropsychological deficits and social issues can occur in some patients, with their neurocognitive levels always appearing to be lower than that of their siblings or peers.1,25,26 Executive function defects (higher-order cognitive abilities controlling and coordinating behavior, and representing the driving force of goal-directed behavior) are the most frequently observed cognitive deficits among treated PKU patients.27 Also, high incidence of attention-deficit hyperactivity disorder (ADHD) in PKU patients has been reported.28,29 The symptoms of ADHD could be attributed to the deficit in executive function and assumption of similarities in brain function between ADHD and PKU, which has been frequently implicated to the increased phenylalanine (Phe) toxic metabolite (phenylethylamine) concentration.30
The psychiatric symptoms and developmental delay being reported in PKU patients could be explained by many mechanisms, such as, myelin abnormalities, defect in the amino acids transport through blood–brain barrier, and reduction of some neurotransmitters levels (dopamine, norepinephrine, and serotonin). These neurotransmitters are involved in mood, emotion, and cognition regulation, causing disturbances in autistic patients.5,31,32
As the national neonatal screening for PKU was started only in 2015, the burden of this disease is still high, especially in Upper Egypt. The main objectives of the current study were to evaluate the pattern of PKU in children, whether early- or late-treated, and to analyze the neuropsychiatric outcomes of PKU in Sohag Province, Upper Egypt.
Patients and methods
Participants and study design
A prospective cohort, hospital-based study was conducted on 113 PKU patients diagnosed during the study period from January 2012 to December 2017, at the Pediatric Neurology Clinic of Sohag University Hospital, Upper Egypt, in collaboration with, Medical Biochemistry Department, Faculty of Medicine, South Valley University, Qena, Egypt. One hundred and ten cases were diagnosed based on clinical suspicion combined with laboratory assay results of their blood phenylalanine levels, and only 3 of them were discovered by neonatal screening. Written informed consent was taken from the caregivers to conduct this research. The study has been approved by the Faculty of Medicine, Sohag University Ethics Committee, and was carried out in accordance with The Code of Ethics of The World Medical Association (Declaration of Helsinki) for experiments in humans.
Biochemical analyses
Phenylalanine blood levels were measured using the Bio-Rad Laboratory (Hercules, CA, USA) products, Microplate Neonatal PHE/GAL (galactose) Assay-(Test Kit 532-6053); for quantitative microplate assay of Phe and galactose in the newborn blood to screen for phenylketonuria, galactosemia on Whatman 903 paper. For older infants and children who presented with clinical suspicion, a 3 cm3 venous blood sample, from each patients, was withdrawn in a heparinized tube and centrifuged at 3,000 rpm for 15 minutes. The separated plasma from each tube was separated immediately, using 1 mL cryotubes for measurement of plasma Phe level using automatic amino acids analyzer (SYKAMS433, Sykam GmbH, Eresing, Germany, Catalog No: 1120001). This analyzer combines the classical method of ion exchange separation with derivatization using ninhydin, with the modern technique of high performance liquid chromatography. Plasma Phe level >3 mg/dL (>180 μmol/L) was considered elevated. Patients were classified as having classic PKU if their Phe levels were >1,200 μmol/L, mild to moderate PKU if Phe levels fall within 600–1,200 μmol/L, while those with Phe level <600 μmol/L were categorized as having mild hyperphenylalaninemia.33,34
Dietetic management
The diagnosed cases were referred to various metabolic centers, located in distant regions, for obtaining low phenylalanine formulas and dietary evaluation by nutritionists. Their dietary regimens were adjusted on daily protein supplementation of 2.5–3.5 g/kg/d in the first year, ≥30 to ≥40 g/d in children (1 to <11 years) and ≥50 to ≥70 g/d in adolescents (11 to ≥19 years). The Phe supplementation was adjusted to 10–70 mg/kg/d in the first year, 200–500 mg/d in children (1 to <11 years) and 220–1,200 mg/d in adolescents (11 to ≥19 years). This was supplied by using the special formulas, in addition to Phe containing foods. Also Tyr supplementation was modified to 250–350 mg/kg/d in the first year, 1.72–4 g/d in children (1 to <11 years) and 3.38–6.5 g/d in adolescents (11 to ≥19 years).35,36 This was interpreted as units and included the use of commonly available foods so as to be easily understood by the parents.
Data collection
All patients were subjected to thorough clinical history, including developmental history and detailed history of the presenting symptoms. Family history of a similar condition, the presence of epilepsy, mental retardation, or global developmental delay was also clarified.
Full clinical examination (general, systematic, and detailed neurological examinations), including developmental assessment was done. Psychometric evaluation, where a battery of psychometric tests were applied on all included children after the age of 2 years, with at least 2 years follow-up period, was done in properly prepared rooms by 2 experts in psychometry who spent a reasonable and standard time for carrying out these tests while the children were in a relaxed and cooperative manner. Language evaluation involving assessment of passive and active vocabularies was done. Evaluation of autistic features and hyperactivity symptoms was done according to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition.37
Intelligent Quotient (IQ) assessment was done using Stanford–Binet Intelligence Scales V as it can be used in children as young as 2 years of age and can be used for assessing both verbal and nonverbal domains. The test publisher classification was set as follows: 145–160 (very gifted or highly advanced), 130–144 (grifted or very advanced), 120–129 (superior), 110–119 (high average), 90–109 (average), 80–89 (low average), 70–79 (borderline impaired or delayed), 55–69 (mildly impaired or delayed), and 40–54 (moderately impaired or delayed).38,39
Childhood Autism Rating Scale (CARS) was used for assessment of the severity of autistic features. The degree of autistic severity was set as no or minimal symptoms if the score range was within 15–29.5 mild to moderate autism if the score was 30–36.5, and severe autism if the score was ≥37.40
Assessment of ADHD was conducted using Children’s Attention and Adjustment Survey (CAAS), subscales: Attention-Deficit Disorder, Attention-Deficit Hyperactivity Disorder, DSM-III-R Attention-Deficit Hyperactivity Disorder. The version used was the child’s parent form. A range of values for the subtest standard scores (hyperactivity, impulsivity, inattention) and the ADHD Quotient is provided for estimating the probability of ADHD. Scores were set as <40 (low probability), 40–49 (below average), 50–59 (average), 60–69 (super average), and ≥70 (severe).41
Neuroimaging (MRI of the brain and/or computed tomography [CT] of the brain) and EEG were done for some patients included in the study.
Statistical analysis
Data were analyzed using IBM SPSS Statistics for Windows version 22 (IBM Corporation, Armonk, NY, USA). Quantitative data were expressed as means ± SD. Qualitative data were expressed as number and percentage. The data were tested for normality using Kolmogorov–Smirnov and Shapiro–Wilk tests. Independent Samples t-test and one-way analysis of variance were used for the normally distributed data. Chi-square (χ2) and Fisher’s exact tests were used for qualitative variables as appropriate. Stepwise binary logistic regression was used to determine factors associated with poor outcome. P-value <0.05 was considered significant.
Results
Sociodemographic and disease-related criteria
The current study was carried out on 113 patients. Their mean age at presentation was 43.91±39.19 months, with a range from 1 to 204 months, and they commonly presented below the age of 2 years (48/42.5%), with male predominance (69/61.1%). A large proportion of cases (86/77%) was from the rural districts. The majority (110 cases/97.3%) were diagnosed after clinical presentation, and only 3 (2.7%) cases were discovered by neonatal screening. Positive consanguinity was reported in 96 patients (85%), while family history of PKU or neurological problems was found in 47 patients (41.6%). Motor developmental delay was encountered in 62 cases (54.9%), delayed speech was found in 111 patients (98.2%), and social development was impaired in 109 patients (96.5%). The mean duration of nutritional therapy was 35.01±27.5 months (Table 1).
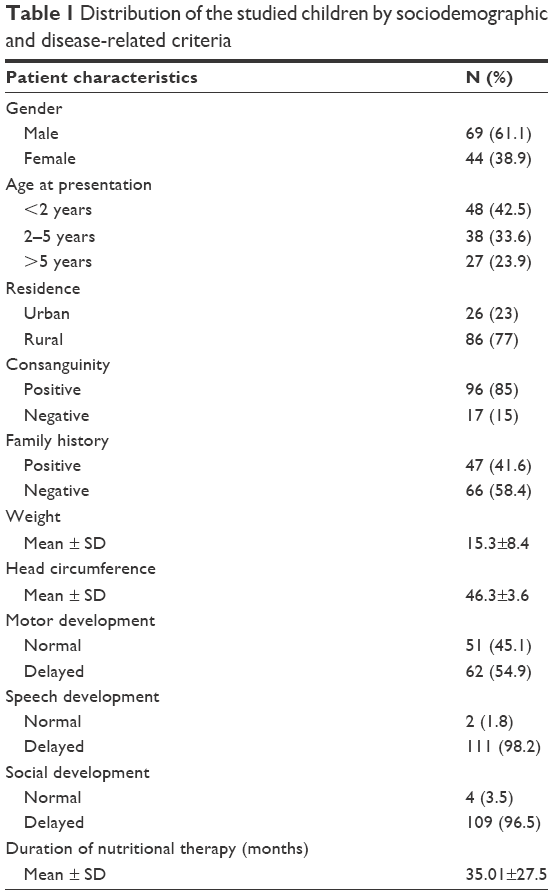 | Table 1 Distribution of the studied children by sociodemographic and disease-related criteria |
Clinical presentations
The most frequent presentations were global developmental delay (affection of 2 or more developmental domains) (62 cases, 54.9%), followed by isolated language delay (33, 29.2%), autistic features (27 cases, 23.9%), hyperactivity and conduct (21 cases, 18.6%), and seizures (11 cases, 9.7%). Three cases (2.7%) had poor scholastic achievement, 3 cases (2.7%) were diagnosed by neonatal screening, and 1 case (0.9%) presented with head nodding (Figures 1 and 2).
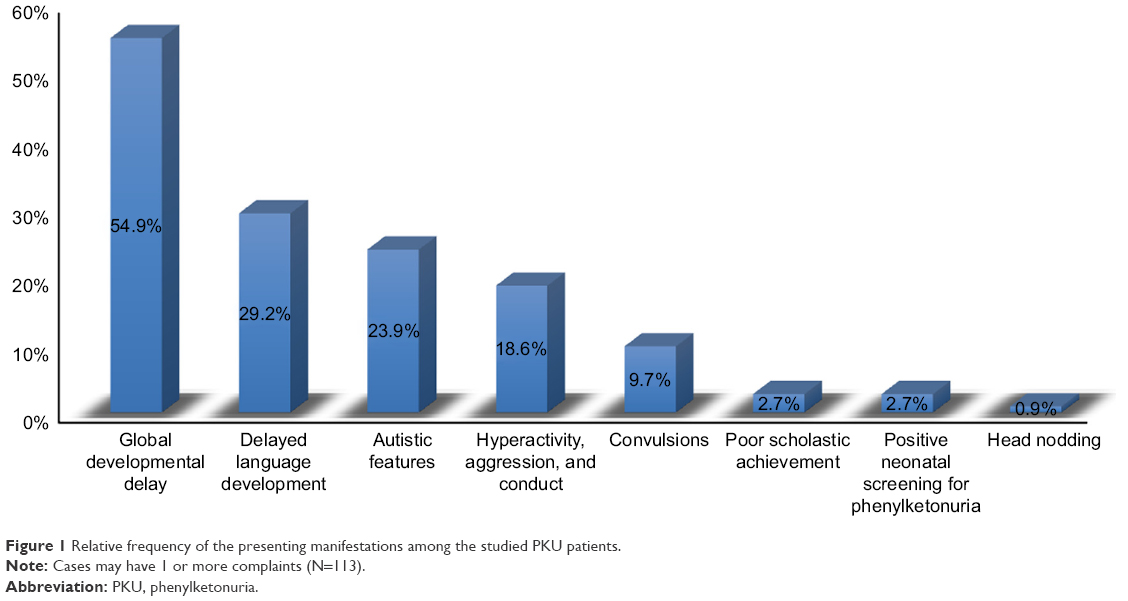 | Figure 1 Relative frequency of the presenting manifestations among the studied PKU patients. |
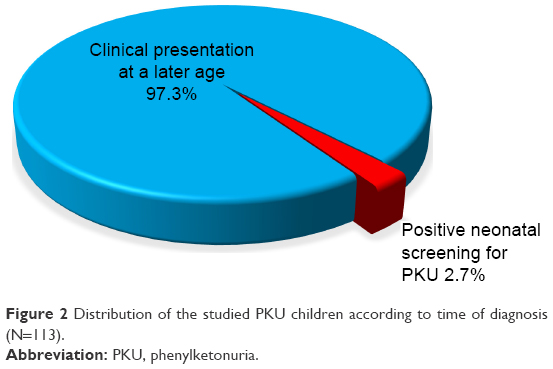 | Figure 2 Distribution of the studied PKU children according to time of diagnosis (N=113). |
Neurological examination, investigations, and developmental outcome
Sixty-one patients showed a normal neurological examination (54%), while, hypotonia and exaggerated stretch reflexes were reported in 50 cases (44.2%). According to the plasma Phe level, patients were classified into hyperphenylalaninemia, which was presented in 18 cases (15.9%), mild to moderate PKU (40 cases, 35.4%), and classic PKU (55 cases, 48.7%). The mean plasma Phe level was 1,212±546 μmol/L, with a range from 246 to 2,940 μmol/L. (Table 2). After follow-up, with at least 2 years of using special formula and dietary regimens, the developmental assessment showed that 79.6% of patients showed an improvement in their motor function, 15% had improved language skills, while 17 cases (15%) did not fit the diagnosis of global developmental delay or intellectual disability any longer.
 | Table 2 Neurological examination and investigations done for the studied children |
The majority of the patients had poor behavioral outcome with persistence of hyperactivity and aggressive behavior and conduct (108/95.6%) (Table 3). Out of 64 cases who were subjected to brain CT, 39 cases (61%) showed brain atrophic changes. On the other hand, 13 cases (65%) out of 20 patients who underwent MRI brain showed white matter disease. Seizures were reported in 24 cases (21.2%), controlled in 16 cases (66.7%), and refractory in 8 cases (33.3%). EEG records revealed that generalized epileptic discharges were frequent in 24 (21.2%) cases, focal epileptic discharges in 10 (8.8%) cases, and hypsarrhythmia pattern in 9 (8%) cases, while 14 (12.4%) cases had normal EEG (Table 2).
 | Table 3 Distribution of the studied children according to the developmental outcome |
Psychometric evaluations (IQ, CARS, and CAAS)
The overall IQ assessment showed that the majority of cases (87/77%) were moderately impaired or delayed, while 18 (18.9%) cases were mildly impaired or delayed, with the mean overall IQ of 49.08±10.04 and range between 39 and 97. Verbal IQ evaluation revealed that 94 (83.2%) cases were moderately impaired or delayed, while 12 (10.6%) cases were mildly impaired or delayed. The mean verbal IQ was 47.88±9.72 with a range between 38 and 93. Regarding CARS test, more than half of the cases had autism (57 cases, 50.4%), with a mean CARS of 30.74±7.002.
Furthermore, CAAS showed that near half of the cases (55 cases [48.7%]) had a superaverage probability of ADHD, while 45 cases (39.8%) had average probability. The mean CAAS was 59.42±5.61 (Table 4).
 | Table 4 Psychometric evaluation of the studied patients |
Predictors of developmental outcomes, IQ, CARS, and CAAS
Those presenting before 2 years of age who were diagnosed early and received dietetic management early (P<0.001) and those with a negative family history of PKU or other neurological disorders (P<0.045) had a good motor outcome. Males tend to have a better motor outcome than females (P=0.052). Cases with abnormal neuroimaging (CT or MRI brain) had a poorer motor outcome than those with normal findings (P<0.001), while epileptic EEG findings, initial Phe level, and duration of nutritional therapy had no significant effect on the outcome (Table 5). Univariate binary logistic regression analysis of predictor variables of poor motor outcome showed that female gender (odds ratio [OR]: 3.1, P=0.019) and abnormal neuroimaging (WMA and brain atrophy) (OR: 6.1, P=0.023) were factors, while, multiple variable logistic regression showed that abnormal neuroimaging (adjusted OR: 6.6, P=0.027) and positive family history (adjusted OR: 3.1, P=0.045) were predictors of poor motor outcome (Table 6), (Figures 3 and 4).
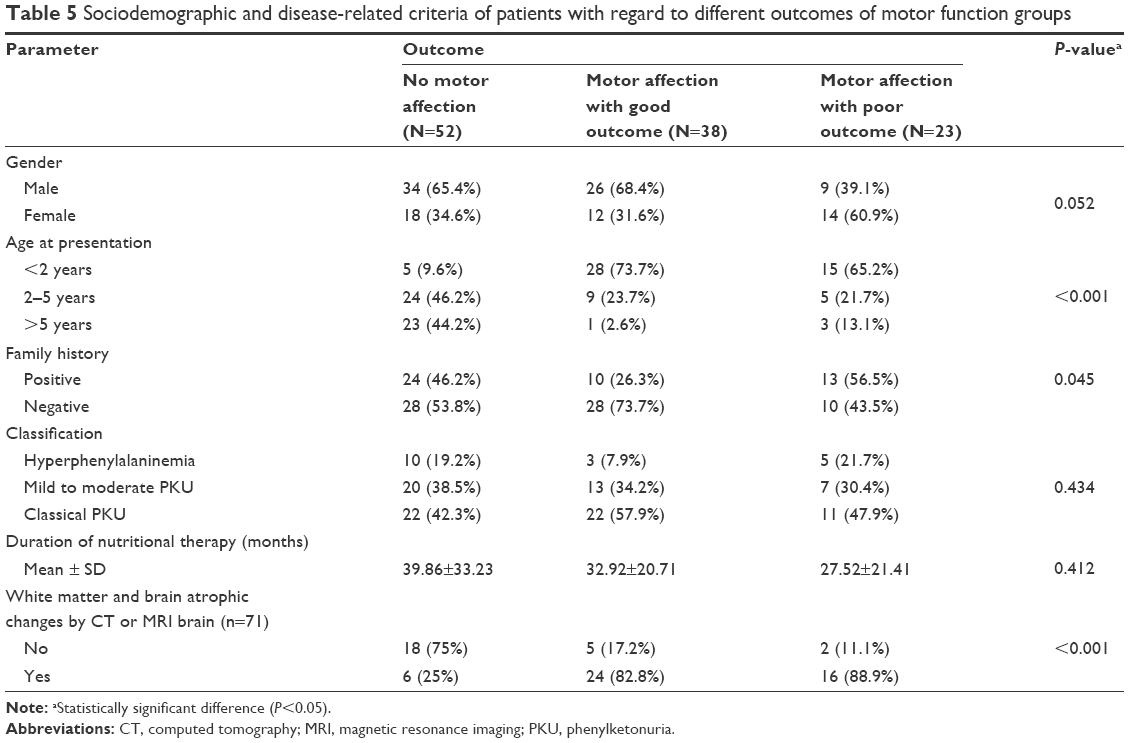 | Table 5 Sociodemographic and disease-related criteria of patients with regard to different outcomes of motor function groups |
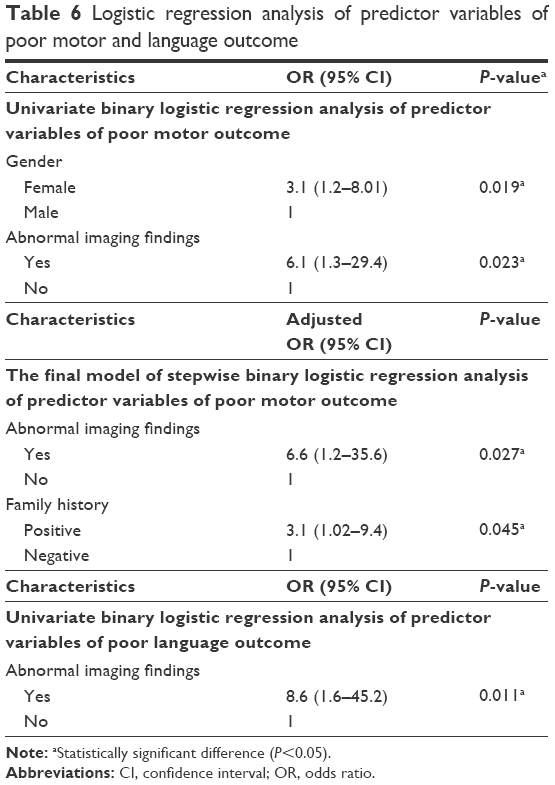 | Table 6 Logistic regression analysis of predictor variables of poor motor and language outcome |
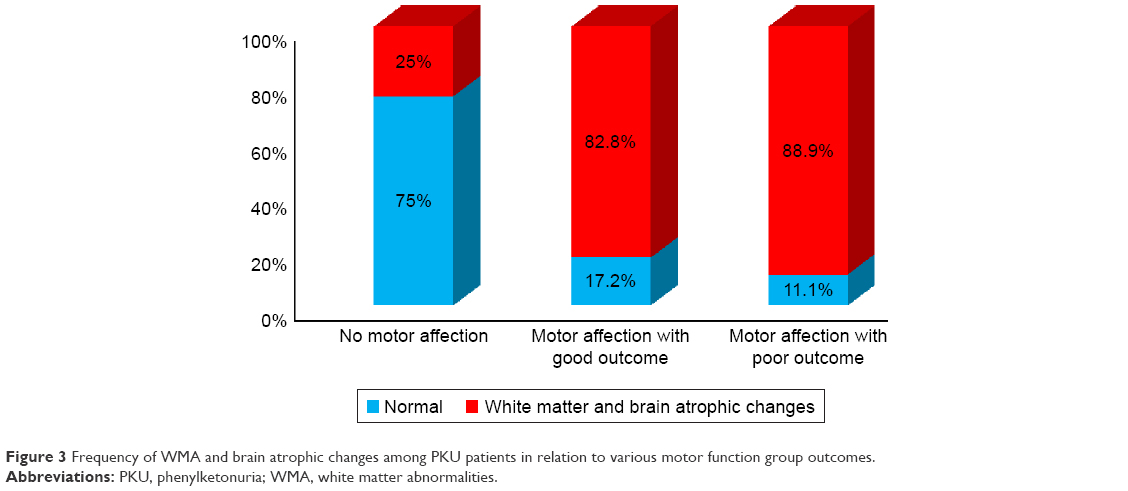 | Figure 3 Frequency of WMA and brain atrophic changes among PKU patients in relation to various motor function group outcomes. |
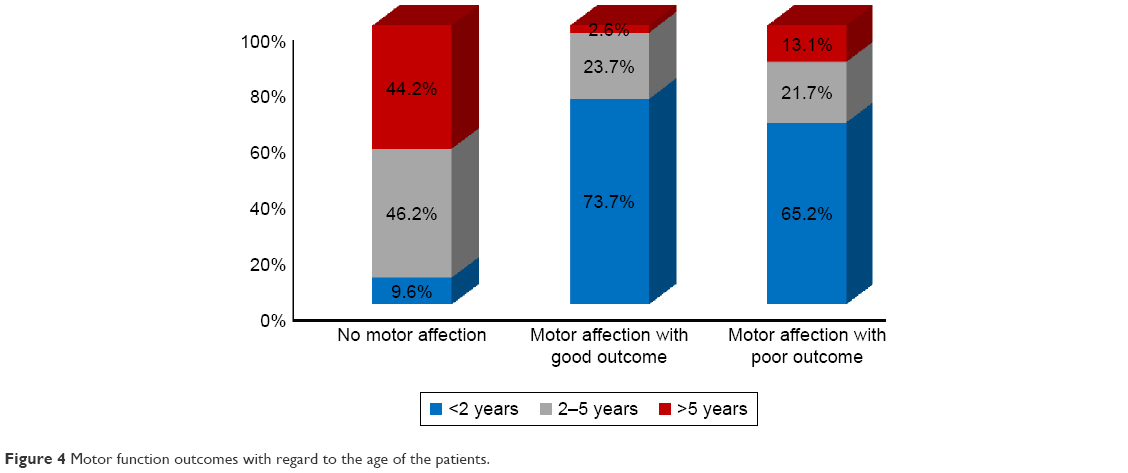 | Figure 4 Motor function outcomes with regard to the age of the patients. |
The predictors of language development showed that mild to moderate PKU (P=0.022) was associated with good language outcome, while abnormal neuroimaging was associated with poor language outcome (P=0.007), and epileptic EEG had no significant effect. Abnormal neuroimaging was a predictor for poor language outcome when univariate binary logistic regression analysis was used (OR: 8.6, P=0.011; Table 6). The overall IQ was higher in males than females (P=0.036), while initial Phe level (P=0.528) and duration of nutritional therapy (P=0.961) had no statistically significant effect. CARS score was slightly higher in those presenting after the age of 5 years (P=0.07) and those with positive consanguinity (P=0.064), while initial Phe level and duration of nutritional therapy had no significant effect. CAAS score tended to be higher in those with mild to moderate PKU (P=0.08), while abnormal neuroimaging, epileptic EEG changes, and duration of nutritional therapy had no statistically significant effect.
Discussion
The neuropsychiatric consequences, behavioral problems and social issues of PKU, even in those who are treated early, are well known. Also, the late initiation (in 2015) of national neonatal screening for PKU in Egypt was an additional factor aggravating the actual burden of the disease on such children. This provides a larger number of cases who were referred for evaluation of various neuropsychiatric and developmental problems, such as those previously described in our previously published pilot study.42 In the current study, we had to clarify the current status of the disease burden and the neuropsychiatric outcome of PKU in our locality. The vast majority of our cases (97.3%) presented beyond the neonatal period and only 2.7% of the cases were discovered during neonatal screening. The mean age of presentation was 43.91±39.19 months, and this was comparable to our previous study (40.44±36 months),42 but higher than that observed by Khemir et al24 (the mean age at diagnosis of PKU was 36 months). Males were predominantly more in the current study (61.1%), and this was in line with other studies.23
In the present study, seizures were reported in 21.2% of the cases, and 33.3% of them were refractory. EEG showed that epileptic discharges, including hypsarrhythmia pattern, were found in 50.7% of the cases in whom EEG was conducted. Our findings were in agreement with another study43 which reported abnormal EEG findings in 66.6% of their cases with higher frequency of seizures (52.3%). Abnormal EEG was reported in 45%, and 50%, respectively, by other investigators (Gross et al,8 Abdel-Salam et al44). There is evidence that psychocognitive impairment and behavioral disturbance can be caused by subclinical discharges.
In our study, the overall IQ and verbal IQ scores showed that the majority of cases (77% and 83.2% respectively) were moderately impaired or delayed, and there was a lag between the mean age of the patients and the mean duration of nutritional therapy (43.91±39.19 vs 35.01±27.5 months). These findings were consistent with a previous meta-analysis, which reported a proportional correlation between blood Phe, during ages 0–12 and 0–18 years and IQ. In early-treated PKU patients, every 100 µM increase in the blood Phe was predictive of an average 2.6 point decrease in IQ.45 Another study found that in children <10 years of age, a 7-point decrease in their IQ for each 300 µM increase in Phe was seen.46 This finding was also explained by another study, which stated that IQ score decreased by nearly 4 points for every 4 weeks delay in treatment initiation.47 Adult studies confirmed the long-term effect of elevated Phe level in childhood on cognitive and mental health outcome, even in those who had undergone early treatment and had a social and socioeconomic environment conducive to good cognitive and psychological development.4 The majority of our cases were diagnosed and treated late. Another longitudinal study including adolescents showed that 2 brain function measurements, one for complex neurocognitive skills and the other for specific information network processing, were more impaired when there is rapid rise of Phe level, and this is also associated with younger age.48 Furthermore, another study reported that IQ remains stable over the second decade of life; however, other patients showed an increase or decrease in their IQ scores that was not explained by the quality of metabolic control and could be related to hidden subjective factors.49
In the present study, more than half of the cases had autism (50.4%), while the mean CARS was 30.74±7.002. Our findings were in accordance with the study conducted by Khemir et al;24 they found that 15 children (79%) in their study had autism. Also, 5.7% of late-diagnosed PKU children had autism in an Italian study.50 Fombonne and du Mazaubrun,51 found a significantly higher frequency of untreated PKU patients in the autistic group. Other studies also showed results that were consistent with our findings.23,52–55 The variation in the percentages between studies could be attributed to the difference in PKU populations, whether early-treated, late-treated or mixed.
Furthermore, CAAS showed that nearly half of the cases (48.7%) had a superaverage probability of ADHD, while 45 cases (39.8%) had average probability (88.5% of cases had ADHD symptoms) with mean CAAS of 59.42±5.61. Our findings were comparable to many studies; Karimzadeh et al43 showed that ADHD symptoms were found in 85.7% of their cases in comparison with 40% in the controls. Also, another study reported significant attention symptoms in 50% of their children with PKU, and 26% of them (n=38), received a stimulant medication for attention difficulties.28 Lower percentages were obtained by other studies; 1 study found ADHD symptoms in 13% of the 46 children with PKU in their study.56 Furthermore, the study done by González et al57 found that 22.8% of their cases had ADHD. This could be attributed to the PKU patient characteristics, whether early-treated, late-treated, or mixed, as in our study. The evaluation of ADHD symptoms in PKU children was supported by Wyrwich et al,58 who found the appropriateness of using ADHD rating scale IV (ADHD RS-IV) for assessment of ADHD symptoms.
Furthermore, in the current study, univariate binary logistic regression analysis of predictor variables for poor motor outcome were female gender and abnormal neuroimaging (WMA and brain atrophy), while multiple variable logistic regression showed that abnormal neuroimaging and positive family history were predictors of poor motor developmental outcome. Also, univariate binary logistic regression analysis showed that a predictor variable for poor language outcome was abnormal neuroimaging. Thus, abnormal neuroimaging, whether brain atrophic changes or white matter changes, appeared to significantly affect both motor and language outcome in our patients. Our findings were in agreement with other researchers reporting that both early- and late-treated PKU patients could have WMA.9–11,59 Many researchers concluded that that the extent and severity of WMA could be modified by patient age and/or strict dietary control (blood Phe level). Also, WMA could be increased with older age patients and/or in those with higher Phe levels.12–15 Our study did not find any significant relation between the initial Phe level and overall IQ, CARS, and CAAS scores, except mild tendency of CAAS test score to be higher in those with mild to moderate PKU.
Study limitations
The major limitation in our study is the lack of data about the mean serum Phe levels during the follow-up period. Also, not all participants underwent neuroimaging or EEG records, so their data are missing.
Conclusion
The current study proved that the delayed initiation of neonatal screening for PKU in Egypt (2015) and delayed diagnosis have resulted in a high burden of the disease. The majority of PKU children had intellectual disability (delayed or impaired IQ), with half of them suffering from autism, while 88.5% of them had ADHD symptoms. Our recommendations support the continuity of national screening program for PKU, expansion of metabolic centers, and highlighting community and physician awareness in terms of the disease consequences. Furthermore, integration of a well-trained neuropsychiatric team in the routine evaluation and medical visits of PKU patients is fundamental.
Acknowledgments
We thank the professors and staff in the Pediatric Neurology Unit and Pediatric Departments, Faculty of Medicine, Sohag University, for their kind help during this work. Also, thanks to our patients and their families for participation in the current study.
Disclosure
The authors report no conflicts of interest in this work.
References
Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet. 2010;376(9750):1417–1427. | ||
van Spronsen FJ, van Wegberg AM, Ahring K, et al. Key European guidelines for the diagnosis and management of patients with phenylketonuria. Lancet Diabetes Endocrinol. 2017;5(9):743–756. | ||
Dyer CA. Pathophysiology of phenylketonuria. Ment Retard Dev Disabil Res Rev. 1999;5(2):104–112. | ||
Jahja R, van Spronsen FJ, de Sonneville LMJ, et al. Long-Term Follow-Up of Cognition and Mental Health in Adult Phenylketonuria: A PKU-COBESO Study. Behav Genet. 2017;47(5):486–497. | ||
de Groot MJ, Hoeksma M, Blau N, Reijngoud DJ, van Spronsen FJ. Pathogenesis of cognitive dysfunction in phenylketonuria: review of hypotheses. Mol Genet Metab. 2010;99 Suppl 1:S86–S89. | ||
Jahja R, van Spronsen FJ, de Sonneville LMJ, et al. Social-cognitive functioning and social skills in patients with early treated phenylketonuria: a PKU-COBESO study. J Inherit Metab Dis. 2016;39(3):355–362. | ||
Macleod MD, Munro JF, Ledingham JG, Farquhar JW. Management of the extrapyramidal manifestations of phenylketonuria with L-dopa. Arch Dis Child. 1983;58(6):457–458. | ||
Gross PT, Berlow S, Schuett VE, Fariello RG. EEG in phenylketonuria. Attempt to establish clinical importance of EEG changes. Arch Neurol. 1981;38(2):122–126. | ||
Anderson PJ, Leuzzi V. White matter pathology in phenylketonuria. Mol Genet Metab. 2010;99 Suppl 1:S3–S9. | ||
Camp KM, Parisi MA, Acosta PB, et al. Phenylketonuria Scientific Review Conference: state of the science and future research needs. Mol Genet Metab. 2014;112(2):87–122. | ||
Peng H, Peck D, White DA, Christ SE. Tract-based evaluation of white matter damage in individuals with early-treated phenylketonuria. J Inherit Metab Dis. 2014;37(2):237–243. | ||
Antenor-Dorsey JA, Hershey T, Rutlin J, et al. White matter integrity and executive abilities in individuals with phenylketonuria. Mol Genet Metab. 2013;109(2):125–131. | ||
Cleary MA, Walter JH, Wraith JE, et al. Magnetic resonance imaging of the brain in phenylketonuria. Lancet. 1994;344(8915):87–90. | ||
Hood A, Antenor-Dorsey JA, Rutlin J, et al. Prolonged exposure to high and variable phenylalanine levels over the lifetime predicts brain white matter integrity in children with phenylketonuria. Mol Genet Metab. 2015;114(1):19–24. | ||
Leuzzi V, Tosetti M, Montanaro D, et al. The pathogenesis of the white matter abnormalities in phenylketonuria. A multimodal 3.0 tesla MRI and magnetic resonance spectroscopy (1H MRS) study. J Inherit Metab Dis. 2007;30(2):209–216. | ||
Pietz J, Kreis R, Schmidt H, et al. Phenylketonuria: findings at MR imaging and localized in vivo H-1 MR spectroscopy of the brain in patients with early treatment. Radiology. 1996;201(2):413–420. | ||
Thompson AJ, Tillotson S, Smith I, et al. Brain MRI changes in phenylketonuria. Associations with dietary status. Brain. 1993;116(Pt 4):811–821. | ||
White DA, Connor LT, Nardos B, et al. Age-related decline in the microstructural integrity of white matter in children with early- and continuously-treated PKU: a DTI study of the corpus callosum. Mol Genet Metab. 2010;99(Suppl 1):S41–S46. | ||
Ghaziuddin M, Al-Owain M. Autism spectrum disorders and inborn errors of metabolism: an update. Pediatr Neurol. 2013;49(4):232–236. | ||
Schiff M, Benoist JF, Aïssaoui S, et al. Should metabolic diseases be systematically screened in nonsyndromic autism spectrum disorders? PLoS One. 2011;6(7):e21932. | ||
Moss J, Howlin P. Autism spectrum disorders in genetic syndromes: implications for diagnosis, intervention and understanding the wider autism spectrum disorder population. J Intellect Disabil Res. 2009;53(10):852–873. | ||
Caglayan AO. Genetic causes of syndromic and non-syndromic autism. Dev Med Child Neurol. 2010;52(2):130–138. | ||
Saad K, Elserogy Y, Abdel Rahman AA, et al. ADHD, autism and neuroradiological complications among phenylketonuric children in Upper Egypt. Acta Neurol Belg. 2015;115(4):657–663. | ||
Khemir S, Halayem S, Azzouz H, et al. Autism in Phenylketonuria Patients: From Clinical Presentation to Molecular Defects. J Child Neurol. 2016;31(7):843–849. | ||
Enns GM, Koch R, Brumm V, et al. Suboptimal outcomes in patients with PKU treated early with diet alone: revisiting the evidence. Mol Genet Metab. 2010;101(2–3):99–109. | ||
Jahja R, Huijbregts SC, de Sonneville LM, van der Meere JJ, van Spronsen FJ. Neurocognitive evidence for revision of treatment targets and guidelines for phenylketonuria. J Pediatr. 2014;164(4):e2–899. | ||
Christ SE, Huijbregts SC, de Sonneville LM, White DA. Executive function in early-treated phenylketonuria: profile and underlying mechanisms. Mol Genet Metab. 2010;99(Suppl 1):S22–S32. | ||
Arnold GL, Vladutiu CJ, Orlowski CC, Blakely EM, Deluca J. Prevalence of stimulant use for attentional dysfunction in children with phenylketonuria. J Inherit Metab Dis. 2004;27(2):137–143. | ||
Burton B, Grant M, Feigenbaum A, et al. A randomized, placebo-controlled, double-blind study of sapropterin to treat ADHD symptoms and executive function impairment in children and adults with sapropterin-responsive phenylketonuria. Mol Genet Metab. 2015;114(3):415–424. | ||
Feillet F, Macdonald A, Hartung Perron D, Burton B, Hartung D. Outcomes beyond phenylalanine: an international perspective. Mol Genet Metab. 2010;99 Suppl 1:S79–S85. | ||
Surtees R, Blau N. The neurochemistry of phenylketonuria. Eur J Pediatr. 2000;159 Suppl 2:S109–S113. | ||
Stahl SM. Essential Psychopharmacology: Neuroscientific Basis and Practical Applications. 2nd ed. New York, NY: Cambridge University Press; 2000. | ||
Guldberg P, Rey F, Zschocke J, et al. A European multicenter study of phenylalanine hydroxylase deficiency: classification of 105 mutations and a general system for genotype-based prediction of metabolic phenotype. Am J Hum Genet. 1998;63(1):71–79. | ||
Guldberg P, Güttler F. Mutations in the phenylalanine hydroxylase gene: methods for their characterization. Acta Paediatr. 1994;83(S407):27–33. | ||
Vockley J, Andersson HC, Antshel KM, et al. Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet Med. 2014;16(2):188–200. | ||
Singh RH, Rohr F, Frazier D, et al. Recommendations for the nutrition management of phenylalanine hydroxylase deficiency. Genet Med. 2014;16(2):121–131. | ||
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Washington, DC: American Psychiatric Association; 2013. | ||
Roid GH. Stanford-Binet Intelligence Scales. 5th ed. Rolling Meadows, IL: Riverside Publishing; 2003. | ||
Bain SK, Allin JD. Book Review: Stanford-Binet Intelligence Scales, Fifth Edition. J Psychoeduc Assess. 2005;23(1):87–95. | ||
Echo Armman Childhood Autism Rating Scale (CARS). Autism World Retrieved. 2007;3:28. | ||
Lambert N, Hartsough C, Sandoval J. Manual for the Children’s Attention and Adjustment Survey. Circle Pines, MN: American Guidance Service; 1990. | ||
Sadek AA, Emam AM, Alhaggagy MY. Clinicolaboratory profile of phenylketonuria (PKU) in Sohag University Hospital-Upper Egypt. Egyptian J Med Human Genet. 2013;14(3):293–298. | ||
Karimzadeh P, Alaee MR, Zarafshan H. The Association between EEG Abnormality and Behavioral Disorder: Developmental Delay in Phenylketonuria. ISRN Pediatr. 2012;2012:976206. | ||
Abdel-Salam GMH, Abdel-Kader AA, Inical LE. Electroencephalographic (EEG), neuroradiological and molecular (PKU) patients. Egypt J Neurol Psychiatry Neurosurg. 2005;42(2):390–406. | ||
Waisbren SE, Noel K, Fahrbach K, et al. Phenylalanine blood levels and clinical outcomes in phenylketonuria: a systematic literature review and meta-analysis. Mol Genet Metab. 2007;92(1–2):63–70. | ||
Burgard P. Development of intelligence in early treated phenylketonuria. Eur J Pediatr. 2000;159(Suppl 2):S74–S79. | ||
Smith I, Beasley MG, Ades AE. Intelligence and quality of dietary treatment in phenylketonuria. Arch Dis Child. 1990;65(5):472–478. | ||
Leuzzi V, Mannarelli D, Manti F, et al. Age-related psychophysiological vulnerability to phenylalanine in phenylketonuria. Front Pediatr. 2014;2(2):57. | ||
Manti F, Nardecchia F, Paci S, et al. Predictability and inconsistencies in the cognitive outcome of early treated PKU patients. J Inherit Metab Dis. 2017;40(6):793–799. | ||
Baieli S, Pavone L, Meli C, Fiumara A, Coleman M. Autism and phenylketonuria. J Autism Dev Disord. 2003;33(2):201–204. | ||
Fombonne E, du Mazaubrun C. Prevalence of infantile autism in four French regions. Soc Psychiatry Psychiatr Epidemiol. 1992;27(4):203–210. | ||
Chen CH, Hsiao KJ. A Chinese classic phenylketonuria manifested as autism. Br J Psychiatry. 1989;155:251–253. | ||
Steiner CE, Acosta AX, Guerreiro MM, Marques-de-Faria AP. Genotype and natural history in unrelated individuals with phenylketonuria and autistic behavior. Arq Neuropsiquiatr. 2007;65(2A):202–205. | ||
Zafeiriou DI, Ververi A, Vargiami E. Childhood autism and associated comorbidities. Brain Dev. 2007;29(5):257–272. | ||
Okado N. Mechanisms for formation and maintenance of synapses mediated by biogenic amines: pathogenesis and therapy of mental retardation and developmental disabilities by genetic and epigenetic factors. Kaibogaku Zasshi. 1999;74(3):351–362. | ||
Antshel KM, Waisbren SE. Developmental timing of exposure to elevated levels of phenylalanine is associated with ADHD symptom expression. J Abnorm Child Psychol. 2003;31(6):565–574. | ||
González MJ, Gutiérrez AP, Gassió R, et al. Neurological complications and behavioral problems in patients with phenylketonuria in a follow-up unit. Mol Genet Metab. 2011;104 Suppl:S73–S79. | ||
Wyrwich KW, Auguste P, Yu R, et al. Evaluation of Neuropsychiatric Function in Phenylketonuria: Psychometric Properties of the ADHD Rating Scale-IV and Adult ADHD Self-Report Scale Inattention Subscale in Phenylketonuria. Value Health. 2015;18(4):404–412. | ||
Anderson PJ, Wood SJ, Francis DE, et al. Are neuropsychological impairments in children with early-treated phenylketonuria (PKU) related to white matter abnormalities or elevated phenylalanine levels? Dev Neuropsychol. 2007;32(2):645–668. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.