")
Back to Journals » International Medical Case Reports Journal » Volume 10
Choroidal extranodal marginal zone lymphoma diagnosed by full-thickness retinochoroidal biopsy: case report and review of the literature
Authors Dirani A , Allaire G, Callejo S, Belair ML, Biernacki K, Corriveau C, Hammamji K
Received 1 December 2016
Accepted for publication 14 March 2017
Published 4 May 2017 Volume 2017:10 Pages 153—158
DOI https://doi.org/10.2147/IMCRJ.S129171
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Ali Dirani,1,2 Guy Allaire,3,4 Sonia Callejo,1,2 Marie Lyne Belair,1,2 Katarzyna Biernacki,1,2 Christine Corriveau,1,2 Karim Hammamji1,2
1Department of Ophthalmology, University of Montreal, 2Department of Ophthalmology, University of Montreal Health Center (CHUM), 3Department of Pathology, University of Montreal, 4Department of Pathology, University of Montreal Health Center (CHUM), Montreal, Canada
Abstract: The case of an 89-year-old man who was referred for a painless decrease of vision in his right eye (RE) is reported. Fundus examination of the RE showed an elevated amelanotic lesion located in the posterior pole with an adjacent focal round pigmented lesion. There was also a more peripheral amelanotic lesion extending from 6 to 9 o’clock clockwise inferotemporally. Uveitis workup and imaging studies of brain and orbits were normal. A retinochoroidal biopsy was done and showed the presence of choroidal extranodal marginal zone lymphoma. The patient was treated with external beam radiotherapy. This report presents a review of the literature of all reported cases of choroidal extranodal marginal zone lymphoma.
Keywords: choroidal lymphoma, extranodal marginal zone lymphoma, retinochoroidal biopsy
Introduction
Primary choroidal lymphoma (CL) is a rare condition, and the diagnosis can be challenging since presentation can mimic many other ophthalmic diseases including diffuse or amelanotic uveal melanoma, uveal metastases, various uveitis (such as sarcoidosis and birdshot chorioretinopathy), posterior scleritis, and uveal effusion syndrome.1,2 In general, it presents as a low-grade lymphoma with a prolonged, indolent course, and patients frequently go undiagnosed or misdiagnosed as uveitis for extended periods before establishment of the correct diagnosis and proper initiation of treatment.1,2
This report presents a case of a patient who was diagnosed with a choroidal extranodal marginal zone lymphoma based on a full-thickness retinochoroidal biopsy and a review of the literature of all reported cases of CL.
Case report
An 89-year-old man with painless decrease of vision in his right eye (RE) was referred to an ophthalmic oncology clinic for the evaluation of choroidal lesions in his RE. His past ophthalmic history was significant for cataract surgery in both eyes 12 years before presentation with good visual recovery after surgery. The patient was treated for high blood pressure, and he has a pacemaker for cardiac arrhythmia. A written informed consent has been provided by the patient to have the case details and any accompanying images published for this report.
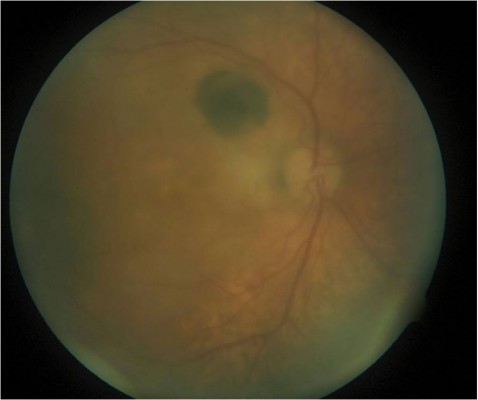
At presentation, the visual acuity was 20/400 in his RE and 20/40 in his left eye (LE). Slit lamp examination (SLE) showed small keratic precipitates in the RE with no cells or flare in the anterior chamber; dilated fundus examination (DFE) of the RE showed an acellular vitreous, an elevated amelanotic yellow-creamy lesion located in the posterior pole (greater diameter of 4 disc diameter [DD]) with an adjacent focal round pigmented lesion of 1 DD located superotemporally to the optic nerve. There was also a more peripheral amelanotic lesion extending from 6 to 9 o’clock clockwise inferotemporally (Figure 1). SLE and DFE of the LE were normal. Macular optical coherence tomography (OCT) examination revealed an elevated choroidal lesion associated with exudative changes (subretinal fluid and intraretinal cysts) (Figure 2). An ultrasound of the RE showed diffuse choroidal thickening and a small elevated lesion (<2 mm in height) located at the macula (Figure 3). A computed tomography scan of the brain and orbits as well as a laboratory workup for uveitis showed no abnormalities. Complete blood count, urea, creatinine, electrolytes, lipid profile, glycemia, glycated hemoglobin, calcium, total protein, albumin, angiotensin-converting enzyme levels, liver enzyme, protein electrophoresis, C-reactive protein, and electrocyte sedimentation rate were assessed and Quantiferon test was performed. The patient then refused to undergo further investigation (especially bone marrow aspiration) and was lost to follow-up. After 6 months, the patient consulted for a substantial decrease of his vision in his RE (hand motion). DFE of the RE showed a bullous exudative retinal detachment involving the macula and most of the inferior and temporal parts of the retina. There was also an increase in the diameter of previously described amelanotic posterior and peripheral lesions. He then consented to undergo full-thickness retinochoroidal biopsy, and the surgery consisted of pars plana vitrectomy, full-thickness retinochoroidal biopsy (dimension 1.5 × 1.5 mm, biopsy done in the infero-nasal quadrant at more than 3 DD from the macula using 23-gauge vertical scissors [Alcon Surgical, Inc., Houston, TX, USA], externalized through an enlarged sclerotomy, then fixated with formaldehyde), endodrainage of the subretinal fluid, air–fluid exchange and flattening of the retina, endolaser, and tamponade with silicone oil (Labtician Ophthalmics, Inc., Oakville, ON, Canada).
 | Figure 1 Image of the dilated fundus of the right eye shows an elevated amelanotic yellow-creamy lesion located in the posterior pole (greater diameter of 4 disc diameter [DD]) with focal round pigmented lesion of 1 DD located superotemporally to the optic nerve. There is also a more peripheral amelanotic lesion extending from 6 to 9 o’clock clockwise inferotemporally. |
 | Figure 2 Macular optical coherence tomography (OCT) examination shows the elevated choroidal lesion (*) associated with exudative changes (subretinal fluid [#] and intraretinal cysts [^]). |
 | Figure 3 An ultrasound of the right eye shows diffuse choroidal thickening (*). There is also a small elevated lesion (#) (<2 mm) located at the macula. |
The biopsy showed a normal segment of neurosensory retina. The choroid was occupied by a dense lymphoplasmacytic infiltrate composed of a majority of small lymphocytes with a slightly irregular nucleus, occasional blasts, and a minor component of cells with plasmacytic features, For example, eccentric nuclei and perinuclear halo (Figure 4). Immunohistochemical studies were performed. The vast majority of lymphocytes were positive for CD79a, CD20, and Bcl2 and negative for CD43, CD10, MUMI, bcl1, and bcl6. A minor component of reactive T lymphocytes showed a positive and similar staining for CD3 and CD5. The plasma cells were positive for CD79a, MUMI, CD138, and kappa light chain and negative for lambda light chain. The proliferation index was low (Ki67 < 5%). Based on histopathologic and immunohistochemical studies, the diagnosis of choroidal extranodal marginal zone lymphoma was confirmed.
 | Figure 4 The choroid biopsy shows a dense lymphoplasmacytic infiltrate composed of a majority of small lymphocytes with a slightly irregular nucleus, occasional blasts, and a minor component of cells with plasmacytic features, for example, eccentric nuclei and perinuclear halo. |
In view of the histopathologic findings, a systemic workup including CT scans of the thorax and abdomen was performed, and the results were normal. Considering the localized nature of the disease, the patient was treated with external beam radiotherapy (EBRT) (total dose of 4 Gray divided in two fractions). The patient was followed for 6 months after the biopsy. The retina remained flat, and his visual acuity was count fingers at his last follow-up appointment.
Discussion
This case report presents a patient with choroidal extranodal marginal zone lymphoma in his RE that was diagnosed with full-thickness chorioretinal biopsy and then treated successfully with EBRT.
CLs are rare tumors that affect the eye, and they can be categorized as primary or secondary when associated with systemic disease.
The largest series of CL was reported from Wills Eye Hospital and consisted of 59 patients (73 eyes) with CL; primary CL with absent systemic lymphoma was present in 41 of patients (69%), and secondary CL was present in 18 patients (31%).1 The mean age at presentation was 63 years (range 32–87), and male-to-female (M/F) ratio was 1.36. Approximately 73% of patients underwent some form of ocular/periocular tissue biopsy, and the most commonly used technique was anterior epibulbar biopsy (50%) followed by transvitreal fine-needle aspiration biopsy (FNAB) of the choroid (36%). In contrast with secondary CL, retrospective data suggested that primary CL is predominantly low grade.11 Patients with primary CL had a mean duration of ocular symptoms before presentation of 17 months and have bilateral ocular involvement in 20%. Poor vision (20/200 and less) was present in 12% of eyes. Iris and ciliary body involvements were reported in 4% and 8% of eyes, respectively. Anterior chamber reaction and vitreous cellular infiltration were present in 14% and 4% of eyes, respectively. Subretinal fluid (SRF) was present in 33 eyes (48%), and only 19% of eyes had SRF involving more than 1 quadrant. None of the patients with primary CL developed systemic disease during follow-up.1 Mashayekhi et al1 recommended that patients with primary CL and no known systemic lymphoma at presentation should undergo systemic evaluation to rule out the possibility of undiagnosed concurrent systemic lymphoma. For the treatment, 29% of eyes were initially observed, without active treatment. The most common form of initial treatment was EBRT (41% of eyes), followed by some form of systemic chemotherapy treatment (rituximab chemotherapy) for 15% of eyes. After a median follow-up of 27 months, the CL regressed in 71%, remained stable in 25%, and worsened in 4%. Among the patients for whom systemic follow-up information was available, 12% died because of complications of systemic lymphoma and 4% died because of other causes after a mean follow-up of 37 months.
Aronow et al reported a cohort of 22 patients with CL; 21 out of 22 patients had primary CL.2 The median age at diagnosis was 68 years, and M/F ratio was 2.1. The choroid was involved in 21 cases (95.5%), and 1 case (4.5%) had ciliochoroidal disease. Twelve patients (54.5%) had bilateral disease. The most common presenting symptom was decreased vision (68.2%), and the median delay before diagnosis was 4.0 months. Fundus examination showed yellow-white choroidal infiltrates. Twenty-two patients (75.9%) had extrascleral extension diagnosed with B-scan ultrasonography, and a strong correlation was observed between CL and coexisting ocular adnexal lymphoma (OAL): other ocular adnexal structures were involved in 13 patients including the conjunctiva in 4 (18.2%), the orbit in 7 (31.8%), and both in 2 (9.1%). The most common biopsy sites were orbit and conjunctiva (68%). The biopsy technique was either incisional or FNAB and extranodal marginal zone lymphoma was the predominant histologic subtype accounting for 76% of cases.2 EBRT was most commonly chosen for primary treatment (72.7% of patients, median dose 30.6 Gy). The remaining patients were treated with systemic chemotherapy (rituximab or combination chemotherapy with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone [RCHOP]). Systemic imaging at the time of diagnosis revealed that the majority of cases (77.3%) were localized to the eye; none of the patients developed new systemic disease. All patients treated with radiotherapy (localized disease) achieved complete remission (median follow-up, 28.7 months). The three remaining patients achieved a state of stable/asymptomatic disease (one patient with 10 months follow-up) or partial remission (two patients with a median follow-up of 19.7 months).
Coupland et al reviewed 13 cases of lymphoproliferative lesions of the uvea.3 Their histological and immunohistochemical findings suggested a diagnosis of extranodal marginal zone lymphoma in all cases. Twelve were primary lesions and one was a secondary manifestation of a primary pulmonary lymphoma. The mean age was 58 (range 33–88), and the M/F ratio was 3.3. Visual acuity was between no light perception and 20/200. Enucleation was performed in all patients either due to difficulties in excluding a malignant uveal melanoma or to pain as a result of malignant glaucoma. Two out of 13 patients received concomitant radiotherapy. Concurrent disease was seen in one patient (pulmonary manifestation), and subsequent systemic disease was diagnosed in two cases, 3 and 7 years after the initial diagnosis of uveal lymphoma. Table 1 summarizes the three major series of patients with primary CL.
 | Table 1 Review of major series of patients with primary CL Abbreviations: CL, choroidal lymphoma; EBRT, external beam radiotherapy; EMZL, external marginal zone lymphoma; FNAB, fine-needle aspiration biopsy; FU, follow-up; M/F, male-to-female ratio; RCHOP, rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone. |
In addition to these three large series, 26 previously reported cases of choroidal extranodal marginal zone lymphoma with adequate and detailed demographic, clinical, and histological data were found.4–19 There were 16 men and 10 women. The age of the patients ranged from 40 to 85 years (mean 58 years). The main initial complaint was blurred vision in all patients except three (one asymptomatic, second had a red and painful eye, and the third patient had a conjunctival mass).8,14,19 The delay before diagnosis ranged from 1 to 48 months (mean 17 months). There was bilateral disease in only two patients.11,17 The initial visual acuity varied from 20/20 to hand motion. Fundus exam disclosed white yellowish creamy choroidal lesion(s) in 20 patients, retinal detachment in 15, and uveal effusion in 3. Concomitant ocular adnexal involvement occurred in 18 patients: orbit (7), conjunctiva (6), and orbit + conjunctiva (5). B-scan ultrasound findings were reported in 24 cases: diffuse choroidal thickening (10), choroidal and scleral thickening (5), posterior pole thickening (4), choroidal tumor (1), subretinal collection (1), choroidal detachment and scleral thickening (1), scleral thickening (1), and unremarkable (1). Orbital extension was found in 12 cases. When performed, fluorescein angiography showed early hypofluorescence with late hyperfluorescence of the choroidal lesions. The biopsied sites were the conjunctiva (6), conjunctiva + tenon (2), conjunctiva + orbit (3), deep sclera (1), orbit (5), tenon + sclera (1), choroid (3), and FNAB of choroid (3). Two patients underwent a diagnostic enucleation. None of the patients had systemic involvement prior to treatment. Nine patients were treated by chemotherapy alone, 13 by radiotherapy, and 2 by a combination of chemotherapy and radiotherapy. Two patients underwent a diagnostic enucleation. In all patients but one, there was complete or near-complete resolution of the fundus anomalies. The follow-up period ranged from 4 to 156 months (mean 41 months). Posttreatment visual acuity was reported in 16 cases. It improved in nine patients, was stable in one, and deteriorated in the remaining six patients. One patient developed a gastric mucosa-associated lymphoid tissue lymphoma, 1 year after the treatment.
Other ophthalmic diseases can mimic the presentation of CL and should be excluded. These diseases include diffuse or amelanotic uveal melanoma, uveal metastases, various uveitis, and posterior scleritis.20–22 A thorough review of systemic signs and symptoms with careful past medical and surgical history of the patient should be done. A laboratory workup is crucial to rule out the possible cause of uveitis. Sometimes, imaging studies, like brain magnetic resonance imaging, are needed to rule out possible masquerade syndromes. Vitreous biopsy may be done prior to retinochoroidal biopsy in case of suspicion of vitreoretinal lymphoma.20–22
In summary, the case of primary choroidal extranodal marginal zone lymphoma is presented. It is a slowly progressive lymphoproliferative lesion which can mimic an inflammatory disease, thus delaying the diagnosis. Involvement of ocular adnexae was present in more than half of the patients reported in the literature. As in our case and previously reported ones, it can be associated with retinal detachment. There is usually no systemic involvement in these cases, but imaging studies are recommended at presentation to exclude systemic association. Although responsive to radiotherapy and/or chemotherapy, in one-third of the cases the posttreatment visual acuity deteriorated.
Disclosure
The authors report no conflicts of interest in this work.
References
Mashayekhi A, Shukla SY, Shields JA, Shields CL. Choroidal lymphoma: clinical features and association with systemic lymphoma. Ophthalmology. 2014;121:342–351. | ||
Aronow ME, Portell CA, Sweetenham JW, Singh AD. Uveal lymphoma: clinical features, diagnostic studies, treatment selection, and outcomes. Ophthalmology. 2014;121:334–341. | ||
Coupland SE, Foss H-D, Hidayat AA, Cockerham GC, Hummel M, Stein H. Extranodal marginal zone B cell lymphomas of the uvea: an analysis of 13 cases. J Pathol. 2002;197:333–340. | ||
Baryla J, Allen LH, Kwan K, Ong M, Sheidow T. Choroidal lymphoma with orbital and optic nerve extension: case and review of literature. Can J Ophthalmol. 2012;47:79–81. | ||
Coupland SE, Joussen A, Anastassiou G, Stein H. Diagnosis of a primary uveal extranodal marginal zone B-cell lymphoma by chorioretinal biopsy: case report. Giaefes Arch Clin Exp Ophthalmol. 2005;243:482–486. | ||
Coutinho AB, Muccioli C, Martins MC, Belfort Jr R, Sant’ Anna AE, Bumier Jr MN. Extranodal B-cell lymphoma of the uvea: a case report. Can J Ophthalmol. 2005;40:623–626. | ||
Erickson B, Mantopoulos D, Schoenfield L, Cebulla CM. Multimodal imaging and clinicopathologic correlation in primary uveal lymphoma. Case Rep Ophthalmol. 2016;7:39–43. | ||
Fuller ML, Sweetenham J, Schoenfield L, Singh AD. Uveal lymphoma: a variant of ocular adnexal lymphoma. Leuk Lymphoma. 2008;49:2393–2397. | ||
Gao X, Li B, You Q, Peng X. Primary extranodal marginal zone B-cell lymphoma with diffuse uveal involvement and focal infiltration of the trabecular meshwork: a case report and review of the literature. BMC Ophthalmol. 2015;15:48. | ||
Garcia-Alvarez C, Saomil MA, Blanco G, Mendez MC, Lopez-Lara F. Extranodal B-cell uveal lymphoma with extraocular involvement. Can J Ophthalmol. 2009;44:213–214. | ||
Gaucher D, Bodaghi B, Charlotte F, et al. MALT-type B-cell lymphoma masquerading as scleritis or posterior uveitis. J Fr Ophtalmol. 2005;28(1):31–38. | ||
Hoang-Xuan T, Bodaghi B, Toublanc M, Delmer A, Schwartz L, D’Hermies F. Scleritis and mucosa-associated lymphoid tissue lymphoma. A new masquerade syndrome. Ophthalmology. 1996;103:631–635. | ||
Loriaut P, Charlotte F, Bodaghi B, et al. Choroidal and adnexal extranodal marginal zone B-cell lymphoma: presentation, imaging findings, and therapeutic management in a series of nine cases. Eye (Lond). 2013;27:828–835. | ||
Ramulu P, Iliff NT, Green WR, Kuo IC. Asymptomatic conjunctival mucosa-associated lymphoid tissue-type lymphoma with presumed intraocular involvement. Cornea. 2007;26(4):484–486. | ||
Rasic DM, Stankovic Z, Terzic T, Kovacevic D, Koturovic Z, Markovic V. Primary extranodal marginal zone lymphoma of the uvea associated with massive diffuse epibulbar extension and focal infiltration of the optic nerve and meninges, clinically presented as uveitis masquerade syndrome: a case report. Med Oncol. 2010;27(3):1010–1016. | ||
Rospond-Kubiak I, Kociecki J, Stopa M. Primary uveal lymphoma effectively managed with oral chlorambucil: a case report. J Med Case Rep. 2013;7:173. | ||
Sarraf D, Jain A, Dubovy S, Kreiger A, Fong D, Paschal J. Mucosa-associated lymphoid tissue lymphoma with intraocular involvement. Retina. 2005;25(1):94–98. | ||
Tagami M, Nagai T, Sekimukai D, Hara R, Azumi A. Uveal extranodal marginal zone B-cell lymphoma of the mucosa-associated lymphoid tissue type with concomitant extraocular lesions in a Japanese man. Jpn J Ophthalmol. 2011;55:585–587. | ||
Williams Jr BK, Tsui I, McCannel TA. Spectral-domain optical coherence tomography of conjunctiva mucosa-associated lymphoid tissue lymphoma with presumed choroidal involvement. Graefes Arch Clin Exp Ophthalmol. 2010;248(12):1837–1840. | ||
Brown SM, Jampol LM, Cantrill HL. Intraocular lymphoma presenting as retinal vasculitis. Surv Ophthalmol. 1994;39:133–140. | ||
Gill MK, Jampol LM. Variations in the presentation of primary intraocular lymphoma: case reports and a review. Surv Ophthalmol. 2001;45:463–471. | ||
Rothova A, Ooijman F, Kerkhoff F, Van Der Lelij A, Lokhorst HM. Uveitis masquerade syndromes. Ophthalmology. 2001;108:386–399. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.