")
Back to Journals » OncoTargets and Therapy » Volume 9
Cholesterol biosynthesis inhibitor RO 48-8071 suppresses growth of hormone-dependent and castration-resistant prostate cancer cells
Authors Liang Y, Mafuvadze B , Aebi J, Hyder S
Received 3 February 2016
Accepted for publication 14 March 2016
Published 30 May 2016 Volume 2016:9 Pages 3223—3232
DOI https://doi.org/10.2147/OTT.S105725
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Faris Farassati
Yayun Liang,1 Benford Mafuvadze,1 Johannes D Aebi,2 Salman M Hyder1
1Dalton Cardiovascular Research Center and Department of Biomedical Sciences, University of Missouri-Columbia, Columbia, MO, USA; 2Medicinal Chemistry, Roche Pharma Research and Early Development (pRED), Roche Innovation Center Basel, F Hoffmann-La Roche Ltd., Basel, Switzerland
Abstract: Standard treatment for primary prostate cancer includes systemic exposure to chemotherapeutic drugs that target androgen receptor or antihormone therapy (chemical castration); however, drug-resistant cancer cells generally emerge during treatment, limiting the continued use of systemic chemotherapy. Patients are then treated with more toxic standard therapies. Therefore, there is an urgent need for novel and more effective treatments for prostate cancer. The cholesterol biosynthetic pathway is an attractive therapeutic target for treating endocrine-dependent cancers because cholesterol is an essential structural and functional component of cell membranes as well as the metabolic precursor of endogenous steroid hormones. In this study, we have examined the effects of RO 48-8071 (4'-[6-(allylmethylamino)hexyloxy]-4-bromo-2'-fluorobenzophenone fumarate; Roche Pharmaceuticals internal reference: RO0488071) (RO), which is an inhibitor of 2, 3-oxidosqualene cyclase (a key enzyme in the cholesterol biosynthetic pathway), on prostate cancer cells. Exposure of both hormone-dependent and castration-resistant human prostate cancer cells to RO reduced prostate cancer cell viability and induced apoptosis in vitro. RO treatment reduced androgen receptor protein expression in hormone-dependent prostate cancer cells and increased estrogen receptor β (ERβ) protein expression in both hormone-dependent and castration-resistant prostate cancer cell lines. Combining RO with an ERβ agonist increased its ability to reduce castration-resistant prostate cancer cell viability. In addition, RO effectively suppressed the growth of aggressive castration-resistant human prostate cancer cell xenografts in vivo without any signs of toxicity to experimental animals. Importantly, RO did not reduce the viability of normal prostate cells in vitro. Our study is the first to demonstrate that the cholesterol biosynthesis inhibitor RO effectively suppresses growth of human prostate cancer cells. Our findings suggest that cholesterol biosynthesis inhibitors such as RO, when used in combination with commonly used chemotherapeutic drugs or ERβ specific ligands, could represent a novel therapeutic approach to prevent the growth of prostate cancer tumors.
Keywords: prostate cancer, cholesterol biosynthesis inhibitor, cell viability, xenograft, castration resistant
Introduction
Despite concerted efforts to develop new strategies for preventing and treating prostate cancer, almost 240,000 new cases are reported each year in the US and more than 28,000 males die of the disease annually.1 In addition, prostate cancer is associated with significant physical burden, including bowel, urinary, and sexual dysfunction in early-stage disease and painful bony lesions in more advanced cancers.2,3 Most deaths from prostate cancer occur due to complications that arise following metastasis from the primary tumor to other tissues and organs, a process dependent upon increased angiogenesis.4 Human prostate cancer cells often proliferate in response to endogenous or exogenous androgens and estrogens, which also inhibit cell death and promote metastasis.5 Whereas chemical castration, in the form of systemic exposure to chemotherapeutic drugs or antihormones, is the standard treatment for primary prostate cancer, drug-resistant cancer cells often emerge, limiting the usefulness of continued chemotherapy.5 In addition, many patients suffering from prostate cancer fail to respond to any form of hormonal therapy, leading to poor clinical prognosis. As a consequence, novel, less toxic, and more effective treatments for prostate cancer are urgently needed.
Cholesterol is an essential structural and functional component of cell membranes and also the metabolic precursor of endogenous steroid hormones. In addition, cholesterol is associated with increased angiogenesis in prostate tumors.4,6 Consequently, the pathway leading to cholesterol biosynthesis is an attractive therapeutic target through which endocrine-dependent cancers might be treated. Historically, statins have been used to inhibit 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase, an enzyme essential for cholesterol biosynthesis to treat cancer.7 However, statins cause a number of undesirable side effects attributed to reduced levels of isoprenoids, defective posttranslational modification of membrane proteins, and impaired membrane structure and function.8 Alternative approaches using different cholesterol biosynthesis inhibitors are currently being considered to lower cholesterol levels. 2,3-oxidosqualene cyclase (OSC), an enzyme that acts downstream of HMG-CoA reductase to convert 2, 3-monoepoxysqualene to lanosterol (a key step in the biosynthesis of cholesterol) has emerged as a possible new target by which to inhibit cholesterol biosynthesis.9 RO 48-8071 (4′-[6-(allylmethylamino)hexyloxy]-4-bromo-2′-fluorobenzophenone fumarate; Roche Pharmaceuticals internal reference: RO0488071) (RO) is a potent inhibitor of OSC.10,11 For this reason, we conducted a series of studies to examine the anticancer properties of RO. In the studies reported here, we found that RO exerts a number of novel effects that might be exploited to treat or prevent prostate cancer. RO reduced viability of both hormone-dependent and castration-resistant prostate cancer cells, but not normal prostate cells, in vitro; in addition, RO induced apoptosis of both hormone-dependent and castration-resistant prostate cancer cells in vitro. RO also simultaneously lowered levels of androgen receptor (AR), a pro-proliferative protein, in hormone-dependent prostate cancer cells and induced estrogen receptor β (ERβ), a potent antiproliferative protein,12–16 in both hormone-dependent and castration-resistant prostate cancer cells in vitro.
Finally, RO arrested the growth of human castration-resistant prostate cancer cell xenografts in vivo. These biological effects of RO suggest that it may represent a suitable therapeutic agent by which to arrest human prostate cancer cell growth in vivo.
Materials and methods
Cell lines and culture
The ARα-positive prostate cancer cell lines LNCaP and the castration-resistant cell lines PC-3 and DU145 were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). A castration-resistant derivative cell line C4-2, derived from LnCaP cells, was obtained from UROCOR (Urosciences Group, Oklahoma City, OK, USA). Cells were grown in Dulbecco’s Modified Eagle’s Medium or Roswell Park Memorial Institute 1640 medium (RPMI-1640 medium) (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10%–20% fetal bovine serum (Sigma-Aldrich Co., St Louis, MO, USA) as indicated. RWPE-1, a normal prostate cell line, was also obtained from ATCC and grown in complete growth medium according to instructions provided by the vendor. The University of Missouri Ethics Committee/Institutional Review Board does not require that ethical approval be obtained for the use of commercially available cell lines.
Reagents
RO was purchased from Sigma-Aldrich for in vitro experiments and obtained from Roche Pharmaceuticals (Basel, Switzerland, provided by Dr Johannes Aebi) for in vivo studies. ERβ (H-150, Cat# sc-8974) and AR (N-20, Cat# sc-816) antibodies were obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). β-Actin antibody was obtained from Sigma-Aldrich. The ERβ agonist diarylpropionitrile (DPN)17 was obtained from Tocris Biosciences (Ellisville, MO, USA).
Cell viability assay
The sulforhodamine B assay was used to measure cell viability, as previously described.18
Cell apoptosis assay
The Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection Kit (Biovision Research Products, Mountain View, CA, USA) was used to measure apoptosis and cell death in prostate cancer cells, as we have previously described.19
In vivo prostate cancer tumor growth inhibition assays
All animal experiments were approved by the University of Missouri Animal Care Institutional Review Committee. This study adhered to the guidelines of the US Government Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research, and Training. Six-week-old male athymic nude mice (nu/nu, Foxn1) weighing 20–22 g were purchased from Harlan Sprague Dawley, Inc. (Indianapolis, IN, USA). Castration-resistant PC-3 cells (5×106 in 0.15 mL solution) were mixed with matrigel and RPMI-1640 medium (1/1, v/v) and injected subcutaneously into both flanks of each mouse (n=6 animals/group) and tumors allowed to develop. The tumors were measured twice per week with a digital caliper as described previously.20 Tumor volumes were calculated by the formula (L × W × H) × π/6. Drug treatment was started when tumor volumes reached ~100 mm3. Mice were given daily tail vein injections of 0.1 mL solution of either 5 or 20 mg/kg RO for 5 days. This was followed by an injection every other day for six additional treatments and then a final injection 2 hours prior to sacrifice. Control mice received the same volume of phosphate-buffered saline on the same schedule. The animals were weighed and tumor volumes were measured twice weekly throughout the drug treatment period.
Western blots
Whole-cell extracts were prepared with a nuclear extraction TransAm Kit (Active Motif, Carlsbad, CA, USA) and Western blotting was carried out as previously described.21,22 Briefly, samples were subjected to Western blot analysis (30–40 μg) in a NuPAGE 10% Bis-Tris Gel (Thermo Fisher Scientific). Blots were blocked with 5% nonfat dry milk in Tris-buffered saline, 0.1% Tween 20 for 1 hour at room temperature (RT), and then the blots were treated with a 1:200 dilution of primary antibody for 2 hours at RT. Blots were then incubated with secondary antibody (1:3,000 dilution) for 1 hour at RT and washed seven times with Tris-buffered saline, 0.1% Tween 20. Immunoreactive bands were visualized using an ECL Plus Detection Kit (Amersham, Pharmacia Biotech, Arlington Heights, IL, USA). Membranes were stripped and reprobed for β-actin, which was used as a protein loading control.
Statistical analysis
Differences between groups or among groups were tested by either paired t-test or by using one-way analysis of variance (ANOVA) with repeated measures over time. The assumption of the ANOVA was examined, and a nonparametric measure based on ranks was used if needed. When ANOVA indicated a significant effect (F-ratio, P<0.05), the Student–Newman–Keuls multirange test was used to compare the means of the individual groups. Statistical analyses were conducted using SigmaPlot software, version 12.5 (San Jose, CA, USA). For all comparisons, P<0.05 was regarded as statistically significant. Values are reported as mean ± standard error of the mean.
Results
RO reduces prostate cancer cell viability in vitro
We initially determined the dose–response and time-dependent effect of RO on prostate cancer cells in vitro using a cell viability assay.18 After 24 or 48 hours exposure to RO, the viability of both hormone-dependent (LNCaP) cells and castration-resistant (PC-3 and DU145) prostate cancer cells was reduced, with an half maximal inhibitory concentration (IC50) value of ~10 μM (Figure 1A and Table 1). We also conducted studies using normal RWPE-1 prostate cells to determine whether RO specifically affects prostate cancer cells. When RWPE-1 cells were exposed to concentrations of RO of up to 10 μM, no effect was observed, whereas PC-3 prostate cancer cells exhibited significantly reduced viability (Figure 1B). This suggests that RO may be used against prostate cancer cells with little or no toxicity to normal prostate cells.
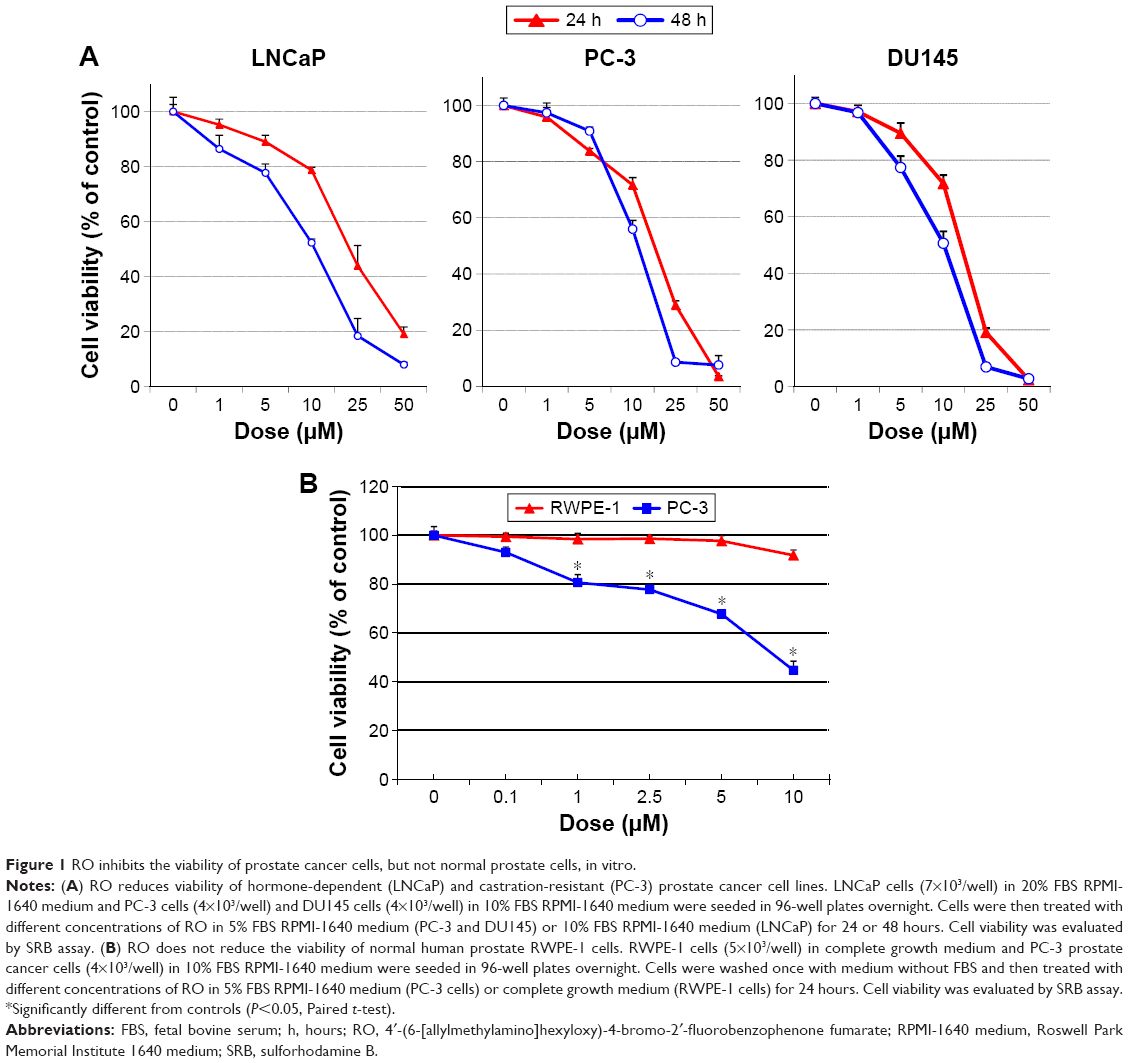 | Figure 1 RO inhibits the viability of prostate cancer cells, but not normal prostate cells, in vitro. |
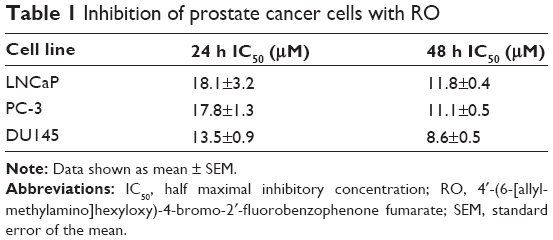 | Table 1 Inhibition of prostate cancer cells with RO |
The affinity of RO for OSC is in the nM range.10,11 With this in mind, we performed studies over an extended period of time (7 days) to determine whether a range of low doses of RO would affect cell viability in a manner comparable with shorter term (24–48 hours) exposure to higher doses. Concentrations of RO as low as 1 nM significantly reduced the viability of both LNCaP (hormone-dependent) and PC-3 (castration-resistant) cells in a 7-day assay (Figure 2).
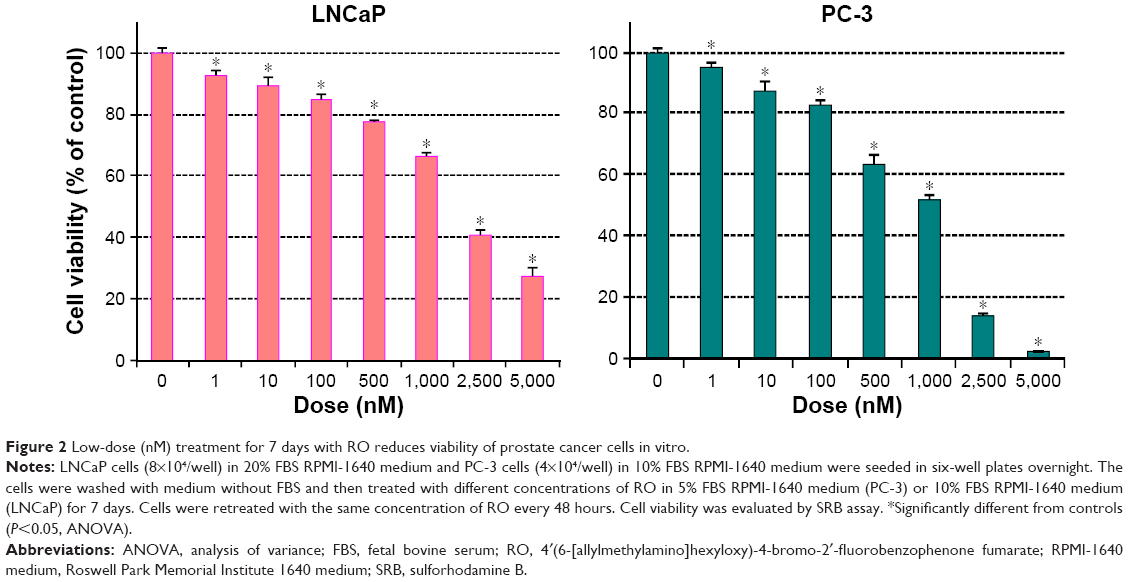 | Figure 2 Low-dose (nM) treatment for 7 days with RO reduces viability of prostate cancer cells in vitro. |
RO induces apoptosis in prostate cancer cells in vitro
We next studied the mechanism of action through which RO reduces prostate cancer cell viability. The hormone-dependent prostate cancer cell lines LNCaP and its castration-resistant variant C4-2 were incubated with 10–30 μM RO for 24 hours, after which apoptosis was measured by Annexin V-FITC-based fluorescence-activated cell sorting analysis. RO induced apoptosis of both cell lines in a dose-dependent manner (Figure 3A). Interestingly, castration-resistant PC-3 and DU145 cells also demonstrated significant levels of apoptosis following 24-hour treatment with RO (Figure 3B).
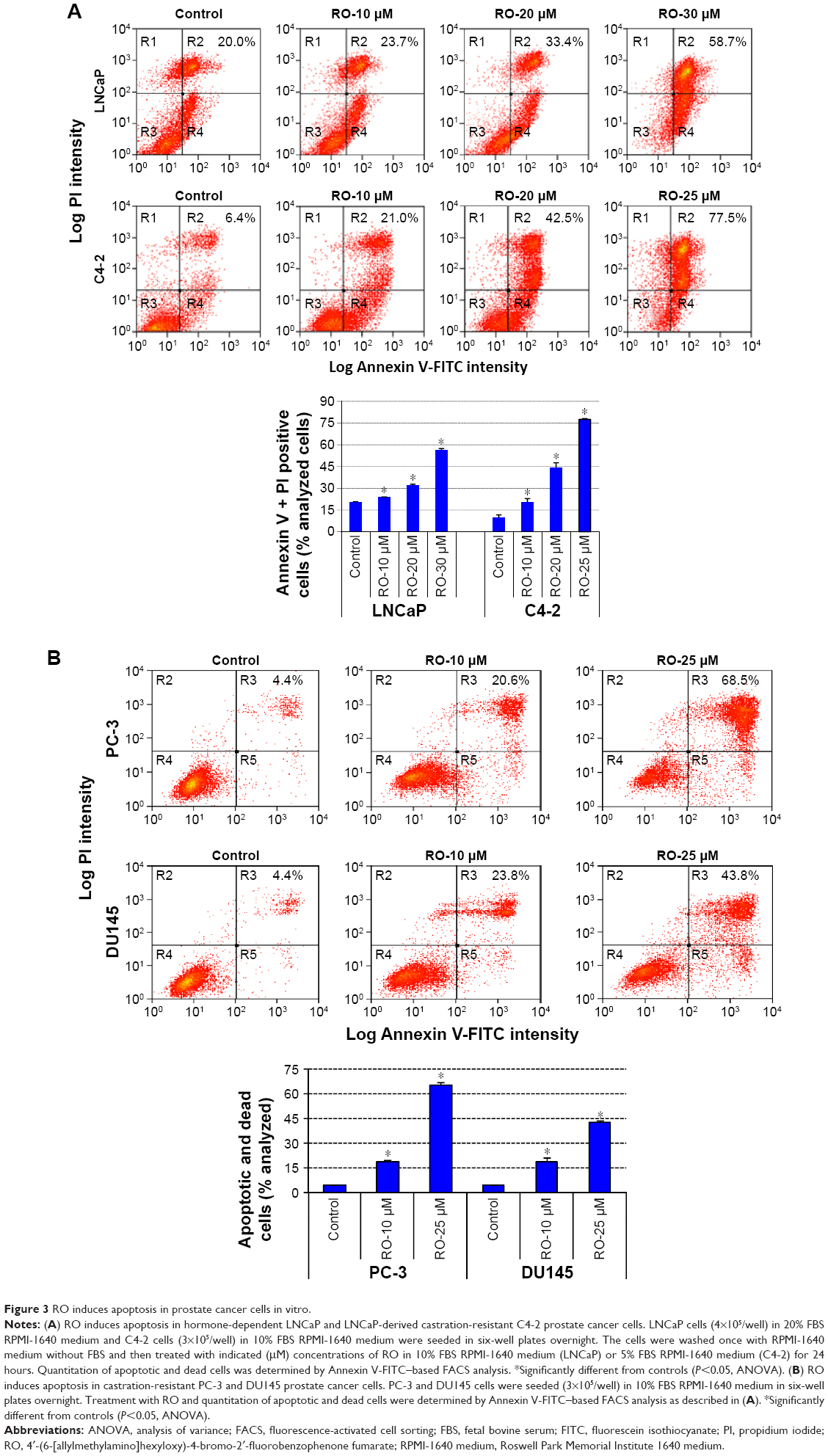 | Figure 3 RO induces apoptosis in prostate cancer cells in vitro. |
RO reduces AR protein expression and increases ERβ protein expression in prostate cancer cells in vitro
Because AR is the main driving force for growth of both hormone-dependent and castration-resistant prostate cancer cells,3–5 we performed studies in vitro to determine whether AR levels are affected by RO in these cells. In addition, we examined whether RO might also influence levels of ERβ, which is a suppressor of tumor cell growth.15,16 LNCaP prostate cancer cells (AR positive) were exposed to pharmacological (10–25 μM) doses of RO in a short-term (6 hours) study; long-term studies were also performed using low (0.1–1 μM) RO doses for 7 days. In both short- and long-term studies, treatment with RO reduced AR protein expression in a dose-dependent manner (Figure 4A and B). Importantly, RO increased ERβ protein expression dose-dependently in both hormone-dependent LNCaP and castration-resistant PC-3 cells in short-term studies (Figure 4C). Our findings therefore indicate that treatment of prostate cancer cells with RO reduces levels of pro-proliferative AR while simultaneously increasing antiproliferative ERβ. We have previously reported such off-target effects of RO on ERα and ERβ in breast cancer cells in vitro.23,24
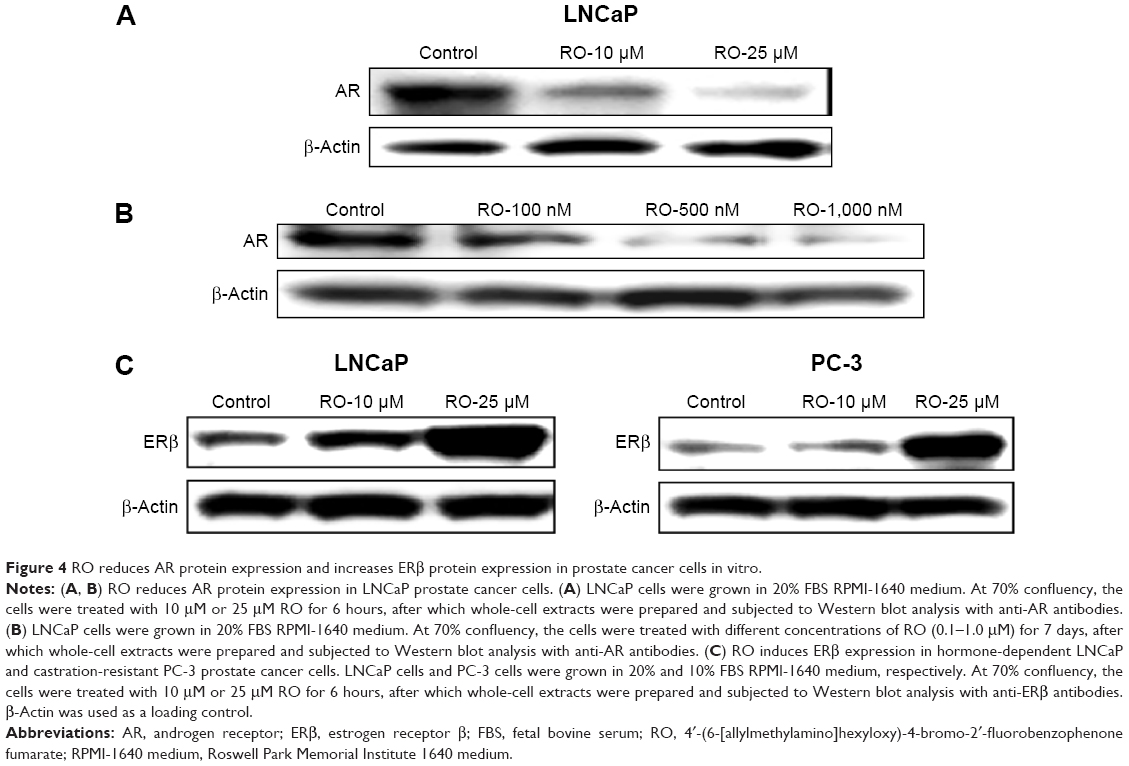 | Figure 4 RO reduces AR protein expression and increases ERβ protein expression in prostate cancer cells in vitro. |
Stimulation of ERβ activity enhances the ability of RO to reduce prostate cancer cell viability in vitro
ERβ plays an antiproliferative role in prostate cancer cells.25–27 In order to determine whether induction of ERβ protein potentiates the antiproliferative effects of RO, we treated castration-resistant PC-3 prostate cancer cells with RO in the presence of DPN, an ERβ agonist. Exposure of PC-3 cells to both DPN and RO reduced cell viability synergistically (Figure 5), suggesting that activation of ERβ is partially responsible for RO-mediated effects on prostate cancer cell viability.
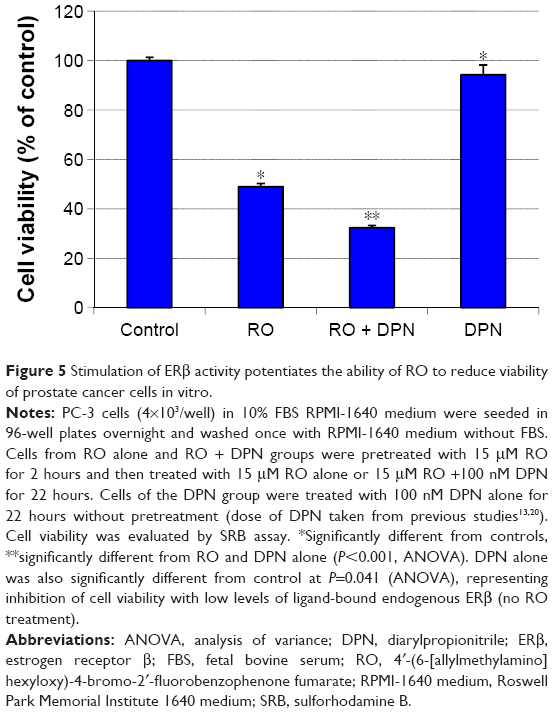 | Figure 5 Stimulation of ERβ activity potentiates the ability of RO to reduce viability of prostate cancer cells in vitro. |
RO suppresses growth of prostate cancer cell xenografts in vivo
Having demonstrated the effectiveness of RO in suppressing prostate cancer cell growth in vitro, we conducted studies to establish whether RO had the same effect in vivo. Castration-resistant PC-3 prostate cancer cells were used to establish xenografts in 6-week-old mice. Subsequent treatment with either 5 or 20 mg/kg RO significantly reduced in vivo tumor growth compared with vehicle-treated controls (Figure 6A). Furthermore, animal weights were unaffected by RO treatment compared with vehicle-treated controls (Figure 6B), indicating that the compound was nontoxic at the doses administered. In addition, two of the 12 tumors being monitored in the mice administered with 20 mg/kg RO were completely eradicated in the timeframe tested (Figure 6C).
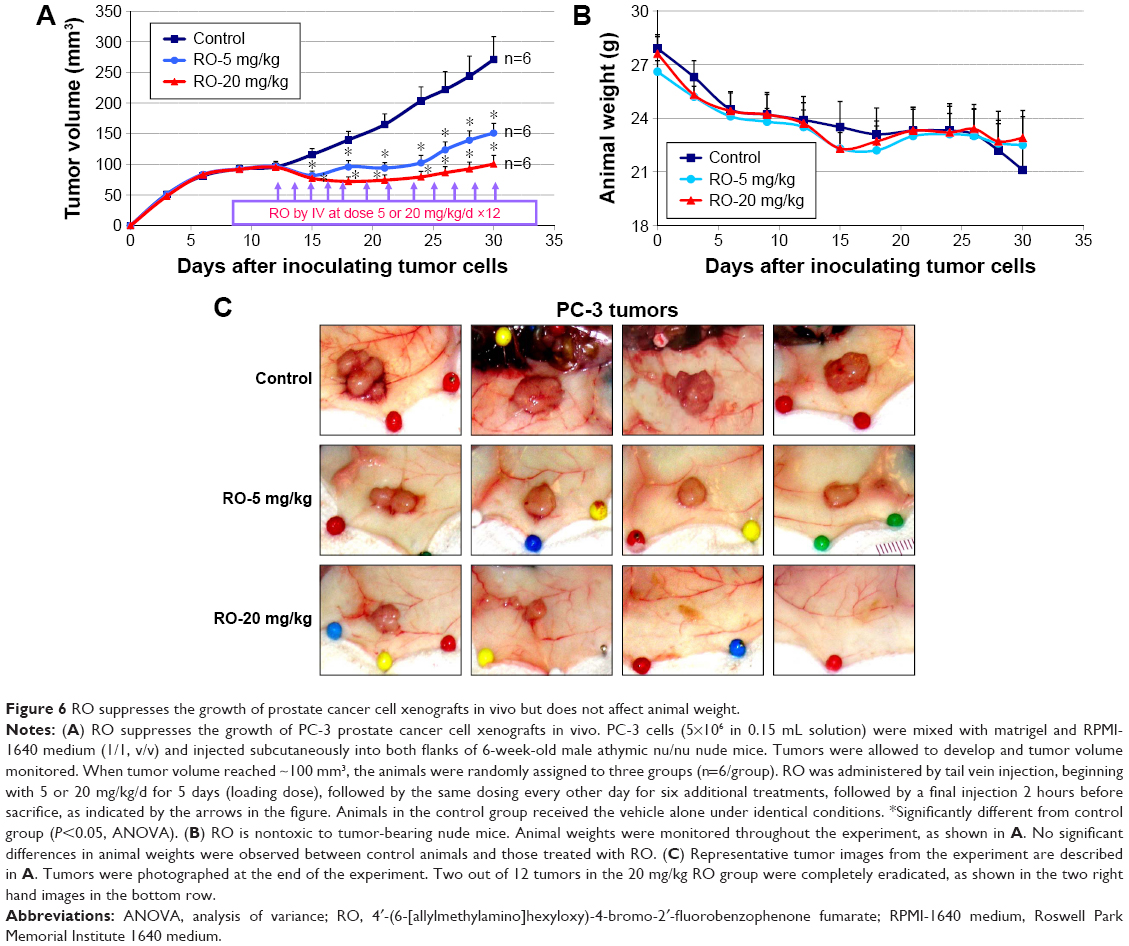 | Figure 6 RO suppresses the growth of prostate cancer cell xenografts in vivo but does not affect animal weight. |
Discussion
The risk of prostate cancer increases with a males age, though other risk factors, such as race and family history, may predispose certain individuals to developing the disease earlier.1–3 Prostate cancer is a deadly disease that, in its early stages, expresses functional AR and is consequently hormone dependent. At this point, patients respond to anti-androgen treatment. However, although a number of antihormonal treatment strategies are currently employed to control progression of the disease, drug-resistant tumors frequently emerge.4,5 Cancers that become nonresponsive to antihormone therapy are termed castration-resistant, even though AR still plays a role in these tumors.28 Patients with antihormone-resistant tumors have a poor prognosis,29,30 and, because treating such tumors is so challenging, new ways to control this type of disease are urgently sought.
In recent reports, we have shown that RO, an inhibitor of OSC that prevents cholesterol biosynthesis, arrests the growth of breast cancer cells.23,24 We further showed in vitro that RO also abolished AR-dependent transcriptional activity,31 leading us to conduct the present studies investigating the ability of RO to suppress the growth of prostate cancer cells. In the studies reported here, RO reduced the viability of three prostate cancer cell lines: hormone-dependent LNCaP cells, as well as castration-resistant PC-3 and DU145 cells. While the growth of LNCaP cells is AR-dependent and responsive to androgens; the other cell lines are resistant to antihormone therapy. We also determined that concentrations of RO up to 10 μM had no effect on the viability of normal prostate cells, suggesting that its in vitro effects are specific to cancerous prostate cells. We therefore propose that because RO appears to be nontoxic to normal cells, it might be used to target tumors with little risk of patient toxicity. Our subsequent in vivo studies discussed later support this notion, further indicating that the use of RO as a therapeutic agent is a possibility with little or no risk of toxic side effects.
In order to better understand how RO exerts its antitumor properties, we first determined whether RO induces apoptosis in prostate cancer cells. Indeed, RO did induce apoptosis in a dose-dependent manner in both hormone-dependent and castration-resistant cells. It therefore appears that induction of apoptosis is likely the major mechanism of action of RO in vitro, a finding in accord with RO effects that we have previously reported in breast cancer cells.23,24
To further explore the mechanism of action of RO responsible for its effects in prostate cancer cells in vitro, we examined its influence on AR and ERβ protein expression. We observed a dramatic, dose-dependent reduction in AR protein expression in response to RO. Future studies will determine whether RO reduces AR transcription or reduces AR protein expression independent of effects on mRNA, directly affecting AR at the protein level. Additional studies will also include competition assays to ascertain whether or not RO binds directly to AR, given that there are strict structural requirements that facilitate the binding of AR to a ligand, and our previous data does not show transcriptional inhibition effect of RO on AR.31,32
We also observed that RO induced ERβ, which has been associated with reduced viability of prostate cancer cells,13–16 in both hormone-dependent LNCaP cells and castration-resistant PC-3 cells. This is particularly important as ERβ may potentially be targeted by specific drugs to potentiate its anticancer effects. Furthermore, the ERβ-specific agonist DPN potentiated the effects of RO, further confirming the important role played by ERβ in reducing cell viability. We propose therefore that drugs such as DPN, which increase ERβ activity in prostate cancer cells, could be made even more effective when administered in conjunction with RO. The development of therapeutic regimens using a combination of two drugs might make it possible to manage disease using lower levels of both agents, reducing the likelihood of toxic side effects that arise from current therapeutic modalities.33 In future studies, we will examine in more detail the mechanism by which RO affects cellular ERβ levels by analyzing both RNA transcription and protein stability. We will also compare the growth inhibitory effects of RO with other cholesterol-lowering agents, such as statins.
Because castration-resistant prostate cancers are notoriously difficult to treat using current therapeutic protocols and respond poorly to clinical intervention, in this study, we selected castration-resistant PC-3 cells to examine the ability of RO to suppress prostate cancer cell xenografts in vivo. Compared with controls that did not receive RO, growth of tumors in animals receiving RO was significantly suppressed, and two out of 12 tumors were completely eradicated in mice receiving the higher dose of RO in the timeframe tested. No RO-mediated toxicity was observed in mice, as we have previously reported.23 These findings suggest that RO could be an effective means by which to suppress the in vivo growth of cells that are resistant to antihormones. We plan to test this possibility in several other cell lines, as well as in patient-derived xenografts.
Conclusion
In summary, the data presented here strongly suggest that, in addition to its ability to suppress cholesterol biosynthesis, the OSC inhibitor RO has powerful antitumor properties. As well as inducing apoptosis of prostate cancer cells, RO also reduces levels of pro-proliferative protein AR and increases antiproliferative protein ERβ in these cells. Together, these activities may lead to a cellular milieu that favors reduced proliferation of prostate cancer cells, as has previously been shown with other agents.34,35 Certainly, when RO was given in combination with an ERβ agonist, inhibition of prostate cancer cell viability was even further enhanced, strongly suggesting that ERβ plays a central role in RO-mediated tumor cell growth suppression. We therefore propose that combination therapy using inhibitors of cholesterol biosynthesis such as RO, together with commonly used chemotherapeutic drugs or agents that specifically target and activate ERβ protein, could prove beneficial as a means of preventing the growth of prostate cancer tumors.
Acknowledgments
The studies reported were supported by the Department of Defense Prostate Cancer Program grant W81XWH-14-1-0246, by a peer-reviewed Committee on Research grant from the College of Veterinary Medicine, University of Missouri-Columbia, and by generous support from donors of Ellis Fischel Cancer Center, University of Missouri-Columbia. We would also like to thank Dr Carolyn Henry for her invaluable support during the completion of this project. SMH is the Zalk Missouri Professor of Tumor Angiogenesis.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. | ||
Penson DF, Litwin MS. The physical burden of prostate cancer. Urol Clin North Am. 2003;30:305–313. | ||
O’Shaughnessy JA, Kelloff GJ, Gordon GB, et al. Treatment and prevention of intraepithelial neoplasia: an important target for accelerated new agent development. Clin Cancer Res. 2002;8: 314–346. | ||
Pelton K, Freeman MR, Solomon KR. Cholesterol and prostate cancer. Curr Opin Pharmacol. 2012;12:751–759. | ||
Ali S, Ali S. Multidimensional approaches in dealing with prostate cancer. Gene. 2008;410:1–8. | ||
Solomon KR, Pelton K, Boucher K, et al. Ezetimibe is an inhibitor of tumor angiogenesis. Am J Pathol. 2009;174:1017–1026. | ||
Garcia-Ruiz C, Morales A, Fernandez-Checa JC. Statins and protein prenylation in cancer cell biology and therapy. Anticancer Agents Med Chem. 2012;12:303–315. | ||
McTaggart SJ. Isoprenylated proteins. Cell Mol Life Sci. 2006;63:255–267. | ||
Charlton-Menys V, Durrington PN. Squalene synthase inhibitors: clinical pharmacology and cholesterol-lowering potential. Drugs. 2007;67:11–16. | ||
Morand OH, Aebi JD, Dehmlow H, et al. RO 48-8.071, a new 2,3-oxidosqualene: lanosterol cyclase inhibitor lowering plasma cholesterol in hamsters, squirrel monkeys, and minipigs: comparison to simvastatin. J Lipid Res. 1997;38:373–390. | ||
Abe I, Zheng YF, Prestwich GD. Photoaffinity labeling of oxidosqualene cyclase and squalene cyclase by a benzophenone-containing inhibitor. Biochemistry. 1998;37:5779–5784. | ||
Dondi D, Piccolella M, Biserni A, et al. Estrogen receptor beta and the progression of prostate cancer: role of 5alpha-androstane-3beta,17beta-diol. Endocr Relat Cancer. 2010;17:731–742. | ||
Pravettoni A, Mornati O, Martini PG, et al. Estrogen receptor beta (ERbeta) and inhibition of prostate cancer cell proliferation: studies on the possible mechanism of action in DU145 cells. Mol Cell Endocrinol. 2007;263:46–54. | ||
Singh V, Sharma V, Verma V, et al. Apigenin manipulates the ubiquitin- proteasome system to rescue estrogen receptor-β from degradation and induce apoptosis in prostate cancer cells. Eur J Nutr. 2015;54:1255–1267. | ||
Dey P, Barros RP, Warner M, Ström A, Gustafsson JÅ. Insight into the mechanisms of action of estrogen receptor β in the breast, prostate, colon, and CNS. J Mol Endocrinol. 2013;51:T61–T74. | ||
Dey P, Ström A, Gustafsson JÅ. Estrogen receptor β upregulates FOXO3a and causes induction of apoptosis through PUMA in prostate cancer. Oncogene. 2014;33:4213–4225. | ||
Harrington WR, Sheng S, Barnett DH, Petz LN, Katzenellenbogen JA, Katzenellenbogen BS. Activities of estrogen receptor alpha- and beta-selective ligands at diverse estrogen responsive gene sites mediating transactivation or transrepression. Mol Cell Endocrinol. 2003;206:13–22. | ||
Rubinstein LV, Shoemaker RH, Paull KD, et al. Comparison of in vitro anticancer–drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J Natl Cancer Inst. 1990;82:1113–1118. | ||
Liang Y, Brekken RA, Hyder SM. VEGF induces proliferation of breast cancer cells and counteracts the anti-proliferative activity of anti-hormones. Endocr Relat Cancer. 2006;13:905–919. | ||
Liang Y, Besch-Williford C, Brekken RA, Hyder SM. Progestin-dependent progression of human breast tumor xenografts: a novel model for evaluating anti-tumor therapeutics. Cancer Res. 2007;67:9929–9936. | ||
Liang Y, Besch-Williford C, Benakanakere I, Hyder SM. Re-activation of p53 pathway inhibits growth of hormone-dependent human breast cancer cells in vitro and in vivo. Int J Oncol. 2007;31:777–784. | ||
Liang Y, Besch-Williford C, Benakanakere I, Thorpe PE, Hyder SM. Targeting mutant p53 protein and the tumor vasculature: an effective combination therapy for advanced breast tumors. Breast Cancer Res Treat. 2011;125:407–420. | ||
Liang Y, Besch-Williford C, Aebi JD, et al. Cholesterol biosynthesis inhibitors as potent novel anti-cancer agents: suppression of hormone-dependent breast cancer by the oxidosqualene cyclase inhibitor RO 48-8071. Breast Cancer Res Treat. 2014;146:51–62. | ||
Grinter SZ, Liang Y, Huang SY, Hyder SM, Zou X. An inverse docking approach for identifying new potential anti-cancer targets. J Mol Graph Model. 2011;29:795–799. | ||
Cheng J, Lee EJ, Madison LD, Lazennec G. Expression of estrogen receptor beta in prostate carcinoma cells inhibits invasion and proliferation and triggers apoptosis. FEBS Lett. 2004;566:169–172. | ||
Sharma V, Verma V, Lal N, et al. Disulfiram and its novel derivative sensitize prostate cancer cells to the growth regulatory mechanisms of the cell by re-expressing the epigenetically repressed tumor suppressor-estrogen receptor β. Mol Carcinog. Epub 2015 Nov 24. | ||
Hussain S, Lawrence MG, Taylor RA, et al. Estrogen receptor β activation impairs prostatic regeneration by inducing apoptosis in murine and human stem/progenitor enriched cell populations. PLoS One. 2012;7:e40732. | ||
Shen HC, Shanmugasundaram K, Simon NI, et al. In silico discovery of androgen receptor antagonists with activity in castration resistant prostate cancer. Mol Endocrinol. 2012;26:1836–1846. | ||
Di Lorenzo G, Bracarda S, Buonerba C, Aieta M, Mirone V. Poor survival in prostate cancer patients with primary refractoriness to docetaxel. Eur Urol. 2014;65:505–507. | ||
Rajan P, Sudbery IM, Villasevil ME, et al. Next-generation sequencing of advanced prostate cancer treated with androgen-deprivation therapy. Eur Urol. 2014;66:32–39. | ||
Mafuvadze B, Liang Y, Hyder SM. Cholesterol synthesis inhibitor RO 48-8071 suppresses transcriptional activity of human estrogen and androgen receptor. Oncol Rep. 2014;32:1727–1733. | ||
Tan MH, Li J, Xu HE, Melcher K, Yong EL. Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol Sin. 2015;36:3–23. | ||
Boegemann M, Schrader AJ, Krabbe LM, Herrmann E. Present, emerging and possible future biomarkers in castration resistant prostate cancer (CRPC). Curr Cancer Drug Targets. 2015;15:243–255. | ||
Shazer RL, Jain A, Galkin AV, et al. Raloxifene, an oestrogen-receptor-beta-targeted therapy, inhibits androgen-independent prostate cancer growth: results from preclinical studies and a pilot phase II clinical trial. BJU Int. 2006;97:691–697. | ||
Lai KP, Huang CK, Chang YJ, et al. New therapeutic approach to suppress castration-resistant prostate cancer using ASC-J9 via targeting androgen receptor in selective prostate cells. Am J Pathol. 2013;182:460–473. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.