")
Back to Journals » International Journal of Nanomedicine » Volume 16
Chitosan-Poly(Acrylic Acid) Nanoparticles Loaded with R848 and MnCl2 Inhibit Melanoma via Regulating Macrophage Polarization and Dendritic Cell Maturation
Authors Liu X, Xu Y, Yin L, Hou Y , Zhao S
Received 1 May 2021
Accepted for publication 11 August 2021
Published 21 August 2021 Volume 2021:16 Pages 5675—5692
DOI https://doi.org/10.2147/IJN.S318363
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Xinghan Liu,1 Yujun Xu,1 Lijie Yin,1 Yayi Hou,1,2 Shuli Zhao3
1The State Key Laboratory of Pharmaceutical Biotechnology, Division of Immunology, Medical School, Nanjing University, Nanjing, 210093, People’s Republic of China; 2Jiangsu Key Laboratory of Molecular Medicine, Nanjing University, Nanjing, 210093, People’s Republic of China; 3General Clinical Research Center, Nanjing First Hospital, Nanjing Medical University, Nanjing, 210006, People’s Republic of China
Correspondence: Yayi Hou
The State Key Laboratory of Pharmaceutical Biotechnology, Division of Immunology, Medical School, Nanjing University, Nanjing, 210093, People’s Republic of China
, Tel/Fax +86-25-8968-8441
Email [email protected]
Shuli Zhao
General Clinical Research Center, Nanjing First Hospital, Nanjing Medical University, Nanjing, 210006, People’s Republic of China
, Tel/Fax +86-25-8968-8441
Email [email protected]
Purpose: Since immune cells in the tumor microenvironment (TME) can affect the development and progression of tumors, strategies modulating immune cells are considered to have an important therapeutic effect. As a TLR7/8 agonist, R848 effectively activates the innate immune cells to exert an anti-tumor effect. Mn2+ has been reported to strongly promote the maturation of antigen-presenting cells (APCs), thereby enhancing the cytotoxicity of CD8+ T cells. Thus, we tried to investigate whether chitosan-poly(acrylic acid) nanoparticles (CS-PAA NPs) loaded with R848 and MnCl2 (R-M@CS-PAA NPs) could exert an anti-tumor effect by regulating the function of immune cells.
Methods: R-M@CS-PAA NPs were prepared, and their basic characteristics, anti-tumor effect, and potential mechanisms were explored both in vitro and in vivo.
Results: R-M@CS-PAA NPs easily released MnCl2 and R848 at low pH. In B16F10 mouse melanoma model, R-M@CS-PAA NPs exerted the most significant anti-melanoma effect compared with the control group and CS-PAA NPs loaded with R848 or MnCl2 alone. FITC-labeled R-M@CS-PAA NPs were displayed to be accumulated at the tumor site. R-M@CS-PAA NPs significantly increased the infiltration of M1 macrophages and CD8+ T cells but reduced the number of suppressive immune cells in the TME. Moreover, in vitro experiments showed that R-M@CS-PAA NPs polarized macrophages into the M1 phenotype to inhibit the proliferation of B16F10 cells. R-M@CS-PAA NPs also enhanced the killing function of CD8+ T cells to B16F10 cells. Of note, R-M@CS-PAA NPs not only promoted the maturation of APCs such as dendritic cells and macrophages by STING and NF-кB pathways, but also enhanced the ability of dendritic cells to present ovalbumin to OT-I CD8+ T cells to enhance the cytotoxicity of OT-I CD8+ T cells to ovalbumin-expressing B16F10 cells.
Conclusion: These data indicate that the administration of R-M@CS-PAA NPs is an effective therapeutic strategy against melanoma.
Keywords: T cell, STING, NF-кB, antigen-presenting cell
Introduction
The tumor microenvironment (TME), consisting of various cells such as tumor cells and immune cells, and non-cellular components such as chemokines and growth factors, plays a pivotal role during the occurrence, growth, and metastasis of tumors.1 Some immune cells in the TME promote the formation of an immunosuppressive microenvironment, which facilitates tumor growth.2 In solid tumors, macrophages can account for up to 50% of all immune cells, representing one of the most abundant cells in the TME.3 Macrophages are divided into two types: M1 (classically activated macrophages) and M2 (alternatively activated macrophages). Among them, M1 macrophages exert a pro-inflammatory effect to slow down tumor progression.4 While tumor-associated macrophages (TAMs), mainly considered as M2 macrophages, promote tumor invasion by producing matrix metalloproteinases and contribute to tumor angiogenesis by releasing platelet-derived growth factor and vascular endothelial growth factor.5 Additionally, some studies have shown that TAM-derived cytokines and proteins, such as CCL-17, arginase 1, and galectin 3, contribute to the formation of an immunosuppressive TME.6,7 In contrast, infiltrated CD8+ T cells in the TME recognize and destroy tumor cells. After stimulation by professional antigen-presenting cells (APCs), CD8+ T cells proliferate and differentiate into effector cells, namely cytotoxic T lymphocytes (CTLs).8 As the most effective type of APCs, dendritic cells (DCs) can internalize and present tumor-associated antigens, and trigger the expansion of tumor-specific CD8+ T cells.9–12 Activated CD8+ T cells destroy tumor cells by releasing perforin and granzyme or producing Fas ligand and TNF-related apoptosis-inducing ligand.13 It is evident that promoting the polarization of macrophages to the M1 type or enhancing the function of CD8+ T cells and DCs may be a reasonable strategy for the treatment of tumors. However, there are some limitations and obstacles in the current development of tumor immunotherapies, such as the focus on the regulation of T cells, the selection of molecules that do not efficiently affect M1 macrophage polarization, and the unsatisfactory effects of monotherapy.
To address these challenges, we tried to design nanoparticles (NPs) that can active innate immunity to response against tumors. Rodell CB et al selected 38 drugs related to macrophage polarization from the literature and found that the Toll-like receptor (TLR)7/8 agonist R848 (Resiquimod) was the most effective driver for the M1 polarization of TAMs through morphometric analysis.14,15 Studies have shown that R848 not only promote the switching of TAMs from the M2 to M1 type, but also act as an effective adjuvant for DC maturation.16 In vitro stimulation and intravenous injection of R848 increase the number of CD11c+ DCs in the spleen of mice.17 These activated DCs secrete tumor necrosis factor (TNF)-α and interleukin (IL)-12 and migrate to the T-cell area in vivo.18 However, oral or intravenous administration of R848 leads to the rapid clearance of R848 from plasma and induces side effects such as systemic inflammation and autoimmune reactions.19,20 Therefore, it necessary to seek alternative ways to control the release of R848 and reduce its toxicity.
Manganese (Mn), a nutrient inorganic trace element, participates in the regulation of the immune system.21 Recent studies have reported that mice insufficient in Mn exhibit a higher tumor burden and reduced number of tumor-infiltrating CD8+ T cells than normal mice. Moreover, Mn2+ strongly promotes the maturation and antigen-presenting abilities of DCs and macrophages, thereby enhancing the activation of CD8+ T cells and natural killer cells,22 and up-regulating type I interferon (IFN) expression.23 In vivo experiments have shown that type I IFN is essential for spontaneous tumor-specific CD8+ T-cell priming, thereby controlling the growth of tumors.24,25 Recently, a new type of nano-Mn compound adjuvant has been developed for activating humoral, cellular, and mucosal immunity, especially the cytotoxic T cell-mediated immune response.26,27 However, excess exposure to Mn2+ can cause cognitive impairment.28 The delivery of Mn2+ with NPs may help in its controlled release, thereby reducing neurotoxicity. However, Mn2+-loaded NPs have been mostly used in studies involving cancer imaging,29 and few studies have demonstrated the role of Mn2+-loaded NPs alone or in combination with other substances in tumor treatment and immune cell regulation.
Chitosan, an abundant polysaccharide extracted from chitin, is used in the modification of heart patches to improve the efficacy for stem-cell-based therapy and in the preparation of NPs for drug delivery into intestinal organoids due to its non-toxicity, low allergenicity, and excellent biodegradability and biocompatibility.30–32 By conjugating 6-mercaptopurine to carboxymethyl chitosan, the synthesized NPs can target folate receptors and enhance intracellular drug accumulation in tumor cells.33 In addition, chitosan-coated NPs significantly increase the hepatic accumulation and bioavailability of insoluble drugs like zedoary turmeric oil and present with pH sensitivity; therefore, chitosan-coated NPs can release anti-tumor drugs such as doxorubicin and daunorubicin in the acidic TME.34–36 Notedly, chitosan-poly(acrylic acid) NPs (CS-PAA NPs) are water-soluble hollow nanospheres prepared from chitosan and poly(acrylic acid) that present with low toxicity and good biodegradability.37 In this study, CS-PAA NPs were selected to delivery R848 and MnCl2 based on their following properties. First, R848 is a hydrophobic drug, which can be loaded into the hydrophobic cores of CS-PAA NPs via hydrophilic reaction.38 Second, CA-PAA NPs have numerous carboxylic acid groups, making them suitable for Mn2+ loading.39 Finally, CS-PAA NPs display pH-dependent dissolution behavior, which may facilitate the release of R848 and MnCl2 in the acidic TME.40 Thus, we prepared novel CS-PAA NPs loaded with R848 and MnCl2 (R-M@CS-PAA NPs) and explored their effects on melanoma growth and regulation of immune cells, including macrophages, T cells, and DCs, and the potential underlying mechanisms.
Materials and Methods
Materials
Chitosan and acrylic acid were obtained from China National Pharmaceutical Group Corporation (Beijing, China) and Aladdin Biochemical Technology Co., Ltd. (Shanghai, China), respectively. R848 and MnCl2 were procured from Yuanye Bio-Technology Co., Ltd. (Shanghai, China) and Sigma-Aldrich (MO, USA), respectively.
Preparation of NPs
Preparation of CS-PAA NPs
To prepare CS-PAA NPs, 0.4 g of chitosan, 0.2 mL of an analytically pure acrylic acid solution, and 50 mL of deionized water were mixed in a 100 mL flask and heated to 80 °C under magnetic stirring (400 rpm), followed by the addition of potassium persulfate (0.03 g). The mixture was allowed to react for 1 h and then cooled down to room temperature. Finally, the compound was centrifuged at 12,000 rpm for 30 min, and the precipitate was ultrasonically dispersed in deionized water. After repeating the last step for three times, the precipitate was lyophilized for use.
Preparation of R-M@CS-PAA NPs
To prepare R-M@CS-PAA NPs, 2 mL of R848 solution (2.5 mg/mL) and 5 mL of MnCl2 solution (10 mg/mL) were added dropwise into 10 mL of CS-PAA NP solution (10 mg/mL). Then, the suspension was centrifuged at 12,000 rpm for 30 min. Finally, the precipitate was collected and re-dispersed for later use.
Preparation of FITC-Labeled R-M@CS-PAA NPs
To prepare FITC-labeled R-M@CS-PAA NPs, 0.2 mL of FITC solution (0.1 g of FITC dissolved in dimethyl sulfoxide [DMSO]) was mixed with 10 mL of CS-PAA NP solution (2 mg/mL), and the mixture was magnetically stirred for 12 h in the dark. Then, the compound was dialyzed for 72 h in a dialysis bag (14 kDa) to obtain the FITC-labeled R-M@CS-PAA NPs.
Characterization of R-M@CS-PAA NPs
The size distribution, zeta potential, and polymer dispersion coefficient (PDI) of CS-PAA NPs and R-M@CS-PAA NPs were examined via a dynamic light scattering (DLS; Nanosizer ZS90, Malvern Zetasizer, UK). The morphologies of CS-PAA NPs, R848@CS-PAA NPs, MnCl2@CS-PAA NPs, and R-M@CS-PAA NPs were observed under a transmission electron microscope (TEM; JEM-1200EX, JEOL, Japan). To evaluate their stability in serum, R-M@CS-PAA NPs were diluted with 1640 medium containing 10% fetal bovine serum (FBS; Grand Island, NY, USA) and then incubated at 37 °C. The particle sizes and zeta potentials of the synthesized NPs were detected by DLS at the indicated time points.
In vitro Drug Release
The release kinetics of R848 and MnCl2 from R-M@CS-PAA NPs was evaluated by the dialysis method. Ten milliliters of R-M@CS-PAA NPs (2 mg/mL) were placed into a dialysis bag (MWCO = 14 kDa) and then transferred into 10 mL of 0.1M phosphate-buffered saline (PBS) solution with different pH values (5.5, 6.8, and 7.4). After incubation in a 37 °C shaker without light for the indicated time points, the dialysate outside the dialysis bag was regularly collected for testing, and 10 mL of fresh 0.01 M PBS solution was re-added. Finally, the liquid in the dialysis bag was taken out, and 1 mL of hydrochloric acid solution (1 M) was added to ultrasonically destroy the NPs. The collected solutions were divided into two groups. One group was used to measure the Mn2+ content by the salicylic hydroxamic acid method, and the absorption values were assessed by a microplate reader at 580 nm; the other group was used to measure the R848 content by a microplate reader at an emission wavelength of 490 nm and an excitation wavelength of 350 nm.
Cell Culture
RAW 264.7 cells, B16F10 melanoma cells, and ovalbumin (OVA)-expressing B16F10 (B16F10-OVA) cells were obtained from Ruibowen Company (Nanjing, China). RAW 264.7 and B16F10 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Carlsbad, CA, USA) or RPMI-1640 with 10% FBS (Gibco) and 1% v/v penicillin-streptomycin (Gibco) at 37 °C in a 5% CO2 incubator. B16F10-OVA cells were cultured in DMEM medium containing 10% FBS, 100 μg/mL hygromycin B, and 1% v/v penicillin-streptomycin at 37 °C in a 5% CO2 incubator.
Tumor Mouse Model
All of the animal experiments were approved by the Institutional Animal Care and Use Committee of Nanjing First Hospital, Nanjing Medical University (DW20201204) and were performed in accordance with the guidelines published by the National Institutes of Health. Specific pathogen-free (SPF)-grade C57BL/6 male mice (5–6 weeks old, 18–20g) were obtained from Nanjing Model Animal Institute and maintained in an SPF-grade animal room with a temperature range of 20–22 °C and a humidity of 40–60% under a 12 h light/dark alternate.
To establish the melanoma mouse model, PBS (100 μL) containing 5×105 B16F10 cells was subcutaneously administrated into the right flank of the mice. When the tumor volume reaching approximately 100 mm3 (on day 9), the mice were randomly divided into five groups and treated with PBS, CS-PAA NPs (0.5 mg CS-PAA NPs per mice), R848@CS-PAA NPs (20 μg R848 and 0.5 mg CS-PAA NPs per mice), MnCl2@CS-PAA NPs (200 μg MnCl2 and 0.5 mg CS-PAA NPs per mice), and R-M@CS-PAA NPs (20 μg R848, 200 μg MnCl2 and 0.5 mg CS-PAA NPs per mice), respectively, via tail vein injection every day for seven days. Mice were weighted, and their tumor sizes were measured using calipers on alternate days. Tumor volume was calculated according to the formula: volume (V) = length (L) x width (W)2/2.
Tracking of R-M@CS-PAA NPs
FITC-labeled R-M@CS-PAA NPs were prepared to study their distribution in vivo and uptake by cells in vitro. Hundred microliters of R-M@CS-PAA NPs containing 20 μg R848, 200 μg MnCl2, and 0.5 mg CS-PAA NPs was intravenously injected into melanoma-bearing mice. The distribution of R-M@CS-PAA NPs in vivo was visualized using IVIS Lumina XR (Caliper Life Science, Hopkinton, MA, USA) and detected by flow cytometry (FCM). The uptake of R-M@CS-PAA NPs by bone marrow-derived DCs (BMDCs) in vitro was confirmed by FCM after BMDCs were treated with FITC-labeled R-M@CS-PAA NPs for 6 h.
Analysis of Tumor-Infiltrating Immunocytes
Tumor tissues were harvested and digested with 2 mL of 1640 medium containing 5% FBS, 2 mg/mL collagenase IV (Sigma), and 5 U/mL DNase I (Sigma) for 20 min in a 37 °C incubator. Single cell suspensions were obtained after lysing the tissues with 2 mL of blood cell lysis buffer for 2 min and filtering through a 70 μm mesh. The cells were then incubated with the following anti-mouse antibodies: CD45-PerCP (45–0451-80, eBioscience), CD11b-PE/Dazzle 594 (101255, BioLegend), Gr-1-APC (17–5931, eBioscience), CD3-apccy7 (10221, BioLegend), CD4-percy7 (25–0041, eBioscience), CD8-FITC (11–0081, eBioscience), Ly6C-FITIC (128015, BioLegend), Ly6G-PE, F4/80-FITC (11–4801, eBioscience), iNOS-PE (12–5920, eBioscience), CD206-APC (17–2061, eBioscience), CD25-APC (102011, BioLegend), FOXP3-PE (126403, BioLegend), CD69-af700 (104539, BioLegend), granzyme B-PE (12–8898, eBioscience), IFN-γ-PE (12–7311, eBioscience), TNF-α-percy 7 (25–7321, eBioscience), and perforin-APC (17–9392, eBioscience). FCM was performed using CYTEK Aurora (Cytek Biosciences, USA) to assess the immunocyte populations. Before detecting the ratio of IFN-γ+ and TNF-α+CD8+ T cells in tumor tissues, cell suspensions were stimulated with 1640 medium containing 10% FBS, PMA (20 ng/mL), BFA (20 μg/mL), and ionomycin (1 μg/mL) in a 37°C incubator for 4 h. The flow cytometric data were analyzed using the FlowJo software (Treestar, Ashland, OR, USA).
Immunohistochemistry
Tumor tissues (thickness: 4 μm) from mice with different treatment groups were fixed with 4% paraformaldehyde (ServiceBio, Wuhan, China) and embedded in paraffin. The proliferation of tumors was evaluated using an antibody against Ki67. Images were captured using a light microscope and then quantified by Image J software (National Institutes of Health, USA).
Enzyme-Linked Immunosorbent Assay (ELISA)
The expression levels of IL-12, IFN-β, IFN-γ, and TNF-α in the supernatants of cultured cells or sera of mice were assayed using commercially available mouse IL-12 ELISA kit (Elabscience, Wuhan, China), IFN-β ELISA kit (Elabscience), IFN-γ ELISA kit (Elabscience), and TNF-α ELISA kit (4A Biotech Co., Ltd., Beijing, China), respectively, according to the manufacturer’s instructions.
Cell Viability Assay
RAW 264.7 cells, BMDMs, and B16F10 cells were seeded in 96-well plates at appropriate densities in quintuplicate and treated with CS-PAA NPs (25 μg/mL), R848@CS-PAA NPs (1 μg/mL R848 + 25 μg/mL CS-PAA NPs), MnCl2@CS-PAA NPs (10 μg/mL MnCl2 + 25 μg/mL CS-PAA NPs), and R-M@CS-PAA NPs (1 μg/mL R848 + 10 μg/mL MnCl2 + 25 μg/mL CS-PAA NPs) for 48 h. Then, the cell viability was assessed using Cell Counting Kit-8 (CCK-8) assay according the manufacturer’s instructions.
Apoptosis Assay
B16F10 cells were treated with CS-PAA NPs (25 μg/mL), R848@CS-PAA NPs (1 μg/mL R848 + 25 μg/mL CS-PAA NPs), MnCl2@CS-PAA NPs (10 μg/mL MnCl2 + 25 μg/mL CS-PAA NPs), and R-M@CS-PAA NPs (1 μg/mL R848 + 10 μg/mL MnCl2 + 25 μg/mL CS-PAA NPs) for 48 h. Then, the cells were collected and resuspended in 100 μL of binding buffer solution containing 5 μL of Annexin V and 2.5 μL of propidium iodide (PI). After incubating for 15 min at 25°C in the dark, the cells were diluted with binding buffer solution and subjected to FCM analysis.
Generation of Bone Marrow-DCs (BMDCs)
Male C57BL/6 mice (6–8 weeks old) were sacrificed by cervical vertebra dislocation and were sterilized by soaking them in a solution containing 75% alcohol for 5 min. To obtain the bone marrow cells, the femurs and tibias were separated using scissors and 1 mL sterile syringes. Then, the cells were lysed with 1 mL of red blood cell lysis buffer for 2 min, filtered through a gauze, and cultured in complete RPMI-1640 medium containing 40 ng/mL recombinant mouse granulocyte-macrophage colony-stimulating factor (GM-CSF; PeproTech, USA) and 20 ng/mL recombinant mouse IL-4 (PeproTech, USA) to obtain BMDCs. During this period, the medium was replaced with fresh complete medium containing cytokines on day 3 and 5, and the cells were used for subsequent experiments on day 6.
Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
Total RNA from treated cells was extracted using TRIzol reagent (Invitrogen, Grand Island, NY, USA). Then, 1 μg of the collected RNA was reverse-transcribed to cDNA using HiScript II Q RT SynthesisMix Kit (Vazyme, Nanjing, China) according to the manufacturer’s instructions, and cDNA was subjected for PCR quantification by SYBR Green SuperMix reagent (Bio-Rad, CA, USA). The expression levels of target genes were analyzed using the 2−ΔΔCt method using GAPDH as an endogenous control. The sequences of primers used in qRT-PCR are listed in Table 1.
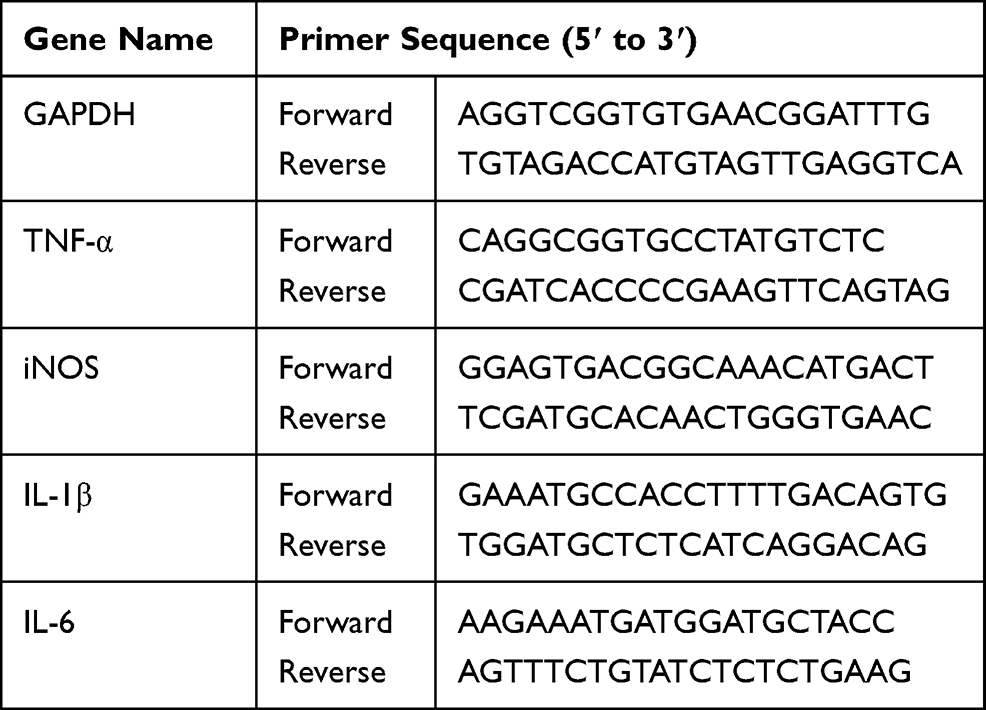 |
Table 1 Primer Sequences Used in qRT-PCR |
Isolation of CD8+ T Cells
C57BL/6 mice were obtained from Nanjing Model Animal Institute, and OT-I mice were purchased from Ziyuan Experimental Animal Technology Co., Ltd. (Hangzhou, China). To isolate CD8+ T cells, spleens were placed in PBS and milled by a sterile syringe. Then, the cell suspensions were collected and filtered into a centrifuge tube through a cell sieve. The cells were centrifuged and lysed in 2 mL of red blood cell lysis buffer for 2 min. After washing with PBS for three times, the cells were resuspended in an appropriate amount of magnetic-activated cell sorting (MACS) running buffer (107 cells/40 μL) and biotin-antibody cocktail (107 cells/10 μL), followed by incubation at 4 °C for 5 min. An appropriate amount of MACS running buffer (107 cells/30 μL) and anti-biotin microbeads (107 cells/30 μL) was sequentially added to the cells, followed by incubation at 4 °C for 10 min. Finally, the cells were sorted using MACS separation columns (Miltenyi Biotec, Germany) to obtain CD8+ T or OT-I CD8+ T cells.
Co-Culture of Cells
Co-Culture of B16F10 and RAW 264.7 Cells
First, B16F10 cells were pre-stained with the cell proliferation dye carboxyfluorescein diacetate succinimidyl ester (CFSE; Dojindo, Shanghai, China). Then, B16F10 and RAW 264.7 cells were indirectly co-cultured at a ratio of 1:2 and treated with CS-PAA NPs (25 μg/mL), R848@CS-PAA NPs (1 μg/mL R848 + 25 μg/mL CS-PAA NPs), MnCl2@CS-PAA NPs (10 μg/mL MnCl2 + 25 μg/mL CS-PAA NPs), and R-M@CS-PAA NPs (1 μg/mL R848 + 10 μg/mL MnCl2 + 25 μg/mL CS-PAA NPs) for 48 h and then subjected to FCM analysis. The proliferation index (PI) of tumor cells was assessed using Mod Fit LT 3.0 software and the cell growth inhibition rate was calculated according to the following equation: Cell growth inhibition rate = (1-(PI [groups with different treatment]/PI [control group])) × 100%.
Co-Culture of BMDCs and Spleen Cells
Spleen cells obtained from mice were stained with CFSE. BMDCs and CFSE-labeled spleen cells at a ratio of 1:2 were directly co-cultured and treated with CS-PAA NPs (25 μg/mL), R848@CS-PAA NPs (1 μg/mL R848 + 25 μg/mL CS-PAA NPs), MnCl2@CS-PAA NPs (10 μg/mL MnCl2 + 25 μg/mL CS-PAA NPs), and R-M@CS-PAA NPs (1 μg/mL R848 + 10 μg/mL MnCl2 + 25 μg/mL CS-PAA NPs). After 96 h, the proliferation ability of CD8+ T cells was detected by FCM.
Co-Culture of BMDCs, OT-I CD8+ T Cells and B16F10-OVA Cells
BMDCs, OT-I CD8+ T and B16F10-OVA cells (labeled with CFSE) at a ratio of 1:2:2 were co-cultured with a mixture of 1 μg/mL mouse recombinant CD3 and 2 μg/mL CD28 cytokines in the 1640 medium. Then, the co-cultured cells were treated with CS-PAA NPs (25 μg/mL), R848@CS-PAA NPs (1 μg/mL R848 + 25 μg/mL CS-PAA NPs), MnCl2@CS-PAA NPs (10 μg/mL MnCl2 + 25 μg/mL CS-PAA NPs), and R-M@CS-PAA NPs (1 μg/mL R848 + 10 μg/mL MnCl2 + 25 μg/mL CS-PAA NPs). After 48 h of treatment, the cells in the supernatant and the adherent B16F10-OVA cells were collected and analyzed by FCM.
Western Blotting (WB) Analysis
Total proteins were extracted with radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime, Shanghai, China), and the protein concentration was quantified using a bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA). Then, the membranes were incubated with Tris-buffered saline with 0.1% Tween 20 (TBST) containing 5% bovine serum albumin (BSA) solution for 1.5 h at 25°C. After treating with primary antibodies at a dilution ratio of 1:1000 at 4°C overnight and secondary antibodies at a dilution ratio of 1:5000 for 1 h at 25°C, the protein bands in the membranes were observed using ECL Plus WB detection reagents (Affinity Biosciences, OH, USA). The following primary antibodies were used: rabbit anti-GAPDH (Cell Signaling Technology [CST]), rabbit anti-phosphorylated (p)-TBK1 (CST), and rabbit anti-TBK1 (CST), rabbit anti-p-IKK α (CST), rabbit anti-IKK α (CST), α rabbit anti-p-P65 (CST), and rabbit anti-P65 (CST). Anti-rabbit IgG, HRP-linked antibody (CST) was used as the secondary antibody.
Statistical Analysis
Student’s t-test or one-way analysis of variance (ANOVA) was used to analyze the data in Prism 6 (GraphPad Software, Inc., San Diego, CA, USA). Statistically significant differences were indicated as*p < 0.05 and **p < 0.01.
Results
Synthesis and Characterization of R-M@CS-PAA NPs
The particle sizes of CS-PAA NPs and R-M@CS-PAA NPs were 176.5 ± 9.3 nm and 195.8 ± 11.7 nm, respectively. CS-PAA NPs showed a positive zeta potential of 21.47 ± 2.98 mV, while the zeta potential of R-M@CS-PAA NPs showed a positive zeta potential of 28.40 ± 2.80 mV. The PDI signifies the uniformity of NPs. The PDI of CS-PAA NPs and R-M@CS-PAA NPs were 0.043 and 0.082, respectively, indicating that the surface system of NPs was well dispersed and had a low tendency to agglomerate. The morphologies of CS-PAA NPs, R848@CS-PAA NPs, MnCl2@CS-PAA NPs, and R-M@CS-PAA NPs were spherical, as revealed by TEM (Figure 1A).
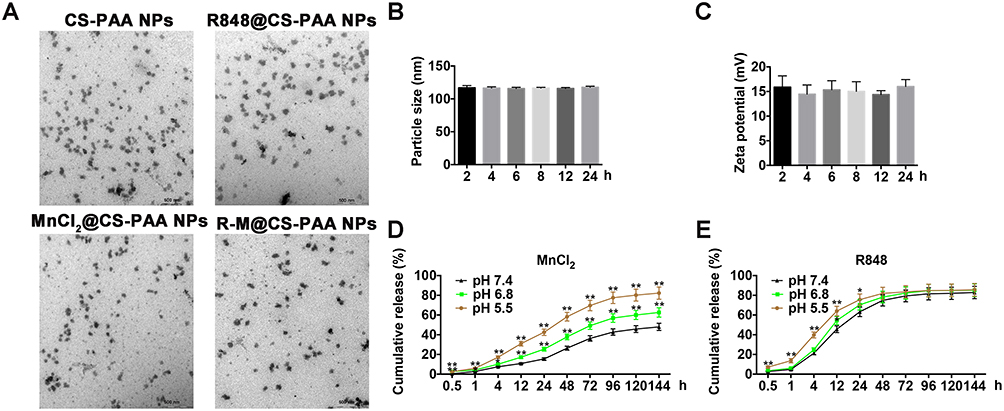 |
Figure 1 Characteristics of R-M@CS-PAA NPs. (A) The morphologies of CS-PAA NPs, R848@CS-PAA NPs, MnCl2@CS-PAA NPs, and R-M@CS-PAA NPs were observed by TEM. (B and C) Particle sizes (B) and zeta potentials (C) of R-M@CS-PAA NPs in 1640 medium containing 10% FBS at the indicated time points. (D and E) Release profiles of MnCl2 (D) and R848 (E) from R-M@CS-PAA NPs. *p < 0.05, **p < 0.01. |
The stability of NPs in serum is a key parameter for systemic administration. Therefore, we incubated the NPs in 1640 medium containing 10% FBS for different times and found that the particle sizes and zeta potentials of R-M@CS-PAA NPs did not change significantly, showing good stability (Figure 1B and C). Furthermore, the release profiles of R-M@CS-PAA NPs were evaluated. In the presence of PBS with pH 7.4, approximately 26.34% of MnCl2 was released from R-M@CS-PAA NPs after 48 h, while at lower pH values (6.8 and 5.4), the release of MnCl2 was accelerated (release rates of 37.83% and 58.33%, respectively) (Figure 1D). In contrast, R848 was released more efficiently from the R-M@CS-PAA NPs. After 48 h of incubation in the PBS medium (pH = 7.4), 74.89% of R848 was released from R-M@CS-PAA NPs. Under the acidic environments of pH 5.5 and 6.8, the accumulated release of R848 increased to 78.25% and 81.93%, respectively (Figure 1E). The above results suggested that R-M@CS-PAA NPs were more sensitive to cleavage at lower pH, thereby exhibiting higher drug release rates.
R-M@CS-PAA NPs Inhibited the Growth of Mouse Melanoma in vivo
We established a subcutaneous melanoma mouse model by subcutaneously injecting 5×105 B16F10 cells mixed in 100 μL of PBS solution. The mice were subjected to different treatments (control, CS-PAA NPs, R848@CS-PAA NPs, MnCl2@CS-PAA NPs, and R-M@CS-PAA NPs) via tail vein injection. We found that both R848@CS-PAA NPs and MnCl2@CS-PAA NPs slowed down the growth of tumors. R-M@CS-PAA NPs had the strongest inhibitory effect on tumor growth as evidenced by the smaller tumor size, weight, and volume (Figure 2A–C). Moreover, the results of the Ki67 staining experiment showed that tumor cells of mice in the R-M@CS-PAA NP group demonstrated the lowest proliferation level (Figure 2D and E).
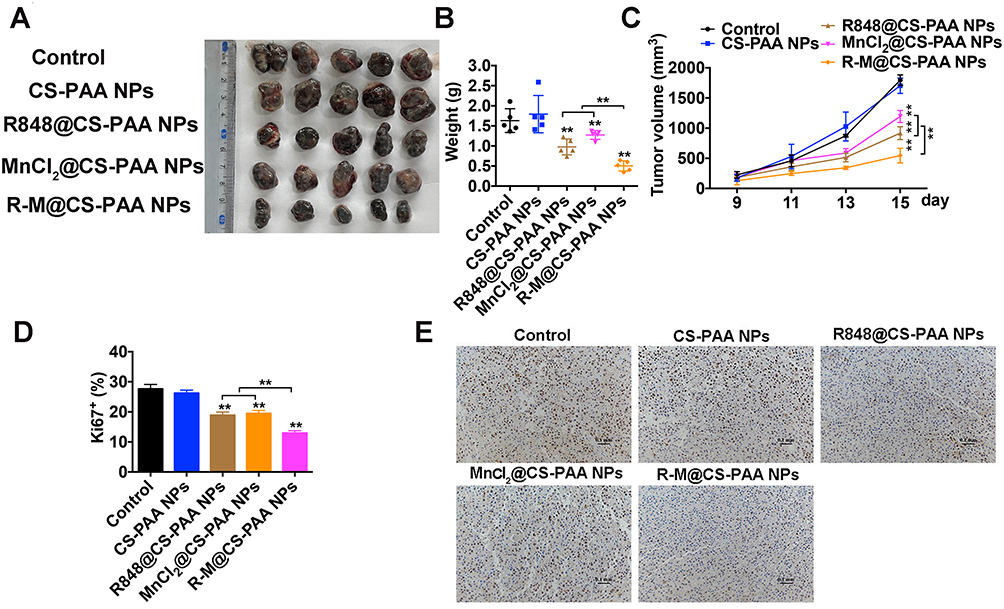 |
Figure 2 R-M@CS-PAA NPs suppressed tumor growth in vivo. (A) Photograph of tumor tissues from different mice groups. (B, C) Tumor weight (B) and tumor volume (C) of tumor tissues from different mice groups. (D, E) Quantitative data (D) and histopathologic photograph (E) of Ki67 staining of tumor tissues from different mice groups. **p < 0.01. |
To investigate the distribution of R-M@CS-PAA NPs in vivo, we labeled R-M@CS-PAA NPs with FTIC. By using a small animal live imaging device, we found that after 24 h of intravenous injection, FITC-labeled R-M@CS-PAA NPs were accumulated at the tumor site (Figure 3A). Then, we harvested different organs and tumor tissues from B16F10-bearing mice and prepared single-cell suspensions of different tissues. FCM results showed that after 3 h of intravenous injection, the accumulation of R-M@CS-PAA NPs was the highest in the kidney, followed by tumor tissues (Figure 3B and C). However, with an increase in the injection time, the accumulation of R-M@CS-PAA NPs in the tumor site was increased (Figure 3D and E). After 24 h of intravenous injection, the accumulation of R-M@CS-PAA NPs in tumor tissues was higher than that in the kidney and other tissues (Figure 3F and G). The results demonstrated that the systemic administration of R-M@CS-PAA NPs facilitated targeted drug delivery at the tumor site.
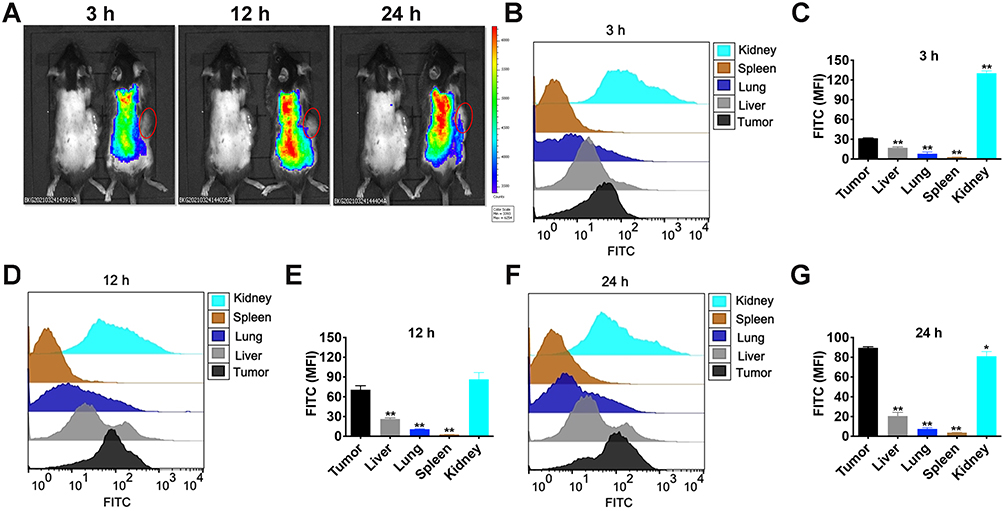 |
Figure 3 R-M@CS-PAA NPs were accumulated at the tumor site. (A) The distribution of R-M@CS-PAA NPs in vivo was observed by IVIS Lumina XR. (B) The distribution of R-M@CS-PAA NPs in vivo after 3 h of intravenous injection was detected FCM. (C) Quantitative data of (B). (D) The distribution of R-M@CS-PAA NPs in vivo after 12 h of intravenous injection was detected FCM. (E) Quantitative data of (D). (F) The distribution of R-M@CS-PAA NPs in vivo after 24 h of intravenous injection was detected FCM. (G) Quantitative data of (F). *p < 0.05, **p < 0.01. |
R-M@CS-PAA NPs Increased the Number of M1 Macrophages in TME
Tumor tissues from different groups of mice were harvested to explore the distributions of marrow-derived suppressive cells (MDSCs), macrophages, and their subgroups. Compared with the control, R848@CS-PAA NP, and MnCl2@CS-PAA NP groups, the infiltration of MDSCs in the tumor tissues of the R-M@CS-PAA NPs-treated group was significantly reduced, while the ratio of G-MDSCs and M-MDSCs was not affected (Figure 4A). Further analysis showed that compared with the control group, although R-M@CS-PAA NPs did not affect the number of total macrophages in tumor tissues, they significantly increased the numbers of M1-type macrophages and significantly reduced the ratio of M2 macrophages (Figure 4B).
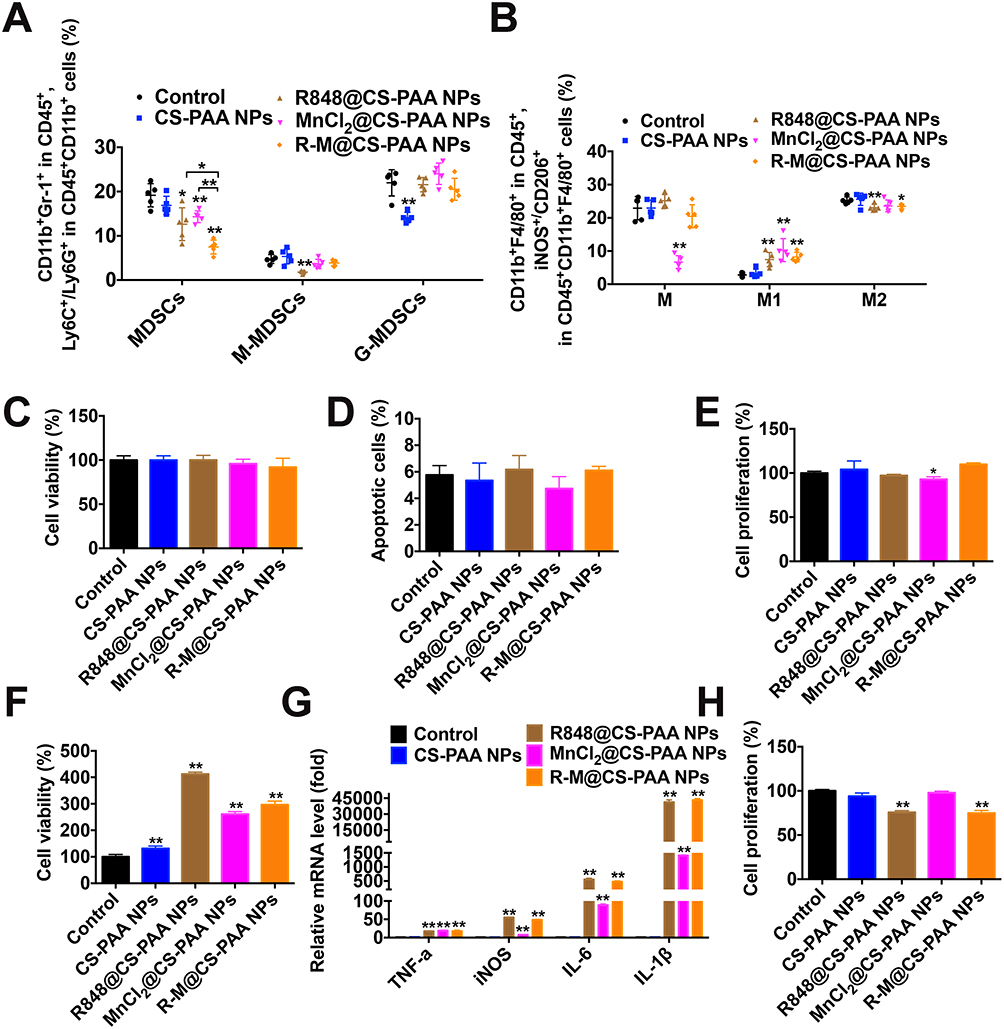 |
Figure 4 R-M@CS-PAA NPs increased the number of M1 macrophages. (A) Quantitative data from FCM analysis of MDSCs, M-MDSCs, and G-MDSCs in the tumor tissues from different mouse groups. (B) Quantitative data from FCM analysis of macrophages, M1 macrophages, and M2 macrophages in the tumor tissues from different mice groups. (C-E) B16F10 cells were treated with CS-PAA NPs, R848@CS-PAA NPs, MnCl2@CS-PAA NPs, and R-M@CS-PAA NPs for 48 h, and the cell viability (C), apoptotic rate (D), and proliferation ability (E) of B16F10 cells were examined by CCK-8, Annexin V/PI staining, and FCM, respectively. (F) RAW 264.7 cells were treated with CS-PAA NPs, R848@CS-PAA NPs, MnCl2@CS-PAA NPs, and R-M@CS-PAA NPs for 48 h, and the cell viability was examined by CCK-8 assay. (G) RAW 264.7 cells were treated with CS-PAA NPs, R848@CS-PAA NPs, MnCl2@CS-PAA NPs, and R-M@CS-PAA NPs for 6 h, and the mRNA expression levels of TNF-α, iNOS, IL-6, and IL-1β were examined by qRT-PCR. (H) In the presence of CS-PAA NPs, R848@CS-PAA NPs, MnCl2@CS-PAA NPs, and R-M@CS-PAA NPs, B16F10 cells were indirectly co-cultured with RAW 264.7 cells for 48 h, and the proliferation of B16F10 cells was evaluated by FCM. *p < 0.05, **p < 0.01. |
Given that R-M@CS-PAA NPs regulated the function of macrophages in vivo, we further analyzed their role in macrophages in vitro. We treated B16F10 or CFSE-labeled B16F10 cells with CS-PAA NPs, R848@CS-PAA NPs, MnCl2@CS-PAA NPs, and R-M@CS-PAA NPs for 48 h. The results showed that R-M@CS-PAA NPs treatment had no obvious effect on the viability, apoptosis, and proliferation ability of B16F10 cells (Figure 4C–E), indicating that R-M@CS-PAA NPs did not directly destroy B16F10 cells. R848@CS-PAA NPs, MnCl2@CS-PAA NPs, and R-M@CS-PAA NPs promoted the proliferation of RAW 264.7 cells and significantly upregulated the expression of M1 type macrophage markers like TNF-α, INOS, IL-6, and IL-1β (Figure 4F and G). However, there was no statistically significant difference between the effects of R848@CS-PAA NPs and R-M@CS-PAA NPs on macrophage polarization (Figure 4G). To confirm the effect of NPs-regulated macrophages on the proliferation of B16F10 cells, we indirectly co-cultured B16F10 and RAW 264.7 cells. As shown in Figure 4H, incubation with R848@CS-PAA NPs and R-M@CS-PAA NPs significantly inhibited the proliferation of B16F10 cells by 24.28% and 25.24%, respectively, but there was no statistical difference between the two groups. The above results illustrated that R-M@CS-PAA NPs reduced the proliferation of tumor cells by inducing the polarization of macrophages to the M1 type.
R-M@CS-PAA NPs Affected the Distribution and Function of T Cells
Since the distribution and function of T cells also affect the efficacy of drug treatment, similar to macrophages, we examined the distribution of T cells by FCM. Figure 5A showed that R848@CS-PAA NPs and MnCl2@CS-PAA NPs significantly increased the proportion of CD8+ T cells and significantly decreased the proportion of regulatory T cells (Tregs) in tumor tissues of mice, although the effect of R-M@CS-PAA NPs was more obvious. Additionally, the ratio of CD69+CD8+ T cells was significantly increased after treatment with R-M@CS-PAA NPs, suggesting the activation of CD8+ T cells (Figure 5B). Meanwhile, an increase in the number of CD8+ T cells expressing IFN-γ, TNF-α, and granzyme B was observed in the tumor tissues of mice treated with R-M@CS-PAA NPs, indicating that R-M@CS-PAA NPs may promote an anti-tumor immune response by activating CD8+ T cells (Figure 5C–E).
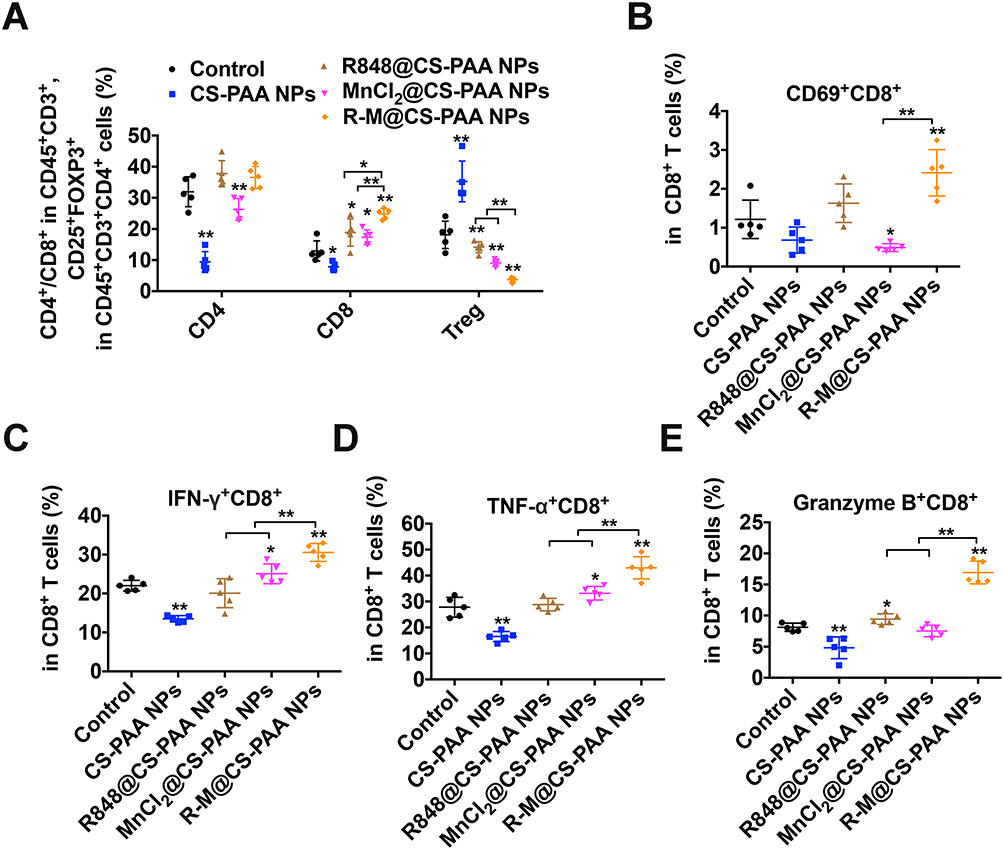 |
Figure 5 R-M@CS-PAA NPs affected the distribution of T cells in vivo. (A) Quantitative data of CD4+ T, CD8+ T, and Treg cells in tumor tissues with indicated treatments. (B-E) Quantitative data of the ratio of CD69+ (B), IFN-γ+ (C), TNF-α+ (D), and granzyme B+ (E) CD8+ T cells. *p < 0.05, **p < 0.01. |
Next, we evaluated the effects of CS-PAA NPs, R848@CS-PAA NPs, MnCl2@CS-PAA NPs, and R-M@CS-PAA NPs on the proliferation of CD8+ T cells. Spleen cells labeled with CFSE were stimulated with these NPs for 96 h. FCM results showed that except R848@CS-PAA NPs, other stimuli had no obvious effect on the proliferation of CD8+ T cells in the spleen (Figure 6A). Then, CD8+ T cells were isolated from the mouse spleen, and the effects of different NP treatments on CD8+ T cells were examined. After 48 h of exposure, the proportions of IFN-γ+, granzyme B+, and perforin+ CD8+ T cells in each group were significantly increased, of which R-M@CS-PAA NPs had the most significant effect (Figure 6B–D). However, the results of the ELISA experiment showed that after stimulation with CS-PAA NPs, R848@CS-PAA NPs, and MnCl2@CS-PAA NPs, the secretion of TNF-α, IFN-β, and IFN-γ in the supernatant of CD8+ T cells did not change significantly. Only R-M@CS-PAA NP treatment obviously up-regulated the expression of TNF-α and IFN-γ in the supernatant of CD8+ T cells (Figure 6E). Furthermore, we co-cultured CD8+ T cells and CFSE-labeled B16F10 cells with different NPs for 48 h. The data showed that the growth inhibition rate of B16F10 cells increased to 59.31% after incubation with R-M@CS-PAA NPs (Figure 6F).
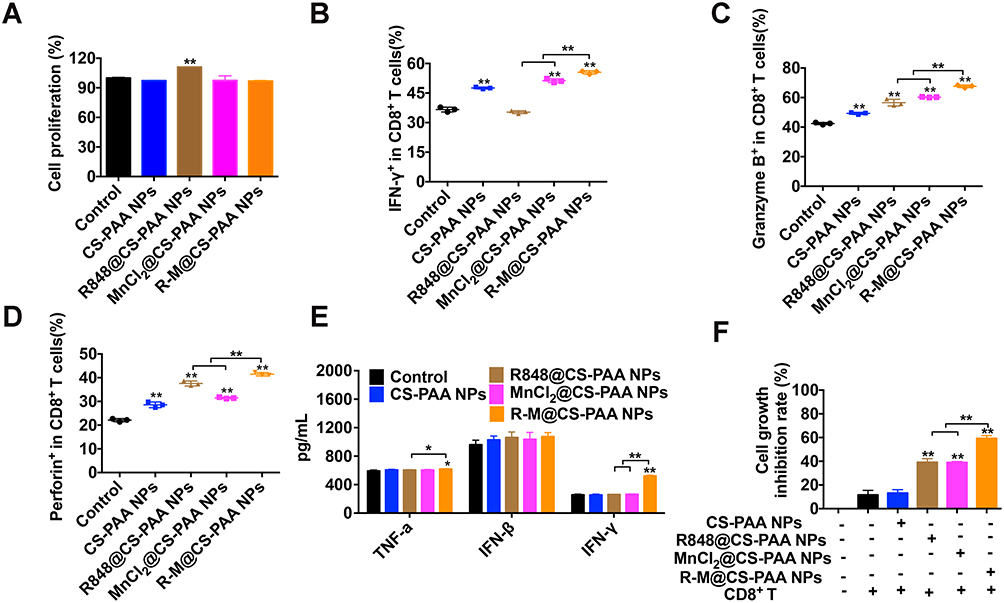 |
Figure 6 R-M@CS-PAA NPs affected the function of CD8+ T cells in vitro. (A) The effects of different NPs on the proliferation of CD8+ T cells. (B-D) CD8+ T cells were treated with different NPs for 48 h, and the proportion of IFN-γ+ (B), granzyme B+ (C), and perforin+(D) CD8+ T cells were examined by FCM. (E) CD8+ T cells were treated with different NPs for 48 h, and the expression levels of TNF-α, IFN-β, and IFN-γ in the supernatant were detected by ELISA assay. (F) In the presence of different NPs, B16F10 cells were co-cultured with CD8+ T cells for 48 h, and the proliferation of B16F10 cells was examined by FCM. *p < 0.05, **p < 0.01. |
R-M@CS-PAA NPs Induced DC Maturation via STING and NF-kB Pathways
Type I IFNs activate DCs, which in turn promote CD8+ T-cell activation.41 Therefore, we studied the effect of NPs on DC maturation. First, we treated BMDCs with FITC-labeled R-M@CS-PAA NPs and found R-M@CS-PAA NPs could be phagocytosed by 29.73% of the BMDCs after 6 h of incubation, as evidenced by the FCM results (Figure 7A). After 48 h of stimulation with different NPs, almost all BMDCs were still alive, indicating that these NPs did not affect the viability of BMDCs (Figure 7B). The further results of FCM showed that the expression levels of co-stimulatory molecules (CD80 and CD86) on the surface of BMDCs were upregulated after incubation with R848@CS-PAA NPs and MnCl2@CS-PAA NPs for 24 h; however, the expression levels of CD80 and CD86 reached a peak after treatment with R-M@CS-PAA NPs (Figure 7C–F).
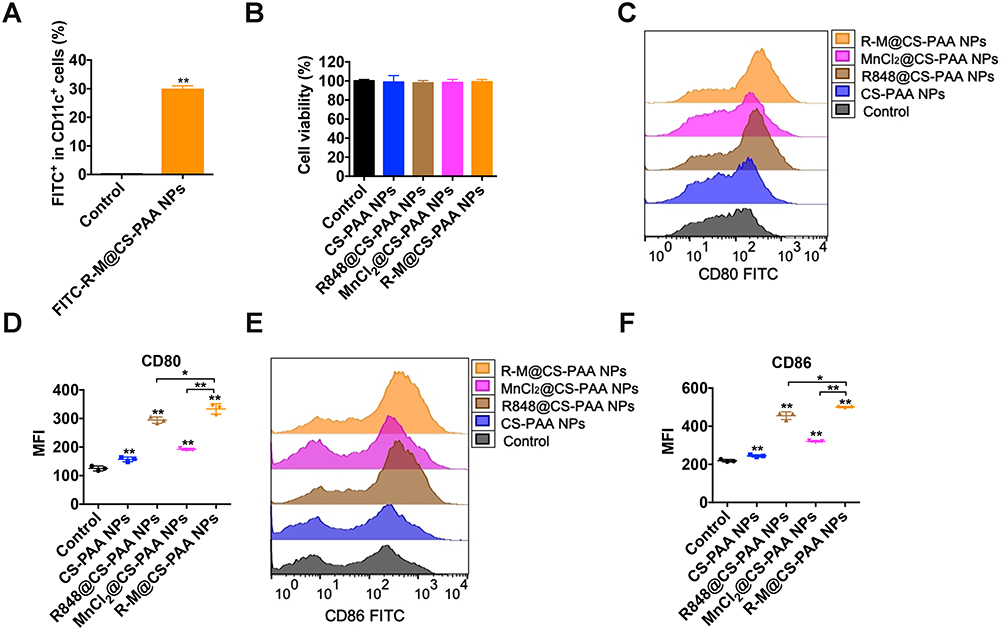 |
Figure 7 R-M@CS-PAA NPs induced the maturation of DCs. (A) BMDCs were stimulated with FITC-labeled R-M@CS-PAA NPs for 6 h, and the percentage of FITC+ BMDCs was examined by FCM. (B) BMDCs were treated with different NPs for 24 h, and the viability of BMDCs was detected by CCK-8 assay. (C) BMDCs were treated with different NPs for 24h, and the expression of CD80 was detected by FCM. (D) Quantitative data of (C). (E) BMDCs were treated with different NPs for 24h, and the expression of CD86 was detected by FCM. (F) Quantitative data of (E). *p < 0.05, **p < 0.01. |
Mature DCs trigger specific immune response via the major histocompatibility complex (MHC) I or MHC II antigen presentation pathway. Moreover, R848@CS-PAA NPs and R-M@CS-PAA NPs promoted the expression of MHC I and MHC II (Figure 8A and B). Several cytokines (IL-1β, IL-4, and TNF-α) stimulate the maturation of DCs; mature DCs secrete cytokines (IL-12 and TNF-α), which can further promote the maturation of DCs.42 IL-12, mainly secreted by APCs, mediates the production of TNF-α and IFN-γ, and promotes the proliferation and killing activity of T cells.43 TNF-α induces apoptosis and inhibits tumor growth.44 The results of ELISA showed that both R848@CS-PAA NPs and MnCl2@CS-PAA NPs increased the secretion of IL-12P70, TNF-α, and IFN-β by DCs, but not that of IFN-γ. Compared with R848@CS-PAA NPs and MnCl2@CS-PAA NPs, R-M@CS-PAA NPs significantly enhanced the secretion of these cytokines in the supernatants of DCs (Figure 8C). Studies have reported that Mn2+ activates the cGAS-STING pathway,45 and that R848 can activate the NF-кB signaling pathway via TLR7/8. Therefore, we speculated that R-M@CS-PAA NPs might exert their effects through STING and NF-кB signaling pathways. By WB analyses, we found that R-M@CS-PAA NPs significantly upregulated p-TBK1 expression via the STNG signaling pathway and p-IKKα and p-P65 expression via the NF-кB signaling pathway (Figure 8D), indicating that R-M@CS-PAA NPs might promote the maturation of BMDCs, at least in part, through STING and NF-кB pathways. Since macrophages also function as APCs, we detected the maturation-related molecules in RAW 264.7 cells after treatment with different NPs. FCM results found that the surface molecules including CD80, CD86, MHC I, and MHC II in RAW 264.7 cells were also upregulated by R-M@CS-PAA NPs (Figure 8E–H). These data displayed that R-M@CS-PAA NPs promoted the maturation and antigen presentation ability of professional APCs.
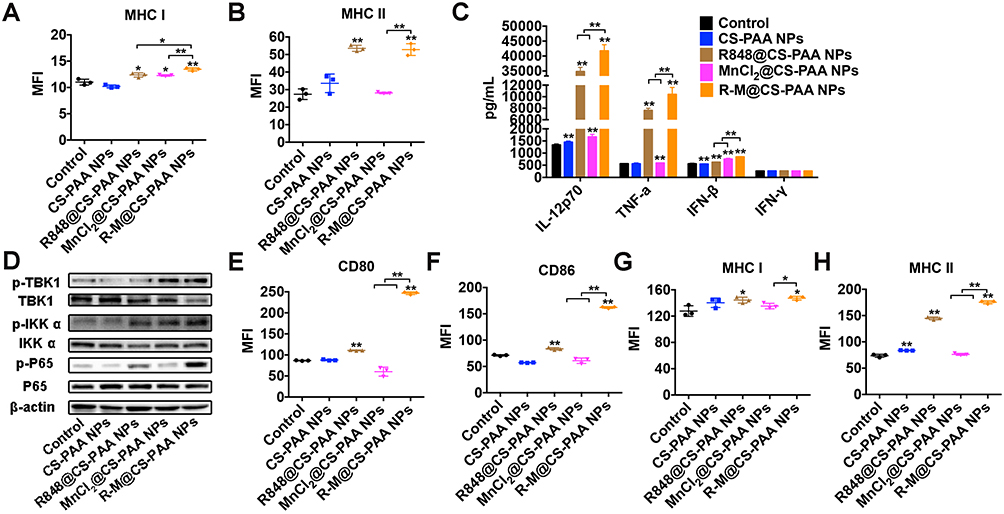 |
Figure 8 R-M@CS-PAA NPs induced the maturation of DCs via MAPK and STING pathways. (A, B) BMDCs were treated with different NPs for 24 h, and the expression of MHC I (A) and MHC II (B) was detected by FCM. (C) BMDCs were treated with different NPs for 24 h, and the expression levels of IL-12p70, TNF-α, IFN-β, and IFN-γ in the supernatant were detected by ELISA assay. (D) BMDCs were treated with different NPs for 24 h, and the expression of target genes was detected by WB assay. (E-H) RAW 264.7 cells were treated with different NPs for 24 h, and the expression of CD80 (E), CD86 (F), MHC I (G), and MHC II (H) was detected by FCM. *p < 0.05, **p < 0.01. |
R-M@CS-PAA NP-Treated DCs Enhanced the Cytotoxicity of OT-I CD8+ T Cells to B16F10-OVA Cells
Mature DCs can present antigens to CD8+ T cells to enhance the killing activity of CD8+ T cells by MHC I. Therefore, we constructed B16F10-OVA cells and co-cultured them with BMDCs to investigate the antigen-presenting ability of BMDCs. As displayed in Figure 9A, after 48 h, the proportions of BMDCs expressing OVA was significantly increased, indicating the successful presentation of OVA by BMDCs. Then, we isolated OT-I CD8+ T cells from the spleen of OT-I mice, labeled B16F10-OVA cells with CFSE, and co-cultured BMDCs, OT-I CD8+ T cells, and CFSE-labeled B16F10-OVA cells at a ratio of 1:2:2. After stimulation with different NPs for 48 h, the cells in the supernatant (mainly BMDCs and OT-I CD8+ T cells) and adherent B16F10-OVA cells were collected. The proportion of cells in the supernatant was detected by FCM, which showed that the proportions of TNF-α+, IFN-γ+, granzyme B+, and perforin+ OT-I CD8+ T cells were significantly increased in the R-M@CS-PAA NP group (Figure 9B–E). As shown in Figure 9F, R848@CS-PAA NPs and MnCl2@CS-PAA NPs resulted in an inhibition of proliferation rates of B16F10-OVA cells being 49.13% and 84.46%, respectively, while R-M@CS-PAA NPs treatment resulted in an inhibition rate of 91.61%. The above data indicated that R-M@CS-PAA NPs indeed promoted the maturation of DCs to enhance the function of CD8+ T cells and ultimately exert anti-tumor effects. At the same time, we also evaluated the secretion of IL-12P70, TNF-α, IFN-β, and IFN-γ in the mouse serum by ELISA. The results showed that the expression levels of IL-12P70, TNF-α, and IFN-β, but not IFN-γ, in the serum of mice administrated with R-M@CS-PAA NPs were significantly increased (Figure 9G). These data verified that R-M@CS-PAA NP-treated DCs enhanced the killing efficiency of OT-I CD8+ T cells to B16F10-OVA cells.
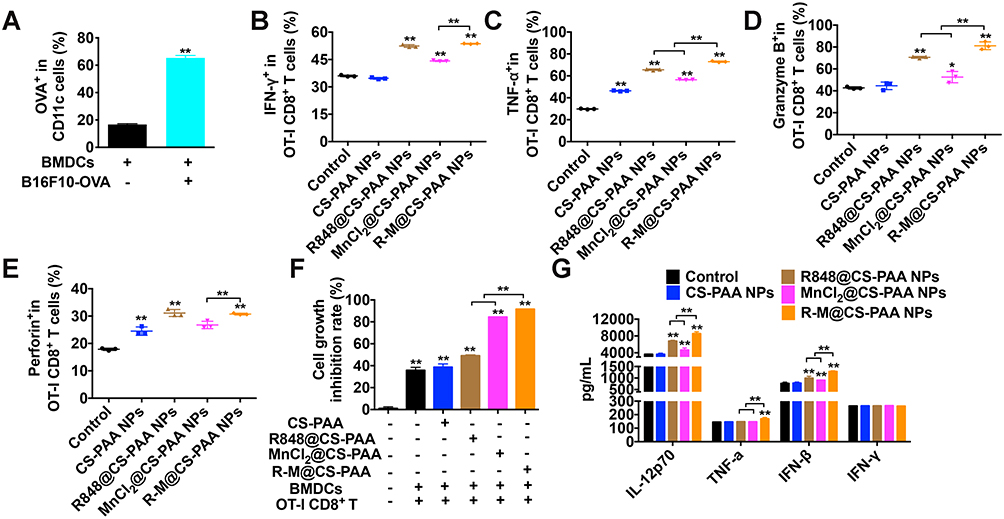 |
Figure 9 R-M@CS-PAA NPs-treated DCs enhanced the cytotoxicity of OT-I CD8+ T cells to B16F10-OVA cells. (A) BMDCs were co-cultured with B16F10-OVA cells for 48 h, and the percentage of OVA+ BMDCs was detected by FCM. (B-F) In the presence of different NPs, BMDCs, OT-I CD8+ T cells, and B16F10-OVA cells were co-cultured for 48 h. The ratio of IFN-γ+ (B), TNF-α+ (C), granzyme B+ (D), and perforin+ (E) OT-I CD8+ T cells, and the proliferation of B16F10-OVA cells (F) were examined by FCM. (G) The expression of IL-12p70, TNF-α, IFN-β, and IFN-γ in the serum of mice treated with different NPs was detected by ELISA assay. *p < 0.05, **p < 0.01. |
Discussion
CD8+ T cells exert an anti-tumor effect through their effector cytotoxic functions and an increase of the number of infiltrating T cells (mainly CD8+ T cells) in the TME are associated with a better prognosis of melanoma.46,47 Therefore, the development of strategies to simultaneously stimulate innate and acquired immune responses and enhance the cytotoxic ability of CD8+ T cells has aroused the interest of many researchers. Studies have used immunostimulants as adjuvants for tumor treatment, such as the use of Bacille Calmette Guerin (BCG) for the treatment of bladder cancer,48 and the use of infiltrating lymphocytes for the elimination of melanoma.49 However, due to the lack of an effective and long-lasting CD8+ T cell-mediated cytotoxicity, current immunotherapeutic drugs fail to achieve sustained efficacy in the body. Even in the presence of antigen-specific CD8+ T cells, tumors can continue to grow by suppressing T cell effector and memory functions, thereby evading immune surveillance.50 To overcome this limitation, it is necessary to prompt the innate immune to generate CD8+ T cells in a sustained manner using appropriate effector epitopes. Therefore, researchers have developed vaccines against DCs. As the most effective type of APCs, DCs play a key role in initiating and guiding CD8+ T cell cytotoxicity. Kantoff et al developed a DC-based Sipuleucel-T immunotherapy, which significantly prolonged the patient’s overall survival but did not affect tumor progression. The vaccine has been approved for the treatment of prostate cancer.51 Based on the previous studies, we have developed R-M@CS-PAA NPs aiming to enhance the function of macrophages and DCs to promote the anti-tumor immune response mediated by macrophages and CD8+ T cells.
In our study, the synthesized R-M@CS-PAA NPs exerted an obvious anti-melanoma effect in a subcutaneous melanoma mouse model. In addition to its effects on MDSCs and macrophages, R-M@CS-PAA NPs significantly reduced the infiltration of Tregs in the TME of mice, while they significantly increased the infiltration of CD8+ T cells in the TME. CD69, which is secreted at an early stage of T-cell activation, not only participates in inducing T-cell proliferation, but also enhances the activation of T cells and CD3 monoclonal antibody, leading to the production of TNF and IFN-γ.52 The results of FCM showed that the ratio of activated (CD69+) and killer (IFN-γ+, TNF-α+, or granzyme B+) CD8+ T cells was significantly increased. Studies have shown that DCs play an important role in the homeostasis of CD8+ T cell-mediated immunity and tolerance to tumor antigens; therefore, we explored the effect of R-M@CS-PAA NPs on the functions of DCs. In vitro experiments showed that R-M@CS-PAA NPs promoted the maturation of DCs through STING and NF-кB pathways. STING is an important signal adaptor that mediates the innate immune response induced by cytoplasmic DNA. The binding of cGAS to dsDNA binding leads to the conformational in cGAS, which in turn induces cGAMP activity.53 cGAMP binds to the STING dimer,54 and promotes the transport of STING from the endoplasmic reticulum to the perinuclear microsome through the Golgi apparatus.55 Subsequently, STING recruits and activates TBK1 and IRF3 through phosphorylation,56 during which NF-кB pathway is also activated by STING in a TBK1-dependent manner.57 At the same time, we found that R-M@CS-PAA NPs induced the secretion of IFN-β by DCs, but they did not affect the production of type II IFN-γ. IFN-β is secreted by DCs in a STING-dependent manner and is related to the uptake of tumor-derived DNA.58 BMDCs lacking cGAS or STING will recognize defects in the tumor DNA and impair the production of tumor-infiltrating CD8+ T cells.59 Therefore, we believe that R-M@CS-PAA NPs may activate the cGAS-STING pathway in DCs and thus induce the production of type I IFN, which may promote the cross-presentation of tumor antigens leading to T-cell activation.
The body can mount CD8+ T cell-mediated anti-tumor responses through the following consecutive steps: first, DCs uptake tumor-associated antigens and present them to T cells for recognition; second, cytotoxic CD8+ T cells proliferate; finally, effector CTLs eliminate tumor cells.60 In our study, we found that in the presence of R-M@CS-PAA NPs, DCs increased the ratio of IFN-γ+CD8+ T and TNF-α+CD8+ T cells. IFN-γ, a pro-inflammatory cytokine released by tumor suppressor cells, such as CTLs, M1 macrophages, and Th1 cells, can inhibit tumor cell angiogenesis and promote adaptive immune responses.13 IFN-γ induces the transformation of CD8+ T cells into antigen-specific CTLs with enhanced tumor-killing efficiency. Furthermore, IFN-γ can also promote the M1 type polarization of macrophages, producing an anti-tumor effect.61 TNF-α, another pro-inflammatory cytokine produced by tumor suppressor cells such as CTLs and M1 macrophages, which can inhibit angiogenesis and directly induce tumor cell apoptosis.62 We co-cultured DCs, CD8+ T cells, and tumor cells and found that compared with that in the control group, the proliferation of tumor cells was significantly reduced in the group treated with R-M@CS-PAA NPs. The above results suggest that R-M@CS-PAA NPs may promote the generation of CTLs with killing function from CD8+ T cells by promoting the maturation and antigen-presenting ability of DCs to control tumor growth (Figure 10).
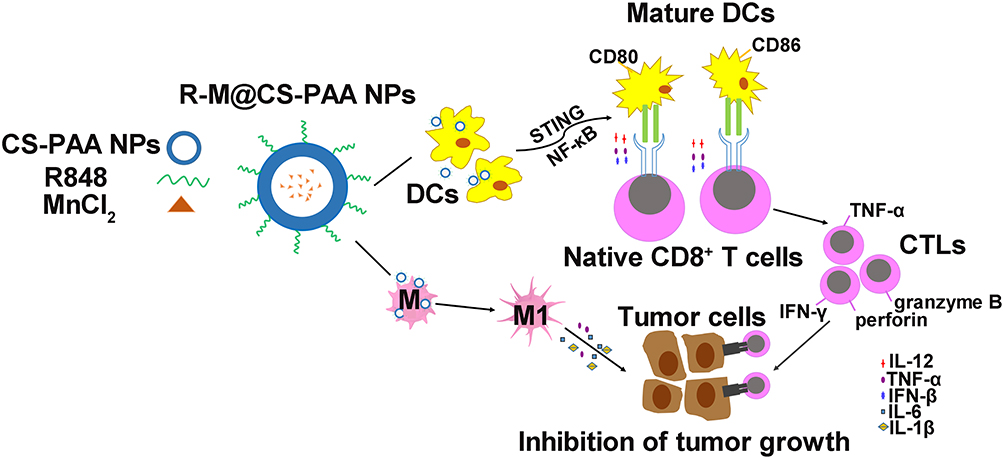 |
Figure 10 Schematic illustration of macrophage polarization and DC maturation induced by R-M@CS-PAA NPs for the treatment of melanoma. |
Conclusion
In the present study, novel R-M@CS-PAA NPs were developed and tested for their anti-melanoma effect. R-M@CS-PAA NPs displayed excellent anti-melanoma efficacy; in the TME, R-M@CS-PAA NPs induced the polarization of macrophages to the M1 type and significantly promoted the maturation of DCs by upregulating the expression of co-stimulatory molecules and antigen-presenting ability via STING and NF-кB pathways. Mature DCs enhanced the tumor-killing function of CD8+ T cells by activating the MHC I antigen presentation pathway. Our results suggest that the administration of R-M@CS-PAA NPs may be a potential therapeutic strategy for the treatment of melanoma.
Acknowledgments
This work was supported by the Jiangsu Provincial Special Program of Medical Science (grant number: BE2019617) and the National Natural Science Foundation of China (grant number: 81872114).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–1437. doi:10.1038/nm.3394
2. Lei X, Lei Y, Li JK, et al. Immune cells within the tumor microenvironment: biological functions and roles in cancer immunotherapy. Cancer Lett. 2020;470:126–133. doi:10.1016/j.canlet.2019.11.009
3. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61. doi:10.1016/j.immuni.2014.06.010
4. Belardelli F, Ferrantini M. Cytokines as a link between innate and adaptive antitumor immunity. Trends Immunol. 2002;23(4):201–208. doi:10.1016/S1471-4906(02)02195-6
5. Dirkx AE, Oude Egbrink MG, Wagstaff J, Griffioen AW. Monocyte/macrophage infiltration in tumors: modulators of angiogenesis. J Leukoc Biol. 2006;80(6):1183–1196. doi:10.1189/jlb.0905495
6. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi:10.1016/j.cell.2010.03.014
7. Biragyn A, Longo DL. Neoplastic “Black Ops”: cancer’s subversive tactics in overcoming host defenses. Semin Cancer Biol. 2012;22(1):50–59. doi:10.1016/j.semcancer.2012.01.005
8. Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427(6970):154–159. doi:10.1038/nature02238
9. Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol. 2006;6(6):476–483. doi:10.1038/nri1845
10. Jego G, Pascual V, Palucka AK, Banchereau J. Dendritic cells control B cell growth and differentiation. Curr Dir Autoimmun. 2005;8:124–139. doi:10.1159/000082101
11. Münz C, Dao T, Ferlazzo G, de Cos MA, Goodman K, Young JW. Mature myeloid dendritic cell subsets have distinct roles for activation and viability of circulating human natural killer cells. Blood. 2005;105(1):266–273. doi:10.1182/blood-2004-06-2492
12. Fujii S, Shimizu K, Kronenberg M, Steinman RM. Prolonged IFN-gamma-producing NKT response induced with alpha-galactosylceramide-loaded DCs. Nat Immunol. 2002;3(9):867–874. doi:10.1038/ni827
13. Farhood B, Najafi M, Mortezaee K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: a review. J Cell Physiol. 2019;234(6):8509–8521. doi:10.1002/jcp.27782
14. Islam MA, Rice J, Reesor E, et al. Adjuvant-pulsed mRNA vaccine nanoparticle for immunoprophylactic and therapeutic tumor suppression in mice. Biomaterials. 2021;266:120431. doi:10.1016/j.biomaterials.2020.120431
15. Rodell CB, Arlauckas SP, Cuccarese MF, et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng. 2018;2(8):578–588. doi:10.1038/s41551-018-0236-8
16. Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3(2):196–200. doi:10.1038/ni758
17. Yrlid U, Milling SW, Miller JL, Cartland S, Jenkins CD, MacPherson GG. Regulation of intestinal dendritic cell migration and activation by plasmacytoid dendritic cells, TNF-alpha and type 1 IFNs after feeding a TLR7/8 ligand. J Immunol. 2006;176(9):5205–5212. doi:10.4049/jimmunol.176.9.5205
18. Asselin-Paturel C, Brizard G, Chemin K, et al. Type I interferon dependence of plasmacytoid dendritic cell activation and migration. J Exp Med. 2005;201(7):1157–1167. doi:10.1084/jem.20041930
19. Pockros PJ, Guyader D, Patton H, et al. Oral resiquimod in chronic HCV infection: safety and efficacy in 2 placebo-controlled, double-blind phase IIa studies. J Hepatol. 2007;47(2):174–182. doi:10.1016/j.jhep.2007.02.025
20. Zhang H, Tang WL, Kheirolomoom A, et al. Development of thermosensitive resiquimod-loaded liposomes for enhanced cancer immunotherapy. J Control Release. 2021;330:1080–1094. doi:10.1016/j.jconrel.2020.11.013
21. Kwakye GF, Paoliello MM, Mukhopadhyay S, Bowman AB, Aschner M. Manganese-Induced Parkinsonism and Parkinson’s Disease: shared and Distinguishable Features. Int J Environ Res Public Health. 2015;12(7):7519–7540. doi:10.3390/ijerph120707519
22. Manome H, Aiba S, Tagami H. Simple chemicals can induce maturation and apoptosis of dendritic cells. Immunology. 1999;98(4):481–490. doi:10.1046/j.1365-2567.1999.00916.x
23. Lv M, Chen M, Zhang R, et al. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res. 2020;30(11):966–979. doi:10.1038/s41422-020-00395-4
24. Diamond MS, Kinder M, Matsushita H, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208(10):1989–2003. doi:10.1084/jem.20101158
25. Dunn GP, Bruce AT, Sheehan KC, et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol. 2005;6(7):722–729. doi:10.1038/ni1213
26. Zhang R, Wang C, Guan Y, et al. The manganese salt (MnJ) functions as a potent universal adjuvant. bioRxiv. 2019;1:783910. doi:10.1101/783910
27. Zhang R, Wang C, Guan Y, et al. Manganese salts function as potent adjuvants. Cell Mol Immunol. 2021;18(5):1222–1234. doi:10.1038/s41423-021-00669-w
28. Yang Y, Zhang J, Yang X, et al. Dysregulated APP expression and α-secretase processing of APP is involved in manganese-induced cognitive impairment. Ecotoxicol Environ Saf. 2021;220:112365. doi:10.1016/j.ecoenv.2021.112365
29. Wang M, Zhou B, Wang L, et al. Biodegradable pH-responsive amorphous calcium carbonate nanoparticles as immunoadjuvants for multimodal imaging and enhanced photoimmunotherapy. J Mater Chem B. 2020;8(36):8261–8270. doi:10.1039/D0TB01453B
30. Anitha A, Sowmya S, Kumar PTS, et al. Chitin and chitosan in selected biomedical applications. Prog Polym Sci. 2014;39(9):1644–1667. doi:10.1016/j.progpolymsci.2014.02.008
31. Chen J, Zhan Y, Wang Y, et al. Chitosan/silk fibroin modified nanofibrous patches with mesenchymal stem cells prevent heart remodeling post-myocardial infarction in rats. Acta Biomater. 2018;80:154–168. doi:10.1016/j.actbio.2018.09.013
32. Davoudi Z, Peroutka-Bigus N, Bellaire B, Jergens A, Wannemuehler M, Wang Q.Gut organoid as a new platform to study alginate and chitosan mediated PLGA nanoparticles for drug delivery. Mar Drugs. 2021;19(5):282. doi:10.3390/md19050282
33. Wei X, Liao J, Davoudi Z, et al. Folate receptor-targeted and GSH-responsive carboxymethyl chitosan nanoparticles containing covalently entrapped 6-mercaptopurine for enhanced intracellular drug delivery in Leukemia. Mar Drugs. 2018;16(11):439. doi:10.3390/md16110439
34. Yang B, Jiang J, Jiang L, et al. Chitosan mediated solid lipid nanoparticles for enhanced liver delivery of zedoary turmeric oil in vivo. Int J Biol Macromol. 2020;149:108–115. doi:10.1016/j.ijbiomac.2020.01.222
35. Hu R, Zheng H, Cao J, Davoudi Z, Wang Q. Synthesis and in vitro characterization of carboxymethyl chitosan-CBA-doxorubicin conjugate nanoparticles as pH-sensitive drug delivery systems. J Biomed Nanotechnol. 2017;13(9):1097–1105. doi:10.1166/jbn.2017.2407
36. Zhang X, Zhang H, Yin L, et al. A pH-Sensitive nanosystem based on carboxymethyl chitosan for tumor-targeted delivery of daunorubicin. J Biomed Nanotechnol. 2016;12(8):1688–1698. doi:10.1166/jbn.2016.2278
37. Guo R, Wilson LD. Synthetically engineered chitosan-based materials and their sorption properties with methylene blue in aqueous solution. J Colloid Interface Sci. 2012;388(1):225–234. doi:10.1016/j.jcis.2012.08.010
38. Hu Y, Jiang X, Ding Y, Ge H, Yuan Y, Yang C. Synthesis and characterization of chitosan-poly(acrylic acid) nanoparticles. Biomaterials. 2002;23(15):3193–3201. doi:10.1016/S0142-9612(02)00071-6
39. Hu Y, Ding Y, Ding D, et al. Hollow chitosan/poly(acrylic acid) nanospheres as drug carriers. Biomacromolecules. 2007;8(4):1069–1076. doi:10.1021/bm0608176
40. Wang Y, Wang J, Yuan Z, et al. Chitosan cross-linked poly(acrylic acid) hydrogels: drug release control and mechanism. Colloids Surf B Biointerfaces. 2017;152:252–259. doi:10.1016/j.colsurfb.2017.01.008
41. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15(7):405–414. doi:10.1038/nri3845
42. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13(4):227–242. doi:10.1038/nri3405
43. Floros T, Tarhini AA. Anticancer Cytokines: biology and Clinical Effects of Interferon-α2, Interleukin (IL)-2, IL-15, IL-21, and IL-12. Semin Oncol. 2015;42(4):539–548. doi:10.1053/j.seminoncol.2015.05.015
44. Balkwill F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev. 2006;25(3):409–416. doi:10.1007/s10555-006-9005-3
45. Wang C, Guan Y, Lv M, et al. Manganese Increases the Sensitivity of the cGAS-STING Pathway for double-stranded dna and is required for the host defense against DNA Viruses. Immunity. 2018;48(4):675–687.e677. doi:10.1016/j.immuni.2018.03.017
46. Johansson J, Siarov J, Kiffin R, et al. Presence of tumor-infiltrating CD8(+) T cells and macrophages correlates to longer overall survival in patients undergoing isolated hepatic perfusion for uveal melanoma liver metastasis. Oncoimmunology. 2020;9(1):1854519. doi:10.1080/2162402X.2020.1854519
47. Yang S, Liu T, Nan H, et al. Comprehensive analysis of prognostic immune-related genes in the tumor microenvironment of cutaneous melanoma. J Cell Physiol. 2020;235(2):1025–1035. doi:10.1002/jcp.29018
48. Sylvester RJ, Van der MA, Lamm DL. Intravesical bacillus Calmette-Guerin reduces the risk of progression in patients with superficial bladder cancer: a meta-analysis of the published results of randomized clinical trials. J Urol. 2002;168(5):1964–1970. doi:10.1016/S0022-5347(05)64273-5
49. Rosenberg SA, Packard BS, Aebersold PM, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319(25):1676–1680. doi:10.1056/NEJM198812223192527
50. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi:10.1038/nrc3239
51. Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. doi:10.1056/NEJMoa1001294
52. Ziegler SF, Ramsdell F, Alderson MR. The activation antigen CD69. Stem Cells. 1994;12(5):456–465. doi:10.1002/stem.5530120502
53. Gao P, Ascano M, Wu Y, et al. Cyclic [G(2ʹ,5ʹ)pA(3ʹ,5ʹ)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. 2013;153(5):1094–1107. doi:10.1016/j.cell.2013.04.046
54. Gao P, Ascano M, Zillinger T, et al. Structure-function analysis of STING activation by c[G(2ʹ,5ʹ)pA(3ʹ,5ʹ)p] and targeting by antiviral DMXAA. Cell. 2013;154(4):748–762. doi:10.1016/j.cell.2013.07.023
55. Saitoh T, Fujita N, Hayashi T, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A. 2009;106(49):20842–20846. doi:10.1073/pnas.0911267106
56. Liu S, Cai X, Wu J, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347(6227):aaa2630. doi:10.1126/science.aaa2630
57. Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol. 2014;88(10):5328–5341. doi:10.1128/JVI.00037-14
58. Woo SR, Fuertes MB, Corrales L, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41(5):830–842. doi:10.1016/j.immuni.2014.10.017
59. Khoo LT, Chen LY. Role of the cGAS-STING pathway in cancer development and oncotherapeutic approaches. EMBO Rep. 2018;19(12):e46935. doi:10.15252/embr.201846935
60. Fu C, Jiang A. Dendritic Cells and CD8 T Cell Immunity in Tumor Microenvironment. Front Immunol. 2018;9:3059. doi:10.3389/fimmu.2018.03059
61. Xiang W, Shi R, Kang X, et al. Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat Commun. 2018;9(1):2574. doi:10.1038/s41467-018-04999-8
62. Bertrand F, Montfort A, Marcheteau E, et al. TNFα blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat Commun. 2017;8(1):2256. doi:10.1038/s41467-017-02358-7
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.