")
Back to Journals » International Journal of Nanomedicine » Volume 16
Chitosan Nanovaccines as Efficient Carrier Adjuvant System for IL-12 with Enhanced Protection Against HBV
Authors Zhao H, Wang H, Hu Y, Xu D, Yin C, Han Q, Zhang J
Received 28 April 2021
Accepted for publication 5 July 2021
Published 21 July 2021 Volume 2021:16 Pages 4913—4928
DOI https://doi.org/10.2147/IJN.S317113
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Video abstract presented by Huajun Zhao.
Views: 217
Huajun Zhao,1 Haigang Wang,1 Yifei Hu,1 Dongqing Xu,1 Chunlai Yin,2 Qiuju Han,1 Jian Zhang1
1Institute of Immunopharmaceutical Sciences, School of Pharmaceutical Sciences, Shandong University, Jinan, People’s Republic of China; 2Department of Immunology, Dalian Medical University, Dalian, People’s Republic of China
Correspondence: Jian Zhang
Institute of Immunopharmaceutical Sciences, School of Pharmaceutical Sciences, Shandong University, Jinan, People’s Republic of China
Tel +86-531-8838-3781
Fax +86-531-8838-3782
Email [email protected]
Purpose: Alum adjuvant in HBV prophylactic vaccines is poor in inducing cellular immunity with the inhibition of IL-12 secretion, and approximately 5– 10% of immunised individuals fail to clear HBV upon infection. IL-12 plasmids (pIL-12) as adjuvants enhance significant humoral and cellular immune response in vaccines. However, finding a novel delivery system to protect pIL-12 from enzymatic degradation and achieve efficient delivery remains a major challenge.
Methods: We prepared the chitosan nanovaccine-loaded IL-12 expression plasmid (termed as “Ng(-)pIL-12”) and analysed the physicochemical properties, encapsulation efficiency and safety. Then, we evaluated the efficiency of Ng(-)pIL-12 for prophylactic HBV vaccine. Serum samples were collected and analysed for IL-12, HBsAg, anti-HBs IgG, IgG1 and IgG2b. Liver tissues were collected and analysed for HBV DNA and RNA. BMDCs and lymphocytes were collected and analysed for HBV-specific immune responses. To further confirm the long-term protective immune response against HBV, these immunised mice were challenged with hydrodynamic injection of pAAV/HBV 1.2 plasmid on day 56 after the initiation of immunisation.
Results: Chitosan nanovaccine prepared with CS and γ-PGA could load pIL-12 effectively and safely, and IL-12 was efficiently produced in vivo. Interestingly, Ng(-)pIL-12 adjuvant combined with HBsAg induced higher levels of anti-HBs IgG, IgG1 and IgG2b, promoted maturation and presentation capacity of DCs, especially CD8α+/CD103+ DCs. Meanwhile, Ng(-)pIL-12 adjuvant generated robust HBV-specific CD8+ T and CD4+ T cell responses. More importantly, Ng(-)pIL-12 adjuvant triggered terminally differentiated effector memory responses with strong anti-HBV effects.
Conclusion: Chitosan nanovaccines as an efficient carrier adjuvant system for pIL-12 combined with HBsAg induced protective anti-HBs IgG and enhanced HBV-specific CD8+ T and CD4+ T cell responses, and achieved long-term memory response against HBV, making it a promising candidate for prophylactic HBV vaccines.
Keywords: nanovaccines, hepatitis B, prophylactic vaccine, CD8+ T cells, long-term memory
Introduction
Currently, chronic hepatitis B virus (CHB) infection affects about 240 million people around the world.1 Patients with hepatitis B virus (HBV) have a higher risk of developing liver cirrhosis and hepatocellular carcinoma.1 The current treatment options for HBV infection include two classes of antivirals: Pegylated interferon alpha2a (PegIFN) and nucleoside/nucleotide analogue.2 However, these antiviral therapies achieve hepatitis B surface antigen (HBsAg) loss in only a limited number of patients, resulting in a functional but not virological cure.3 Although commercially available hepatitis B prophylactic vaccines are widely used to prevent HBV infection, the overall success rate of hepatitis B vaccination is approximately 90%,4 and several million people are still at risk of HBV infection.5 Furthermore, alum adjuvant in hepatitis B prophylactic vaccines is poor in inducing cellular immunity and not an optimal adjuvant for vaccines, since alum selectively inhibits the interleukin-12 (IL-12) secretion by dendritic cells (DCs),6 which in turn led to the impaired ability of IFN-γ production and cytolytic activity of NK cells as well as inducing Th1-type immune responses.7–9 Accordingly, IL-12 has been employed as a potential adjuvant in prophylactic and therapeutic vaccines.10,11 Additionally, some immunopotentiors such as CpG ODN could also induce IL-12 production, indirectly contributing to humoral and cellular immune responses against pathogens.12–15
Exogenous IL-12 combined with HBV antigens facilitates the secretion of IFN-γ in peripheral blood mononuclear cells by augmenting the frequency of central memory CD8+ T cells.16 However, clinical trials have shown that the administration of recombinant IL-12 proteins results in serious potential systemic toxicity with instability and short half-life, which limits their therapeutic potential.17–19 Therefore, designing new strategies to achieve durable, local, nontoxic clinical responses to IL-12 remains a challenge. Instead of immunising directly with IL-12 adjuvant, administration of naked plasmid DNA encoding IL-12 is nontoxic and more beneficial, because the expression of IL-12 can be maintained at low levels and will eventually taper progressively to basal levels.19,20 IL-12 plasmids (pIL-12) have shown promising results in preclinical and early phase clinical studies.21,22 Application of IL-12 plasmid in an HBV DNA vaccine exhibited a significant enhancement of Th1 cells, as well as a marked inhibition of Th2 cells.23 However, a major challenge in plasmid immunisation is the delivery of biomolecules and protection from enzymatic biodegradation until they reach the target cells.
Many biocompatible polymeric nanoparticle platforms have been employed as immunomodulators and vaccine carriers to improve immunotherapy.24–26 The tuned physicochemical properties can promote the interaction between nanoparticles and innate immune cells including dendritic cells and macrophages.27 Additionally, nanoparticle delivery systems can further improve undesirable drug properties, including better biocompatibility and chemical stability in vivo, improved body distribution, longer plasma half-lives, and controlled drug release.28–30 Nanoparticle-based targeted delivery can also diminish off-target toxicity and immune-related adverse events.31,32 Currently, nanomedicines delivering immunological agents for cancer immunotherapy have already shown promising results in preclinical and clinical studies.24,33 For instance, folate-modified chitosan nanoparticles-mediated delivery of plasmid DNA encoding interferon-inducible protein-10 (IP-10) promoted the expression of IP-10 and efficiently enhanced the activity of adoptive pMAGE-A1(278–286) specific CTLs against transplanted human hepatocellular carcinoma.34
Here, we prepared a chitosan nanovaccine as efficient carrier adjuvant system for IL-12 expression plasmid (Ng(-)pIL-12) and evaluated the adjuvant effect of Ng(-)pIL-12 as a prophylactic HBV vaccine against pAAV/HBV1.2 challenge. Interestingly, Ng(-)pIL-12 adjuvant promoted the maturation and antigen-presenting ability of DCs, and induced robust HBV-specific CD8+ and CD4+ T cell responses. More importantly, Ng(-)pIL-12 adjuvant triggered the terminally differentiated effector memory responses with strong anti-HBV effects against HBV infection.
Materials and Methods
Animals and Reagents
Five- to six-week-old male C57BL/6J mice were purchased from Beijing HFK Bioscience Co. Ltd (Beijing, China). All animals were kept under specific pathogen-free conditions. All animal experiments were approved by the Institutional Animal Care and Use Committee of the Shandong University, and performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of the Ethical Committee of Shandong University. CS (molecular weight: 5 kDa; deacetylation: 85%) was purchased from Jinan Haidebei Marine Bioengineering Co., Ltd (Jinan, China). γ-PGA (molecular weight: 700 kDa) was purchased from Shandong Mingren Freda Pharmaceutical Co., Ltd. (Jinan, China). Recombinant HBsAg (Hansenula polymorpha) was purchased from Dalian Hissen Bio-pharm. Co., Ltd. (Dalian, China). Mouse colorectal cancer cells (CT-26) were purchased from the cell bank of the Chinese Academy of Sciences (Shanghai, China). A recombinant plasmid encoding the entire pIL-12, comprising p40 and p35, was constructed previously in our laboratory. pAAV/HBV1.2 plasmid containing the full-length HBV DNA was kindly provided by Pei-Jer Chen (NTU, Taiwan).
Preparation and Characterisation of the Chitosan Nanoparticle
Ng(-)pIL-12 chitosan nanoparticle was prepared using the ionic gelation method as described previously35 with some modifications. Briefly, 400 µL of IL-12 plasmid (760 µg/mL in sterile ddH2O) was added dropwise into 1 mL of γ-PGA solution (2 mg/mL in sterile ddH2O). After stirring for 20 min, an equal volume of CS solution (0.5 mg/mL which was adjusted to pH 5.5 by 10% HAc) was added dropwise into the solution. Another 20 min later, 1% NaOH was used to modify the pH of this mixture to neutral. Then, the suspension was centrifuged at 10,000 × rpm for 10 min, and the harvested nanoparticles (Ng(-)pIL-12) were re-dispersed in 1 mL of sterile ddH2O for the further experiments.
The particle size distribution and zeta potential of Ng(-)pIL-12 were measured using the Malvern Zetasizer Nano-ZS90 dynamic light scattering (Malvern Instruments, Malvern, UK). The morphology of the Ng(-)pIL-12 was examined using TEM (JEM-100CX II, Jeol, Tokyo, Japan). The pIL-12 encapsulation efficiency was measured directly by gel retardation analysis.
Vaccination and HBV Challenge
C57 BL/6J mice were injected intramuscularly with three doses of PBS, 2 μg HBsAg alone, 5 μg naked pIL-12 alone, 2 μg HBsAg combined with 40 μg blank Ng(-) or 2 μg HBsAg combined with 45 μg Ng(-)pIL-12 (40 μg Ng(-) containing 5 μg pIL-12) at two-week intervals, separately (Table 1). To further evaluate the efficiency of Ng(-)pIL-12 adjuvant for the prophylaxis of HBV, these immunised mice were challenged with hydrodynamic injection of 8 μg pAAV/HBV 1.2 plasmid on day 56 after the initiation of immunisation. Blood samples were drawn from the lateral tail vein, and the serum was stored at −80 °C until further analysis.
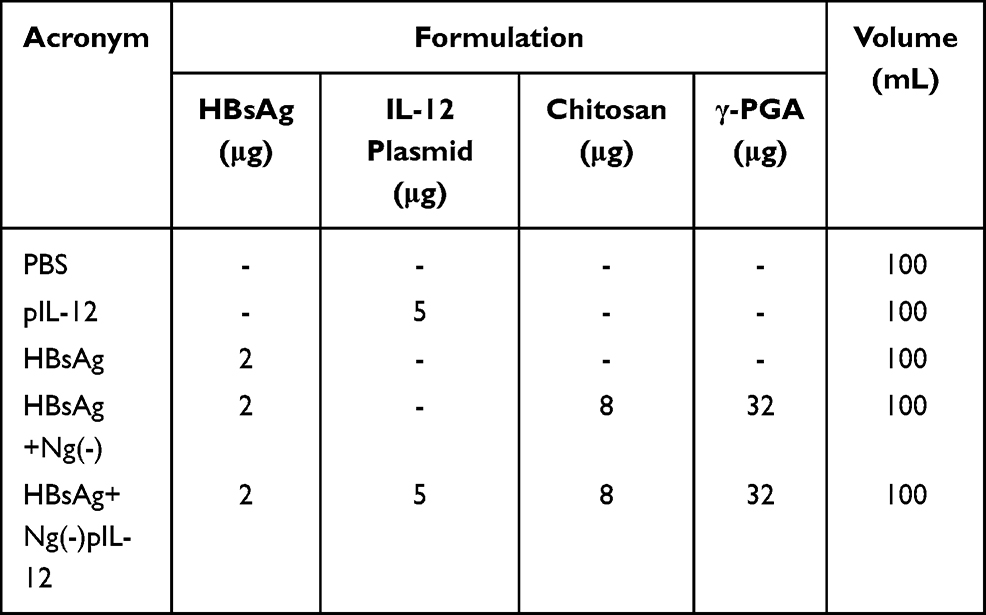 |
Table 1 Vaccination Groups and the Formulations |
The Generation and Stimulation of Bone Marrow-Derived Dendritic Cells (BMDCs) in vitro
Murine BMDCs were generated as previously described.36 Briefly, bone marrow (BM) cells were cultured in RPMI 1640 with 10 ng/mL rmGM-CSF (E. coli, BioLegend, San Diego, USA) and 5 ng/mL rmIL-4 (E. coli, BioLegend, San Diego, USA), and the medium was replaced on day 3 and 5. Non-adherent cells were harvested on day 7, and CD11c+ BMDCs were identified and enriched using FACS (> 90%). The isolated BMDCs were incubated with naked pIL-12, blank Ng(-), and Ng(-)pIL-12 (containing 1 μg/mL pIL-12) for 24 or 36 h, and the surface expression of costimulatory molecules CD40 and CD86, and MHC class II (MHC II) was analysed using flow cytometry (Table 2).
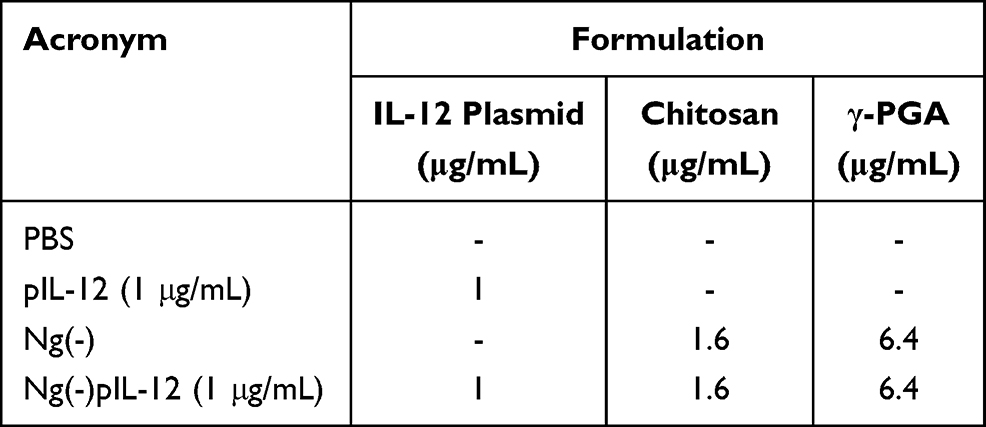 |
Table 2 The Formulations Tested on CT-26 for IL-12 Assay and BMDCs for Maturation in vitro |
Analysis of IL-12 in CT-26 Cells
For intracellular IL-12 analysis, CT-26 cells (mouse colorectal cancer cells, purchased from the cell bank of the Chinese Academy of Sciences, Shanghai, China) were stimulated with naked pIL-12, Ng(-), and Ng(-)pIL-12 (containing 1 μg/mL pIL-12) for 24 or 36 h, followed by further incubation with BFA (Biolegend, San Diego, USA) (5 μg/mL) for 4 h (Table 2). Then these cells were harvested, and the IL-12 expression in CT-26 cells was analysed using flow cytometry.
Serum IL-12 Analysis by ELISA
C57 BL/6J mice were intramuscularly injected with three doses of 5 μg naked pIL-12 alone and 45 μg Ng(-)pIL-12 (40 μg Ng(-) containing 5 μg pIL-12) at two-week intervals, separately. Serum levels of IL-12 p70 were determined on day 5, 10, and 15 after intramuscular injection (Table 3) using IL-12 ELISA kit (Multi-Science, Hangzhou, China) according to the manufacturer’s instructions. Briefly, 100 μL of serum samples was added to the wells followed by the adding of 50 μL HRP-conjugate. Then, plates were incubated at room temperature for 90 min. After extensive washing, the plates were incubated with 100 μL of the TMB substrate for 30 min in the dark at room temperature. The reaction was then stopped with stop solution, and the absorbance of the solution was measured using the Synergy 2 Multi-Mode Microplate Reader (BioTek, Vermont, USA) by dual wavelength 450 nm/630 nm for detection.
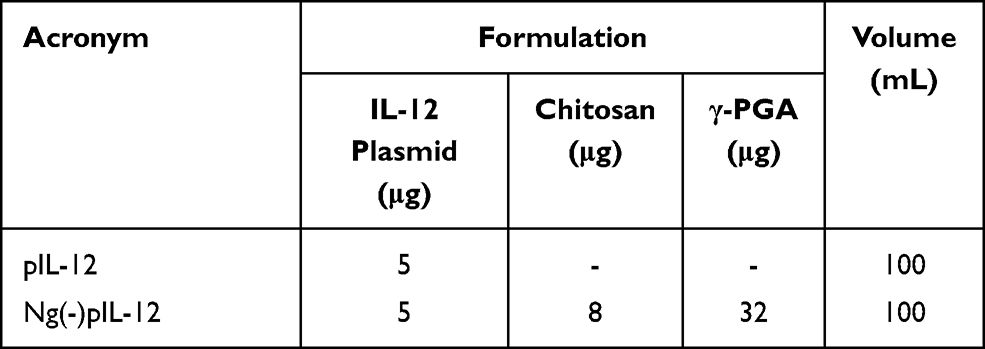 |
Table 3 The Formulations Tested for the IL-12 Assay in vivo |
Serum HBsAg Analysis by CLIA
Serum samples were analysed using HBsAg chemiluminescent enzyme immunoassay (CLIA) kit (Autobio, Zhengzhou, China) according to the manufacturer’s instructions. Briefly, 50 μL of serum samples was added to the wells followed by the adding of 50 μL detection reagent. Then, plates were incubated at 37 °C for 60 min. After extensive washing, the plates were incubated with 50 μL of the chemiluminescent substrate solution for 10 min in the dark at room temperature. The plates were measured using the Synergy 2 Multi-Mode Microplate Reader (BioTek, Vermont, USA).
Serum Hepatitis B Surface Antibody (Anti-HBs) IgG Analysis by ELISA
Serum samples were analysed using anti-HBs IgG ELISA kit (Wantai Bio-pharm, Beijing, China) according to the manufacturer’s instructions. Briefly, 50 μL of serum samples was added to the wells followed by the adding of 50 μL HRP-conjugate. Then, plates were incubated at 37 °C for 60 min. After extensive washing, the plates were incubated with 100 μL of the TMB substrate for 30 min in the dark at 37 °C. The reaction was then stopped with stop solution, and the absorbance of the solution was measured using the Synergy 2 Multi-Mode Microplate Reader (BioTek, Vermont, USA) by dual wavelength 450 nm/630 nm for detection.
Serum Anti-HBs IgG Isotypes Analysis by ELISA
This determination was performed by an anti-HBs IgG kit (Wantai Bio-pharm, Beijing, China) with goat anti-mouse IgG1 (SA00012-1, Proteintech, Wuhan, China) and IgG2b (SA00012-3, Proteintech, Wuhan, China) labeled with HRP. Briefly, 50 μL of serum samples diluted 1:20,000 in PBS–Tween was added to the wells. Goat anti-mouse IgG1 and IgG2b were diluted 1:5000 with the ELISA buffer diluent and added to the plates, respectively. Plates were incubated at 37 °C for 60 min followed by extensive washing. Then, the plates were incubated with 100 μL of the TMB substrate for 30 min in the dark at 37 °C. The reaction was stopped with stop solution, and the absorbance of the solution at 450 nm was measured using the Synergy 2 Multi-Mode Microplate Reader (BioTek, Vermont, USA). The adjusted OD450 value was calculated by subtracting the OD450 value of the negative wells.
HBV DNA and RNA Detection
Liver HBV genomic DNA was extracted using a gDNA kit (Tiangen Biotech, Beijing, China). To specifically analyse HBV covalently closed circular DNA (cccDNA) over the different HBV DNA forms, one microgram of extracted DNA was digested at 25 °C for 15 min with 10 U of SwaI (New England Biolabs, Beijing, China), and then 1 U of ATP dependent DNase (Takara Biomedical Technology, Beijing, China) was added, followed by incubation at 37 °C for 16 h.37 Total RNA was extracted from livers of pAAV-HBV 1.2-infected mice using TRIzol reagent (CW Biotech, Beijing, China). For cDNA synthesis, the RNA was reverse transcribed (RT-PCR) with a cDNA Synthesis Kit (CW Biotech, Beijing, China). Samples were then analysed using q-PCR with the following primers: HBV-cccDNA-real-F, 5ʹ-CGTCTG TGC CTT CTC ATC TGC-3ʹ; HBV-cccDNA-real-R, 5ʹ GCA CAG CTT GGA GGC TTG AA-3ʹ; HBV-DNA-real-F, 5ʹ-CAC ATC AGG ATT CCT AGG ACC-3ʹ; HBV-DNA-real-R, 5ʹ-GGT GAGTGA TTG GAG GTT G-3ʹ; HBV-total-real-F, 5ʹ-TCA CCA GCA CCA TGC AAC-3ʹ; HBV-total real-R, 5ʹ-AAG CCA CCC AAG GCA CAG-3; HBV-3.5kb-real-F, 5ʹ-GAG TGT GGA TTC GCACTC C-3ʹ; HBV-3.5kb-real-R, 5ʹ-GAG GCG AGG GAG TTC TTC T-3ʹ; m-GAPDH-real-F, 5ʹ-AGG TCG GTG TGA ACG GAT TTG-3ʹ; m-GAPDH-real-R, 5ʹ-TGT AGA CCA TGT AGT TGA GGT CA-3ʹ. Real-time PCR was performed using the UltraSYBR Mixture (CW Biotech, Beijing, China) with Lightcycler® 96 (Roche).
Cell Isolation
Hepatic mononuclear cells (MNCs) were isolated as described previously.38 Briefly, the livers were excised, disrupted, and passed through a 200 μm nylon cell strainer. The resulting single-cell suspensions were centrifuged at 700 × rpm for 1 min, and the supernatants were layered over 40% Percoll (GE Healthcare, Uppsala, Sweden). After centrifugation at 1200 × rpm for 10 min, the MNCs were pelleted, and the contaminating erythrocytes were lysed. The spleens and dLNs were passed through a 200 μm nylon cell strainer, and the precipitated cells were harvested, followed by RBC lysis and washing.
Cytotoxicity Assay (CCK-8 Assay)
The CCK-8 assay was used to investigate the in vitro cytotoxicity of Ng(-)pIL-12. Briefly, 1 × 105 splenocytes and 1 × 104 BMDCs were seeded into 96-well plates in RPMI 1640 and incubated for 24 h at 37 °C, separately. Then, different concentrations of Ng(-)pIL-12 (with pIL-12 concentrations of 1, 5, and 10 μg/mL) were added and incubated for 48 h (Table 4). All samples were tested in triplicate. Then, CCK-8 reagents were added to each well, and the cells were incubated at 37 °C for another 2 h. The absorbance of the solution at 450 nm was measured using the Synergy 2 Multi-Mode Microplate Reader (BioTek, Vermont, USA). The results were expressed as the relative expression, and the O.D. of PBS-treated group was identified as 1.
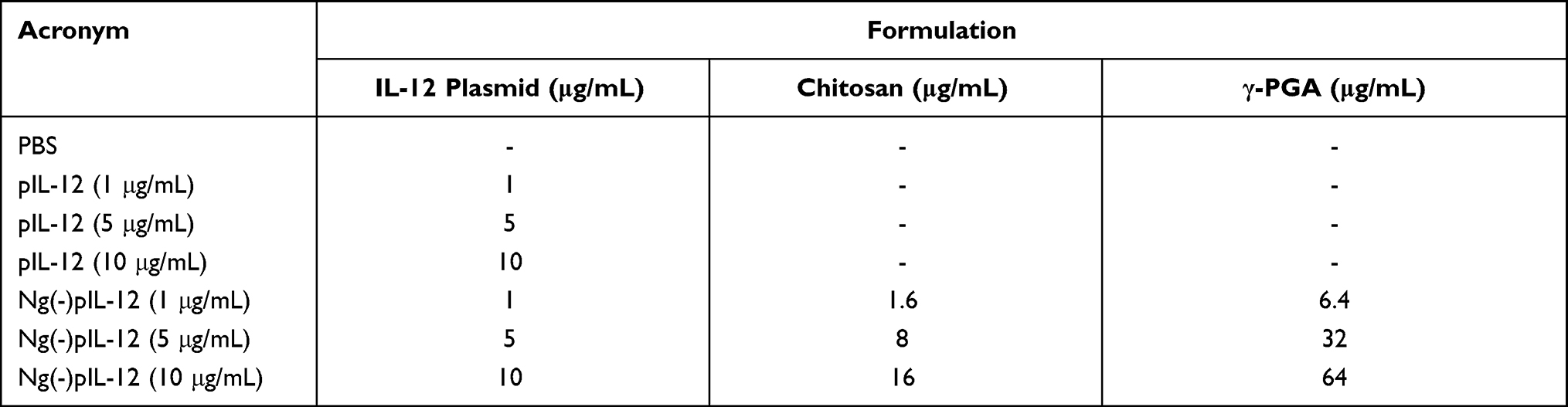 |
Table 4 The Formulations Tested on Splenocytes and BMDCs for Cytotoxicity Assay in vitro |
Flow Cytometry
Cell suspensions were pre-incubated with Fc block (anti-mouse CD16/32, #14-0161-82, eBioscience, California, USA) and stained with the indicated mAb conjugates for 1 h at 4 °C. The following fluorochrome-conjugated antibodies were used: FITC-anti-CD4 (the marker of CD4+ T cell subset, #4313007), FITC-anti-CD8α (the marker of CD8+ T cell subset, #4271604), FITC-anti-CD11c (αX integrin, primarily expressed on DCs, #E00155-1631), PE-anti-CD69 (early activation antigen expressed on T cells, #E01333-1634), PE-anti-CD8α (the marker of CD8+ T cell subset, #E01038-1633), PE-anti-CD86 (a costimulatory molecule on DCs, #E01369-1634), PE-anti-CD49d (α4 integrin, expressed on antigen-specific T cells, #E01278-1635), Percp-Cy5.5-anti-CD3e (the marker of T cells, #4304569), Percp-Cy5.5-anti-CD40 (a costimulatory molecule on DCs, #124624), APC-anti-CD80 (a costimulatory molecule on DCs, #4329685), APC-anti-PD-1 (an inhibitory receptor expressed on T cells, #4344425), and e450-anti-IL-12 (a proinflammatory cytokine which can induce the activation of T cells, #2095501) from eBioscience (California, USA); PE-anti-MHC-II (a useful marker associated with antigen presentation, #107608), PE-anti-KLRG1 (a useful marker for identifying memory and effector T cells, #138408), Percp-Cy5.5-anti-CD11a (αL integrin, expressed on antigen-specific T cells, #101124), APC-anti-CD127 (a useful marker for identifying memory and effector T cells, #135012), BV421-anti-LAG3 (an inhibitory receptor expressed on T cells, #125221), PE-Cy7-anti-TIGIT (an inhibitory receptor expressed on T cells, #142107), and APC-Cy7-anti-CD107a (a useful marker associated with CD8+ T cell cytotoxicity, #121615) from BioLegend (San Diego, USA); PE-CF594-anti-CD8α (the marker of CD8+ T cell subset, #562283) from BD Bioscience (Bedford, USA). All data were acquired on BD FACSCalibur or BD FACSCelesta flow cytometer and analysed with FlowJo software.
Statistical Analysis
Data were analysed using GraphPad Prism 7 and compared between groups using an unpaired two-tailed t test. A p value less than 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Results
Preparation and Physicochemical Properties of Ng(-)pIL-12
The chitosan nanovaccine Ng(-)pIL-12 that loaded IL-12 expression plasmid was prepared as shown in Figure 1A. Physicochemical properties of Ng(-)pIL-12, such as transmission electron microscopy (TEM) image, particle size, zeta potential, and encapsulation efficiency were measured. Ng(-)pIL-12 was observed as homogeneous globular particulates (Figure 1B). The average diameter of Ng(-)pIL-12 was approximately 178.7 nm, and the zeta potential of Ng(-)pIL-12 was around −15.1 mV (Figure 1C). The gel retardation analysis showed that Ng(-) could efficiently encapsulate pIL-12 at different nanogel/pIL-12 mass ratios (Figure 1D). BMDCs and splenocytes were used to evaluate the cytotoxicity of Ng(-)pIL-12 using CCK-8 assay in vitro. As shown in Figure 1E, the viabilities of cells treated with different concentrations of Ng(-)pIL-12 were similar to those of the PBS group; and Ng(-)pIL-12 exhibited no significant cytotoxic effects on BMDCs and splenocytes. These data indicated that the chitosan nanovaccine prepared with CS and γ-PGA could load pIL-12 efficiently and safely.
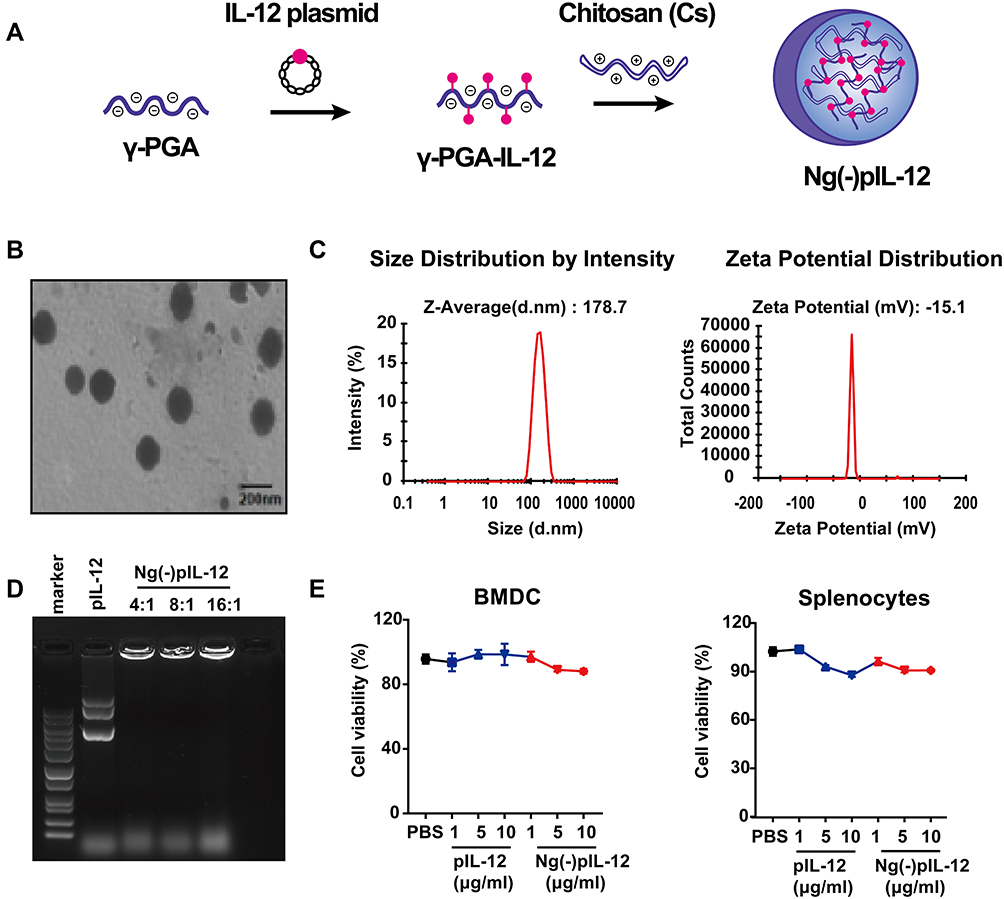 |
Figure 1 Physicochemical properties of Ng(-)pIL-12. (A) The preparation of Ng(-)pIL-12 chitosan nanoparticle. (B) Transmission electron microscopy (TEM) images of Ng(-)pIL-12. Scale bar represents 200 nm. (C) Particle size and Zeta potential of Ng(-)pIL-12. (D) Gel retardation analysis of Ng(-)pIL-12 at different nanogel/pIL-12 mass ratios as 4:1, 8:1, 16:1, respectively. (E) In vitro toxicity of Ng(-)pIL-12 and naked pIL-12 at different indicated concentrations in BMDCs and splenocytes, and the O.D. of PBS-treated group (PBS) was identified as 1. pIL-12, naked pIL-12; Ng(-)pIL-12, Ng(-) containing pIL-12. One representative from three independent experiments. |
Ng(-)pIL-12 Adjuvant Induced Successful Protective Effects Against HBV Infection
First, we evaluated whether Ng(-)-delivered pIL-12 could efficiently express IL-12 in vitro and in vivo. Compared to the naked pIL-12 and blank Ng(-) groups, Ng(-)pIL-12 transfection exhibited an increased IL-12 expression in CT-26 cells (Figure 2A). Consistent with this, compared to the naked pIL-12 group, serum IL-12p70 levels were increased in mice intramuscularly injected with Ng(-)pIL-12 (Figure 2B). We then evaluated the adjuvant effect of Ng(-)pIL-12 as a prophylactic HBV vaccine. Compared to PBS and Ng(-) groups, Ng(-)pIL-12 combined with HBsAg induced higher anti-HBs seroconversion after the second and third vaccinations, while anti-HBs IgG levels were nearly undetectable in HBsAg- or pIL-12-immunised mice (Figure 2C). To further evaluate the efficacy of prophylactic effects against HBV, these mice were challenged with pAAV/HBV 1.2 plasmid on day 56 after the initiation of immunisation. As shown in Figure 2D, Ng(-)pIL-12 combined with HBsAg-immunised mice exhibited significantly higher anti-HBs IgG levels on day 3 and 7 after HBV challenge compared to the other groups, accompanied with higher levels of anti-HBs IgG1 and IgG2b, indicating the Ng(-)pIL-12 significantly enhanced the Th1 and Th2 immune responses against HBV infection (Figure 2E). Meanwhile, the serum HBsAg or intrahepatic HBV RNA, HBV cccDNA, and HBV DNA were nearly undetectable after Ng(-)pIL-12 vaccination (Figure 2F and G). Although Ng(-) combined with HBsAg slightly stimulated the generation of protective anti-HBs IgG on day 7 after HBV challenge and reduced serum HBsAg levels compared to PBS, Ng(-) and naked pIL-12 alone groups (Figure 2D–F), Ng(-) could not efficiently eliminate intrahepatic HBV RNA or DNA (Figure 2G). These findings suggested that using Ng(-)pIL-12 as an adjuvant in the prophylactic HBV vaccine could quickly and successfully protect against HBV infection.
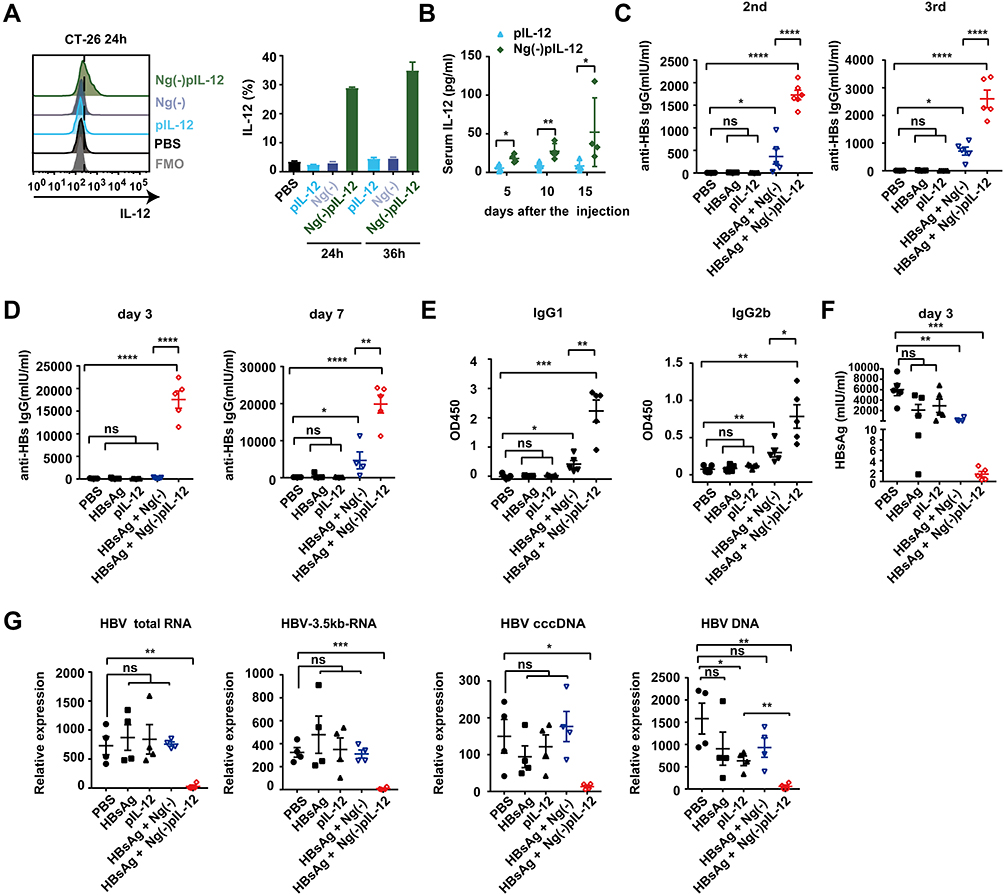 |
Figure 2 Ng(-)pIL-12 adjuvant induced protective effects against HBV infection quickly and successfully. (A) CT-26 cells were treated with PBS, 1 μg/mL pIL-12, Ng(-) and 9 μg/mL Ng(-)pIL-12 (with 1 μg/mL pIL-12) for 24 or 36 h, followed by further incubation with BFA (5 μg/mL) for 4 h. IL-12 expression in CT-26 cells was analysed using FACS. (B) C57 BL/6J mice were intramuscularly injected with three doses of 5 μg pIL-12 and 45 μg Ng(-)pIL-12 at two-week intervals, separately. Serum levels of IL-12 p70 were determined on day 5, 10, and 15 using ELISA after intramuscular injection. (C–F) C57 BL/6J mice were injected intramuscularly with three doses of PBS, 2 μg HBsAg alone, 5 μg pIL-12 alone, 2 μg HBsAg combined with 40 μg Ng(-) and 2 μg HBsAg combined with 45 μg Ng(-)pIL-12 at two-week intervals, separately. Anti-HBs IgG titres were measured two weeks after the second and third vaccination (C). (D–G) These immunised mice were challenged with 8 μg pAAV/HBV 1.2 on day 56 after the initiation of immunisation. Serum levels of anti-HBs IgG were detected using ELISA on day 3 and 7 after HBV challenge (D). Serum levels of anti-HBs IgG1 and IgG2b were detected using ELISA on day 10 after HBV challenge (E). Serum levels of HBsAg were detected using CLIA on day 3 after the HBV challenge (F). Intrahepatic HBV total RNA, intermediate products 3.5 kb RNA, HBV cccDNA, and HBV DNA were analysed using q-PCR and normalised to GAPDH expression on day 10 after the challenge (G). HBsAg, HBsAg alone; pIL-12, naked pIL-12; Ng(-), blank nanogels; Ng(-)pIL-12, 40 μg Ng(-) containing 5 μg pIL-12. All data represent the mean ± SEM (n ≥ 4) from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. |
Ng(-)pIL-12 Induced Robust HBV-Specific CD8+ T and CD4+ T Cell Responses During Vaccination
Since the magnitude and quality of HBV-specific CD8+ T and CD4+ T cell responses correlate with the prophylactic efficacy of HBV vaccine against HBV infection,16,37 we next investigated whether Ng(-)pIL-12 boosted HBV-specific CD8+T and CD4+ T cell responses. As shown in Figure 3A, Ng(-)pIL-12 combined with HBsAg-immunised mice exhibited a significantly higher proportion of HBV-specific CD11ahi CD8αlo cells in peripheral blood compared to the PBS and HBsAg alone groups, accompanied by significantly higher numbers of CD4+ CD11ahi CD49dhi HBV-specific CD4+ T cells10,39 (Figure 3B) and increased expression levels of the activation antigen CD69 (Figure 3C). However, HBsAg alone did not enhance the HBV-specific T cell response. These results suggested that using Ng(-)pIL-12 as an adjuvant in the prophylactic HBV vaccine could augment HBV-specific cellular responses.
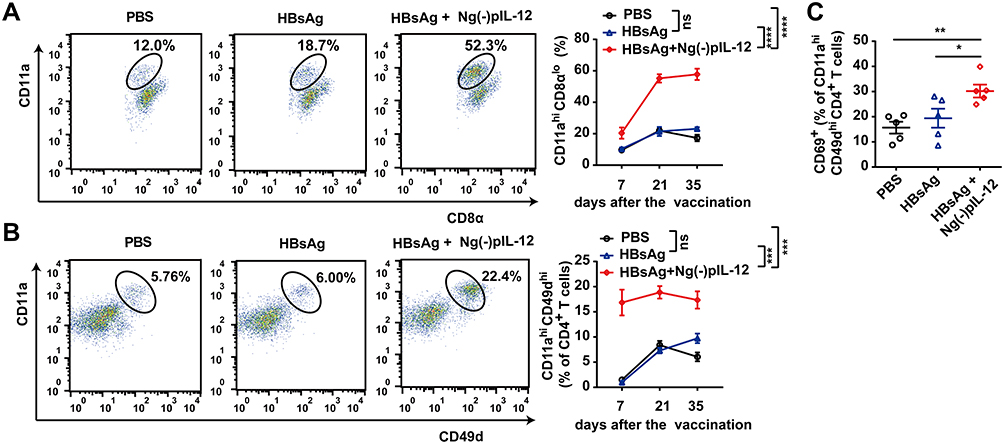 |
Figure 3 Ng(-)pIL-12 adjuvant induced robust HBV-specific CD8+ and CD4+ T cell responses during vaccination. C57 BL/6J mice were injected intramuscularly with three doses of PBS, 2 μg HBsAg alone, 2 μg HBsAg combined with 45 μg Ng(-)pIL-12 at two-week intervals, separately. (A) The proportion of CD11ahi CD8αlo cells among CD8+ T cells in the peripheral blood on day 7, 21 and 35 after the initiation of immunisation. (B) The proportion of CD11ahi CD49dhi cells among CD4+ T cells in the peripheral blood on day 7, 21 and 35 after the initiation of immunisation. (C) The expression of activation antigen CD69 on CD4+ CD11ahi CD49dhi cells on day 7 after the initiation of immunisation. HBsAg, HBsAg alone; Ng(-)pIL-12, 40 μg Ng(-) containing 5 μg pIL-12. All data are expressed as the mean ± SEM (n ≥ 5) from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. |
Ng(-)pIL-12 Adjuvant Promoted the Maturation and Antigen Presenting Ability of DCs, Especially CD8α+/CD103+ DCs
An effective prophylactic HBV vaccine should be potential to activate DCs and then induce HBV-specific T-cell response during HBV infection.40,41 Therefore, we investigated whether Ng(-)pIL-12 adjuvant could enhance the function of DCs after HBV challenge. First, BMDCs were generated in vitro and used to test whether Ng(-)pIL-12 could improve the functions of DCs. Compared to PBS group, naked pIL-12 and blank Ng(-) alone increased the expression of MHC-II, CD86, and CD40 on the surface of BMDCs, but the enhancement of Ng(-)pIL-12 was stronger and showed time-dependent manner (Figure 4A). Subsequently, we found that the proportion of CD11c+ DCs in peripheral blood from the Ng(-)pIL-12 combined with HBsAg-immunised mice was significantly higher than the PBS- and HBsAg-treated mice (Figure 4B), accompanied by the upregulation of co-stimulatory molecule CD80 (Figure 4C). Meanwhile, we found Ng(-)pIL-12 combined with HBsAg-immunised mice increased the expression of MHC-II on CD11c+ DCs during the vaccination, indicating the enhanced ability to present antigen (Figure 4D).
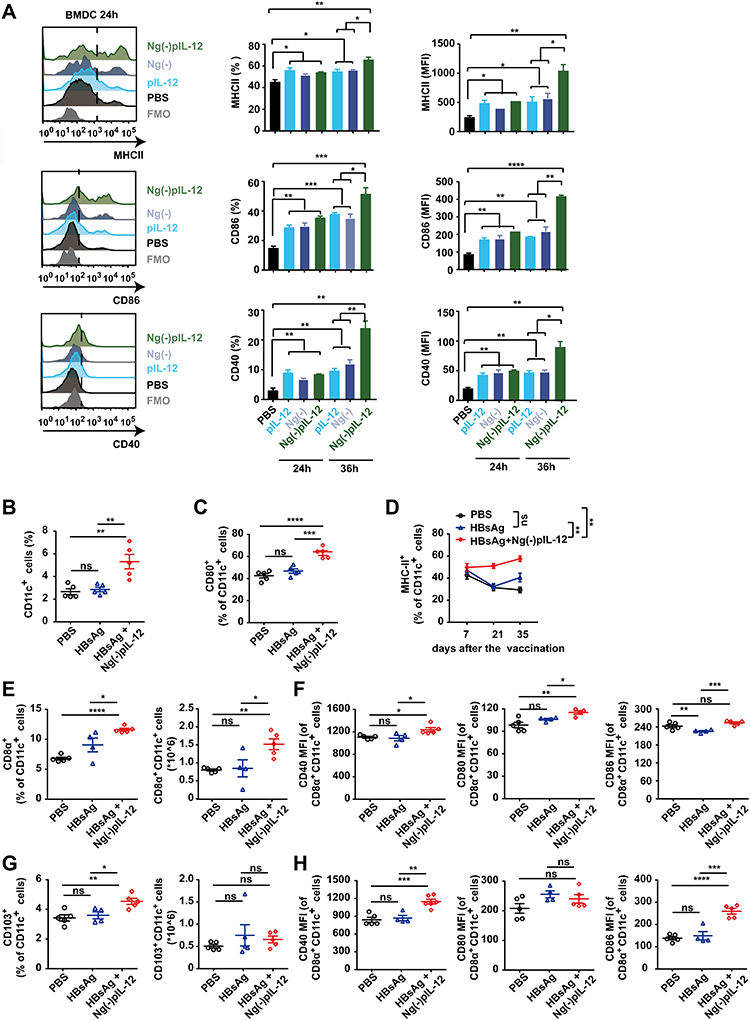 |
Figure 4 Ng(-)pIL-12 adjuvant promoted the maturation and antigen-presenting capacity of the CD8α+/CD103+ DCs after HBV challenge. (A) MHC-II, CD86 and CD40 expression on BMDCs incubated with PBS, pIL-12, Ng(-), Ng(-)pIL-12 for 24 or 36 h. (B–G) C57 BL/6J mice were injected intramuscularly with three doses of PBS (PBS), 2 μg HBsAg alone, 2 μg HBsAg combined with 45 μg Ng(-)pIL-12 at two-week intervals, separately, then these immunised mice were challenged with 8 μg pAAV/HBV 1.2 on day 56 after the initiation of immunisation. The proportion of CD11c+ DCs (B) and the expression of CD80 (C) on CD11c+ DCs in the peripheral blood on day 7 after the initiation of immunisation. Expression of MHC-II on CD11c+ DCs from peripheral blood on days 7, 21 and 35 after the initiation of immunisation (D). The percentage and absolute numbers of splenic CD8α+ DCs, and CD40 (MFI), CD80 (MFI) and CD86 (MFI) on CD8α+ DCs were measured on day 5 after HBV challenge (E–F). The percentage and absolute numbers of splenic CD103+ DCs, CD40 (MFI), CD80 (MFI), and CD86 (MFI) on CD103+ DCs were measured on day 5 after HBV challenge (G and H). HBsAg, HBsAg alone; pIL-12, naked pIL-12; Ng(-), blank nanogels; Ng(-)pIL-12, 40 μg Ng(-) containing 5 μg pIL-12. All data are expressed as the mean ± SEM (n ≥ 4) from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. |
Given their cross-presenting abilities, lymphoid-resident CD8α+ DCs and migratory CD103+ DCs are considered the most important subsets for priming CD8+ T cell responses against pathogens and tumors.42–44 Therefore, we analyzed the effects of Ng(-)pIL-12 on these DC subsets. Compared to the PBS group, the percentage and an absolute number of CD8α+ DCs were increased in the spleen after HBsAg-combined Ng(-)pIL-12 vaccination, accompanied by the elevation of CD40, CD80, and CD86 (Figure 4E and F). A similar phenomenon was observed in splenic CD103+ DCs (Figure 4G and H). However, HBsAg alone did not enhance the functions of DCs (Figure 4B–H). These results indicated that Ng(-)pIL-12 adjuvant promoted the maturation and antigen presentation capacity of DCs in vitro and in vivo.
Ng(-)pIL-12 Adjuvant Generated HBV-Specific CD4+ T and CD8+ T Cell Response to HBV Infection
An effective vaccine should generate appreciable numbers of high-quality memory CD8+ T cells, which can be immediately activated upon exposure to the pathogen.45,46 Most antigen-specific T cells undergo apoptosis, while only few effector cells turn into long-lived memory T cells after the vaccination.47,48 Compared to the PBS group, the absolute numbers of CD8+ T cells and HBV-specific CD11ahi CD8αlo cells in the spleen and liver of Ng(-)pIL-12 combined with HBsAg-immunised mice increased significantly after HBV challenge (Figure 5A and B). In addition, the expression of immune checkpoint molecules that induce T cell exhaustion such as LAG-3 (lymphocyte activation gene 3) and TIGIT (T-cell immunoreceptor with Ig and ITIM domains) on HBV-specific CD11ahi CD8αlo cells was downregulated (Figure 5C). Moreover, the percentage and the absolute number of splenic CD4+ T cells and HBV-specific CD4+ T cells were also increased in Ng(-)pIL-12 combined with HBsAg-immunised challenged mice (Figure 5D and E). These data suggested that Ng(-)pIL-12 adjuvant augmented HBV-specific CD8+ T cell immune response and induced long-term immune memory against HBV infection.
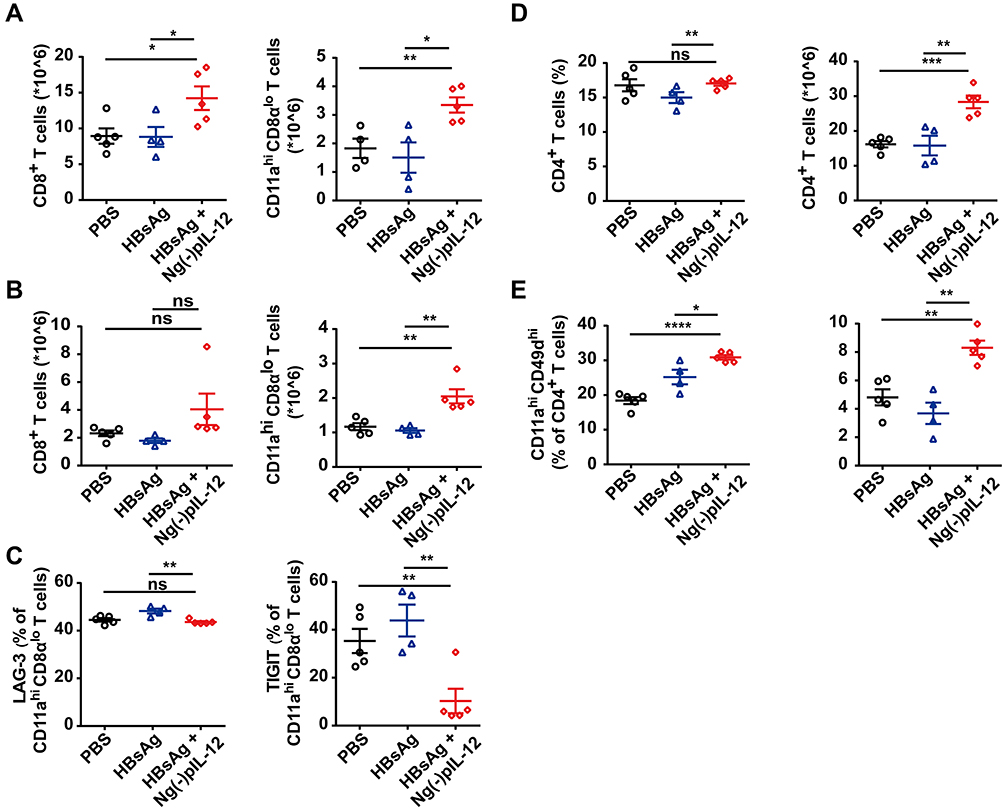 |
Figure 5 Ng(-)pIL-12 adjuvant induced long-lasting antiviral CD4+ T and CD8+ T cell response against HBV infection. C57 BL/6J mice were injected intramuscularly with three doses of PBS, 2 μg HBsAg alone, 2 μg HBsAg combined with 45 μg Ng(-)pIL-12 at two-week intervals, separately, then these immunised mice were challenged with 8 μg pAAV/HBV 1.2 on day 56 after the initiation of immunisation. The absolute numbers of CD8+ T cells and CD11ahi CD8αlo cells from the spleen (A) and the liver (B) on day 5 after HBV challenge. (C) LAG-3 and TIGIT expression on HBV-specific CD11ahi CD8αlo cells in the spleen on day 5 after HBV challenge. The proportion and absolute numbers of CD4+ T cells (D) and CD4+ CD11ahi CD49dhi cells (E) in the spleen on day 5 after HBV challenge. HBsAg, HBsAg alone; Ng(-)pIL-12, 40 μg Ng(-) containing 5 μg pIL-12. All data are expressed as the mean ± SEM (n ≥ 4) from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. |
Ng(-)pIL-12 Adjuvant Triggered the Terminally Differentiated Effector Memory Response with Strong Anti-HBV Effects
During pathogen infection, the effector CD8+ T cells can differentiate into short-lived effector cells (SLECs) with potent effector functions dependent on the inflammatory environment.49,50 Our recent study showed SLECs form the predominant secondary memory subset and are optimal for protective immunity against HBV re-challenge.37 Here, we observed that Ng(-)pIL-12 promoted the differentiation of memory CD8+ T cell, exhibiting a high frequency of SLECs (CD127lo KLRG1hi) in the peripheral blood during vaccination (Figure 6A). Moreover, compared to the PBS group, the frequency of SLECs was increased in the spleen of Ng(-)pIL-12 combined with HBsAg-immunised mice by HBV infection (Figure 6B), with higher levels of CD107a and lower levels of immune regulatory molecules TIGIT (Figure 6C and D). These data indicate that Ng(-)pIL-12 adjuvant-induced generation of SLECs might play a critical role in protecting against HBV infection.
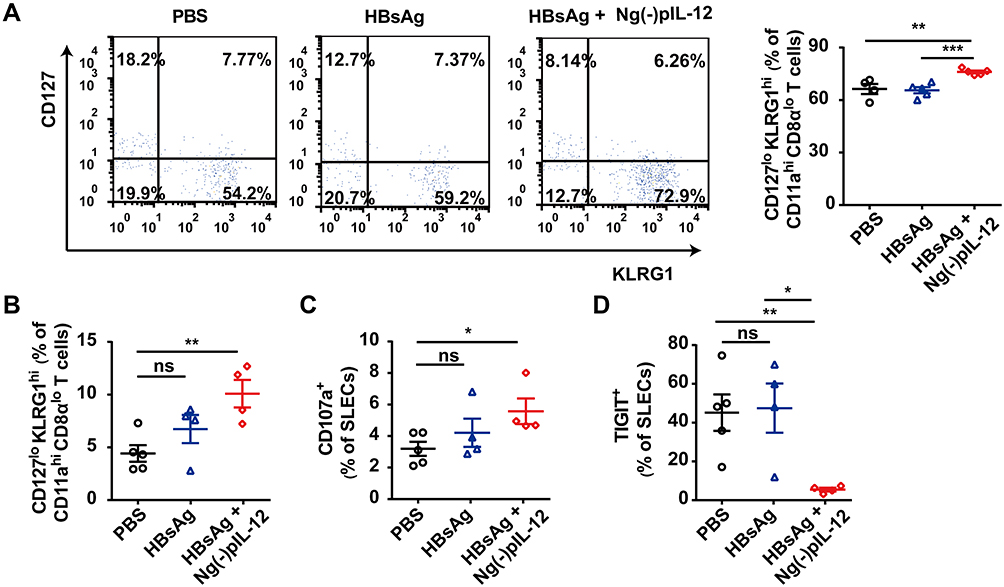 |
Figure 6 Ng(-)pIL-12 adjuvant triggered the terminally differentiated effector memory responses against HBV infection. C57 BL/6J mice were injected intramuscularly with three doses of PBS, 2 μg HBsAg alone, 2 μg HBsAg combined with 45 μg Ng(-)pIL-12 at two-week intervals, separately, then these immunised mice were challenged with 8 μg pAAV/HBV 1.2 on day 56 after the initiation of immunisation. (A) The percentage of SLECs among HBV-specific CD11ahi CD8αlo cells was detected on day 35 after the initiation of immunisation. (B) The percentage of splenic SLECs among HBV-specific CD11ahi CD8αlo cells on day 5 after HBV challenge. The expression of CD107a (C) and TIGIT (D) on splenic SLECs on day 5 after HBV challenge. HBsAg, HBsAg alone; Ng(-)pIL-12, 40 μg Ng(-) containing 5 μg pIL-12. All data are expressed as the mean ± SEM (n ≥ 4) from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001. |
Discussion
In this study, we prepared a chitosan nanovaccine to improve the delivery of IL-12 plasmid and protect it from enzymatic biodegradation. Interestingly, Ng(-)pIL-12 could efficiently load pIL-12 and produce IL-12 both in vitro and in vivo. Additionally, Ng(-)pIL-12 significantly enhanced HBV-specific cellular and humoral immune responses. Since IgG isotypes correlate with cellular immune response, we also measured the expression of anti-HBs IgG1 and IgG2b in our study. Compared to Ng(-) group, Ng(-)pIL-12 vaccination induced higher levels of anti-HBs IgG1 and IgG2b, indicating Ng(-)pIL-12 could significantly enhance the HBV-specific cellular immune response against HBV infection. Although the delivery system Ng(-) slightly increased the anti-HBs levels during vaccination, it could not quickly and successfully induce protective anti-HBs after HBV infection nor eliminate HBV cccDNA, HBV DNA, and HBV RNA, which might be due to insufficient activation of DCs compared to the Ng(-)pIL-12 adjuvant group.
HBV particles and purified HBsAg significantly inhibit IL-12 production through mDCs and monocytes/macrophages, resulting in the dysfunction of these cells.51–53 The impaired IL-12 secretion contributes to chronic HBV infection with high viral load and insufficient HBV-specific adaptive T cell response.54 Exogenous IL-12 rescues the exhausted HBV-specific CD8+ T cell response with more cytotoxicity and polyfunctionality.55 Additionally, a combination treatment using HBsAg vaccine and IL-12 cytokine elicits robust hepatic HBV-specific CD8+ T and CD4+ T cell response, and facilitates the generation of anti-HBs, leading to effective HBV clearance.10
IL-12 is an ideal adjuvant, mainly in protein and plasmid DNA forms. The application of the protein is limited because of the side effects of systemic administration, such as fever, vomiting, headache, and sometimes even life-threatening situations, while the DNA can reduce the occurrence of adverse reactions.17–19 In addition, the DNA more stable, easy to handle, and enables mass production. Meanwhile, the application of nanoparticle delivery systems could combine high biocompatibility with biodegradability and high affinity for nucleic acids.
LPS, as a ubiquitous contaminant in nature, exists in the chemicals and glassware used during the preparation of nanoparticles due to their intrinsic heat stability and resistance to sterilisation.56–60 Since LPS could mediate the activation of the inflammasome and induce the inflammatory cytokine secretion,61,62 this could be an important mechanism by which nanoparticles can contribute to inducing the activation of immune cells. In this study, we prepared the Ng(-) and Ng(-)pIL-12 with same methods, and the pIL-12 used for Ng(-)pIL-12 vaccine was extracted by the E.Z.N.A.® Endo-Free Plasmid Maxi Kit. We found blank Ng(-) control group could not induce the significant immune effects, while Ng(-)pIL-12 vaccination could obviously induce the activation of immune cells in vivo and in vitro and exhibited strong anti-HBV effects. We think the existence of LPS during the preparation of nanoparticles Ng(-) and Ng(-)pIL-12 was insufficient to induce effective influence on immune cells and anti-HBV effects in this study.
DCs play an important role in priming antigen-specific T cells against pathogen infection.63 We found that Ng(-)pIL-12 adjuvant was beneficial for promoting the maturation and antigen presentation ability of DCs. Ng(-)pIL-12 adjuvant could quickly increase the proportion of peripheral blood DCs, accompanied by the augmented expression of co-stimulatory molecules. In particular, Ng(-)pIL-12 adjuvant increased the percentage and the absolute number of lymphoid-resident CD8α+ DCs and migratory CD103+ DCs, accompanied with more cross-presenting abilities after HBV challenge, which could in turn induce the generation of cytotoxic T lymphocyte (CTL) response for HBV clearance.
Rai et al devised a strategy to monitor antigen-experienced CD8+ T cells in response to vaccination or pathogen infection by tracking the changes in CD11a and CD8α expression (CD11ahi CD8αlo),64 while these changes were not driven by inflammatory triggers, such as IL-12 stimulation. Effector CD8+ T cells predict the efficacy of vaccines against pathogens. However, the T cells in patients with chronic HBV infection are generally exhausted, characterised by impaired cytotoxic activity, poor cytokine secretion, and increased expression of multiple co-inhibitory receptors, such as PD-1, LAG-3, CTLA-4, TIGIT, and TIM-3.65,66 Blocking these receptors restores the exhausted CD8+ T cells by increasing their proliferation and the secretion of IFN-γ and IL-2, making it a promising immunotherapeutic strategy to eliminate HBV.67–69 Interestingly, Ng(-)pIL-12 adjuvant increased HBV-specific CD11ahi CD8αlo cells and downregulated the expression of immune checkpoint molecules LAG-3 and TIGIT on CD8+ T cells, indicating strong anti-HBV effects. CD4+ T cells also contribute to HBV clearance by generating and maintaining both neutralizing antibodies and CD8+ T cells.70,71 The “Ag-experienced” CD4+ T cells are characterised by the co-expression of CD11a and CD49d.30 The frequency of HBV-specific CD11ahi CD49dhi CD4+ T cells significantly increased after Ng(-)pIL-12 adjuvant vaccination, indicating the activation of systemic HBV-specific CD4+ T cell response. Therefore, the augmented HBV-specific CD8+ T and CD4+ T cell response induced by the Ng(-)pIL-12 adjuvant vaccination contributed to protecting against HBV infection.
Killer cell lectin-like receptor subfamily G member 1 (KLRG1), together with CD127, has been used as markers to identify effector CD8+ T cell subsets into memory precursor effector cells (MPECs) and SELCs.49,50 SLECs exhibit potent effector functions but are susceptible to death compared to MPECs.45,46 Our recent study showed that vaccination-induced SLECs formed the predominant effector memory subset for HBV elimination.37 Consistently, Ng(-)pIL-12 adjuvant also induced the generation of SLECs with stronger anti-HBV effects, which may be an important mechanism for Ng(-)pIL-12 adjuvant in protecting against HBV infection.
Conclusion
To summarise, chitosan nanovaccine can efficiently load pIL-12, and Ng(-)pIL-12 adjuvant vaccination promotes the maturation and presentation capacity of DCs in vivo and in vitro. Furthermore, Ng(-)pIL-12 acts as an adjuvant for prophylactic HBV vaccine and induces high anti-HBs levels and robust HBV-specific CD4+ T and CD8+ T cell responses. More importantly, Ng(-)pIL-12 adjuvant vaccination triggers the terminally differentiated effector memory responses against HBV infection. Therefore, Ng(-)pIL-12 may also be a promising adjuvant candidate in HBV therapeutic vaccines in the future.
Abbreviations
HBV, hepatitis B virus; CHB, chronic hepatitis B virus; HBsAg, hepatitis B surface antigen; IL-12, interleukin-12; anti-HBs, hepatitis B surface antibody; DCs, dendritic cells; BMDCs, Bone Marrow-Derived Dendritic Cells; MNCs, mononuclear cells; MHC II, MHC class II; CTL, cytotoxic T lymphocyte; IFN-γ, interferon-γ; TIGIT, T-cell immunoreceptor with Ig and ITIM domains; LAG-3, lymphocyte activation gene 3; SLECs, short-lived effector cells.
Acknowledgments
This work was supported by grants from the National Postdoctoral Programme for Innovative Talents (No. BX20190192), China Postdoctoral Science Foundation (No. 2020M672064), the National Science Foundation for Young Scientists of China (No. 82001687), and the National Basic Research Programme of China (No. 2013CB531503).
Disclosure
The authors declare no commercial or financial conflict of interest.
References
1. Tang LSY, Covert E, Wilson E, Kottilil S. Chronic Hepatitis B Infection: a Review. JAMA. 2018;319(17):1802–1813. doi:10.1001/jama.2018.3795.
2. Sundaram V, Kowdley K. Management of chronic hepatitis B infection. BMJ. 2015;351:h4263. doi:10.1136/bmj.h4263.
3. Gill US, Bertoletti A. Clinical Trial Design for Immune-Based Therapy of Hepatitis B Virus. Semin Liver Dis. 2017;37(2):85–94. doi:10.1055/s-0037-1600522.
4. Bruce MG, Bruden D, Hurlburt D, et al. Antibody Levels and Protection After Hepatitis B Vaccine: results of a 30-Year Follow-up Study and Response to a Booster Dose. J Infect Dis. 2016;214(1):16–22. doi:10.1093/infdis/jiv748.
5. Lee MH, Yang HI, Liu J, et al. Prediction models of long-term cirrhosis and hepatocellular carcinoma risk in chronic hepatitis B patients: risk scores integrating host and virus profiles. Hepatology. 2013;58(2):546–554. doi:10.1002/hep.26385.
6. Mori A, Oleszycka E, Sharp FA, et al. The vaccine adjuvant alum inhibits IL-12 by promoting PI3 kinase signaling while chitosan does not inhibit IL-12 and enhances Th1 and Th17 responses. Eur J Immunol. 2012;42(10):2709–2719. doi:10.1002/eji.201242372
7. Vignali DA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol. 2012;13(8):722–728. doi:10.1038/ni.2366.
8. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3(2):133–146. doi:10.1038/nri1001.
9. Tait Wojno ED, Hunter CA, Stumhofer JS. The Immunobiology of the Interleukin-12 Family: room for Discovery. Immunity. 2019;50(4):851–870. doi:10.1016/j.immuni.2019.03.011.
10. Zeng Z, Kong X, Li F, Wei H, Sun R, Tian Z. IL-12-based vaccination therapy reverses liver-induced systemic tolerance in a mouse model of hepatitis B virus carrier. J Immunol. 2013;191(8):4184–4193. doi:10.4049/jimmunol.1203449
11. Jacobson JM, Zheng L, Wilson CC, et al. The Safety and Immunogenicity of an Interleukin-12-Enhanced Multiantigen DNA Vaccine Delivered by Electroporation for the Treatment of HIV-1 Infection. J Acquir Immune Defic Syndr. 2016;71(2):163–171. doi:10.1097/QAI.0000000000000830.
12. Klinman DM, Yi AK, Beaucage SL, Conover J, Krieg AM. CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon gamma. Proc Natl Acad Sci U S A. 1996;93:2879–2883. doi:10.1073/pnas.93.7.2879
13. Klinman DM. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat Rev Immunol. 2004;4:249–258. doi:10.1038/nri1329.
14. Bode C, Zhao G, Steinhagen F, Kinjo T, Klinman DM. CpG DNA as a vaccine adjuvant. Expert Rev Vaccines. 2011;10:499–511. doi:10.1586/erv.10.174
15. Kobiyama K, Temizoz B, Kanuma T, et al. Species-dependent role of type I IFNs and IL-12 in the CTL response induced by humanized CpG complexed with beta-glucan. Eur J Immunol. 2016;46:1142–1151. doi:10.1002/eji.201546059
16. Xiong SQ, Lin BL, Gao X, Tang H, Wu CY. IL-12 promotes HBV-specific central memory CD8+ T cell responses by PBMCs from chronic hepatitis B virus carriers. Int Immunopharmacol. 2007;7(5):578–587. doi:10.1016/j.intimp.2006.12.007.
17. Leonard JP, Sherman ML, Fisher GL, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90(7):2541–2548.
18. Atkins MB, Robertson MJ, Gordon M, et al. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin Cancer Res. 1997;3(3):409–417.
19. Tugues S, Burkhard SH, Ohs I, et al. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015;22(2):237–246. doi:10.1038/cdd.2014.134.
20. Liu X, Gao X, Zheng S, et al. Modified nanoparticle mediated IL-12 immunogene therapy for colon cancer. Nanomedicine. 2017;13(6):1993–2004. doi:10.1016/j.nano.2017.04.006.
21. Chong SY, Egan MA, Kutzler MA, et al. Comparative ability of plasmid IL-12 and IL-15 to enhance cellular and humoral immune responses elicited by a SIVgag plasmid DNA vaccine and alter disease progression following SHIV(89.6P) challenge in rhesus macaques. Vaccine. 2007;25(26):4967–4982. doi:10.1016/j.vaccine.2006.11.070.
22. Sersa G, Teissie J, Cemazar M, et al. Electrochemotherapy of tumors as in situ vaccination boosted by immunogene electrotransfer. Cancer Immunol Immunother. 2015;64(10):1315–1327. doi:10.1007/s00262-015-1724-2.
23. Chow YH, Chiang BL, Lee YL, et al. Development of Th1 and Th2 populations and the nature of immune responses to hepatitis B virus DNA vaccines can be modulated by codelivery of various cytokine genes. J Immunol. 1998;160(3):1320–1329.
24. Irvine DJ, Dane EL. Enhancing cancer immunotherapy with nanomedicine. Nat Rev Immunol. 2020;20(5):321–334. doi:10.1038/s41577-019-0269-6.
25. Yan S, Luo Z, Li Z, et al. Improving Cancer Immunotherapy Outcomes Using Biomaterials. Angew Chem Int Ed Engl. 2020;59(40):17332–17343. doi:10.1002/anie.202002780
26. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17(1):20–37. doi:10.1038/nrc.2016.108.
27. Liu Y, Hardie J, Zhang X, Rotello VM. Effects of engineered nanoparticles on the innate immune system. Semin Immunol. 2017;34:25–32. doi:10.1016/j.smim.2017.09.011.
28. Seth A, Heo MB, Lim YT. Poly (gamma-glutamic acid) based combination of water-insoluble paclitaxel and TLR7 agonist for chemo-immunotherapy. Biomaterials. 2014;35(27):7992–8001. doi:10.1016/j.biomaterials.2014.05.076.
29. Pradhan P, Qin H, Leleux JA, et al. The effect of combined IL10 siRNA and CpG ODN as pathogen-mimicking microparticles on Th1/Th2 cytokine balance in dendritic cells and protective immunity against B cell lymphoma. Biomaterials. 2014;35(21):5491–5504. doi:10.1016/j.biomaterials.2014.03.039.
30. Kuai R, Sun X, Yuan W, et al. Dual TLR agonist nanodiscs as a strong adjuvant system for vaccines and immunotherapy. J Control Release. 2018;282:131–139. doi:10.1016/j.jconrel.2018.04.041.
31. Song Q, Yin Y, Shang L, et al. Tumor Microenvironment Responsive Nanogel for the Combinatorial Antitumor Effect of Chemotherapy and Immunotherapy. Nano Lett. 2017;17(10):6366–6375. doi:10.1021/acs.nanolett.7b03186.
32. Kong M, Tang J, Qiao Q, et al. Biodegradable Hollow Mesoporous Silica Nanoparticles for Regulating Tumor Microenvironment and Enhancing Antitumor Efficiency. Theranostics. 2017;7(13):3276–3292. doi:10.7150/thno.19987.
33. Grippin AJ, Sayour EJ, Mitchell DA. Translational nanoparticle engineering for cancer vaccines. Oncoimmunology. 2017;6(10):e1290036. doi:10.1080/2162402X.2017.1290036.
34. Duan S, Song M, He J, et al. Folate-Modified Chitosan Nanoparticles Coated Interferon-Inducible Protein-10 Gene Enhance Cytotoxic T Lymphocytes’ Responses to Hepatocellular Carcinoma. J Biomed Nanotechnol. 2016;12(4):700–709. doi:10.1166/jbn.2016.2216.
35. Lim YT, Shim SM, Noh YW, et al. Bioderived polyelectrolyte nanogels for robust antigen loading and vaccine adjuvant effects. Small. 2011;7(23):3281–3286. doi:10.1002/smll.201101836.
36. Labeur MS, Roters B, Pers B, et al. Generation of tumor immunity by bone marrow-derived dendritic cells correlates with dendritic cell maturation stage. J Immunol. 1999;162(1):168–175.
37. Zhao HJ, Han QJ, Wang G, et al. Poly I:C-based rHBVvac therapeutic vaccine eliminates HBV via generation of HBV-specific CD8(+) effector memory T cells. Gut. 2019;68(11):2032–2043. doi:10.1136/gutjnl-2017-315588.
38. Beldi G, Banz Y, Kroemer A, et al. Deletion of CD39 on natural killer cells attenuates hepatic ischemia/reperfusion injury in mice. Hepatology. 2010;51(5):1702–1711. doi:10.1002/hep.23510.
39. McDermott DS, Varga SM. Quantifying antigen-specific CD4 T cells during a viral infection: CD4 T cell responses are larger than we think. J Immunol. 2011;187(11):5568–5576. doi:10.4049/jimmunol.1102104.
40. Wang X, Dong A, Xiao J, et al. Overcoming HBV immune tolerance to eliminate HBsAg-positive hepatocytes via pre-administration of GM-CSF as a novel adjuvant for a hepatitis B vaccine in HBV transgenic mice. Cell Mol Immunol. 2016;13(6):850–861. doi:10.1038/cmi.2015.64.
41. Gehring AJ, Ann D’Angelo J. Dissecting the dendritic cell controversy in chronic hepatitis B virus infection. Cell Mol Immunol. 2015;12(3):283–291. doi:10.1038/cmi.2014.95.
42. Ng SL, Teo YJ, Setiagani YA, Karjalainen K, Ruedl C. Type 1 Conventional CD103(+) Dendritic Cells Control Effector CD8(+) T Cell Migration, Survival, and Memory Responses During Influenza Infection. Front Immunol. 2018;9:3043. doi:10.3389/fimmu.2018.03043.
43. Salmon H, Idoyaga J, Rahman A, et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity. 2016;44(4):924–938. doi:10.1016/j.immuni.2016.03.012.
44. Wang L, Wang Z, Qin Y, Liang W. Delivered antigen peptides to resident CD8alpha(+) DCs in lymph node by micelle-based vaccine augment antigen-specific CD8(+) effector T cell response. Eur J Pharm Biopharm. 2020;147:76–86. doi:10.1016/j.ejpb.2019.12.013.
45. Olson JA, McDonald-Hyman C, Jameson SC, Hamilton SE. Effector-like CD8(+) T cells in the memory population mediate potent protective immunity. Immunity. 2013;38(6):1250–1260. doi:10.1016/j.immuni.2013.05.009.
46. Ye F, Turner J, Flano E. Contribution of pulmonary KLRG1(high) and KLRG1(low) CD8 T cells to effector and memory responses during influenza virus infection. J Immunol. 2012;189(11):5206–5211. doi:10.4049/jimmunol.1200137.
47. Kim C, Fang F, Weyand CM, Goronzy JJ. The life cycle of a T cell after vaccination - where does immune ageing strike? Clin Exp Immunol. 2017;187(1):71–81. doi:10.1111/cei.12829.
48. Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol. 2007;25:171–192. doi:10.1146/annurev.immunol.25.022106.141548.
49. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12(11):749–761. doi:10.1038/nri3307.
50. Plumlee CR, Sheridan BS, Cicek BB, Lefrancois L. Environmental cues dictate the fate of individual CD8+ T cells responding to infection. Immunity. 2013;39(2):347–356. doi:10.1016/j.immuni.2013.07.014.
51. Yonejima A, Mizukoshi E, Tamai T, et al. Characteristics of Impaired Dendritic Cell Function in Patients With Hepatitis B Virus Infection. Hepatology. 2019;70(1):25–39. doi:10.1002/hep.30637.
52. Woltman AM, Boonstra A, Janssen HL. Dendritic cells in chronic viral hepatitis B and C: victims or guardian angels? Gut. 2010;59(1):115–125. doi:10.1136/gut.2009.181040.
53. Wang S, Chen Z, Hu C, et al. Hepatitis B virus surface antigen selectively inhibits TLR2 ligand-induced IL-12 production in monocytes/macrophages by interfering with JNK activation. J Immunol. 2013;190(10):5142–5151. doi:10.4049/jimmunol.1201625.
54. Beckebaum S, Cicinnati VR, Zhang X, et al. Hepatitis B virus-induced defect of monocyte-derived dendritic cells leads to impaired T helper type 1 response in vitro: mechanisms for viral immune escape. Immunology. 2003;109(4):487–495. doi:10.1046/j.1365-2567.2003.01699.x.
55. Schurich A, Pallett LJ, Lubowiecki M, et al. The third signal cytokine IL-12 rescues the anti-viral function of exhausted HBV-specific CD8 T cells. PLoS Pathog. 2013;9(3):e1003208. doi:10.1371/journal.ppat.1003208.
56. Gorbet MB, Sefton MV. Endotoxin: the uninvited guest. Biomaterials. 2005;26:6811–6817. doi:10.1016/j.biomaterials.2005.04.063.
57. Vallhov H, Qin J, Johansson SM, et al. The importance of an endotoxin-free environment during the production of nanoparticles used in medical applications. Nano Lett. 2006;6:1682–1686. doi:10.1021/nl060860z.
58. Oostingh GJ, Casals E, Italiani P, et al. Problems and challenges in the development and validation of human cell-based assays to determine nanoparticle-induced immunomodulatory effects. Part Fibre Toxicol. 2011;8:8. doi:10.1186/1743-8977-8-8.
59. Murali K, Kenesei K, Li Y, Demeter K, Kornyei Z, Madarasz E. Uptake and bio-reactivity of polystyrene nanoparticles is affected by surface modifications, ageing and LPS adsorption: in vitro studies on neural tissue cells. Nanoscale. 2015;7(9):4199–4210. doi:10.1039/c4nr06849a.
60. Poh TY, Ali N, Mac Aogain M, et al. Inhaled nanomaterials and the respiratory microbiome: clinical, immunological and toxicological perspectives. Part Fibre Toxicol. 2018;15(1):46. doi:10.1186/s12989-018-0282-0.
61. Boraschi D, Italiani P, Palomba R, et al. Nanoparticles and innate immunity: new perspectives on host defence. Semin Immunol. 2017;34:33–51. doi:10.1016/j.smim.2017.08.013.
62. Li Y, Fujita M, Boraschi D. Endotoxin Contamination in Nanomaterials Leads to the Misinterpretation of Immunosafety Results. Front Immunol. 2017;8:472. doi:10.3389/fimmu.2017.00472.
63. Walsh KP, Mills KH. Dendritic cells and other innate determinants of T helper cell polarisation. Trends Immunol. 2013;34(11):521–530. doi:10.1016/j.it.2013.07.006.
64. Rai D, Pham NL, Harty JT, Badovinac VP. Tracking the total CD8 T cell response to infection reveals substantial discordance in magnitude and kinetics between inbred and outbred hosts. J Immunol. 2009;183(12):7672–7681. doi:10.4049/jimmunol.0902874.
65. Shih C, Chou SF, Yang CC, Huang JY, Choijilsuren G, Jhou RS. Control and Eradication Strategies of Hepatitis B Virus. Trends Microbiol. 2016;24(9):739–749. doi:10.1016/j.tim.2016.05.006.
66. Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–499. doi:10.1038/ni.2035.
67. Fisicaro P, Valdatta C, Massari M, et al. Antiviral intrahepatic T-cell responses can be restored by blocking programmed death-1 pathway in chronic hepatitis B. Gastroenterology. 2010;138(2):
68. Cox MA, Nechanitzky R, Mak TW. Check point inhibitors as therapies for infectious diseases. Curr Opin Immunol. 2017;48:61–67. doi:10.1053/j.gastro.2009.09.052.
69. Fisicaro P, Valdatta C, Massari M, et al. Combined blockade of programmed death-1 and activation of CD137 increase responses of human liver T cells against HBV, but not HCV. Gastroenterology. 2012;143(6):1576–1585 e1574. doi:10.1053/j.gastro.2012.08.041.
70. Chisari FV, Ferrari C. Hepatitis B virus immunopathogenesis. Annu Rev Immunol. 1995;13:29–60. doi:10.1146/annurev.iy.13.040195.000333.
71. Zhu D, Liu L, Yang D, et al. Clearing Persistent Extracellular Antigen of Hepatitis B Virus: an Immunomodulatory Strategy To Reverse Tolerance for an Effective Therapeutic Vaccination. J Immunol. 2016;196(7):3079–3087. doi:10.4049/jimmunol.1502061.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.