")
Back to Journals » Journal of Inflammation Research » Volume 14
Chinese Herbs and Repurposing Old Drugs as Therapeutic Agents in the Regulation of Oxidative Stress and Inflammation in Pulmonary Diseases
Received 24 November 2020
Accepted for publication 14 January 2021
Published 4 March 2021 Volume 2021:14 Pages 657—687
DOI https://doi.org/10.2147/JIR.S293135
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Chien-Chung Yang,1,2 Chuen-Mao Yang3– 5
1Department of Traditional Chinese Medicine, Chang Gung Memorial Hospital at Tao-Yuan, Kwei-San, Tao-Yuan, 33302, Taiwan; 2School of Traditional Chinese Medicine, College of Medicine, Chang Gung University, Kwei-San, Tao-Yuan, 33302, Taiwan; 3Department of Pharmacology, College of Medicine, China Medical University, Taichung, 40402, Taiwan; 4Ph.D. Program for Biotech Pharmaceutical Industry, China Medical University, Taichung, 40402, Taiwan; 5Department of Post-Baccalaureate Veterinary Medicine, College of Medical and Health Science, Asia University, Taichung, 41354, Taiwan
Correspondence: Chuen-Mao Yang No. 91, Hsueh-Shih Road, Taichung, 40402, Taiwan
Tel +886-4-22053366 (ext. 2229)
Email [email protected]
Abstract: Several pro-inflammatory factors and proteins have been characterized that are involved in the pathogenesis of inflammatory diseases, including acute respiratory distress syndrome, chronic obstructive pulmonary disease, and asthma, induced by oxidative stress, cytokines, bacterial toxins, and viruses. Reactive oxygen species (ROS) act as secondary messengers and are products of normal cellular metabolism. Under physiological conditions, ROS protect cells against oxidative stress through the maintenance of cellular redox homeostasis, which is important for proliferation, viability, cell activation, and organ function. However, overproduction of ROS is most frequently due to excessive stimulation of either the mitochondrial electron transport chain and xanthine oxidase or reduced nicotinamide adenine dinucleotide phosphate (NADPH) by pro-inflammatory cytokines, such as interleukin-1β and tumor necrosis factor α. NADPH oxidase activation and ROS overproduction could further induce numerous inflammatory target proteins that are potentially mediated via Nox/ROS-related transcription factors triggered by various intracellular signaling pathways. Thus, oxidative stress is considered important in pulmonary inflammatory processes. Previous studies have demonstrated that redox signals can induce pulmonary inflammatory diseases. Thus, therapeutic strategies directly targeting oxidative stress may be effective for pulmonary inflammatory diseases. Therefore, drugs with anti-inflammatory and anti-oxidative properties may be beneficial to these diseases. Recent studies have suggested that traditional Chinese medicines, statins, and peroxisome proliferation-activated receptor agonists could modulate inflammation-related signaling processes and may be beneficial for pulmonary inflammatory diseases. In particular, several herbal medicines have attracted attention for the management of pulmonary inflammatory diseases. Therefore, we reviewed the pharmacological effects of these drugs to dissect how they induce host defense mechanisms against oxidative injury to combat pulmonary inflammation. Moreover, the cytotoxicity of oxidative stress and apoptotic cell death can be protected via the induction of HO-1 by these drugs. The main objective of this review is to focus on Chinese herbs and old drugs to develop anti-inflammatory drugs able to induce HO-1 expression for the management of pulmonary inflammatory diseases.
Keywords: inflammatory mediators, tracheal smooth muscle cells, pulmonary alveolar epithelial cells, ROS, Nrf2, HO-1
Introduction
Role of Reactive Oxygen Species in Pulmonary Inflammatory Diseases
Reactive oxygen species (ROS) can be internalized in the human body by direct inhalation or can be derived from the chemical processes of various enzymatic reactions. ROS are important for killing invading microorganisms and are also essential for many physiological functions. Increased generation of ROS, including hydroxyl radicals, hydrogen peroxide, and superoxide anion, have been found in individuals with chronic obstructive pulmonary disease (COPD) and asthma.1,2 Among lung diseases, many types of cells, including eosinophils, neutrophils, macrophages, and antigen-presenting cells (APCs), have exhibited increased ROS production.3 In our studies, we found that exposure to cytokines, endotoxins, or cigarette smoke extract in respiratory resident cells such as pulmonary alveolar epithelial cells and tracheal smooth muscle cells leads to the expression of various inflammatory mediators via induction of ROS.4–9 These studies indicate that mitochondria, microsomes, and enzymes are all sources of ROS production, especially phagocytic cells, which produce large amounts of ROS when they are activated.10 Therefore, ROS play a prominent role in the pathogenesis of various pulmonary disorders, in particular when the cells and tissues are exposed to environmental pollutants, infections, inflammatory reactions, or decreased levels of antioxidants lead to oxidative stress. A variety of deleterious effects within the lungs are induced by enhanced levels of ROS and lead to pathophysiological conditions such as COPD and asthma.11–13 Moreover, findings in studies using the canine lung or bovine trachea as models have determined that contractions of smooth muscle can be triggered by hydrogen peroxide and increased levels of environmental oxygen.14 Enhanced ROS levels have been shown to impair cellular functions and enhance inflammatory reactions mediated through damage-inducing carbohydrates, proteins, lipids, and DNA. These cells are stimulated by various factors leading to the activation of the membrane-bound NADPH oxidase (Nox) complex and the generation of superoxide anion when encountering microorganisms, inhaled particles, or other mediators.10 Nox-derived ROS can specifically and reversibly alter the half-life, localization, and activity of proteins. The role of the Nox/ROS pathway is associated with the regulation of cellular signaling.15 Moreover, in the airway alveolar epithelium, ROS have been shown to induce the expression of inflammatory mediators, such as interleukin (IL)-6, tumor necrosis factor (TNF)-α, and IL-1β.16 Serval studies have indicated that oxidants can promote inflammation via the upregulation of transcription factors such as nuclear factor-kappaB (NF-κB) and activator protein 1(AP-1), which are redox-sensitive and orchestrate the expression of multiple inflammatory genes recognized to be implicated in COPD, such as TNF-α, intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), matrix metalloproteinase 9 (MMP-9), cyclooxygenase-2 (COX-2), and cytosolic phospholipases A2 (cPLA2).17 Therefore, ROS play a crucial role during COPD and asthma attacks. Heme oxygenase-1 (HO-1), also known as heat shock protein 32, is a member of the heat-shock family of proteins that protect against oxidative stress and inflammation. HO-1 quenches ROS by the following mechanisms. Heme is catalyzed by HO-1 to iron, carbon monoxide (CO), and biliverdin-IXα.18 Biliverdin-IXα is converted to a potent endogenous antioxidant known as bilirubin-IXα, with the function of anti-inflammation,19 while the iron is sequestered by ferritin, leading to additional anti-apoptotic19 and antioxidant20 effects. CO has abundant biological roles, besides its anti-inflammatory activity,21 and it also shares similar effects of nitric oxide (NO), such as modulation of the vascular tone by increasing cGMP levels and inhibition of smooth muscle cell proliferation and platelet aggregation.19
Mitogen-Activated Protein Kinases (MAPKs) Involved in Pulmonary Inflammatory Diseases
MAPKs are important signaling modules activated by neurotransmitters, growth factors, cytokines as well as mechanical and chemical stressors. Three classes of MAPKs have been identified in mammals: the p38 MAPK, the c-Jun NH2-terminal kinases (JNKs), and the extracellular signal-regulated kinases (ERKs). Asthmatic patients demonstrated increased levels of phospho (p)-JNK1/2, p-p38 MAPK, and p-ERK1/2 in smooth muscle cells or the airway epithelium.22 The pathogenesis of COPD also appears to be associated with the activation of MAPK pathways.23 These activated MAPK signals modify both smooth muscle contraction in acute responses and airway structures in chronic conditions.24,25 In bronchial asthma, MAPKs may control airway remodeling and inflammation.26 In a paraquat-induced mouse model of acute lung injury (ALI), the p38 MAPK signaling pathway was shown to be an important regulator of TNF-α and IL-1β production, and treatment with the p38 MAPK inhibitor SB203580 improved mortality and pathological scores.27 A transgenic mouse model developed to overexpress transforming growth factor (TGF)-α in the lung epithelium showed that treatment with a specific MEK inhibitor, ARRY-142886, prevented the progression of pulmonary fibrosis.28 Another mouse model of pulmonary fibrosis using house dust mite (HDM) demonstrated that CC-930, a JNK1/2 inhibitor, had a protective effect on lung collagen deposition causing inhibition of pulmonary fibrosis.29 In addition, our studies in A549 cells and human airway smooth muscle cells, also found that IL-1β or lipoteichoic acid (LTA) could induce MMP-9, COX-2, or cPLA2 mediated by MAPKs.30,31 Therefore, MAPKs are important in mediating inflammatory responses in the lung and airway. Several lines of evidence have shown that several agents including herbal ingredients can target MAPKs, these include peroxisome proliferator-activated receptor (PPAR)-α agonists (ciprofibrate,32 fenofibrate,33 WY14,64334), PPAR-γ agonists (rosiglitazone,35 pioglitazone,36 LPSF/GQ-237), statins,38,39 and herb-derived substances (salvianolic acid B,40 asiatic acid,41 celastrol,42 fisetin,43 galangin,44 kaempferol,45 luteolin,46 madecassoside,47 oleanolic acid,48 and pristimerin49). We focused on these agents to discuss the molecular mechanisms involved in their activities in the following sections.
Roles of NF-κB and AP-1 in Pulmonary Inflammatory Diseases
NF-κB has been recognized as an important regulator of inflammatory responses due to its importance in mediating the evolution of inflammation. NF-κB controls a wide spectrum of biological effects including tissue remodeling, tumorigenesis, differentiation, apoptosis, and proliferation, in response to immune and stress insults.13 While cells are at rest, NF-κB is bound to inhibitory κB (IκB) an inhibitor protein in the cytoplasm which masks the nuclear translocation signal and thus prevents NF-κB from nuclear translocation. Upon stimulation with various inducers of NF-κB, including several extracellular stimuli, such as oxidative stress, viruses, and environmental particulates [particulate matter (PM10)], TNF-α, and IL-1β, two serine residues of IκBα are rapidly phosphorylated, which target the inhibitor protein for subsequent ubiquitination and degradation, by the E3 ubiquitin-ligases (E3RSIκB, also named β-transducin repeat-containing protein) and the 26S proteasome.50 The NF-κB dimers are released, as either hetero- or homo-dimers, and translocate into the nucleus where they bind to specific DNA elements and κB enhancers to activate target genes. ROS have been known to induce the release of cytokines and chemokines such as IL-6, IL-8, TNF-α, IL-1β, and IL-2 and cause ALI in vivo via the activation of NF-κB.51 Thus, targeting NF-κB could be a potential strategy in the management of lung injury. A growing number of studies have proven that agents having NF-κB inhibitory activity can block lung inflammation in in vivo models. For example, treatment with emodin effectively prevents E-selectin expression, pulmonary edema, monocyte chemoattractant protein-1 (MCP-1) expression, and pulmonary inflammation in a mouse model of lung injury induced by lipopolysaccharides (LPS) and the inactivation of NF-κB may be the major mechanism involved in these effects.52 In the LPS-induced ALI model, NF-κB knockdown using intratracheal instillation of small interfering RNA (siRNA) targeting NF-κB p65 also exerts anti-inflammatory effects, which may be due, in part, to the reduction in the levels of the proinflammatory cytokine TNF-α.53 The upregulation of cytokines is mostly mediated through NF-κB activation. Further, several lines of evidence indicate that lung inflammatory diseases are linked with transcription factor NF-κB activity,54 by alternatively activating macrophages and regulatory T cells, resulting in the generation and maintenance of a pro-inflammatory environment.55 In addition, COPD patients express significantly higher amounts of mRNA of NF-κB family genes and elevated levels of inflammatory molecules, IL-1β, IL-8, and COX-2, than those of healthy controls.56 Clinical evidence has also shown that the severity of COPD is associated with increased NF-κB expression in the epithelia.57 Furthermore, our previous studies also demonstrated that overexpression of HO-1 can downregulate tumor necrosis factor receptor 1 (TNFR1)-dependent oxidative stress and NF-κB activation to protect against TNF-α-mediated airway inflammation.58 Thus, in airway inflammation and lung injury, the expression of inflammatory proteins is mediated through NF-κB signaling, which can be a potential target in managing pulmonary inflammatory disorders such as asthma and COPD.59,60
Fos (c-Fos, Fos B, Fra-1, Fra-2) and Jun (c-Jun, Jun B, Jun D) family members are subunits of the AP-1 transcription factor and are typically responsible for the transcriptional activation of various genes via binding to the promoters of target genes.61 For DNA binding, the “leucine zipper” domain of fos-jun or jun-jun dimers is required to regulate the expression of a wide variety of genes.62 AP-1 binding sites exist in the promoter region of many genes related to inflammatory responses, especially in the promoter region encoding cytokines and chemokines. AP-1 may be activated by various cytokines, including IL-1β and TNF-α via several types of MAPKs and protein kinase C (PKC), which in turn can activate a cascade of intracellular kinases.61 Thus, many inflammatory mediators can be transcriptionally regulated by AP-1. In the lungs of smokers, AP-1 and its components c-fos and c-jun are upregulated.63 Increased AP-1 activities in lung tissues were detected after initiation of the inflammatory response, which was demonstrated by up-regulating mRNAs and proteins of c-jun, jun-B, jun-D, and c-fos, in whole lung tissues and alveolar macrophages.64 Moreover, in a rat lung fibrosis model induced by intratracheal administration of bleomycin, alveolar macrophages and type II pneumocytes expressed high levels of c-jun and c-fos.65 Thus, AP-1 has a critical effect on mediating pulmonary inflammatory responses in lung disorders. Moreover, there is growing evidence indicating that in asthmatic airways, a specific inflammatory response may be triggered by the cooperative interaction of these transcription factors (eg NF-κB and AP-1) leading to the optimal expression of specific genes.66 Thus, in addition to NF-κB, in respiratory diseases, AP-1 is also an important factor.
Roles of Cytokines and Endotoxins in Pulmonary Inflammatory Diseases
In many diseases, chronic inflammation is an important component of pathogenesis. In the orchestration of chronic inflammation, cytokines exert a critical role in many diseases. Multiple chemokines and cytokines have been implicated in the pathophysiology of asthma and COPD. For example, in asthma, TNF-α has been shown to be highly expressed in the airways, and asthmatic inflammation may be amplified by TNF-α-mediated activation of transcription factors such as NF-κB and AP-1.67 A high level of TNF-α has also been shown in COPD patients.68 In asthmatic airways, TNF-α contributes to the dysregulation of inflammatory responses of patients with asthma and COPD who demonstrated increased TNF-α mRNA69 and protein levels.68,70 Moreover, when normal subjects were treated with inhaled recombinant TNF-α, they developed airway neutrophilia and airway hyper-responsiveness.71 However, the detailed mechanisms underlying these observations have not been fully clarified. These responses could be caused by either a direct effect of TNF-α on airway smooth muscle72 or the release of cysteinyl-leukotrienes LTC4 and LTD4.73 In asthma patients, mast cells of the airways release mediators that have been suggested to be involved in the pathogenesis of bronchoconstriction and airway hyper-responsiveness.74 In addition, bacterial infections are involved in several lung and airway inflammatory diseases. The characteristics of these diseases include pathogen-evoked inflammatory responses in the host. These phenomena have been well validated for LTA in gram-positive bacteria and LPS of gram-negative bacteria.75 The LPS of gram-negative bacteria and the LTA of gram-positive bacteria are considered to have analogous characteristics in both physiological and biochemical properties.75 LTA, like LPS, is an amphiphile that is formed by linking a glycolipid to a hydrophilic polyphosphate polymer.75 Highly purified preparations of LTA from Staphylococcus aureus have been used to efficiently activate monocytes via Toll-like receptor 2 (TLR2) to produce TNF-α.76 LTA is an antigen characterized by inflammatory changes in the lungs exacerbating the severity of respiratory disorders. The inflammatory response is thought to be triggered by the shedding of the epithelial barriers that allow LTA to have relatively easy access to the tracheal smooth muscle cells (TSMCs). Airway inflammation can be due to the expression of a variety of inflammatory proteins in airway smooth muscle, such as IL-6, PLA2, COX-2, VCAM-1, and ICAM-1. LTA and TNF-α have been indicated to regulate inflammatory responses by inducing the expression of these inflammatory proteins.
Roles of Heme Oxygenase-1/Carbon Monoxide in Pulmonary Inflammatory Diseases
Tenhunen et al first identified heme oxygenase (HO) in 1968 when they described the catabolizing of heme.77 HO has been associated with apoptosis, cell growth, and vascular tone in a variety of pulmonary diseases.19 The characteristics of the three HO isoforms (HO-1, 2, and 3) have been described previously.78 HO-1 alone is inducible whereas HO-2 and −3 are constitutively expressed. Although the inducible form, HO-1, is known to be part of the integrated response to oxidative stress and inflammation, only recently has it been linked to the regulation of inflammatory lung disorders.21 However, we still have not fully understood how HO-1 performs as an anti-inflammatory and cytoprotective protein. HO-1 is expressed in various types of cells, in the lung, including alveolar macrophages and type II pneumocytes, HO-1 is induced by endotoxins, proinflammatory cytokines, NO, heme, hypoxia, or hyperoxia.19 HO-1 expression is up-regulated, in several pulmonary diseases, including acute respiratory distress syndrome, rejection following lung transplantation, idiopathic pulmonary fibrosis, COPD, asthma, and cystic fibrosis.78
Asthma
Asthma is an inflammatory disease and presents with limited generalized airflow due to bronchoconstriction. The airways of asthmatic patients feature an accumulation of inflammatory cells and mucus. An imbalance between oxidants and antioxidants contributes to airway inflammation, a key component of asthma, which activates redox-sensitive transcription factors such as AP-1 and NF-κB to upregulate the expression of proinflammatory mediators and cause airway epithelium damage.66 Accumulating evidence has supported a role for HO-1 in airway inflammation and asthma. Exhaled CO levels were found to be elevated in asthma due to HO-1 induction in alveolar macrophages in most untreated patients experiencing a recent asthmatic attack compared with controls.18,79 These findings imply that HO-1 exerts a protective role in asthma. Moreover, Almolki et al demonstrated that hemin up-regulates HO-1 to reduce airway responsiveness to histamine, mucus secretion, and airway inflammation in ovalbumin-sensitized guinea pigs.80 In experiments in mouse models, CO has been found to reduce airway hyperresponsiveness and inflammation.21 In addition, a recent study also implied that in asthma, the protective effects of HO-1 on airway smooth muscle remodeling were mediated by bilirubin.19 Recent data have revealed that CO has abundant effects on regulating intracellular signaling processes, which culminate in anti-coagulative, anti-apoptotic, anti-proliferative, and anti-inflammatory effects.19 Conversely, HO-1 has also been reported to impede cell rolling, adhesion, and migration of immune cells from the vessel, possibly due to its down-regulating abilities on the expression and function of adhesion molecules in the endothelium.81
Chronic Obstructive Pulmonary Disease
COPD is a progressive disorder characterized by irreversible airflow limitation caused by chronic inflammation affecting primarily the lung peripheral airways and parenchyma. In the pathogenesis of COPD-related inflammation, oxidative stress plays a crucial role by activating proinflammatory transcription factors such as NF-κB to secrete a variety of proinflammatory mediators, including growth factors, cytokines, chemokines, and lipid mediators in inflammatory cells and structural cells, including epithelial and endothelial cells and fibroblasts. ROS can oxidize different biomolecules such as lipids, proteins, and DNA leading to impaired physiological functions including inactivation of antiproteases, which in turn cause epithelial injury and death as well as aging and other diseases. An imbalance between the oxidant/antioxidant status and increased ROS exposure such as through cigarette smoke and chronic infections are risk factors for the development of COPD. Cells and tissues possess endogenous antioxidant defense systems including the tripeptide glutathione (GSH). The HO-1/CO system also acts as another anti-oxidative stress system. Moreover, a reciprocal regulation exists between GSH concentration and HO-1. HO-1 is transcriptionally upregulated by depletion of GSH.82 A previous study found that alveolar macrophages isolated from bronchoalveolar fluid (BALF) in patients with COPD have reduced levels of HO-1 expression.83 Furthermore, humans express two potentially functional polymorphisms in the HO-1 gene promoter region, which induce different HO-1 transcriptional responses. Polymorphisms of the HO-1 promoter associated with a strong ability to induce HO-1 expression may be an important endogenous protective factor to reduce susceptibility to COPD.84 Moreover, HO-1 induction may attenuate senescence, including reduced replicative capacity, and the inflammatory profile in fibroblasts isolated from COPD patients by restoring mitophagy and protecting against mitochondrial dysfunction.85 In a rat model of smoke-induced emphysema, injection with protoporphyrin IX upregulated HO-1, which in turn attenuated the development of smoke-induced emphysema, the levels of inflammatory mediators, inflammatory cell infiltration as well as oxidative damage.86 Thus, upregulation of HO-1 may possibly decrease the susceptibility of developing COPD.
The clinical significance of HO-1 expression during asthma is still an open issue. HO-1 or its products may be beneficial as both therapeutic and diagnostic targets. Defining not only the regulation of HO-1 during lung and airway inflammation but also the diagnostic and therapeutic roles of the HO-1 pathway in COPD and asthma will remain an important avenue of research in the near future. Below we will discuss the protective mechanisms of HO-1/CO in respiratory inflammatory diseases. We particularly focus on herbal medicines with the potential to induce HO-1 expression, including salvianolic acid A,87 asiatic acid,41 celastrol,88 fisetin,89 galangin,90,91 kaempferol,92–95 luteolin,96 madecassoside,97,98 oleanolic acid,99,100 saikosaponin A,101 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid (CDDO),102 and pristimerin.49,103 In the following sections, the detailed pharmacological mechanisms of these herb ingredients will be further discussed.
Redox-Dependent Transcription Factors Mediate Inducible HO-1 Gene Expression
Cytokines, oxidative stress signals, growth factors, and bacterial compounds, can all act as inducers of HO-1 expression in different species. In particular, HO-1 is regulated at the transcriptional level and the HO-1 promoter contains multiple cis-acting regulatory elements, which have been shown to control basal and inducible HO-1 gene expression.19,104,105 E1 and E2 (two upstream enhancers regulating HO-1 induction) are located in the upstream enhancer regions, which play key roles for the redox-dependent induction of HO-1.105 Several antioxidant response elements (AREs) have been found to exist in both E1 and E2 enhancer regions, as well as in the promoter regions of other stress-inducible antioxidant and Phase 2 detoxifying genes.106,107 The GT-microsatellite polymorphism localized in the proximal human HO-1 gene promoter region is an important difference between the rodent and human HO-1 genes having major biological relevance. In response to stress stimuli, a lower number of GT repeats within this polymorphic sequence leads to higher inducibility of the HO-1 gene108 and this allele seems to be associated with protective ability against cardiovascular disorders.84 Below, we will discuss the critical role of the NF-E2-related factor 2 (Nrf2) a major redox-dependent transcription factor in regulating inducible HO-1 gene expression by statins, PPAR agonists, and Chinese herbal medicines in pulmonary resident cells.
Nrf2 Signaling in Coordinated Activation of Antioxidant Gene Expression
Electrophiles and oxidants impose persistent stresses and exacerbate many chronic diseases. For example, aging and age-related diseases, such as chronic inflammation, cancer, neurodegenerative diseases, and cardiovascular disease are induced by oxidative stresses. In these processes, oxidative stress increases the levels of oxidized DNA, phospholipids, and proteins. At the same time, cells have developed adaptive mechanisms to dynamically offset intrinsic and extrinsic electrophile and oxidant levels imposed by environmental stresses. These systems involve four categories of mechanisms: (1) redox reactions often catalyzed by cytochrome p-450 enzymes, through functional groups that are exposed or introduced onto largely hydrophobic organic molecules; (2) efflux transporters that export toxic metabolites; (3) nucleophilic trapping processes that engage cellular nucleophiles or GSH and electrophilic xenobiotics to facilitate excretion, such as conjugations of cofactors containing electrophilic adenosine with nucleophilic xenobiotics and those catalyzed by GSH S-transferases (GSTs), as well as enzymes, such as catalase,109 GSH peroxidase, and, superoxide dismutases (SOD) which detoxify ROS; and (4) intracellular GSH and thioredoxin, which contain thiol-molecules to maintain reducing conditions. When a cell encounters a potentially toxic agent, the outcome is often largely determined by the balance between reactive intermediates activated by the activities of enzymes from substrates, and these reactive species are detoxified by enzymatic activities. At least three essential components are required to induce this protective response: (a) Nrf2, the transcription factor binds to the ARE through heterodimerization with members of the small Maf family of transcription factors, and expresses ARE-regulated genes by recruiting the general transcriptional machinery; (b) AREs, present on each gene in either single or multiple copies, are upstream regulatory sequences; and (c) Kelch-like ECH-associated protein 1 (Keap1), a repressor protein located in the cytosolic compartment binds to Nrf2, sequesters it in the cytoplasm and thus allows its degradation via the proteasome-ubiquitin protein degradation pathway. For stress signals, several critical cysteine residues on Keap1 serve as primary sensors and conformational changes on Keap1, which are modified by these oxidative stresses, thereby leading to the release of Nrf2. In addition, the detoxification of xenobiotics via conjugation and trapping processes catalyzed by the classical environmental stress response, and genomic analyses indicate that this transcription factor induces gene families to provide antioxidative and anti-inflammatory reactions.
HO-1 Inducers
Pharmacological Induction of HO-1 as an Anti-Inflammatory Therapeutic Target
Cobalt protoporphyrin IX, commonly used in in vitro experimental cell models and animal models, is a type of metalloporphyrin and the prototypical inducer of HO-1. Metalloporphyrins are severely toxic and lack cell specificity and are not suitable for clinical interventions. Similarly, heme has been approved for the treatment of acute intermittent porphyria (heme arginate), which is one of the most potent inducers of HO-1 and only has limited potential for the management of inflammation. A growing body of literature has indicated that HO-1 induction can exert protective effects via decreasing inflammation, apoptosis, vascular remodeling, fibrosis, and improving survival rate in lung injury models including COPD, asthma, pulmonary hypertension, adult respiratory distress syndrome, and pulmonary fibrosis.21,78 In contrast, several pharmacological compounds are currently available, which act as HO-1 inducers and are used in standard therapies, and might also be effective for the clinical intervention of inflammatory disorders. For example, statins, with cholesterol-lowering effects, were initially introduced for the prevention of arteriosclerosis but have also been recognized to induce HO-1 and to exhibit anti-inflammatory effects.110,111 HO-1 induced by protoporphyrin IX can attenuate the development of smoke-triggered emphysema mediated through anti-inflammatory and antioxidant effects.86 Our recent studies have demonstrated that HO-1 is induced by mevastatin mediated via a Nox/ROS-dependent c-Src/PDGFRα/PI3K/Akt/Nrf2/ARE cascade or via c-Jun activation of PKCα/Pyk2/p38α MAPK or JNK1/2 to suppress proinflammatory mediators-mediated inflammatory responses in human pulmonary alveolar epithelial cells (HPAEpiCs).112,113 Carbon monoxide-releasing molecule (CORM-2) induces HO-1 expression via Src, epidermal growth factor receptor (EGFR), and PI3K/Akt to increase the formation of the Nrf2 and AREs complex in HPAEpiCs to suppress TNF-α-mediated inflammatory responses.114 CORM-2 also activates a PYK2/PKCα/ERK1/2/AP-1 pathway leading to HO-1 expression in HPAEpiCs.115 We determined that CORM-2-induced HO-1 expression in human tracheal smooth muscle cells (HTSMCs) was mediated through the PKCα/Pyk2-dependent Nox/ROS/ERK1/2/AP-1 pathway, which can mitigate lipopolysaccharide-induced airway inflammation.116 CORM-2 also activates the c-Src/EGFR/PI3K/Akt/JNK1/2 and p38 MAPK pathways, which in turn trigger Nrf2 activation to induce HO-1 expression in HTSMCs.117 Interestingly, our previous data also demonstrated that the particle-phase extract of cigarette smoke can activate Nrf2 to induce HO-1 expression via a c-Src/NADPH oxidase/ROS/MAPK pathway in HTSMCs.118 Below, we will review the pharmacological effects of HO-1 expression in protecting against lung inflammation.
A novel therapeutic approach for treating oxidative stress and inflammation involves agonists of PPAR, which were originally used as antidiabetic and dyslipidemia agents. The anti-inflammatory effects of PPAR agonists likely account for these protective roles in these diseases. The capability of PPAR agonists to repress proinflammatory genes is exerted via the antagonism of transcription factors including signal transducer and activator of transcription (STAT), AP-1, ATF-1 and 4, and NF-κB.119 In mixed neuron-glial cultures, PPAR agonists have been demonstrated to impede the transcription of the inducible form of nitric oxide synthase120 and COX-2.121 In addition, PPAR agonists possess anti-oxidative and anti-inflammatory effects through the induction of HO-1.122
Natural herbs and herbal supplements can exert extensive health benefits. Based on traditional Chinese herbal medicines, herbs have been used as far as the first century CE or earlier. Indeed, herbs have a variety of functions including medicinal, culinary, and spiritual in some cases. With regard to medicine, herbs exert a wide range of pharmacological effects including anti-oxidative and anti-inflammatory activities. We will focus on the potential effectiveness of several pure compounds extracted from traditional Chinese herbal medicines.
Due to the immunomodulatory actions of HO-1 in various types of cells, these traditional Chinese herbs might be specifically applicable to HO-1 induction in pulmonary resident cells as medicinal interventions. Significant anti-oxidative and anti-inflammatory therapeutic effects have been described by HO-1 induction in these cells. Therefore, we will characterize and identify the pharmacological mechanisms that upregulate HO-1 expression in various resident cells of the pulmonary system based on in vivo and in vitro studies.
PPARs, ligand-inducible transcription factors, belong to a superfamily of the nuclear receptor superfamily. To date, three PPAR isotypes, encoded by separate genes, including PPAR-α, PPAR-β/δ, and PPAR-γ, have been identified.123 PPARs mainly control inflammation, lipid metabolism, adipogenesis, and the maintenance of metabolic homeostasis through the regulation of the expression of related gene networks. Three PPAR isoforms are activated by fatty acid-derived eicosanoids and fatty acids. They act as lipid sensors that can regulate metabolism. A distinct tissue distribution pattern exists among these three PPAR isotypes, in the regulation of energy metabolism and their associated functions. PPAR-α is expressed in tissues including muscles, kidney, heart, and liver, and mainly regulates the metabolism of lipoproteins and lipids mediated through the regulation of target gene expression.124 Except in the liver, PPAR-β/δ is expressed in all body tissues.124 In skeletal muscle, adipose tissue, and the heart, PPAR-γ has emerged as an important regulator of energy balance and lipid metabolism.125 The PPAR-γ protein exists in two isoforms obtained by utilizing distinct 5ʹ-exons and promoters expressed from the same gene sequence. Compared to PPAR-γ1, PPAR-γ2 exerts higher transcriptional activity, which is determined by the ligand-independent domain at the N-terminal end and by an additional stretch of 30 amino acid residues.126,127 The distribution pattern of these two PPAR-γ isoforms is distinct. PPAR-γ1 is mainly expressed in hematopoietic cells, the large intestine, and in adipose tissue and to a lower extent in the small intestine, liver, muscles, pancreas, and kidney. Under physiological conditions, PPAR-γ2 is restricted to brown and white adipose tissue.124,128 All three PPAR subtypes share an identical gene transcription process. PPARs form heterodimers with the retinoid X receptor (RXR), another ligand-activated nuclear receptor, for ligand binding. The PPAR-RXR heterodimer binds to peroxisome proliferator response elements (PPREs), which are DNA-specific sequences in the promoter area of specific target genes.129 When different transcriptional cofactors are recruited, the transcription process is started.130 In addition to differential distribution patterns, PPAR isoforms possess distinct sensitivities and selectivity to ligands and vary in the recruitment of different coactivator proteins, which result in the regulation of different sets of target genes. PPAR-α generally regulates genes implicated in inflammation, vascular function, fatty acid uptake, and oxidation; whereas, PPAR-γ regulates genes implicated in inflammation, glucose homeostasis, and fatty acid uptake and storage. PPAR-δ regulates genes implicated in macrophage lipid homeostasis, inflammation, and fatty acid metabolism.131
PPAR agonists are used for the treatment of metabolic syndrome and cardiovascular disease (CVD). However, during recent decades, PPAR-α and PPAR-γ have been implicated in the regulation of inflammation, including lung inflammation. We will discuss the recent evidence supporting the effects of PPAR-α and PPAR-γ against inflammatory response and define potential targets as a novel strategy for the management of pulmonary inflammation. PPARs are triggered by both synthetic and natural ligands, which are either non-selective or isotype-selective. A variety of endogenous fatty acids have been identified as the main ligands for PPAR-α, such as the arachidonic acid derivate leukotriene B4 (LTB4) and 8S-hydroxyeicosatetraenoic acid. Dehydroepiandrosterone, a precursor of both estrogens and androgens, and oleoyl ethanolamide, a regulator of body mass and feeding, are other activators of PPAR-α activity derived from natural sources. Other compounds such as fibrates are synthetic molecules used clinically to treat dyslipidemia and include bezafibrate, ciprofibrate, and fenofibrate, as well as GW2331 and Wy-14,643, also act as pharmacological tools in inflammatory studies.
The main PPAR-γ agonists include endogenous ligands, such as the 12/15 lipoxygenase products 13-hydroxyoctadecadienoic acid and 15-hydroxyeicosatetraenoic acid, and the cyclopentenone prostaglandin 15-deoxy-D12,14-prostaglandin J2 (15d-PGJ2), as well as thiazolidinediones (TZDs) belonging to the synthetic compounds used in the clinic for their antidiabetic effects. Our recent studies have indicated that HO-1 expression induced by rosiglitazone is mediated through PKCα/AMPKα/p38 MAPKα/Sirtuin 1 (SIRT1)-dependent deacetylation of Ac-PGC1α and fragmentation of nuclear receptor corepressor (NCoR)/PPAR-γ activation in HPAEpiCs, which protects against the inflammatory responses triggered by LPS.132 Moreover, we also found that rosiglitazone induced-HO-1 expression can be mediated through either NOX/ROS/c-Src/Pyk2/Akt-dependent Nrf2 activation or via PPAR-γ in HPAEpiCs to mitigate LPS-mediated inflammatory responses.122 Ibuprofen and indomethacin are non-steroidal anti-inflammatory drugs that also activate PPAR-γ when used at higher concentrations than those required to attenuate the activity of cyclooxygenase enzyme.133 Among these compounds, 15d-PGJ2 has been widely applied to evaluate the potential effects of PPAR-γ as a mediator of anti-inflammatory activity. However, it must be indicated that 15d-PGJ2 at micromolar concentrations also activates PPAR-α, although it is considered a selective agonist of PPAR-γ. Furthermore, many studies have provided evidence that 15d-PGJ2 may also regulate PPAR-γ-dependent anti-inflammatory activity in the lungs.
Roles of PPARs in Pulmonary Inflammatory Diseases
Among the PPARs, PPAR-α was the first shown to have a role in the control of inflammation.134 Devchand et al (1996) found that direct interaction between LTB4 and PPAR-α increased the catabolism of LTB4 and induced enzymes involved in fatty acid degradation, which further enhanced PPAR-α function by either increasing receptor levels or activating the receptor. Several studies using PPAR agonists in knock-out mice or wild-type animals, as well as clinical trials in human, have provided evidence for the anti-inflammatory properties of PPAR-γ and PPAR-α in various diseases including atherosclerosis;135–139 inflammatory bowel diseases, such as ulcerative colitis and Crohn’s disease;140–142 myocardial infarction and stroke;133,143 rheumatoid arthritis;144–146 psoriasis;147,148 COPD;149 and allergic dermatitis.150,151
PPAR-γ Agonists: Rosiglitazone
Hammad et al, using ovalbumin (OVA)-pulsed dendritic cells transfer into the intratracheal region as a sensitization model, showed that rosiglitazone, a selective PPAR-γ agonist, prevents eosinophilic airway inflammation by draining mediastinal lymph nodes, reducing Ag-specific T cell proliferation, and by increasing IL-10 levels produced by T cells.152 Another study exploring the pathogenesis of asthma also revealed that the protective role of rosiglitazone is partly mediated through a mechanism dependent on IL-10.153 Intranasal administration of rosiglitazone significantly blocked not only airway smooth muscle remodeling and the eosinophilic inflammatory response in mice models of eosinophilic airway inflammation following exposure to OVA, but rosiglitazone also decreased the expression of NF-κB and TLR-4 in the OVA-exposed group.154 Similarly, in a 1% cigarette smoke extract in vitro model, rosiglitazone also down-regulated the expression of NF-κB and TLR-4 in 16HBE cells (bronchial epithelial cells).155 The inhibition of the NF-κB pathway by rosiglitazone treatment also attenuated airway mucus hypersecretion and the inflammatory response induced by acrolein in rats.156 Furthermore, rosiglitazone treatment also attenuated ROS production by reducing myeloperoxidase157 activity157 and expression of ICAM-1, and pulmonary overproduction of cytokine-induced neutrophil chemoattractant-1 (CINC-1) and TNF-α as well as malondialdehyde (MDA) levels via blocking the nuclear translocation of NF-κB in an endotoxemia rat model.158
In a microparticle (MP) in vitro model using human lung epithelial cells and A549 alveolar cells, pre-incubation with 15-deoxy-D12,14-prostaglandin-J2 and rosiglitazone reduced NF-κB activation and the synthesis of MCP-1 and IL-8,159 the effects of rosiglitazone were reversed by GW9662, the specific PPAR-γ antagonist. Momoi et al also demonstrated similar findings in A549 cells, whereby thiazolidinedione inhibited IL-1α and TNF-α induced-endogenous MCP-1 messenger RNA expression and protein secretion.160 The effects of rosiglitazone on inhibition of NF-κB were also indicated by Cheng et al who revealed that the PPAR-γ agonist rosiglitazone inhibited NF-κB expression to ameliorate airway inflammation in asthmatic mice, leading to inhibition of inflammatory corpuscle activity of TLR2/Nod-like receptor with pyrin domain containing 3 (NLRP3).161 In the progression of respiratory diseases, such as COPD, neutrophils are important inflammatory cells. In the LPS-induced acute porcine lung injury model,78 intravenous rosiglitazone significantly controlled local pulmonary inflammation as reflected by a significant reduction in the expression of cytokines and neutrophil activity within the alveolar compartments.162
Other studies have demonstrated that when rosiglitazone is used after or before exposure to the aerosolized LPS insult, which induces neutrophilia and associated survival factors/chemoattractants such as CC chemokine ligand-5 (CCL5) and granulocyte colony-stimulating factor (G-CSF) in the airways, it inhibits airway inflammation.163–166 In addition, several lines of evidence have found that rosiglitazone achieves its anti-inflammatory effects through the upregulation of HO-1 expression.122,132,167,168 Many PPAR-γ ligands modulate multiple cellular pathways via both PPAR-γ-dependent and independent pathways, thus for human diseases including lung disease, these ligands are under evaluation as potential remedies.169 Our recent studies have demonstrated that HO-1 is up-regulated via PPAR-dependent and independent pathways,122,132 and attenuates the LPS-induced inflammatory responses in pulmonary resident cells. Kadam et al also revealed that rosiglitazone treatment significantly elevates the expression of Nrf2 and HO-1, which have a lower expression in macrophages from the animals treated with LPS.170 Rosiglitazone ameliorated the lung injury in a mouse model of ALI and promoted epithelial sodium channel (ENaC)-mediated alveolar fluid clearance (AFC) to alleviate pulmonary edema via PPAR-γ/serum and glucocorticoid-induced kinase-1 (SGK1) signaling pathway dependence.171
Based on the literature, rosiglitazone inhibits inflammation-related lung remodeling. Rosiglitazone reduced the levels of MMP-9 and MMP-2 proteins by attenuating MAPKs and NF-κB activation in lung tissues of COPD rat models.35 Rosiglitazone treatment inhibits lung fibroblasts stimulated by fetal bovine serum (FBS) and growth factor proliferation, migration, and myofibroblast transdifferentiation in vitro, suggesting that PPAR-γ agonists may rescue pulmonary fibrosis.172 Another pulmonary fibrosis murine model induced by bleomycin also demonstrated the therapeutic activity exerted by rosiglitazone in pulmonary fibrosis based on findings revealing normal lung features on micro-computed tomography (CT) scans were achieved in the majority (80%) of the intervention group.173 Interestingly, Ward et al found that in a murine model of allergen-induced inflammation, rosiglitazone reduces airway hyperresponsiveness, which is potentially mediated through an anti-inflammatory action-independent effect.174 These studies have revealed that rosiglitazone possesses a broad range of effects in pulmonary inflammatory disorders.
Other Thiazolidinedione PPAR-γ Agonists
Treatment with ciglitazone significantly reduced IκB kinase175 activity and IκBα degradation and completely inhibited NF-κB DNA binding. This reduction of IKK activity induced by ciglitazone appeared to be a consequence of physical interaction between PPAR-γ and IKK. Ciglitazone may ameliorate lung inflammatory injury following hemorrhagic shock, which appears to be mediated by inhibition of the IKK/NF-κB pathway.176 Pioglitazone ameliorates ALI and following fibrosis induced by bleomycin, at least partly through suppression of the expression of connective tissue growth factor (CTGF), TNF-α, and procollagen I.177 PPAR-γ agonists ciglitazone and troglitazone inhibit myofibroblast differentiation and collagen secretion in human lung fibroblast cells induced by TGF-β1. Thus, in a murine model of bleomycin-induced fibrosis, PPAR-γ agonists also inhibit lung fibrosis.178 The inhibitory effects of pioglitazone, a PPAR-γ agonist, on hyperresponsiveness, airway inflammation,179 and remodeling were shown to be mediated through the upregulation of regulator of G protein signaling 4 (RGS4) via ERK1/2 and Akt/mammalian target of rapamycin (mTOR) signaling in OVA-induced asthma in BALB/c mice.36 Similar to rosiglitazone, PPAR-γ ligands 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) and 15d-PGJ2 may promote HO-1 upregulation in a PPAR-independent mechanism and possess potent antifibrotic effects in vitro in primary human lung fibroblasts. CDDO and 15d-PGJ2 upregulate HO-1 via a GSH-dependent mechanism implicating a complex formation of covalent bonds between CDDO or 15d-PGJ2 and glutathione (GSH) but does not involve Nrf2 or AP-1 activity.180
Like rosiglitazone-induced inhibition of chemotaxis of neutrophils, the PPAR-γ ligands troglitazone and 15d-PGJ2 also inhibit polymorphonuclear leukocytes (PMN) chemotactic responses to N-formyl methionyl-leucylphenylalanine (fMLP) and IL-8 in sepsis patients and mice models of sepsis.181 The PPAR-γ agonist ciglitazone inhibits pro-inflammatory pathways in NIH-A549 cells by blocking the overexpression of nitric oxide synthase (NOS) induced by cytokine and cytokine-induced IL-8 secretion.182 CDDO, a PPAR-γ ligand, is a potent inhibitor of the differentiation of human lung fibroblasts to myofibroblasts stimulated by TGF-β, and attenuates the expression of collagen, fibronectin, and, α-smooth muscle actin via a pathway not related to PPAR-γ.183 The PPAR-γ ligands 15d-PGJ2 and troglitazone virtually abrogate Egr-1 activity and target its inflammatory gene in hypoxic mononuclear phagocytes, which provide a potentially protective effect on ischemic pulmonary injury.184
In the guinea pig model of acute lung inflammation induced by LPS, pioglitazone is effective in attenuating the dysfunction of the lung by abrogating oxidative stress, TNFα release, and neutrophilia.185 Synthesis of thiazolidines derivatives-LPSF/GQ-2 anti-inflammatory action of lung injury could be attributed to the inhibition of NF-κB, ERK1/2, p38 MAPK, and poly(ADP-ribose) polymerase (PARP) pathways.37
PPAR-α Agonists
Fenofibrate is a lipid-modifying agent derived from fibric acid, which can activate the PPAR-α. The most common and serious form of idiopathic interstitial pneumonia is idiopathic pulmonary fibrosis. Typical characteristics include abnormal alveolar structure accompanied by collagen deposition and myofibroblast accumulation in the extracellular matrix. Samah et al revealed that fenofibrate attenuates the severity of lung fibrosis and injury induced by bleomycin by decreasing pulmonary hydroxyproline and TGF-β1 levels as effectively as rosiglitazone.186 Dexamethasone or fenofibrate attenuates the severity of bronchial asthma and airway inflammation induced by OVA/LPS through significant amelioration in lung inflammatory cytokines such as TNF-α, IL-4, IL-13, IL-17, IL-23, and TGF-β1 levels, in the serum immunoglobulin (Ig)E, and leukocytic counts.187 In a mouse model, fenofibrate decreased airway reactivity to methacholine in a dose- and time-dependent manner via an enhanced endothelial NOS (eNOS) phosphorylation to upregulate NO generation.188 Fenofibrate protects fatty acid oxidation-induced alveolar epithelial cells dysfunction, a crucial influencer of the pathogenesis of pulmonary injury.189 Fenofibrate (15 mg/day) triggered T helper type 1 (Th1) switching in the immune response to allergens by overcoming inflammatory activity in allergic asthma models induced by OVA+LPS.190 Fenofibrate was shown to mitigate acute pulmonary injury78 in an intestinal ischemia/reperfusion (I/R) model in mice by decreasing inflammatory factors.191 Fenofibrate reduced the production of the following neutrophilic chemokines induced by IL-1β: ENA-78, G-CSF, GM-CSF, and TNF-α.192 Fenofibrate suppressed the down-regulation of PPAR-α associated with airway inflammation in the lung of mice exposed to LPS or OVA.193 Further, the PPAR-α agonist fenofibrate downregulated chemoattractant production, cell infiltration, and enhanced MMP activity in mouse lung triggered by LPS.194
Ciprofibrate inhibited rat airway smooth muscle cell contraction and proliferation induced by cigarette smoke extract in vitro. Ciprofibrate could attenuate airway remodeling in cigarette smoke-exposed rats model of COPD by inhibiting airway hyper-contraction, ameliorating airway smooth muscle proliferation, and reducing IL-1β, IL-12p70, IL-17A, and IL-18 expression.195 Fenofibrate or ciprofibrate downregulates the binding activity of AP-1 and NF-κB induced by TNFα and protects against VCAM-1 and IL-6 gene expression induced by TNF-α in porcine vascular endothelial cells.196
WY14,643, a synthetic fibrate drug, blocks the elevation of the capillary filtration coefficient (Kfc) in a mice model of ALI induced by LPS.197 WY14,643, in a mouse model of ALI induced by LPS, also inhibited the LPS-stimulated induction of nitroxidative stress and pro-inflammatory cytokine levels.198 Furthermore, WY14643 could also up-regulate TGF-β, IL-4, and IL-10 mRNA expression and also attenuated rejection of lung allografts.199
The Role of Statins in Pulmonary Inflammatory Diseases
Statins, also known as 3-hydroxy-3-methyl- glutaryl-coenzyme A (HMG-CoA) reductase inhibitors, were introduced into clinical practice due to their cholesterol-reducing properties and have a proven benefit in the prognosis of ischemic heart disease by lowering the incidence of illnesses and mortality in patients who are at high risk of CVD.200 A meta-analysis study including a total of 70,388 individuals enrolled in 10 trials found that treatment with statins significantly reduced the risk of major coronary events (odds ratio [OR] 0.70, 95% confidence interval [CI]: 0.61–0.81), major cerebrovascular events (OR 0.81, 95% CI: 0.71–0.93), and all-cause mortality (OR 0.88, 95% CI: 0.81–0.96).201 In addition, retrospective longitudinal dynamic cohort studies found that statins could decrease exacerbation of COPD by reducing the number of hospitalizations or emergency room and outpatient visits.202,203 In COPD patients, statins can also reduce pulmonary hypertension, the level of C-reactive protein (CRP), and the risk of mortality, compared with those not taking statins.204,205 For instance, a review has indicated that COPD patients taking statins have lower cardiovascular and respiratory morbidity/mortality, lower risk of lung cancer, and statins also reverse the decline in forced expiratory volume in one second (FEV1).206 Another study also showed that COPD patients receiving long-term statins (>2 years) were associated with a 39% reduced risk of mortality, and in particular, a 78% reduced mortality was reported for a subgroup of patients with a high level of systemic inflammation with high-sensitivity CRP (hsCRP) higher than 3 mg/L.207 Moreover, with regard to the overall survival of patients with non-small-cell lung cancer, statins potentially enhanced the effects of chemotherapy [(hazard ration (HR) 0.86, 95% CI: 0.81–0.91] and of tyrosine kinase inhibitors (HR 0.86, 95% CI: 0.76–0.98).208 Statins have a beneficial effect on the overall survival rate of lung cancer patients. Moreover, in COPD patients, statins may have a beneficial effect on decreasing the risk of lung cancer.209 A study analyzing patient data from 2002 to 2017 from the Taiwan National Health Insurance program indicated that statins may protect COPD patients from pulmonary hypertension (PH) in a dose- and time-dependent manner: Compared with statin nonusers, statin users had a 22% lower risk of PH (subdistribution HR [sHR] 0.78, 95% CI: 0.65–0.94).210 Moreover, Chalmers et al also revealed that statin users were associated with a significant decrease in the development of complicated pneumonia (adjusted odds ratio [aOR] 0.44, 95% CI, 0.25–0.79, P=0.006), lower 30-day mortality [aOR 0.46, 95% CI 0.25–0.85, P=0.01], and lower CRP levels on admission.211 Therefore, statins could be a potential therapy for the management of lung inflammation.
Statins may exert anti-inflammatory properties in the management of COPD. How do statins achieve anti-inflammatory effects in the lung? Statins may attenuate pulmonary inflammation by modulating neutrophil function and infiltration into the lung, by reducing cytokine expression and release, by preserving epithelial and endothelial integrity, by reversing airflow limitation in the lung through inhibiting fibrotic activity, by exerting antioxidant effects on skeletal muscle, by protecting against disruption of pulmonary integrity in community-acquired pneumonia, by reducing the pulmonary infection-mediated lung inflammatory response, and reverse or inhibit the development of epithelial-mesenchymal transition.205,212 Statins via the mevalonate pathway can affect the development of lung cancer and COPD. The mevalonate pathway mediates important intracellular signaling molecules called guanine phosphate transferases (GTPases) such as Rho-A to achieve these effects. While inhibiting the mevalonate pathway, leading to inhibition of the innate immune response to the inflammatory triggers such as smoking, may play a crucial role in lung remodeling and modifying pulmonary inflammation.213 Statins may also attenuate Nox activity via inhibition of Rac1 geranylgeranylation, protect the endothelial cell barrier, and regulate both mRNA stability and enzyme activity of eNOS to exert their potential therapeutic role in ALI.214 Statins also appear to diminish the stabilization of lipid raft formation and prevent the prenylation of signaling molecules with subsequent downregulation of gene expression and immune activation and regulation, which result in reduced expression of adhesion molecules, chemokines, and cytokines, and effects on cell proliferation or apoptosis.215 Statins can also significantly reduce ROS/NOS generation, cytokine expression such as TNF-α and IL-6, and upregulation of inflammatory mediators such as cyclooxygenase-2 in the pulmonary inflammation model.216
The anti-inflammatory effect of atorvastatin is antagonized by PPAR-γ antagonists which indicates its anti-inflammatory effects are mediated via PPAR-γ receptors. The therapeutic target of atorvastatin may be HO-1. A previous study using the zymosan-injected air pouch to trigger inflammation found that pretreatment with atorvastatin attenuated cell influx and the expression of proinflammatory cytokines and chemokines along with induction of HO-1 in the cells of the exudate of the air pouch, effects which were reversed by exposure to tin protoporphyrin IX (SnPPIX), an heme oxygenase inhibitor.217 Our recent study also revealed that HO-1 induction by mevastatin through the Nrf2/ARE axis was regulated by the p47phox/Nox2/ROS-dependent activation of c-Src/PDGFR/PI3K/Akt112 or by c-Jun activation activated by the PKC/Pyk2/p38 MAPK- or JNK1/2-dependent pathways and led to AP-1-binding on the HO-1 promoter region113 using HPAEpiCs and animal studies. Further, mevastatin can suppress inflammatory responses mediated by TNFα.112,113 Chemotaxis is also attenuated by statins, as demonstrated by the rosuvastatin-induced decrease in the number of macrophages, lymphocytes, neutrophils, eosinophils, and total inflammatory cells recruited into BALF, as well as the lower levels of TNF-α, IL-4, IL-5, and IL-13 in BALF.218 Treatment with another statin, simvastatin, also prevented the recruitment of leukocytes to the lung, abrogated pulmonary endothelial injury, attenuated pulmonary hyperpermeability,219,220 reduced pulmonary cytokine levels such as IL-13 and TGF-β1 in patients with lung fibrosis,221,222 and improved oxygenation in mechanically ventilated mice.223 The effectiveness of simvastatin on pulmonary disorders could also be primarily mediated through an HO-1 related pathway. A study using pulmonary hypertension models induced by chronic hypoxia and monocrotaline administration in rats indicated that exposure to simvastatin significantly improved right ventricular hypertrophy and pulmonary arterial hypertension, effects that were associated with significant induction of HO-1 protein levels and activity, which were abolished by SnPP.224 Another study also demonstrated that simvastatin could inhibit pulmonary artery smooth muscle cells (PASMCs) proliferation induced by serotonin stimulation in a dose-dependent manner, which was accompanied by the parallel induction of HO-1/p21WAF1, an effect that was reversed by treatment with Tin-protoporphyrin (SnPP, a selective inhibitor of HO-1).225 These statins mentioned above including atorvastatin, mevastatin, simvastatin, and rosuvastatin can enhance HO-1 activity in different extravascular tissues including the lungs.226 Thus, HO-1 induction represents a crucial mechanism by which statins can exert anti-oxidative and anti-inflammatory effects on the management of inflammatory diseases including pulmonary disorders, such as ALI/acute respiratory distress syndrome60,227,228 The HO system mitigates the effects induced by oxidative stress and its detrimental effects in pulmonary disorders, which could be mediated by decreasing the levels of heme and increasing levels of bilirubin and CO to achieve anti-inflammatory, anti-oxidant, and anti-apoptotic effects.227
Fessler et al determined that lovastatin inhibited aerosolized LPS-induced lung inflammation and caused impairment of host defenses via the inhibition of the mevalonate pathway, which was associated with the reduction of parenchymal MPO and microvascular permeability, and changes in cytokine levels triggered by LPS. Moreover, lovastatin also could inhibit chemotaxis, bactericidal killing capacity, actin polymerization, and Rac activation.229 Therefore, these properties of statins might reduce the possibility of cancerogenesis in lung tissue and may attenuate the progression of COPD. Indeed, in past decades, several clinical studies evaluating oral statins as a potential treatment for chronic pulmonary disorders such as PH, ALI, COPD, and asthma have been conducted, but the findings and conclusions derived from these trials were inconsistent. For example, Xu et al found that among COPD patients treated with statins, those who were current or former smokers exhibited interstitial lung abnormalities (ILA), and pretreatment with statin enhanced lung inflammation and fibrosis induced by bleomycin in vivo and NLRP3-inflammasome activation and augmented mtROS generation.230 Moreover, a review of randomized controlled trials extracted from the Cochrane library (2019) concluded that statin use resulted in a decrease in IL-6 and CRP levels, but that the findings did not translate into a clinical application for COPD patients because of the lack of a significant statistical difference in mortality, FEV1, functional capacity, the number of exacerbations, or quality of life. The authors concluded that additional randomized controlled trials are necessary to explore this issue.231 Sub-therapeutic levels and low systemic bioavailability of statins in the airways following oral delivery of statins may have contributed to these conflicting findings. Therefore, an inhalation formulation of statins may overcome these problems in bioavailability. In several animal experiments, the inhalation of statins such as simvastatin pravastatin, rosuvastatin, pitavastatin, and atorvastatin induced better effects on anti-pulmonary inflammation.232 Thus, statins could be efficacious in the treatment of lung inflammatory disorders and the development of inhalation formulations is warranted.
Chinese Herbal Medicines
Salvianolic Acid A/B
Both salvianolic acid B (SalB) and SalA,16 major bioactive compounds isolated from Salviae Miltiorrhizae (also called Danshen) belonging to the Chinese herb Radix, have been reported to exhibit anti-oxidative and anti-inflammatory effects.233 Endothelial-to-mesenchymal transition (EndMT) participates in the remodeling of the pulmonary vessel, an effect that is partially attributed to inflammatory and oxidative stress in endothelial cells. SalA,16 a polyphenol compound, stimulates the translocation of Nrf2 and subsequent up-regulation of HO-1 to inhibit EndMT-mediated pulmonary vascular remodeling.87 SalA also prevents pulmonary fibrosis by arresting the cell cycle and promoting apoptosis in fibroblasts via the decreased expression of the anti-apoptotic Bcl-2, cyclin B1, cyclin D1, and cyclin E1 protein, and cleaved caspase-3 protein and increased expression of p53 and p21.234 Furthermore, sortase A activity can be inhibited by SalA (IC50 = 5.75 μg/mL) and bacterial adhesion to fibrinogen is repressed by SalA, which prevents the ability of Staphylococcus aureus to infect A549 cells.235 Zhao et al revealed that besides SalA, pre-treatment with SalB in ALI model rats, attenuated oxidative stress by enhancing the levels of GSH peroxidase, CAT, and SOD, and also blocked lung fibrosis by reducing the protein expression of α‑smooth muscle actin, endogenous TGF‑β1 production, and type I collagen.236 Further, SalB exerted anti-inflammatory roles on pulmonary fibrosis in a bleomycin-treated mouse model by protecting endothelial cells from oxidative stress injury, mediated by inhibiting the expression of pro-inflammatory cytokines and endothelial permeability via MAPK and NF-κB signaling pathways.40 Zhang et al also revealed that SalB inhibited cigarette smoke (CS)-induced inflammatory cells infiltration, MCP-1, IL-6, IL-1β, and TNF-α synthesis, and up-regulation of total GSH production induced by CS, via up-regulation of the Nrf-2/HO-1 axis and also by suppressing the NF-κB activation induced by CS.237 Besides regulating Nrf2/Nox4 redox balance, SalB inhibited the TGF-β1/Smad3 signaling pathway, which led to protection against pulmonary injury induced by paraquat.238
Asiatic Acid (AA)
Asiatic acid (AA) is a triterpenoid compound isolated from Centella Asiatica.239 AA has been reported to exert diverse pharmacological activities, such as anti-oxidative, anti-cancer, hepatoprotective, and anti-inflammatory effects.240 AA has been also been shown to attenuate fibrosis progression.241 In a bleomycin model of pulmonary fibrosis, AA ameliorated pulmonary fibrosis by inhibiting inflammatory and pro-fibrotic signaling pathways, including the expression of TGF-β1 and that of matrix metalloproteinase (MMP)-1, α-SMA, type III collagen, and type II collagen, and the formation of the NLRP3 inflammasome, as well as the inactivation of ERK1/2 and Smads.242 AA also reduced ROS generation and neutrophil elastase activity and attenuated MCP-1 expression, as well as the recruitment of inflammatory cells through decreased MAPKs and NF-κB activation and increased expression of SOD3 and HO-1 in lung tissue.41 Xia et al revealed that AA significantly reduced α‑smooth muscle actin and type I collagen expression by inhibiting ROS generation and the TGF‑β1/Smad2/3 signaling pathway.243 Jiang et al determined that AA might reduce the levels of MPO, inflammatory cytokines, ROS, and MDA, and may inhibit neutrophil infiltration, while it promotes an increase in SOD and CAT levels109 by upregulating Nrf2 levels and downregulating NLRP3 inflammasome protein expression in ALI in rats.244
Celastrol
Celastrol, a pentacyclic triterpenoid, was extracted from the roots of Tripterygium wilfordii, which is a component of traditional Chinese medicine and can significantly ameliorate NF-κB245 and NLRP3 activities246 to induce significant anti-inflammatory activity.247 Exposure to celastrol reduced levels of the cytokines MCP-1, TNF-α, and IL-8 in the BALF and serum and upregulated levels of SOD and CAT, which were accompanied by the attenuation of the Ednrb/Kng1 signaling pathway in COPD mouse models.248 In another bleomycin-induced pulmonary fibrosis animal model, celastrol demonstrated antioxidant and anti-fibrotic effects against pulmonary fibrosis. Furthermore, celastrol decreased the expression of the inflammatory mediators MMP-2/9 and TNF-α and it also induced Nrf2 expression, which rescues the activities of Phase II enzymes including NAD(P)H:quinone oxidoreductase 1 (NQO1), HO-1, and GSTs.88 Celastrol also reduced the protein levels of Bax and caspase-3 activity to induce anti-inflammatory and anti-apoptotic effects in burn-induced lung injury.249
Fisetin
Fisetin (3,7,3′,4′-tetrahydroxy flavone) is a flavonoid commonly found in various types of vegetables and fruits such as strawberries, cucumbers, onions, grapes, persimmons, and apples and plants such as smoke trees. Various studies in vivo and in vitro have demonstrated that fisetin possesses diverse pharmacological activities including anti-inflammatory,250 anticancer,251 and antioxidative effects.252 Pretreatment with fisetin in COPD patients markedly inhibited increases in serum concentrations of TNF-α induced by LPS via inhibition of the nuclear enzyme PARP-1.253 Fisetin also reduced neutrophil levels and infiltration of macrophages and also attenuated MPO activity in the LPS-induced ALI model by inhibiting the activation of NF-κB signaling and the TLR4 expression in pulmonary tissues.254 Huang et al also determined that fisetin treatment decreased the infiltration of neutrophils, monocytes, and eosinophils by inhibiting the MyD88/NF-κB signaling pathway to alleviate airway inflammation.255 Conversely, Hussain et al revealed that in lungs exposed to CS, fisetin attenuated inflammation and oxidative stress to protect the lung from CS-mediated injury via the Nrf2‐dependent expression of antioxidative genes (HO‐1, glutathione peroxidase‐2, reduced GSH, SOD) to reduce the infiltration of inflammatory cells and cytokine expression.89
Galangin
Galangin (3,5,7-trihydroxyflavone) is found in Alpinia officinarum and honey in high concentrations and is a member of the flavonol class of flavonoids, and has been used as a spice and herbal medicine for various diseases.256 Growing evidence shows that galangin exhibits anti-fibrotic, anti-oxidant, and anti-inflammatory activities, which are beneficial for various disorders.44 Galangin has been shown to attenuate oxidative damage and inflammation via upregulation of Nrf2/HO-1 in various tissues including the lungs.90,91 Galangin acts as an anti-remodeling agent in an asthma model, as it has been shown to inhibit the TGF-β1-ROS-MAPK pathway and attenuate α-SMA expression, collagen deposition, and goblet cell hyperplasia, in addition to the suppression of MMP-9 and vascular endothelial growth factor (VEGF) expression.257 A report indicated that in an OVA-induced allergic asthma model, oral administration of galangin notably attenuated goblet cell hyperplasia, inflammation, and airway hyperresponsiveness via the suppression of TNF-α, IL-4, IL-5, IL-13, IL-17, NO, ROS, immunoglobulin E,123 and eosinophil peroxidase, and an increase in interferon (IFN)-γ in a PPAR-γ-dependent manner.258
Kaempferol
Kaempferol (KPF), a flavonol, is present in significant amounts in beans, apples, tea, strawberries, and broccoli259 and is known to be beneficial in diseases such as inflammation, allergies, and cancer. Growing evidence has indicated that KPF exerts anti-inflammatory effects on various experimental disease models in vivo and in vitro.260,261 Several lines of evidence have shown that KPF has protective effects against apoptosis and allergic reactions via HO-1 induction in various types of cells.92–95 Qian et al determined that in ALI induced by LPS, KPF disrupts activation of TGF-β-activated kinase 1 (TAK1) and TNF receptor-associated factor 6 (TRAF6)-mediated polyubiquitination, and the activation of subsequent downstream NF-κB and MAPK signaling to reduce cytokine production and inflammatory injury.45 Zhang et al also found that KPF could significantly attenuate enhancement of NF-κB p65 DNA binding activity by inhibiting the upregulation of TLR4/MyD88/phosphorylation of IκBα/NF-κB p65, and the MAPKs phosphorylation, leading to attenuation of ROS production and cytokine overexpression in an H9N2 swine influenza virus-induced ALI model.262 Expression of inflammatory mediators such as COX-2,133 was alleviated by KPF treatment in allergic pulmonary disorders.263 In another LPS-induced ALI mice model, KPF mitigated the activation of the MAPKs and NF-κB signaling pathways to alleviate oxidative stress, cytokine production, and leukocyte infiltration in lung tissues.264 The flavonoid kaempferol-3-O-glucorhamnoside, derived from the plant Thesium chinense Turcz, is a KPF derivative and also suppresses NF-κB and MAPK phosphorylation to attenuate the production of inflammatory cytokines and overall oxidative stress in the pneumonia model infected by Klebsiella pneumoniae both in vitro and in vivo.265
Luteolin
Luteolin is a natural flavonoid compound (also known as 3′,4′,5,7-tetrahydroxyflavone) and widely distributed in the leaves of many types of plants, for example, celery, thyme, dandelion, and basil.266 Growing evidence has demonstrated that the pharmacological activities of luteolin include anti-oxidative, anti-inflammatory, anti-tumor effects, and anti-ischemic effects in response to vascular injury.267 Luteolin pretreatment can promote the expression of HO-1 to prevent apoptosis,96 and exerts anti-inflammatory and antioxidant effects268 in in vivo and in vitro. Hence, luteolin has been applied to the treatment of various diseases, such as cancer, hypertension, and inflammatory diseases.266 Liu and Meng found that luteolin mitigates the activation of the NF-κB signaling pathway induced by LPS by down-regulating miR-132 in a bronchopneumonia murine model.269 Luteolin, in primary cultured mouse lung fibroblasts, also inhibited expression of vimentin, type I collagen, and α-SMA induced by TGF-β1 in vitro and in a bleomycin-treated C57BL/6J mice model, and also effectively attenuated neutrophil infiltration and expression of IL-6 and TNFR in vivo, leading to alleviation of experimental lung fibrosis.270 In the mercuric chloride-induced lung injury mouse model, luteolin treatment was responsible for the reduction of MPO, inflammatory cytokines, and MDA levels and for the increase of SOD and GSH by preventing NF-κB activation, while also activating the Akt/Nrf2 pathway.271 Glossogyne tenuifolia ethanol extract which contains luteolin as its major component has been demonstrated to possess potent anti-inflammatory and antioxidative activities by blocking the NF-κB signaling pathway.272
Madecassoside
Madecassoside is derived from Centella Asiatica (Umbelliferae) and is a triterpene compound. It possesses pleiotropic bioactivities and is effective in many experimental disease models.273–275 Madecassoside has been shown to exert anti-inflammatory activities by activating Nrf2/HO-1 signaling in different disease models.97,98 Further, madecassoside can ameliorate bleomycin-induced pulmonary fibrosis by attenuating oxidative stress, inflammation, and subsequent overexpression of TGF-β1.276 Treatment with madecassoside can also reduce the expression of TGF-β1 and α-SMA, p-Smad2 and p-Smad3 levels, MPO activity, and MDA levels, as well as increase GSH levels and SOD activity in lung tissues.276
Oleanolic Acid
Oleanolic acid (OA) was isolated from different medicinal plants and is a biologically active natural pentacyclic triterpenoid compound.277 Growing evidence has shown that OA has pleiotropic effects including anticancer, anti-inflammation, anti-diabetes, and antiasthmatic effects and also exerts hepatoprotective effects.278 GATA-binding protein 3 (GATA-3), a mediator of allergic airway inflammation, is a crucial factor in vivo.279 OA has been demonstrated to exert anti-inflammatory and anti-asthmatic activity via inhibition of GATA-3 and retinoic acid receptor-related orphan receptor gamma-t pathways in an OVA-induced airway inflammation model.280 Furthermore, OA has been shown to protect cells from acetaminophen-induced hepatotoxicity via induction of Nrf2-dependent HO-1.99 Studies have also demonstrated that OA induces HO-1 expression and protects from ROS-induced cell death in rat vascular smooth muscle cells.100 Santos et al revealed that OA administration has anti-oxidative effects by attenuating ROS generation and restoring the reduced GSH/oxidized glutathione ratio and CAT activity in experimental ALI induced by paraquat.281 Furthermore, OA also diminished TNF-α, macrophage migration inhibitory factor, IL-6, INF-γ, and TGF-β mRNA expression in lung tissues.281 OA can also modulate oxidative stress by reducing inducible NOS expression and enhancing SOD.282 OA exerts significant antioxidant and anti-inflammatory activities by blocking the NF-κB signaling pathway.272 In addition, in a murine model of pulmonary fibrosis and inflammation induced by polyhexamethylene guanidine phosphate, OA acetate effectively reduced elevation of cytokines and the activation of the NLRP3 inflammasome.283 The Chinese medicinal preparation Eriobotrya japonica which contains OA as one of its six main constituents acts on the actin cytoskeleton, tight junctions, focal adhesion, MAPK pathway, and TGF-β pathway to exert anti-inflammatory effects and cough suppression.48 Lee et al demonstrated that Eriobotrya japonica suppresses κB-α phosphorylation and NF-κB activity to inhibit cytokine production.284 In the N-methyl-D-aspartate (NMDA)-induced ALI mice model, OA attenuated NMDA-induced oxidative stress, cytokine expression, and inflammatory cell infiltration by activating SIRT1 and reducing NF-κB acetylation.285
Oleanolic Acid Derivatives
As indicated above OA has been used to treat liver disorders due to its modest biological activities in humans. Bardoxolone methyl [CDDO-Me; 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid methyl ester] is a derivative of OA; it has broad pharmacological functions including antiproliferative, anti-tumorigenic, antioxidant, anti-tumor, and anti-inflammatory effects.286 CDDO-Me has been shown to induce Nrf2/HO-1 upregulation in in vitro and in vivo.287 CDDO-Me effectively inhibits the ALI induced by LPS in vivo, its underlying anti-inflammatory activity might result from the reduction of NO, IL-6, IL-1β, and TNF-α levels via the attenuation of MAPK, Akt, and NF-κB pathways.288 2-Cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid (CDDO) is another synthetic triterpenoid. A previous study revealed that CDDO, at nanomolar concentrations, increased the expression of HO-1 both in vitro and in vivo.102 Nichols et al found that in in vitro cystic fibrosis cell culture models, the synthetic triterpenoid CDDO limited inflammation by reducing NF-κB activation while increasing Nrf2 activity.289 In another model of lung fibrosis induced by bleomycin, triterpenoid CDDO-Me treatment inhibited levels of cytokines IL-6 and keratinocyte-derived chemokine, the pro-fibrotic cytokine TGFβ, and mRNA expression of fibronectin and α-smooth muscle actin, which led to the attenuation of histological fibrosis and improvement of lung function.290 In addition, Wang et al using a radiation-induced pulmonary inflammation and fibrosis animal model also showed that CDDO-Me exerted anti-inflammatory and anti-fibrotic effects by suppressing the expression and secretion of proinflammatory cytokines IL-6 and TGF-β, and elevating the production of cytokines such as IL-10, which has anti-inflammatory properties, and downregulating the expression of profibrotic genes, including collagen I, α-SMA, and fibronectin mRNA levels.291
Saikosaponin A
Saikosaponin A (SSa), isolated from Radix Bupleuri (RB), is a triterpenoid saponin and exhibits several pharmacological activities, such as antioxidative and anti-inflammatory effects.292 SSa significantly attenuated the infiltration of inflammatory cells, IL-1β, TNF-α, and NO production induced by CS and it also inhibited the MDA and MPO activity induced by CS in lung tissues via upregulating the expression of HO-1 and Nrf2 and inhibiting NF-κB activity.101
Pristimerin (Pris)
Pristimerin (Pris) was isolated from plants that belong to the Hippocrateaceae or Celastraceae families and is a natural quinone-methide triterpenoid derivative.293 Pris possesses numerous biological activities including antioxidant, antibacterial, and anti-cancer effects.294–296 Pris exerts its anti-oxidative, anti-inflammatory, and anti-apoptotic effects via induction of HO-1 in various tissue injury models.49,103 Our recent study also suggested that Pris possesses anti-neuroinflammatory and anti-oxidative effects297 through HO-1 up-regulation in rat brain astrocytes (unpublished data). Further, Pris protected against the LPS-induced ALI in mouse models via anti-oxidant, anti-inflammatory, and anti-apoptotic effects. Furthermore, Pris treatment attenuated the production of pro-inflammatory cytokines including IL-6 and TNF-α, and elevation of pro-apoptotic proteins including Bax and caspase-3. Pris has also been shown to up-regulate Bcl2 inhibited by LPS.298
Conclusions
A growing number of studies have indicated that both exogenous and endogenous ROS participate in the pathogenesis of pulmonary disorders, such as ARDS, COPD, and asthma.60 AP-1 and NF-κB are well known as vital regulators of inflammatory mediators, including enzymes (COX2, MMP-9, and NOS), chemokines, cytokines, mucins, and receptors, which all participate in the pathological mechanisms of these respiratory diseases (Figure 1). NF-κB is activated by several factors, such as ROS and pathogen-associated molecular pattern (PAMP), through downstream signaling pathways (Figure 1). The remodeling of damaged tissue, as occurs in pulmonary fibrosis, also plays an important role in lung injury insults including COPD, asthma, and ARDS. TGF-β, CTGF, and endothelin-1 are important players in these processes. These diseases all have a poor prognosis and a high mortality rate. Therefore, it is crucial to develop specific inhibitors as an effective strategy for the treatment of inflammatory disorders. Currently, the literature has indicated that antioxidants are an effective defense against these diseases. These antioxidants include enzymatic antioxidants, such as CAT, SOD, glutathione peroxidase, and HO-1, and non-enzymatic antioxidants, including vitamins A, C, and E, and GSH. A growing number of studies have indicated that HO-1 is an effective antioxidant, and based on these findings, several medicines and Chinese herbs can also induce the upregulation of its expression. Herein, we reviewed several potential HO-1 inducers, including PPAR agonists (Figure 2), statins, and Chinese herbal medicines (Figure 3 and Table 1). Although these studies support their effects on lung injury in animal models, there is currently no evidence in human respiratory diseases. Further clinical studies are necessary to identify the pharmacological activities of these medicines and herbal compounds in these pulmonary inflammatory diseases.
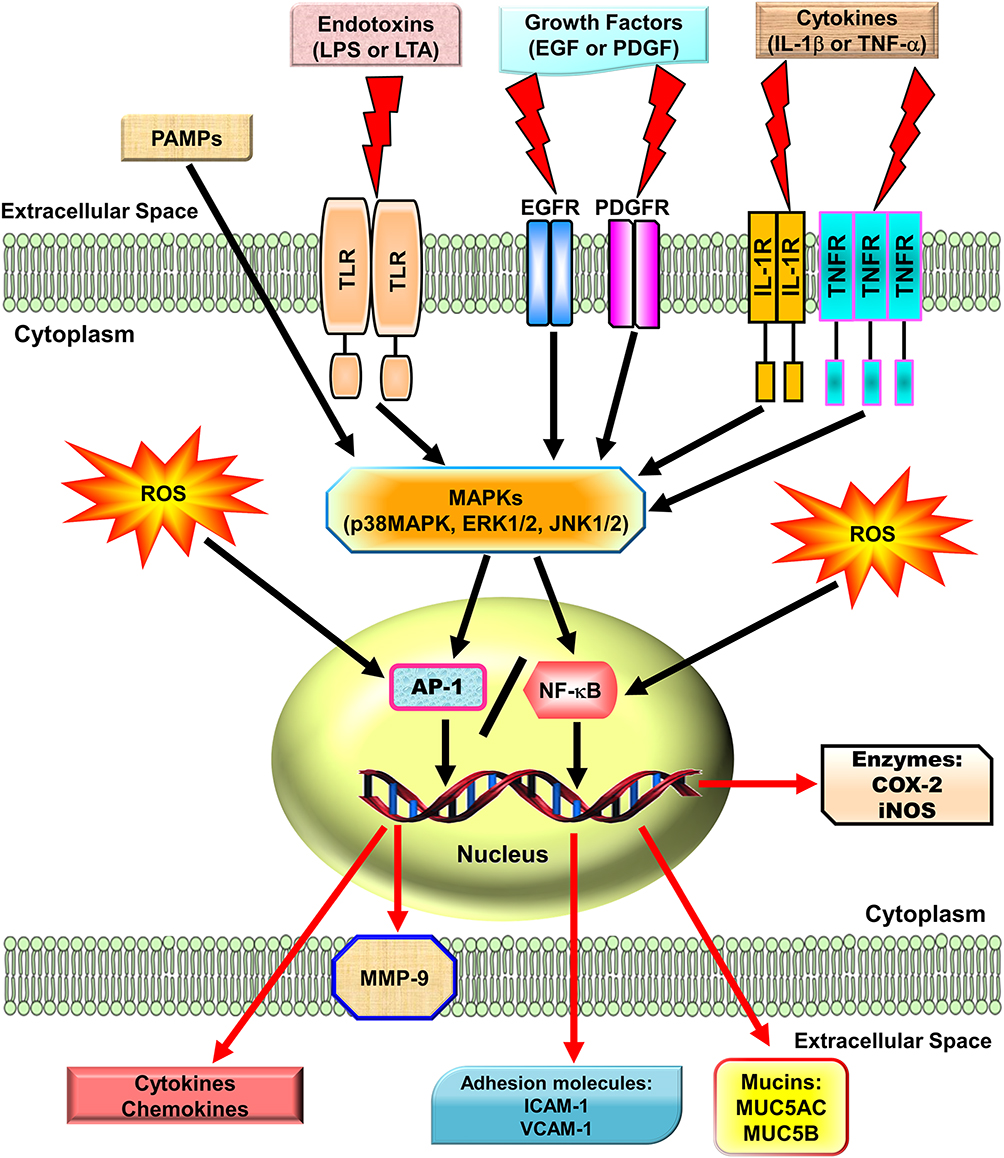 |
Figure 1 The roles of ROS, pro-inflammatory mediators, and transcription factors AP-1 and NF-κB in pulmonary inflammatory diseases. Pathogen-activated molecular patterns (PAMPs), including endotoxins (LPS or LTA), growth factors (EGF or PDGF), and cytokines (IL-1β or TNF-α), activate downstream pathways (p38 MAPK, JNK1/2, and ERK1/2) via their respective receptors (TLR, EGFR, DPGFR, IL-1R, and TNFR) to promote transcription factors NF-κB and AP-1 activities also activated by ROS, leading to genes transcription (including cytokines, chemokines, MMP-9, COX-2, iNOS, ICAM-1, VCAM-1, MUC5AC, and MUC5B) and pulmonary inflammation. Abbreviations: LPS, lipopolysaccharides; LTA, lipoteichoic acid; EGF, epidermal growth factor; TNF-α, tumor necrosis factor α; IL-1β, interleukin 1β; PDGF, platelet-derived growth factor; TLR, Toll-like receptor; AP-1, activator protein 1; MMP-9, matrix metalloproteinases-9, NF-κB, nuclear factor-κB; ROS, reactive oxygen species; iNOS, inducible nitric oxide synthase; COX-2, cyclooxygenase-2; VCAM-1, vascular cell adhesion molecule-1; ICAM-1, intercellular adhesion molecule -1; MUC5AC, mucin 5AC; MUC5B, mucin 5B. |
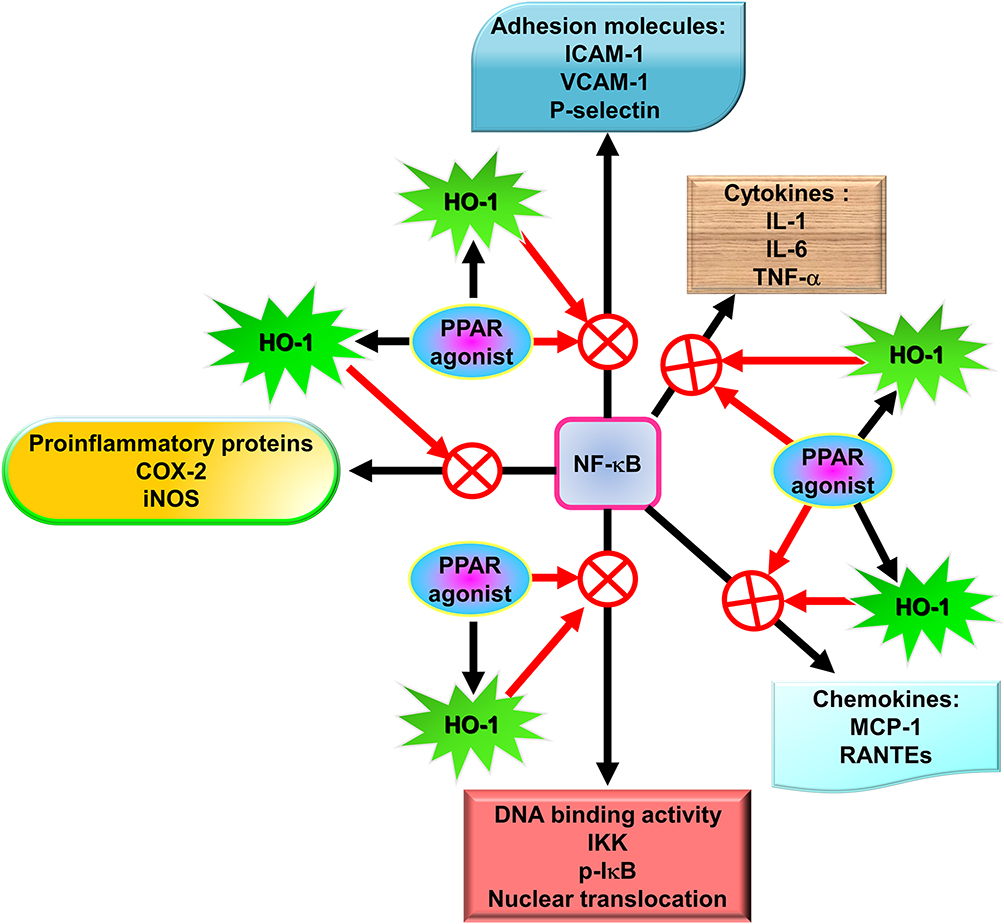 |
Figure 2 The functions of PPARs agonists in pulmonary inflammation. PPARs agonists and PPARs agonist-induced HO-1 upregulation can inhibit NF-κB activity via blocking IKK activity and IκB phosphorylation, leading to suppression of NF-κB nuclear translocation and DNA binding activity and in turn reduction of gene expression including MCP-1, iNOS, VCAM-1, ICAM-1, P-selectin, IL-1, IL-6, TNF-α, COX-2, and RANTEs. Abbreviations: TNF-α, tumor necrosis factor α; ICAM-1, intercellular adhesion molecule-1; VCAM-1, vascular cell adhesion molecule-1; IL, interleukin; iNOS, inducible nitric oxide synthase; COX-2, cyclooxygenase-2; HO, heme oxygenase; NF-κB, nuclear factor-κB; RANTEs, regulated upon activation normal T-cell expressed and secreted; MCP-1, monocyte chemoattractant protein-1; IKK, IκB kinase. |
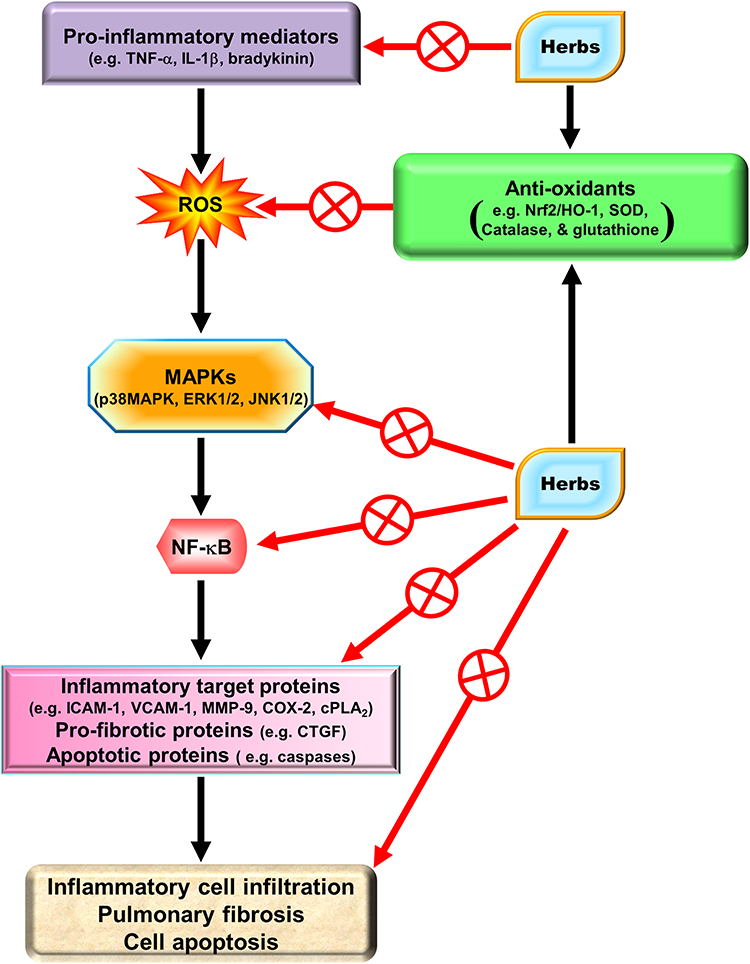 |
Figure 3 The anti-inflammatory, anti-fibrotic, and anti-apoptotic mechanisms of Chinese herbs in the lungs. Herbs can target individual signal molecules including ROS, MAPKs, and NF-κB as well as pro-inflammatory mediators to block the expression of pro-inflammatory proteins, pro-fibrotic proteins, and pro-apoptotic proteins. Abbreviations: COX-2, cyclooxygenase-2; cPLA2, cytosolic phospholipase A2; CTGF, connective tissue growth factor; HO, heme oxygenase; ICAM-1, intercellular adhesion molecule-1; IL-1, interleukin-1; JNKs, c-Jun NH2-terminal kinases; MAPKs, mitogen-activated protein kinases; MMP, matrix metalloproteinase; NF-κB, nuclear factor-κB; Nrf2, NF-E2-related factor 2; ROS, reactive oxygen species; SOD, superoxide dismutase; TNF-α, tumor necrosis factor-α; VCAM-1, vascular cell adhesion molecule-1. |
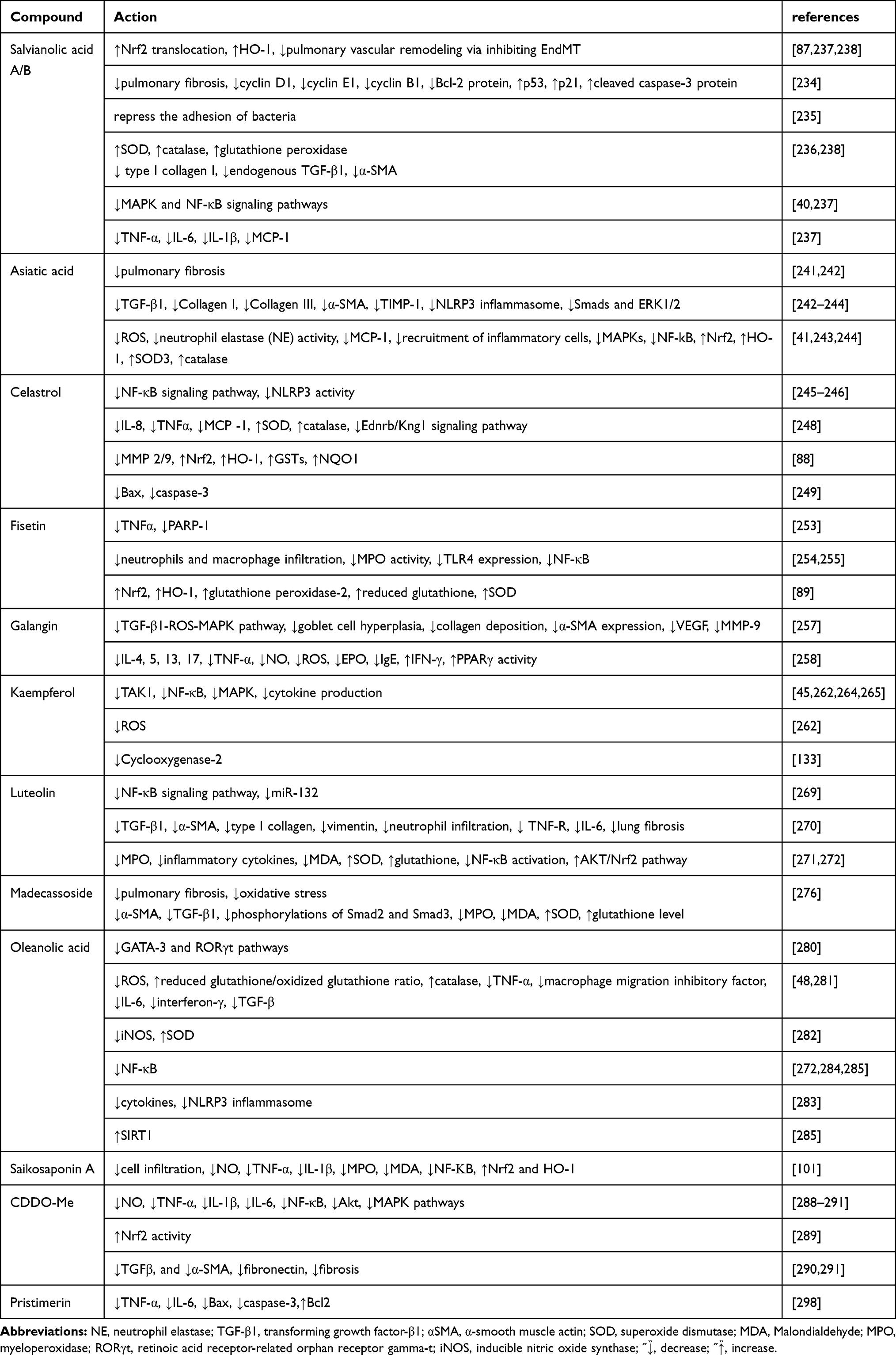 |
Table 1 The Effects of Herbal Compounds in Pulmonary Inflammation |
Abbreviations
AA, asiatic acid; AFC, alveolar fluid clearance; ALI, acute lung injury; AOR, adjusted odds ratio; AP-1, activator protein 1; APCs, antigen-presenting cells; ARDS, acute respiratory distress syndrome; AREs, antioxidant response elements; CAT, catalase; CDDO, 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid; CDDO-Me, 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid methyl ester; CI, confidence interval; CO, carbon monoxide; COPD, chronic obstructive pulmonary disease; COX-2, cyclooxygenase-2; cPLA2, cytosolic phospholipase A2; CTGF, connective tissue growth factor; CVD, cardiovascular disease; 15d-PGJ2, 15-deoxy-D12,14-prostaglandin J2; ENaC, epithelial sodium channel; EndMT, endothelial-to-mesenchymal transition; E3RSIκB, E3 ubiquitin-ligases; ERKs, extracellular signal-regulated kinases; FBS, fetal bovine serum; fMLP, N-formyl methionyl-leucylphenylalanine; G-CSF, granulocyte colony-stimulating factor; GSH, glutathione; GSTs, glutathione S-transferases; HDM, house dust mite; HO, heme oxygenase; ICAM-1, intercellular adhesion molecule-1; IHD, ischaemic heart disease; IκB, inhibitory κB; IL-1β, interleukin-1β; NLRP3, Nod-like receptor with pyrin domain containing 3; NQO1, NAD(P)H: quinone oxidoreductase 1; JNKs, c-Jun NH2-terminal kinases; Keap1, Kelch-like ECH associated protein 1; KPF, kaempferol; LPS, lipopolysaccharide; LTA, lipotechoic acid; LTB4, leukotriene B4; MAPKs, mitogen-activated protein kinases; MCP-1, monocyte chemoattractant protein-1; MDA, malondialdehyde; MMP, matrix metalloproteinase; NF-κB, nuclear factor-kappaB; NO, nitric oxide; Nox, NADPH oxidase; Nrf2, NF-E2-related factor 2; OA, oleanolic acid; OVA, ovalbumin; PARP, poly(ADP-ribose) polymerase; PPARs, peroxisome proliferator-activated receptors; PKC, protein kinase C; Pris, pristimerin; ROS, reactive oxygen species; Sal, salvianolic acid; SGK1, serum and glucocorticoid-induced kinase-1; SOD, superoxide dismutase; Ssa, saikosaponin A; TAK1, TGF-β-activated kinase 1; TGF, transforming growth factor; TNF-α, tumor necrosis factor-α; TSMCs, tracheal smooth muscle cells; TLR, Toll-like receptor; TZDs, Thiazolidinediones; VCAM-1, vascular cell adhesion molecule-1.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Ministry of Science and Technology, Taiwan [Grant numbers: MOST108-2320-B-039-061, MOST109-2320-B-039-061, MOST109-2813-C-039-029-B, and MOST108-2320-B-182-014]; China Medical University, Taiwan [Grant number: CMU109-MF-09]; Chang Gung Medical Research Foundation, Taiwan [Grant numbers: CMRPG5F0203, CMRPG5J0142, and CMRPG5J0143].
Disclosure
The authors report no conflicts of interest in this work.
References
1. McGuinness AJ, Sapey E. Oxidative stress in COPD: sources, markers, and potential mechanisms. J Clin Med. 2017;6(2):21. doi:10.3390/jcm6020021
2. Qu J, Li Y, Zhong W, Gao P, Hu C. Recent developments in the role of reactive oxygen species in allergic asthma. J Thorac Dis. 2017;9(1):E32–e43. doi:10.21037/jtd.2017.01.05
3. Park HS, Kim SR, Lee YC. Impact of oxidative stress on lung diseases. Respirology. 2009;14(1):27–38. doi:10.1111/j.1440-1843.2008.01447.x
4. Cheng SE, Luo SF, Jou MJ, et al. Cigarette smoke extract induces cytosolic phospholipase A2 expression via NADPH oxidase, MAPKs, AP-1, and NF-κB in human tracheal smooth muscle cells. Free Radic Biol Med. 2009;46(7):948–960. doi:10.1016/j.freeradbiomed.2009.01.006
5. Cho RL, Yang CC, Lee IT, et al. Lipopolysaccharide induces ICAM-1 expression via a c-Src/NADPH oxidase/ROS-dependent NF-κB pathway in human pulmonary alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2016;310(7):L639–657. doi:10.1152/ajplung.00109.2014
6. Hsu CK, Lee IT, Lin CC, Hsiao LD, Yang CM. Nox2/ROS-dependent human antigen R translocation contributes to TNF-α-induced SOCS-3 expression in human tracheal smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2014;306(6):L521–533. doi:10.1152/ajplung.00274.2013
7. Lee CW, Lin CC, Lee IT, Lee HC, Yang CM. Activation and induction of cytosolic phospholipase A2 by TNF-α mediated through Nox2, MAPKs, NF-κB, and p300 in human tracheal smooth muscle cells. J Cell Physiol. 2011;226(8):2103–2114. doi:10.1002/jcp.22537
8. Lin CC, Lin WN, Cho RL, Wang CY, Hsiao LD, Yang CM. TNF-α-induced cPLA2 expression via NADPH oxidase/reactive oxygen species-dependent NF-κB cascade on human pulmonary alveolar epithelial cells. Front Pharmacol. 2016;7:447.
9. Luo SF, Chang CC, Lee IT, et al. Activation of ROS/NF-κB and Ca2+/CaM kinase II are necessary for VCAM-1 induction in IL-1β-treated human tracheal smooth muscle cells. Toxicol Appl Pharmacol. 2009;237(1):8–21. doi:10.1016/j.taap.2009.02.025
10. Vallyathan V, Shi X. The role of oxygen free radicals in occupational and environmental lung diseases. Environ Health Perspect. 1997;105(Suppl 1):165–177. doi:10.1289/ehp.97105s1165
11. Antus B, Kardos Z. Oxidative stress in COPD: molecular background and clinical monitoring. Curr Med Chem. 2015;22(5):627–650. doi:10.2174/092986732205150112104411
12. Liu Z, Ren Z, Zhang J, et al. Role of ROS and nutritional antioxidants in human diseases. Front Physiol. 2018;9:477. doi:10.3389/fphys.2018.00477
13. Lee I-T, Yang C-M. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem Pharmacol. 2012;84(5):581–590. doi:10.1016/j.bcp.2012.05.005
14. Stewart RM, Weir EK, Montgomery MR, Niewoehner DE. Hydrogen peroxide contracts airway smooth muscle: a possible endogenous mechanism. Respir Physiol. 1981;45(3):333–342. doi:10.1016/0034-5687(81)90016-5
15. Rahman I, Biswas SK, Kode A. Oxidant and antioxidant balance in the airways and airway diseases. Eur J Pharmacol. 2006;533(1–3):222–239. doi:10.1016/j.ejphar.2005.12.087
16. Haddad JJ, Safieh-Garabedian B, Saadé NE, Kanaan SA, Land SC. Chemioxyexcitation (ΔpO2/ROS)-dependent release of IL-1β, IL-6 and TNF-α: evidence of cytokines as oxygen-sensitive mediators in the alveolar epithelium. Cytokine. 2001;13(3):138–147. doi:10.1006/cyto.2000.0789
17. Lee I-T, Yang C-M. Inflammatory signalings involved in airway and pulmonary diseases. Mediators Inflamm. 2013;2013:1–12. doi:10.1155/2013/791231
18. Harju T, Soini Y, Pääkkö R, Kinnula VL. Up-regulation of heme oxygenase-I in alveolar macrophages of newly diagnosed asthmatics. Respir Med. 2002;96(6):418–423. doi:10.1053/rmed.2001.1283
19. Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86(2):583–650.
20. Primiano T, Kensler TW, Kuppusamy P, Zweier JL, Sutter TR. Induction of hepatic heme oxygenase-1 and ferritin in rats by cancer chemopreventive dithiolethiones. Carcinogenesis. 1996;17(11):2291–2296. doi:10.1093/carcin/17.11.2291
21. Ryter SW, Kim HP, Nakahira K, Zuckerbraun BS, Morse D, Choi AM. Protective functions of heme oxygenase-1 and carbon monoxide in the respiratory system. Antioxid Redox Signal. 2007;9(12):2157–2173. doi:10.1089/ars.2007.1811
22. Liu W, Liang Q, Balzar S, Wenzel S, Gorska M, Alam R. Cell-specific activation profile of extracellular signal-regulated kinase 1/2, Jun N-terminal kinase, and p38 mitogen-activated protein kinases in asthmatic airways. J Allergy Clin Immunol. 2008;121(4):893–902.e892. doi:10.1016/j.jaci.2008.02.004
23. Renda T, Baraldo S, Pelaia G, et al. Increased activation of p38 MAPK in COPD. Eur Respir J. 2008;31(1):62–69. doi:10.1183/09031936.00036707
24. Gerthoffer WT, Singer CA. MAPK regulation of gene expression in airway smooth muscle. Respir Physiol Neurobiol. 2003;137(2–3):237–250. doi:10.1016/S1569-9048(03)00150-2
25. Mossman BT, Lounsbury KM, Reddy SP. Oxidants and signaling by mitogen-activated protein kinases in lung epithelium. Am J Respir Cell Mol Biol. 2006;34(6):666–669. doi:10.1165/rcmb.2006-0047SF
26. Kumasawa F, Hashimoto S, Mizumura K, et al. Mitogen-activated protein kinase (MAPK) regulates leukotriene D4-induced HB-EGF and ADAM12 expression in human airway smooth muscle cells. Asian Pac J Allergy Immunol. 2013;31(1):58–66.
27. Pei YH, Cai XM, Chen J, et al. The role of p38 MAPK in acute paraquat-induced lung injury in rats. Inhal Toxicol. 2014;26(14):880–884. doi:10.3109/08958378.2014.970784
28. Madala SK, Schmidt S, Davidson C, Ikegami M, Wert S, Hardie WD. MEK-ERK pathway modulation ameliorates pulmonary fibrosis associated with epidermal growth factor receptor activation. Am J Respir Cell Mol Biol. 2012;46(3):380–388. doi:10.1165/rcmb.2011-0237OC
29. van der Velden JL, Ye Y, Nolin JD, et al. JNK inhibition reduces lung remodeling and pulmonary fibrotic systemic markers. Clin Transl Med. 2016;5(1):36. doi:10.1186/s40169-016-0117-2
30. Lee I-T, Lee C-W, Tung W-H, et al. Cooperation of TLR2 with MyD88, PI3K, and Rac1 in lipoteichoic acid–induced cPLA2/COX-2–dependent airway inflammatory responses. Am J Pathol. 2010;176(4):1671–1684. doi:10.2353/ajpath.2010.090714
31. Lin -C-C, Kuo C-T, Cheng C-Y, et al. IL-1β promotes A549 cell migration via MAPKs/AP-1-and NF-κB-dependent matrix metalloproteinase-9 expression. Cell Signal. 2009;21(11):1652–1662. doi:10.1016/j.cellsig.2009.07.002
32. Tzeng TF, Tzeng YC, Cheng YJ, Liou SS, Liu IM. The ethanol extract from lonicera japonica thunb. Regresses nonalcoholic steatohepatitis in a methionine- and choline-deficient diet-fed animal model. Nutrients. 2015;7(10):8670–8684. doi:10.3390/nu7105423
33. De Silva DS, Wilson RM, Hutchinson C, et al. Fenofibrate inhibits aldosterone-induced apoptosis in adult rat ventricular myocytes via stress-activated kinase-dependent mechanisms. Am J Physiol Heart Circ Physiol. 2009;296(6):H1983–1993. doi:10.1152/ajpheart.00002.2009
34. Larter CZ, Yeh MM, Van Rooyen DM, Brooling J, Ghatora K, Farrell GC. Peroxisome proliferator-activated receptor-α agonist, Wy 14,643, improves metabolic indices, steatosis and ballooning in diabetic mice with non-alcoholic steatohepatitis. J Gastroenterol Hepatol. 2012;27(2):341–350. doi:10.1111/j.1440-1746.2011.06939.x
35. Hou G, Yin Y, Han D, Wang QY, Kang J. Rosiglitazone attenuates the metalloprotease/anti-metalloprotease imbalance in emphysema induced by cigarette smoke: involvement of extracellular signal-regulated kinase and NF-κB signaling. Int J Chron Obstruct Pulmon Dis. 2015;10:715–724. doi:10.2147/COPD.S77514
36. Meng X, Sun X, Zhang Y, et al. PPARgamma agonist PGZ attenuates OVA-induced airway inflammation and airway remodeling via RGS4 signaling in mouse model. Inflammation. 2018;41(6):2079–2089. doi:10.1007/s10753-018-0851-2
37. Santos L, Rodrigues GB, Mota FVB, et al. New thiazolidinedione LPSF/GQ-2 inhibits NF-κB and MAPK activation in LPS-induced acute lung inflammation. Int Immunopharmacol. 2018;57:91–101. doi:10.1016/j.intimp.2018.02.011
38. Choi M, Rolle S, Rane M, Haller H, Luft FC, Kettritz R. Extracellular signal-regulated kinase inhibition by statins inhibits neutrophil activation by ANCA. Kidney Int. 2003;63(1):96–106. doi:10.1046/j.1523-1755.2003.00718.x
39. Carlin CM, Peacock AJ, Welsh DJ. Fluvastatin inhibits hypoxic proliferation and p38 MAPK activity in pulmonary artery fibroblasts. Am J Respir Cell Mol Biol. 2007;37(4):447–456. doi:10.1165/rcmb.2007-0012OC
40. Liu Q, Shi X, Tang L, et al. Salvianolic acid B attenuates experimental pulmonary inflammation by protecting endothelial cells against oxidative stress injury. Eur J Pharmacol. 2018;840:9–19. doi:10.1016/j.ejphar.2018.09.030
41. Lee JW, Park HA, Kwon OK, et al. Asiatic acid inhibits pulmonary inflammation induced by cigarette smoke. Int Immunopharmacol. 2016;39:208–217. doi:10.1016/j.intimp.2016.07.010
42. Jung HW, Chung YS, Kim YS, Park YK. Celastrol inhibits production of nitric oxide and proinflammatory cytokines through MAPK signal transduction and NF-κB in LPS-stimulated BV-2 microglial cells. Exp Mol Med. 2007;39(6):715–721. doi:10.1038/emm.2007.78
43. Ren Q, Guo F, Tao S, Huang R, Ma L, Fu P. Flavonoid fisetin alleviates kidney inflammation and apoptosis via inhibiting Src-mediated NF-κB p65 and MAPK signaling pathways in septic AKI mice. Biomed Pharmacother. 2020;122:109772. doi:10.1016/j.biopha.2019.109772
44. Yang CC, Lin CC, Hsiao LD, Yang CM. Galangin inhibits thrombin-induced MMP-9 expression in SK-N-SH cells via protein kinase-dependent NF-κB phosphorylation. Int J Mol Sci. 2018;19(12):4084. doi:10.3390/ijms19124084
45. Qian J, Chen X, Chen X, et al. Kaempferol reduces K63-linked polyubiquitination to inhibit nuclear factor-κB and inflammatory responses in acute lung injury in mice. Toxicol Lett. 2019;306:53–60. doi:10.1016/j.toxlet.2019.02.005
46. Yu D, Li M, Tian Y, Liu J, Shang J. Luteolin inhibits ROS-activated MAPK pathway in myocardial ischemia/reperfusion injury. Life Sci. 2015;122:15–25. doi:10.1016/j.lfs.2014.11.014
47. Wang Q, Yao L, Xu K, et al. Madecassoside inhibits estrogen deficiency-induced osteoporosis by suppressing RANKL-induced osteoclastogenesis. J Cell Mol Med. 2019;23(1):380–394. doi:10.1111/jcmm.13942
48. Tao J, Hou Y, Ma X, et al. An integrated global chemomics and system biology approach to analyze the mechanisms of the traditional Chinese medicinal preparation Eriobotrya japonica - Fritillaria usuriensis dropping pills for pulmonary diseases. BMC Complement Altern Med. 2016;16:4. doi:10.1186/s12906-015-0983-y
49. El-Agamy DS, El-Harbi KM, Khoshhal S, et al. Pristimerin protects against doxorubicin-induced cardiotoxicity and fibrosis through modulation of Nrf2 and MAPK/NF-kB signaling pathways. Cancer Manag Res. 2019;11:47–61. doi:10.2147/CMAR.S186696
50. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023. doi:10.1038/sigtrans.2017.23
51. Chow CW, Herrera Abreu MT, Suzuki T, Downey GP. Oxidative stress and acute lung injury. Am J Respir Cell Mol Biol. 2003;29(4):427–431. doi:10.1165/rcmb.F278
52. Xiao M, Zhu T, Zhang W, et al. Emodin ameliorates LPS-induced acute lung injury, involving the inactivation of NF-κB in mice. Int J Mol Sci. 2014;15(11):19355–19368. doi:10.3390/ijms151119355
53. Li N, Song Y, Zhao W, et al. Small interfering RNA targeting NF-κB attenuates lipopolysaccharide-induced acute lung injury in rats. BMC Physiol. 2016;16(1):7. doi:10.1186/s12899-016-0027-y
54. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27. doi:10.1016/j.jaci.2016.05.011
55. Zaynagetdinov R, Sherrill TP, Gleaves LA, et al. Chronic NF-κB activation links COPD and lung cancer through generation of an immunosuppressive microenvironment in the lungs. Oncotarget. 2016;7(5):5470–5482. doi:10.18632/oncotarget.6562
56. Zhou L, Liu Y, Chen X, et al. Over-expression of nuclear factor-κB family genes and inflammatory molecules is related to chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2018;13:2131–2138. doi:10.2147/COPD.S164151
57. Di Stefano A, Caramori G, Oates T, et al. Increased expression of nuclear factor-κB in bronchial biopsies from smokers and patients with COPD. Eur Respir J. 2002;20(3):556–563. doi:10.1183/09031936.02.00272002
58. Lee I-T, Luo S-F, Lee C-W, et al. Overexpression of HO-1 protects against TNF-α-mediated airway inflammation by down-regulation of TNFR1-dependent oxidative stress. Am J Pathol. 2009;175(2):519–532. doi:10.2353/ajpath.2009.090016
59. Schuliga M. NF-κB signaling in chronic inflammatory airway disease. Biomolecules. 2015;5(3):1266–1283. doi:10.3390/biom5031266
60. Edwards MR, Bartlett NW, Clarke D, Birrell M, Belvisi M, Johnston SL. Targeting the NF-κB pathway in asthma and chronic obstructive pulmonary disease. Pharmacol Ther. 2009;121(1):1–13. doi:10.1016/j.pharmthera.2008.09.003
61. Vesely PW, Staber PB, Hoefler G, Kenner L. Translational regulation mechanisms of AP-1 proteins. Mutat Res Rev Mutat Res. 2009;682(1):7–12. doi:10.1016/j.mrrev.2009.01.001
62. Karin M, Liu Z, Zandi E, et al. AP-‐1 function and regulation. Curr Opin Cell Biol. 1997;9(2):240–246. doi:10.1016/S0955-0674(97)80068-3
63. Wodrich W, Volm M. Overexpression of oncoproteins in non-small cell lung carcinomas of smokers. Carcinogenesis. 1993;14(6):1121–1124. doi:10.1093/carcin/14.6.1121
64. Guo RF, Lentsch AB, Sarma JV, et al. Activator protein-1 activation in acute lung injury. Am J Pathol. 2002;161(1):275–282. doi:10.1016/S0002-9440(10)64179-X
65. Haase M, Koslowski R, Lengnick A, et al. Cellular distribution of c-Jun and c-Fos in rat lung before and after bleomycin induced injury. Virchows Archiv. 1997;431(6):441–448. doi:10.1007/s004280050121
66. Barnes PJ, Adcock I. Transcription factors and asthma. Eur Respir J. 1998;12(1):221–234. doi:10.1183/09031936.98.12010221
67. Kips JC, Tavernier JH, Joos GF, Peleman RA, Pauwels RA. The potential role of tumour necrosis factor α in asthma. Clin Exp Allergy. 1993;23(4):247–250. doi:10.1111/j.1365-2222.1993.tb00317.x
68. Kersul AL, Iglesias A, Á R, et al. Molecular mechanisms of inflammation during exacerbations of chronic obstructive pulmonary disease. Arch Bronconeumol. 2011;47(4):176–183. doi:10.1016/j.arbres.2010.12.003
69. Ying S, Robinson DS, Varney V, et al. TNF-α mRNA expression in allergic inflammation. Clin Exp Allergy. 1991;21(6):745–750. doi:10.1111/j.1365-2222.1991.tb03205.x
70. Bradding P, Roberts JA, Britten KM, et al. Interleukin-4, −5, and −6 and tumor necrosis factor-α in normal and asthmatic airways: evidence for the human mast cell as a source of these cytokines. Am J Respir Cell Mol Biol. 1994;10(5):471–480. doi:10.1165/ajrcmb.10.5.8179909
71. Thomas PS, Yates DH, Barnes PJ. Tumor necrosis factor-α increases airway responsiveness and sputum neutrophilia in normal human subjects. Am J Respir Crit Care Med. 1995;152(1):76–80. doi:10.1164/ajrccm.152.1.7599866
72. Adner M, Rose AC, Zhang Y, et al. An assay to evaluate the long-term effects of inflammatory mediators on murine airway smooth muscle: evidence that TNFα up-regulates 5-HT2A-mediated contraction. Br J Pharmacol. 2002;137(7):971–982. doi:10.1038/sj.bjp.0704928
73. Huber M, Beutler B, Keppler D. Tumor necrosis factor α stimulates leukotriene production in vivo. Eur J Immunol. 1988;18(12):2085–2088. doi:10.1002/eji.1830181233
74. Brightling CE, Bradding P, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID. Mast-cell infiltration of airway smooth muscle in asthma. N Engl J Med. 2002;346(22):1699–1705. doi:10.1056/NEJMoa012705
75. Ginsburg I. Role of lipoteichoic acid in infection and inflammation. Lancet Infect Dis. 2002;2(3):171–179. doi:10.1016/S1473-3099(02)00226-8
76. Ellingsen E, Morath S, Flo T, et al. Induction of cytokine production in human T cells and monocytes by highly purified lipoteichoic acid: involvement of Toll-like receptors and CD14. Med Sci Monit. 2002;8(5):Br149–156.
77. Tenhunen R, Marver HS, Schmid R. Microsomal heme oxygenase. Characterization of the enzyme. J Biol Chem. 1969;244(23):6388–6394. doi:10.1016/S0021-9258(18)63477-5
78. Fredenburgh LE, Perrella MA, Mitsialis SA. The role of heme oxygenase-1 in pulmonary disease. Am J Respir Cell Mol Biol. 2007;36(2):158–165. doi:10.1165/rcmb.2006-0331TR
79. Horváth I, Donnelly LE, Kiss A, Paredi P, Kharitonov SA, Barnes PJ. Raised levels of exhaled carbon monoxide are associated with an increased expression of heme oxygenase-1 in airway macrophages in asthma: a new marker of oxidative stress. Thorax. 1998;53(8):668–672. doi:10.1136/thx.53.8.668
80. Almolki A, Taillé C, Martin GF, et al. Heme oxygenase attenuates allergen-induced airway inflammation and hyperreactivity in guinea pigs. Am J Physiol Lung Cell Mol Physiol. 2004;287(1):L26–34. doi:10.1152/ajplung.00237.2003
81. Wagener FA, Eggert A, Boerman OC, et al. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood. 2001;98(6):1802–1811. doi:10.1182/blood.V98.6.1802
82. Yang CC, Hsiao LD, Lin HH, et al. Induction of HO-1 by 5, 8-dihydroxy-4ʹ,7-dimethoxyflavone via activation of ROS/p38 MAPK/Nrf2 attenuates thrombin-induced connective tissue growth factor expression in human cardiac fibroblasts. Oxid Med Cell Longev. 2020;2020:1080168. doi:10.1155/2020/1080168
83. Slebos DJ, Kerstjens HA, Rutgers SR, Kauffman HF, Choi AM, Postma DS. Haem oxygenase-1 expression is diminished in alveolar macrophages of patients with COPD. Eur Respir J. 2004;23(4):
84. Exner M, Minar E, Wagner O, Schillinger M. The role of heme oxygenase-1 promoter polymorphisms in human disease. Free Radic Biol Med. 2004;37(8):1097–1104. doi:10.1016/j.freeradbiomed.2004.07.008
85. Even B, Fayad-Kobeissi S, Gagliolo JM, et al. Heme oxygenase-1 induction attenuates senescence in chronic obstructive pulmonary disease lung fibroblasts by protecting against mitochondria dysfunction. Aging Cell. 2018;17(6):e12837. doi:10.1111/acel.12837
86. Wei J, Fan G, Zhao H, Li J. Heme oxygenase-1 attenuates inflammation and oxidative damage in a rat model of smoke-induced emphysema. Int J Mol Med. 2015;36(5):1384–1392. doi:10.3892/ijmm.2015.2353
87. Chen Y, Yuan T, Zhang H, et al. Activation of Nrf2 attenuates pulmonary vascular remodeling via inhibiting endothelial-to-mesenchymal transition: an insight from a plant polyphenol. Int J Biol Sci. 2017;13(8):1067–1081. doi:10.7150/ijbs.20316
88. Divya T, Dineshbabu V, Soumyakrishnan S, Sureshkumar A, Sudhandiran G. Celastrol enhances Nrf2 mediated antioxidant enzymes and exhibits anti-fibrotic effect through regulation of collagen production against bleomycin-induced pulmonary fibrosis. Chem Biol Interact. 2016;246:52–62. doi:10.1016/j.cbi.2016.01.006
89. Hussain T, Al-Attas OS, Alamery S, Ahmed M, Odeibat HAM, Alrokayan S. The plant flavonoid, fisetin alleviates cigarette smoke-induced oxidative stress, and inflammation in Wistar rat lungs. J Food Biochem. 2019;43(8):e12962. doi:10.1111/jfbc.12962
90. Aladaileh SH, Abukhalil MH, Saghir SAM, et al. Galangin activates Nrf2 signaling and attenuates oxidative damage, inflammation, and apoptosis in a rat model of cyclophosphamide-induced hepatotoxicity. Biomolecules. 2019;9(8):346. doi:10.3390/biom9080346
91. Shu YS, Tao W, Miao QB, Lu SC, Zhu YB. Galangin dampens mice lipopolysaccharide-induced acute lung injury. Inflammation. 2014;37(5):1661–1668. doi:10.1007/s10753-014-9894-1
92. Gao SS, Choi BM, Chen XY, et al. Kaempferol suppresses cisplatin-induced apoptosis via inductions of heme oxygenase-1 and glutamate-cysteine ligase catalytic subunit in HEI-OC1 cell. Pharm Res. 2010;27(2):235–245.
93. Hong JT, Yen JH, Wang L, Lo YH, Chen ZT, Wu MJ. Regulation of heme oxygenase-1 expression and MAPK pathways in response to kaempferol and rhamnocitrin in PC12 cells. Toxicol Appl Pharmacol. 2009;237(1):59–68. doi:10.1016/j.taap.2009.02.014
94. Hirose E, Matsushima M, Takagi K, et al. Involvement of heme oxygenase-1 in kaempferol-induced anti-allergic actions in RBL-2H3 cells. Inflammation. 2009;32(2):99–108. doi:10.1007/s10753-009-9108-4
95. Yao H, Sun J, Wei J, Zhang X, Chen B, Lin Y. Kaempferol protects blood vessels from damage induced by oxidative stress and inflammation in association with the Nrf2/HO-1 signaling pathway. Front Pharmacol. 2020;11:1118. doi:10.3389/fphar.2020.01118
96. Sun GB, Sun X, Wang M, et al. Oxidative stress suppression by luteolin-induced heme oxygenase-1 expression. Toxicol Appl Pharmacol. 2012;265(2):229–240. doi:10.1016/j.taap.2012.10.002
97. Liu S, Li G, Tang H, et al. Madecassoside ameliorates lipopolysaccharide-induced neurotoxicity in rats by activating the Nrf2-HO-1 pathway. Neurosci Lett. 2019;709:134386. doi:10.1016/j.neulet.2019.134386
98. Wang W, Wu L, Li Q, et al. Madecassoside prevents acute liver failure in LPS/D-GalN-induced mice by inhibiting p38/NF-κB and activating Nrf2/HO-1 signaling. Biomed Pharmacother. 2018;103:1137–1145. doi:10.1016/j.biopha.2018.04.162
99. Reisman SA, Aleksunes LM, Klaassen CD. Oleanolic acid activates Nrf2 and protects from acetaminophen hepatotoxicity via Nrf2-dependent and Nrf2-independent processes. Biochem Pharmacol. 2009;77(7):1273–1282. doi:10.1016/j.bcp.2008.12.028
100. Feng J, Zhang P, Chen X, He G. PI3K and ERK/Nrf2 pathways are involved in oleanolic acid-induced heme oxygenase-1 expression in rat vascular smooth muscle cells. J Cell Biochem. 2011;112(6):1524–1531. doi:10.1002/jcb.23065
101. Chen RJ, Guo XY, Cheng BH, Gong YQ, Ying BY, Lin MX. Saikosaponin a inhibits cigarette smoke-induced oxidant stress and inflammatory responses by activation of Nrf2. Inflammation. 2018;41(4):1297–1303. doi:10.1007/s10753-018-0778-7
102. Liby K, Hock T, Yore MM, et al. The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res. 2005;65(11):4789–4798. doi:10.1158/0008-5472.CAN-04-4539
103. El-Agamy DS, Shaaban AA, Almaramhy HH, Elkablawy S, Elkablawy MA. Pristimerin as a novel hepatoprotective agent against experimental autoimmune hepatitis. Front Pharmacol. 2018;9:292. doi:10.3389/fphar.2018.00292
104. Immenschuh S, Ramadori G. Gene regulation of heme oxygenase-1 as a therapeutic target. Biochem Pharmacol. 2000;60(8):1121–1128. doi:10.1016/S0006-2952(00)00443-3
105. Alam J, Camhi S, Choi AM. Identification of a second region upstream of the mouse heme oxygenase-1 gene that functions as a basal level and inducer-dependent transcription enhancer. J Biol Chem. 1995;270(20):11977–11984. doi:10.1074/jbc.270.20.11977
106. Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–260. doi:10.1146/annurev.pharmtox.43.100901.140229
107. Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi:10.1146/annurev.pharmtox.46.120604.141046
108. Yamada N, Yamaya M, Okinaga S, et al. Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to emphysema. Am J Hum Genet. 2000;66(1):187–195. doi:10.1086/302729
109. Pawlinski R, Tencati M, Hampton CR, et al. Protease-activated receptor-1 contributes to cardiac remodeling and hypertrophy. Circulation. 2007;116(20):2298–2306. doi:10.1161/CIRCULATIONAHA.107.692764
110. Lee TS, Chang CC, Zhu Y, Shyy JY. Simvastatin induces heme oxygenase-1: a novel mechanism of vessel protection. Circulation. 2004;110(10):1296–1302. doi:10.1161/01.CIR.0000140694.67251.9C
111. Grosser N, Hemmerle A, Berndt G, et al. The antioxidant defense protein heme oxygenase 1 is a novel target for statins in endothelial cells. Free Radic Biol Med. 2004;37(12):2064–2071. doi:10.1016/j.freeradbiomed.2004.09.009
112. Lin CC, Lin WN, Cho RL, et al. Induction of HO-1 by mevastatin mediated via a nox/ROS-dependent c-Src/PDGFRα/PI3K/Akt/Nrf2/ARE cascade suppresses TNF-α-induced lung inflammation. J Clin Med. 2020;9(1):226. doi:10.3390/jcm9010226
113. Yang CM, Lin CC, Yang CC, Cho RL, Hsiao LD. Mevastatin-induced AP-1-dependent HO-1 expression suppresses vascular cell adhesion molecule-1 expression and monocyte adhesion on human pulmonary alveolar epithelial cells challenged with TNF-α. Biomolecules. 2020;10(3):381. doi:10.3390/biom10030381
114. Lin CC, Hsiao LD, Cho RL, Yang CM. CO-releasing molecule-2 induces Nrf2/ARE-dependent heme oxygenase-1 expression suppressing TNF-α-induced pulmonary inflammation. J Clin Med. 2019;8(4).
115. Lin CC, Chiang YC, Cho RL, et al. Up-regulation of PYK2/PKCα-dependent haem oxygenase-1 by CO-releasing molecule-2 attenuates TNF-α-induced lung inflammation. Br J Pharmacol. 2018;175(3):456–468. doi:10.1111/bph.14094
116. Lin CC, Hsiao LD, Cho RL, Yang CM. Carbon monoxide releasing molecule-2-upregulated ROS-dependent heme oxygenase-1 axis suppresses lipopolysaccharide-induced airway inflammation. Int J Mol Sci. 2019;20(13):3157. doi:10.3390/ijms20133157
117. Yang CM, Lin CC, Lee IT, et al. c-Src-dependent transactivation of EGFR mediates CORM-2-induced HO-1 expression in human tracheal smooth muscle cells. J Cell Physiol. 2015;230(10):2351–2361. doi:10.1002/jcp.24912
118. Cheng SE, Lee IT, Lin CC, Kou YR, Yang CM. Cigarette smoke particle-phase extract induces HO-1 expression in human tracheal smooth muscle cells: role of the c-Src/NADPH oxidase/MAPK/Nrf2 signaling pathway. Free Radic Biol Med. 2010;48(10):1410–1422. doi:10.1016/j.freeradbiomed.2010.02.026
119. Chung JH, Seo AY, Chung SW, et al. Molecular mechanism of PPAR in the regulation of age-related inflammation. Ageing Res Rev. 2008;7(2):126–136. doi:10.1016/j.arr.2008.01.001
120. Makris D, Manoulakas E, Komnos A, et al. Effect of pravastatin on the frequency of ventilator-associated pneumonia and on intensive care unit mortality: open-label, randomized study. Crit Care Med. 2011;39(11):2440–2446. doi:10.1097/CCM.0b013e318225742c
121. Kim EJ, Kwon KJ, Park JY, Lee SH, Moon CH, Baik EJ. Effects of peroxisome proliferator-activated receptor agonists on LPS-induced neuronal death in mixed cortical neurons: associated with iNOS and COX-2. Brain Res. 2002;941(1–2):1–10. doi:10.1016/S0006-8993(02)02480-0
122. Cho RL, Yang CC, Tseng HC, Hsiao LD, Lin CC, Yang CM. Haem oxygenase-1 up-regulation by rosiglitazone via ROS-dependent Nrf2-antioxidant response elements axis or PPARγ attenuates LPS-mediated lung inflammation. Br J Pharmacol. 2018;175(20):3928–3946. doi:10.1111/bph.14465
123. Wang L, Waltenberger B, Pferschy-Wenzig EM, et al. Natural product agonists of peroxisome proliferator-activated receptor gamma (PPARγ): a review. Biochem Pharmacol. 2014;92(1):73–89.
124. Auboeuf D, Rieusset J, Fajas L, et al. Tissue distribution and quantification of the expression of mRNAs of peroxisome proliferator-activated receptors and liver X receptor-α in humans: no alteration in adipose tissue of obese and NIDDM patients. Diabetes. 1997;46(8):1319–1327. doi:10.2337/diab.46.8.1319
125. Seedorf U, Aberle J. Emerging roles of PPARδ in metabolism. Biochim Biophys Acta. 2007;1771(9):1125–1131. doi:10.1016/j.bbalip.2007.04.017
126. Zhu Y, Qi C, Korenberg JR, et al. Structural organization of mouse peroxisome proliferator-activated receptor γ (mPPARγ) gene: alternative promoter use and different splicing yield two mPPARγ isoforms. Proc Natl Acad Sci U S A. 1995;92(17):7921–7925. doi:10.1073/pnas.92.17.7921
127. Werman A, Hollenberg A, Solanes G, Bjorbaek C, Vidal-Puig AJ, Flier JS. Ligand-independent activation domain in the N terminus of peroxisome proliferator-activated receptor γ (PPARγ). Differential activity of PPARγ-1 and −2 isoforms and influence of insulin. J Biol Chem. 1997;272(32):20230–20235. doi:10.1074/jbc.272.32.20230
128. Vidal-Puig AJ, Considine RV, Jimenez-Liñan M, et al. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J Clin Invest. 1997;99(10):2416–2422. doi:10.1172/JCI119424
129. Gearing KL, Göttlicher M, Teboul M, Widmark E, Gustafsson JA. Interaction of the peroxisome-proliferator-activated receptor and retinoid X receptor. Proc Natl Acad Sci U S A. 1993;90(4):1440–1444. doi:10.1073/pnas.90.4.1440
130. Yu S, Reddy JK. Transcription coactivators for peroxisome proliferator-activated receptors. Biochim Biophys Acta. 2007;1771(8):936–951. doi:10.1016/j.bbalip.2007.01.008
131. Staels B, Fruchart JC. Therapeutic roles of peroxisome proliferator-activated receptor agonists. Diabetes. 2005;54(8):2460–2470. doi:10.2337/diabetes.54.8.2460
132. Cho RL, Lin WN, Wang CY, et al. Heme oxygenase-1 induction by rosiglitazone via PKCα/AMPKα/p38 MAPKα/SIRT1/PPARγ pathway suppresses lipopolysaccharide-mediated pulmonary inflammation. Biochem Pharmacol. 2018;148:222–237. doi:10.1016/j.bcp.2017.12.024
133. Dormandy JA, Charbonnel B, Eckland DJ, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366(9493):1279–1289. doi:10.1016/S0140-6736(05)67528-9
134. Devchand PR, Keller H, Peters JM, Vazquez M, Gonzalez FJ, The WW. PPARα-leukotriene B4 pathway to inflammation control. Nature. 1996;384(6604):39–43. doi:10.1038/384039a0
135. Arai H, Yamashita S, Yokote K, Araki E, Suganami H, Ishibashi S. Efficacy and safety of K-877, a novel selective peroxisome proliferator-activated receptor α modulator (SPPARMα), in combination with statin treatment: two randomised, double-blind, placebo-controlled clinical trials in patients with dyslipidaemia. Atherosclerosis. 2017;261:144–152. doi:10.1016/j.atherosclerosis.2017.03.032
136. Marfella R, D’Amico M, Esposito K, et al. The ubiquitin-proteasome system and inflammatory activity in diabetic atherosclerotic plaques: effects of rosiglitazone treatment. Diabetes. 2006;55(3):622–632. doi:10.2337/diabetes.55.03.06.db05-0832
137. Skochko OV, Kaidashev IP. Effect of pioglitazone on insulin resistance, progression of atherosclerosis and clinical course of coronary heart disease. Wiad Lek. 2017;70(5):881–890.
138. van Wijk JP, Cabezas MC, Coll B, Joven J, Rabelink TJ, de Koning EJ. Effects of rosiglitazone on postprandial leukocytes and cytokines in type 2 diabetes. Atherosclerosis. 2006;186(1):152–159. doi:10.1016/j.atherosclerosis.2005.07.001
139. Werner C, Kamani CH, Gensch C, Böhm M, Laufs U. The peroxisome proliferator-activated receptor-γ agonist pioglitazone increases number and function of endothelial progenitor cells in patients with coronary artery disease and normal glucose tolerance. Diabetes. 2007;56(10):2609–2615. doi:10.2337/db07-0069
140. Lewis JD, Lichtenstein GR, Deren JJ, et al. Rosiglitazone for active ulcerative colitis: a randomized placebo-controlled trial. Gastroenterology. 2008;134(3):688–695. doi:10.1053/j.gastro.2007.12.012
141. Liang HL, Ouyang Q. [A clinical trial of rosiglitazone and 5-aminosalicylate combination for ulcerative colitis]. Zhonghua Nei Ke Za Zhi. 2006;45(7):548–551. Chinese.
142. Pedersen G, Brynskov J. Topical rosiglitazone treatment improves ulcerative colitis by restoring peroxisome proliferator-activated receptor-γ activity. Am J Gastroenterol. 2010;105(7):1595–1603. doi:10.1038/ajg.2009.749
143. Kernan WN, Viscoli CM, Furie KL, et al. Pioglitazone after ischemic stroke or transient ischemic attack. N Engl J Med. 2016;374(14):1321–1331. doi:10.1056/NEJMoa1506930
144. Marder W, Khalatbari S, Myles JD, et al. The peroxisome proliferator activated receptor-γ pioglitazone improves vascular function and decreases disease activity in patients with rheumatoid arthritis. J Am Heart Assoc. 2013;2(6):e000441. doi:10.1161/JAHA.113.000441
145. Ormseth MJ, Oeser AM, Cunningham A, et al. Peroxisome proliferator-activated receptor γ agonist effect on rheumatoid arthritis: a randomized controlled trial. Arthritis Res Ther. 2013;15(5):R110. doi:10.1186/ar4290
146. Ormseth MJ, Oeser AM, Cunningham A, et al. Reversing vascular dysfunction in rheumatoid arthritis: improved augmentation index but not endothelial function with peroxisome proliferator-activated receptor γ agonist therapy. Arthritis Rheumatol. 2014;66(9):2331–2338. doi:10.1002/art.38686
147. Bongartz T, Coras B, Vogt T, Schölmerich J, Müller-Ladner U. Treatment of active psoriatic arthritis with the PPARγ ligand pioglitazone: an open-label pilot study. Rheumatology (Oxford). 2005;44(1):126–129. doi:10.1093/rheumatology/keh423
148. Ellis CN, Varani J, Fisher GJ, et al. Troglitazone improves psoriasis and normalizes models of proliferative skin disease: ligands for peroxisome proliferator-activated receptor-γ inhibit keratinocyte proliferation. Arch Dermatol. 2000;136(5):609–616. doi:10.1001/archderm.136.5.609
149. Rinne ST, Liu CF, Feemster LC, et al. Thiazolidinediones are associated with a reduced risk of COPD exacerbations. Int J Chron Obstruct Pulmon Dis. 2015;10:1591–1597. doi:10.2147/COPD.S82643
150. De Belilovsky C, Roo-Rodriguez E, Baudouin C, Menu F, Chadoutaud B, Msika P. Natural peroxisome proliferator-activated receptor-α agonist cream demonstrates similar therapeutic response to topical steroids in atopic dermatitis. J Dermatolog Treat. 2011;22(6):359–365. doi:10.3109/09546634.2010.499932
151. Fukaya M, Kimata H. Topical clofibrate improves symptoms in patients with atopic dermatitis and reduces serum TARC levels: a randomized, double-blind, placebo-controlled pilot study. J Drugs Dermatol. 2014;13(3):259–263.
152. Hammad H, de Heer HJ, Soullie T, et al. Activation of peroxisome proliferator-activated receptor-γ in dendritic cells inhibits the development of eosinophilic airway inflammation in a mouse model of asthma. Am J Pathol. 2004;164(1):263–271. doi:10.1016/S0002-9440(10)63116-1
153. Kim SR, Lee KS, Park HS, et al. Involvement of IL-10 in peroxisome proliferator-activated receptor γ-mediated anti-inflammatory response in asthma. Mol Pharmacol. 2005;68(6):1568–1575. doi:10.1124/mol.105.017160
154. Lee HY, Rhee CK, Kang JY, et al. Effect of intranasal rosiglitazone on airway inflammation and remodeling in a murine model of chronic asthma. Korean J Intern Med. 2016;31(1):89–97. doi:10.3904/kjim.2016.31.1.89
155. Yin Y, Hou G, Li ER, Wang QY, Kang J. Regulation of cigarette smoke-induced toll-like receptor 4 expression by peroxisome proliferator-activated receptor-γ agonists in bronchial epithelial cells. Respirology. 2013;18(Suppl 3):30–39. doi:10.1111/resp.12167
156. Liu DS, Liu WJ, Chen L, et al. Rosiglitazone, a peroxisome proliferator-activated receptor-γ agonist, attenuates acrolein-induced airway mucus hypersecretion in rats. Toxicology. 2009;260(1–3):112–119. doi:10.1016/j.tox.2009.03.016
157. Rossi A, Inciardi RM, Rossi A, et al. Prognostic effects of rosuvastatin in patients with co-existing chronic obstructive pulmonary disease and chronic heart failure: a sub-analysis of GISSI-HF trial. Pulm Pharmacol Ther. 2017;44:16–23. doi:10.1016/j.pupt.2017.03.001
158. Liu D, Zeng BX, Zhang SH, Yao SL. Rosiglitazone, an agonist of peroxisome proliferator-activated receptor γ, reduces pulmonary inflammatory response in a rat model of endotoxemia. Inflamm Res. 2005;54(11):464–470. doi:10.1007/s00011-005-1379-0
159. Neri T, Armani C, Pegoli A, et al. Role of NF-κB and PPAR-γ in lung inflammation induced by monocyte-derived microparticles. Eur Respir J. 2011;37(6):1494–1502. doi:10.1183/09031936.00023310
160. Momoi A, Murao K, Imachi H, et al. Inhibition of monocyte chemoattractant protein-1 expression in cytokine-treated human lung epithelial cells by thiazolidinedione. Chest. 2001;120(4):1293–1300. doi:10.1378/chest.120.4.1293
161. Cheng Y, Li S, Wang M, Cheng C, Liu R. Peroxisome proliferator activated receptor gamma (PPARγ) agonist rosiglitazone ameliorate airway inflammation by inhibiting toll-like receptor 2 (TLR2)/Nod-like receptor with pyrin domain containing 3 (NLRP3) inflammatory corpuscle activation in asthmatic mice. Med Sci Monit. 2018;24:9045–9053. doi:10.12659/MSM.910766
162. Mirakaj V, Mutz C, Vagts D, et al. Rosiglitazone dampens pulmonary inflammation in a porcine model of acute lung injury. Inflammation. 2014;37(4):1102–1110. doi:10.1007/s10753-014-9834-0
163. Birrell MA, Patel HJ, McCluskie K, et al. PPAR-γ agonists as therapy for diseases involving airway neutrophilia. Eur Respir J. 2004;24(1):18–23. doi:10.1183/09031936.04.00098303
164. Morissette MC, Shen P, Thayaparan D, Stämpfli MR. Impacts of peroxisome proliferator-activated receptor-γ activation on cigarette smoke-induced exacerbated response to bacteria. Eur Respir J. 2015;45(1):191–200. doi:10.1183/09031936.00004314
165. Lea S, Plumb J, Metcalfe H, et al. The effect of peroxisome proliferator-activated receptor-γ ligands on in vitro and in vivo models of COPD. Eur Respir J. 2014;43(2):409–420. doi:10.1183/09031936.00187812
166. Gopal R, Mendy A, Marinelli MA, et al. Peroxisome proliferator-activated receptor gamma (PPARγ) suppresses inflammation and bacterial clearance during influenza-bacterial super-infection. Viruses. 2019;11(6). doi:10.3390/v11060505
167. Xu J, Zhu YT, Wang GZ, et al. The PPARgamma agonist, rosiglitazone, attenuates airway inflammation and remodeling via heme oxygenase-1 in murine model of asthma. Acta Pharmacol Sin. 2015;36(2):171–178. doi:10.1038/aps.2014.128
168. Kronke G, Kadl A, Ikonomu E, et al. Expression of heme oxygenase-1 in human vascular cells is regulated by peroxisome proliferator-activated receptors. Arterioscler Thromb Vasc Biol. 2007;27(6):1276–1282. doi:10.1161/ATVBAHA.107.142638
169. Kulkarni AA, Woeller CF, Thatcher TH, Ramon S, Phipps RP, Sime PJ. Emerging PPARγ-independent role of PPARγ ligands in lung diseases. PPAR Res. 2012;2012:705352. doi:10.1155/2012/705352
170. Kadam L, Gomez-Lopez N, Mial TN, Kohan-Ghadr HR, Drewlo S. Rosiglitazone regulates TLR4 and rescues HO-1 and NRF2 expression in myometrial and decidual macrophages in inflammation-induced preterm birth. Reprod Sci. 2017;24(12):1590–1599. doi:10.1177/1933719117697128
171. He J, Qi D, Tang XM, et al. Rosiglitazone promotes ENaC-mediated alveolar fluid clearance in acute lung injury through the PPARγ/SGK1 signaling pathway. Cell Mol Biol Lett. 2019;24:35. doi:10.1186/s11658-019-0154-0
172. Lin Q, Fang LP, Zhou WW, Liu XM. Rosiglitazone inhibits migration, proliferation, and phenotypic differentiation in cultured human lung fibroblasts. Exp Lung Res. 2010;36(2):120–128. doi:10.3109/01902140903214659
173. Jin GY, Bok SM, Han YM, et al. Effectiveness of rosiglitazone on bleomycin-induced lung fibrosis: assessed by micro-computed tomography and pathologic scores. Eur J Radiol. 2012;81(8):1901–1906. doi:10.1016/j.ejrad.2010.12.061
174. Ward JE, Fernandes DJ, Taylor CC, Bonacci JV, Quan L, Stewart AG. The PPARγ ligand, rosiglitazone, reduces airways hyperresponsiveness in a murine model of allergen-induced inflammation. Pulm Pharmacol Ther. 2006;19(1):39–46. doi:10.1016/j.pupt.2005.02.005
175. Racanelli AC, Kikkers SA, Choi AMK, Cloonan SM. Autophagy and inflammation in chronic respiratory disease. Autophagy. 2018;14(2):221–232. doi:10.1080/15548627.2017.1389823
176. Chima RS, Hake PW, Piraino G, Mangeshkar P, Denenberg A, Zingarelli B. Ciglitazone ameliorates lung inflammation by modulating the inhibitor κB protein kinase/nuclear factor-κB pathway after hemorrhagic shock. Crit Care Med. 2008;36(10):2849–2857. doi:10.1097/CCM.0b013e318187810e
177. Aoki Y, Maeno T, Aoyagi K, et al. Pioglitazone, a peroxisome proliferator-activated receptor gamma ligand, suppresses bleomycin-induced acute lung injury and fibrosis. Respiration. 2009;77(3):311–319. doi:10.1159/000168676
178. Milam JE, Keshamouni VG, Phan SH, et al. PPAR-γ agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294(5):L891–901. doi:10.1152/ajplung.00333.2007
179. Jumeau C, Rupin A, Chieng-Yane P, et al. Direct thrombin inhibitors prevent left atrial remodeling associated with heart failure in rats. JACC Basic Transl Sci. 2016;1(5):328–339. doi:10.1016/j.jacbts.2016.05.002
180. Ferguson HE, Thatcher TH, Olsen KC, et al. Peroxisome proliferator-activated receptor-γ ligands induce heme oxygenase-1 in lung fibroblasts by a PPARγ-independent, glutathione-dependent mechanism. Am J Physiol Lung Cell Mol Physiol. 2009;297(5):L912–919. doi:10.1152/ajplung.00148.2009
181. Reddy RC, Narala VR, Keshamouni VG, Milam JE, Newstead MW, Standiford TJ. Sepsis-induced inhibition of neutrophil chemotaxis is mediated by activation of peroxisome proliferator-activated receptor-γ. Blood. 2008;112(10):4250–4258. doi:10.1182/blood-2007-12-128967
182. Wang AC, Dai X, Luu B, Conrad DJ. Peroxisome proliferator-activated receptor-γ regulates airway epithelial cell activation. Am J Respir Cell Mol Biol. 2001;24(6):688–693. doi:10.1165/ajrcmb.24.6.4376
183. Ferguson HE, Kulkarni A, Lehmann GM, et al. Electrophilic peroxisome proliferator-activated receptor-γ ligands have potent antifibrotic effects in human lung fibroblasts. Am J Respir Cell Mol Biol. 2009;41(6):722–730. doi:10.1165/rcmb.2009-0006OC
184. Okada M, Yan SF, Pinsky DJ. Peroxisome proliferator-activated receptor-γ (PPAR-γ) activation suppresses ischemic induction of Egr-1 and its inflammatory gene targets. FASEB J. 2002;16(14):1861–1868. doi:10.1096/fj.02-0503com
185. Sharma R, Kaundal RK, Sharma SS. Amelioration of pulmonary dysfunction and neutrophilic inflammation by PPARγ agonist in LPS-exposed guinea pigs. Pulm Pharmacol Ther. 2009;22(3):183–189. doi:10.1016/j.pupt.2008.11.011
186. Samah M, El-Aidy Ael R, Tawfik MK, Ewais MM. Evaluation of the antifibrotic effect of fenofibrate and rosiglitazone on bleomycin-induced pulmonary fibrosis in rats. Eur J Pharmacol. 2012;689(1–3):186–193. doi:10.1016/j.ejphar.2012.05.026
187. Elaidy SM, Essawy SS, Hussain MA, El-Kherbetawy MK, Hamed ER. Modulation of the IL-23/IL-17 axis by fenofibrate ameliorates the ovalbumin/lipopolysaccharide-induced airway inflammation and bronchial asthma in rats. Naunyn Schmiedebergs Arch Pharmacol. 2018;391(3):309–321. doi:10.1007/s00210-017-1459-z
188. Becker J, Delayre-Orthez C, Frossard N, Pons F. The peroxisome proliferator-activated receptor α agonist fenofibrate decreases airway reactivity to methacholine and increases endothelial nitric oxide synthase phosphorylation in mouse lung. Fundam Clin Pharmacol. 2012;26(3):340–346. doi:10.1111/j.1472-8206.2011.00935.x
189. Cui H, Xie N, Banerjee S, Ge J, Guo S, Liu G. Impairment of fatty acid oxidation in alveolar epithelial cells mediates acute lung injury. Am J Respir Cell Mol Biol. 2019;60(2):167–178. doi:10.1165/rcmb.2018-0152OC
190. Delayre-Orthez C, Becker J, Auwerx J, Frossard N, Pons F. Suppression of allergen-induced airway inflammation and immune response by the peroxisome proliferator-activated receptor-α agonist fenofibrate. Eur J Pharmacol. 2008;581(1–2):177–184.
191. Zhu Q, He G, Wang J, Wang Y, Chen W. Protective effects of fenofibrate against acute lung injury induced by intestinal ischemia/reperfusion in mice. Sci Rep. 2016;6:22044. doi:10.1038/srep22044
192. Stolarz AJ, Farris RA, Wiley CA, O’Brien CE, Price ET. Fenofibrate attenuates neutrophilic inflammation in airway epithelia: potential drug repurposing for cystic fibrosis. Clin Transl Sci. 2015;8(6):696–701. doi:10.1111/cts.12310
193. Becker J, Delayre-Orthez C, Frossard N, Pons F. Regulation of peroxisome proliferator-activated receptor-α expression during lung inflammation. Pulm Pharmacol Ther. 2008;21(2):324–330. doi:10.1016/j.pupt.2007.08.001
194. Delayre-Orthez C, Becker J, Guenon I, et al. PPARα downregulates airway inflammation induced by lipopolysaccharide in the mouse. Respir Res. 2005;6(1):91. doi:10.1186/1465-9921-6-91
195. Ke Q, Yang L, Cui Q, et al. Ciprofibrate attenuates airway remodeling in cigarette smoke-exposed rats. Respir Physiol Neurobiol. 2020;271:103290. doi:10.1016/j.resp.2019.103290
196. Reiterer G, Toborek M, Hennig B. Peroxisome proliferator activated receptors α and γ require zinc for their anti-inflammatory properties in porcine vascular endothelial cells. J Nutr. 2004;134(7):1711–1715. doi:10.1093/jn/134.7.1711
197. Schaefer MB, Pose A, Ott J, et al. Peroxisome proliferator-activated receptor-α reduces inflammation and vascular leakage in a murine model of acute lung injury. Eur Respir J. 2008;32(5):1344–1353. doi:10.1183/09031936.00035808
198. Yoo SH, Abdelmegeed MA, Song BJ. Activation of PPARα by Wy-14643 ameliorates systemic lipopolysaccharide-induced acute lung injury. Biochem Biophys Res Commun. 2013;436(3):366–371. doi:10.1016/j.bbrc.2013.05.073
199. Yanagisawa J, Shiraishi T, Iwasaki A, et al. PPARα ligand WY14643 reduced acute rejection after rat lung transplantation with the upregulation of IL-4, IL-10 and TGFβ mRNA expression. J Heart Lung Transplant. 2009;28(11):1172–1179. doi:10.1016/j.healun.2009.06.016
200. Chou R, Dana T, Blazina I, Daeges M, Jeanne TL. Statins for prevention of cardiovascular disease in adults: evidence report and systematic review for the US preventive services task force. JAMA. 2016;316(19):2008–2024. doi:10.1001/jama.2015.15629
201. Brugts JJ, Yetgin T, Hoeks SE, et al. The benefits of statins in people without established cardiovascular disease but with cardiovascular risk factors: meta-analysis of randomised controlled trials. BMJ. 2009;338:b2376. doi:10.1136/bmj.b2376
202. Ajmera M, Shen C, Sambamoorthi U. Association between statin medications and COPD-specific outcomes: a real-world observational study. Drugs Real World Outcomes. 2017;4(1):9–19. doi:10.1007/s40801-016-0101-6
203. Wang MT, Lo YW, Tsai CL, et al. Statin use and risk of COPD exacerbation requiring hospitalization. Am J Med. 2013;126(7):598–606.e592. doi:10.1016/j.amjmed.2013.01.036
204. Lu Y, Chang R, Yao J, Xu X, Teng Y, Cheng N. Effectiveness of long-term using statins in COPD - a network meta-analysis. Respir Res. 2019;20(1):17. doi:10.1186/s12931-019-0984-3
205. Young RP, Hopkins R, Eaton TE. Pharmacological actions of statins: potential utility in COPD. Eur Respir Rev. 2009;18(114):222–232. doi:10.1183/09059180.00005309
206. Young RP, Hopkins R, Eaton TE. Potential benefits of statins on morbidity and mortality in chronic obstructive pulmonary disease: a review of the evidence. Postgrad Med J. 2009;85(1006):414–421. doi:10.1136/pgmj.2008.078477
207. Lahousse L, Loth DW, Joos GF, et al. Statins, systemic inflammation and risk of death in COPD: the Rotterdam study. Pulm Pharmacol Ther. 2013;26(2):212–217. doi:10.1016/j.pupt.2012.10.008
208. Xia DK, Hu ZG, Tian YF, Zeng FJ. Statin use and prognosis of lung cancer: a systematic review and meta-analysis of observational studies and randomized controlled trials. Drug Des Devel Ther. 2019;13:405–422. doi:10.2147/DDDT.S187690
209. Raymakers A, Sin DD, Sadatsafavi M, FitzGerald JM, Marra CA, Lynd LD. Statin use and lung cancer risk in chronic obstructive pulmonary disease patients: a population-based cohort study. Respir Res. 2020;21(1):118. doi:10.1186/s12931-020-01344-w
210. Wu WT, Chen CY. Protective effect of statins on pulmonary hypertension in chronic obstructive pulmonary disease patients: a nationwide retrospective, matched cohort study. Sci Rep. 2020;10(1):3104. doi:10.1038/s41598-020-59828-0
211. Chalmers JD, Singanayagam A, Murray MP, Hill AT. Prior statin use is associated with improved outcomes in community-acquired pneumonia. Am J Med. 2008;121(11):1002–1007.e1001. doi:10.1016/j.amjmed.2008.06.030
212. Troeman DP, Postma DF, van Werkhoven CH, Oosterheert JJ. The immunomodulatory effects of statins in community-acquired pneumonia: a systematic review. J Infect. 2013;67(2):93–101. doi:10.1016/j.jinf.2013.04.015
213. Young RP, Hopkins RJ. The mevalonate pathway and innate immune hyper-responsiveness in the pathogenesis of COPD and lung cancer: potential for chemoprevention. Curr Mol Pharmacol. 2017;10(1):46–59. doi:10.2174/1874467209666160112130016
214. Singla S, Jacobson JR. Statins as a novel therapeutic strategy in acute lung injury. Pulm Circ. 2012;2(4):397–406. doi:10.4103/2045-8932.105028
215. Hothersall E, McSharry C, Thomson NC. Potential therapeutic role for statins in respiratory disease. Thorax. 2006;61(8):729–734. doi:10.1136/thx.2005.057976
216. Malekinejad H, Khoramjouy M, Hobbenaghi R, Amniattalab A. Atorvastatin attenuates the paraquat-induced pulmonary inflammation via PPARγ receptors: a new indication for atorvastatin. Pestic Biochem Physiol. 2014;114:79–89. doi:10.1016/j.pestbp.2014.06.011
217. El-Achkar GA, Mrad MF, Mouawad CA, et al. Heme oxygenase-1-Dependent anti-inflammatory effects of atorvastatin in zymosan-injected subcutaneous air pouch in mice. PLoS One. 2019;14(5):e0216405. doi:10.1371/journal.pone.0216405
218. Zhu T, Zhang W, Wang DX, et al. Rosuvastatin attenuates mucus secretion in a murine model of chronic asthma by inhibiting the gamma-aminobutyric acid type A receptor. Chin Med J (Engl). 2012;125(8):1457–1464.
219. Leite CF, Marangoni FA, Camargo EA, et al. Simvastatin attenuates neutrophil recruitment in one-lung ventilation model in rats. Acta Cir Bras. 2013;28(4):245–250. doi:10.1590/S0102-86502013000400003
220. Davis BB, Zeki AA, Bratt JM, et al. Simvastatin inhibits smoke-induced airway epithelial injury: implications for COPD therapy. Eur Respir J. 2013;42(2):350–361. doi:10.1183/09031936.00042512
221. Zhang S, Rahman M, Zhang S, Qi Z, Herwald H, Thorlacius H. Simvastatin regulates CXC chemokine formation in streptococcal M1 protein-induced neutrophil infiltration in the lung. Am J Physiol Lung Cell Mol Physiol. 2011;300(6):L930–939. doi:10.1152/ajplung.00422.2010
222. Tulek B, Kiyan E, Kiyici A, Toy H, Bariskaner H, Suerdem M. Effects of simvastatin on bleomycin-induced pulmonary fibrosis in female rats. Biol Res. 2012;45(4):345–350. doi:10.4067/S0716-97602012000400003
223. Müller HC, Hellwig K, Rosseau S, et al. Simvastatin attenuates ventilator-induced lung injury in mice. Crit Care. 2010;14(4):R143. doi:10.1186/cc9209
224. Hsu HH, Ko WJ, Hsu JY, et al. Simvastatin ameliorates established pulmonary hypertension through a heme oxygenase-1 dependent pathway in rats. Respir Res. 2009;10(1):32. doi:10.1186/1465-9921-10-32
225. Li M, Liu Y, Shi H, et al. Statins inhibit pulmonary artery smooth muscle cell proliferation by upregulation of HO-1 and p21WAF1. Naunyn Schmiedebergs Arch Pharmacol. 2012;385(10):961–968. doi:10.1007/s00210-012-0768-5
226. Hsu M, Muchova L, Morioka I, Wong RJ, Schröder H, Stevenson DK. Tissue-specific effects of statins on the expression of heme oxygenase-1 in vivo. Biochem Biophys Res Commun. 2006;343(3):738–744. doi:10.1016/j.bbrc.2006.03.036
227. Drummond GS, Baum J, Greenberg M, Lewis D, Abraham NG. HO-1 overexpression and underexpression: clinical implications. Arch Biochem Biophys. 2019;673:108073.
228. Pereira MLM, Marinho CRF, Epiphanio S. Could heme oxygenase-1 be a new target for therapeutic intervention in malaria-associated acute lung injury/acute respiratory distress syndrome? Front Cell Infect Microbiol. 2018;8:161. doi:10.3389/fcimb.2018.00161
229. Fessler MB, Young SK, Jeyaseelan S, et al. A role for hydroxy-methylglutaryl coenzyme a reductase in pulmonary inflammation and host defense. Am J Respir Crit Care Med. 2005;171(6):606–615. doi:10.1164/rccm.200406-729OC
230. Xu JF, Washko GR, Nakahira K, et al. Statins and pulmonary fibrosis: the potential role of NLRP3 inflammasome activation. Am J Respir Crit Care Med. 2012;185(5):547–556. doi:10.1164/rccm.201108-1574OC
231. Walsh A, Perrem L, Khashan AS, Henry MT, Ni Chroinin M. Statins versus placebo for people with chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2019;7(7):Cd011959. doi:10.1002/14651858.CD011959.pub2
232. Bradbury P, Traini D, Ammit AJ, Young PM, Ong HX. Repurposing of statins via inhalation to treat lung inflammatory conditions. Adv Drug Deliv Rev. 2018;133:93–106. doi:10.1016/j.addr.2018.06.005
233. Chen T, Liu W, Chao X, et al. Salvianolic acid B attenuates brain damage and inflammation after traumatic brain injury in mice. Brain Res Bull. 2011;84(2):163–168. doi:10.1016/j.brainresbull.2010.11.015
234. Pan Y, Fu H, Kong Q, et al. Prevention of pulmonary fibrosis with salvianolic acid a by inducing fibroblast cell cycle arrest and promoting apoptosis. J Ethnopharmacol. 2014;155(3):1589–1596. doi:10.1016/j.jep.2014.07.049
235. Mu D, Luan Y, Wang L, et al. The combination of salvianolic acid A with latamoxef completely protects mice against lethal pneumonia caused by methicillin-resistant Staphylococcus aureus. Emerg Microbes Infect. 2020;9(1):169–179. doi:10.1080/22221751.2020.1711817
236. Zhao DH, Wu YJ, Liu ST, Liu RY. Salvianolic acid B attenuates lipopolysaccharide-induced acute lung injury in rats through inhibition of apoptosis, oxidative stress and inflammation. Exp Ther Med. 2017;14(1):759–764. doi:10.3892/etm.2017.4534
237. Zhang DF, Zhang J, Li R. Salvianolic acid B attenuates lung inflammation induced by cigarette smoke in mice. Eur J Pharmacol. 2015;761:174–179. doi:10.1016/j.ejphar.2015.05.003
238. Liu B, Cao B, Zhang D, et al. Salvianolic acid B protects against paraquat-induced pulmonary injury by mediating Nrf2/Nox4 redox balance and TGF-β1/Smad3 signaling. Toxicol Appl Pharmacol. 2016;309:111–120. doi:10.1016/j.taap.2016.08.004
239. Huang X, Zuo L, Lv Y, et al. Asiatic acid attenuates myocardial ischemia/reperfusion injury via Akt/GSK-3β/HIF-1α signaling in rat H9c2 cardiomyocytes. Molecules. 2016;21(9):1248. doi:10.3390/molecules21091248
240. Wei J, Huang Q, Huang R, et al. Asiatic acid from Potentilla chinensis attenuate ethanol-induced hepatic injury via suppression of oxidative stress and Kupffer cell activation. Biol Pharm Bull. 2013;36(12):1980–1989. doi:10.1248/bpb.b13-00634
241. Adtani PN, Narasimhan M, Punnoose AM, Kambalachenu HR. Antifibrotic effect of Centella asiatica Linn and asiatic acid on arecoline-induced fibrosis in human buccal fibroblasts. J Investig Clin Dent. 2017;8(2):e12208. doi:10.1111/jicd.12208
242. Dong SH, Liu YW, Wei F, Tan HZ, Han ZD. Asiatic acid ameliorates pulmonary fibrosis induced by bleomycin (BLM) via suppressing pro-fibrotic and inflammatory signaling pathways. Biomed Pharmacother. 2017;89:1297–1309. doi:10.1016/j.biopha.2017.03.005
243. Xia X, Dai C, Yu H, et al. Asiatic acid prevents the development of interstitial lung disease in a hypochlorous acid-induced mouse model of scleroderma. Oncol Lett. 2018;15(6):8711–8716. doi:10.3892/ol.2018.8412
244. Jiang W, Li M, He F, et al. Protective effects of asiatic acid against spinal cord injury-induced acute lung injury in rats. Inflammation. 2016;39(6):1853–1861. doi:10.1007/s10753-016-0414-3
245. Chen X, Zhang B, Li J, et al. Celastrol attenuates incision-induced inflammation and pain associated with inhibition of the NF-κB signalling pathway via SARM. Life Sci. 2018;205:136–144. doi:10.1016/j.lfs.2018.05.020
246. Yu X, Zhao Q, Zhang X, et al. Celastrol ameliorates inflammation through inhibition of NLRP3 inflammasome activation. Oncotarget. 2017;8(40):67300–67314. doi:10.18632/oncotarget.18619
247. Zhang X, Wang Y, Ge HY, et al. Celastrol reverses palmitic acid (PA)-caused TLR4-MD2 activation-dependent insulin resistance via disrupting MD2-related cellular binding to PA. J Cell Physiol. 2018;233(10):6814–6824. doi:10.1002/jcp.26547
248. Shi K, Chen X, Xie B, et al. Celastrol alleviates chronic obstructive pulmonary disease by inhibiting cellular inflammation induced by cigarette smoke via the Ednrb/Kng1 signaling pathway. Front Pharmacol. 2018;9:1276. doi:10.3389/fphar.2018.01276
249. Liu J, Liu J, Wang H, Bai M. Protective effect of celastrol for burn-induced acute lung injury in rats. Int J Clin Exp Pathol. 2019;12(2):576–583.
250. Kim SC, Kang SH, Jeong SJ, Kim SH, Ko HS, Kim SH. Inhibition of c-Jun N-terminal kinase and nuclear factor κ B pathways mediates fisetin-exerted anti-inflammatory activity in lipopolysccharide-treated RAW264.7 cells. Immunopharmacol Immunotoxicol. 2012;34(4):645–650. doi:10.3109/08923973.2011.648270
251. Zhang XJ, Jia SS. Fisetin inhibits laryngeal carcinoma through regulation of AKT/NF-κB/mTOR and ERK1/2 signaling pathways. Biomed Pharmacother. 2016;83:1164–1174. doi:10.1016/j.biopha.2016.08.035
252. Khan N, Syed DN, Ahmad N, Mukhtar H. Fisetin: a dietary antioxidant for health promotion. Antioxid Redox Signal. 2013;19(2):151–162. doi:10.1089/ars.2012.4901
253. Weseler AR, Geraets L, Moonen HJ, et al. Poly (ADP-ribose) polymerase-1-inhibiting flavonoids attenuate cytokine release in blood from male patients with chronic obstructive pulmonary disease or type 2 diabetes. J Nutr. 2009;139(5):952–957. doi:10.3945/jn.108.102756
254. Feng G, Jiang ZY, Sun B, Fu J, Li TZ. Fisetin alleviates lipopolysaccharide-induced acute lung injury via TLR4-mediated NF-κB signaling pathway in rats. Inflammation. 2016;39(1):148–157. doi:10.1007/s10753-015-0233-y
255. Huang W, Li ML, Xia MY, Shao JY. Fisetin-treatment alleviates airway inflammation through inhbition of MyD88/NF-κB signaling pathway. Int J Mol Med. 2018;42(1):208–218. doi:10.3892/ijmm.2018.3582
256. Wang X, Gong G, Yang W, Li Y, Jiang M, Li L. Antifibrotic activity of galangin, a novel function evaluated in animal liver fibrosis model. Environ Toxicol Pharmacol. 2013;36(2):288–295. doi:10.1016/j.etap.2013.04.004
257. Liu YN, Zha WJ, Ma Y, et al. Galangin attenuates airway remodelling by inhibiting TGF-β1-mediated ROS generation and MAPK/Akt phosphorylation in asthma. Sci Rep. 2015;5:11758. doi:10.1038/srep11758
258. Henry LJK, Ramar MK, Palanisamy S, Natesan S, Kandasamy R. Mechanistic investigation of PPARγ-facilitated anti-asthmatic effects of Galangin (Norizalpinin): insights from in silico and in vivo analyses. Biochem Biophys Res Commun. 2020;526(3):833–840. doi:10.1016/j.bbrc.2020.03.158
259. Somerset SM, Johannot L. Dietary flavonoid sources in Australian adults. Nutr Cancer. 2008;60(4):442–449. doi:10.1080/01635580802143836
260. Cheng X, Yang YL, Yang H, Wang YH, Du GH. Kaempferol alleviates LPS-induced neuroinflammation and BBB dysfunction in mice via inhibiting HMGB1 release and down-regulating TLR4/MyD88 pathway. Int Immunopharmacol. 2018;56:29–35. doi:10.1016/j.intimp.2018.01.002
261. Chen X, Qian J, Wang L, et al. Kaempferol attenuates hyperglycemia-induced cardiac injuries by inhibiting inflammatory responses and oxidative stress. Endocrine. 2018;60(1):83–94. doi:10.1007/s12020-018-1525-4
262. Zhang R, Ai X, Duan Y, et al. Kaempferol ameliorates H9N2 swine influenza virus-induced acute lung injury by inactivation of TLR4/MyD88-mediated NF-κB and MAPK signaling pathways. Biomed Pharmacother. 2017;89:660–672. doi:10.1016/j.biopha.2017.02.081
263. Kang DR, Belal SA, Choe HS, Shin DK, Shim KS. Effect of kaempferol on cyclooxygenase 2 (Cox2) and cytosolic phospholipase A2 (cPLA2) protein expression in BALB/c mice. Iran J Allergy Asthma Immunol. 2018;17(5):428–435. doi:10.18502/ijaai.v17i5.301
264. Chen X, Yang X, Liu T, et al. Kaempferol regulates MAPKs and NF-κB signaling pathways to attenuate LPS-induced acute lung injury in mice. Int Immunopharmacol. 2012;14(2):209–216. doi:10.1016/j.intimp.2012.07.007
265. Sun Z, Li Q, Hou R, et al. Kaempferol-3-O-glucorhamnoside inhibits inflammatory responses via MAPK and NF-κB pathways in vitro and in vivo. Toxicol Appl Pharmacol. 2019;364:22–28. doi:10.1016/j.taap.2018.12.008
266. Lin Y, Shi R, Wang X, Shen HM. Luteolin, a flavonoid with potential for cancer prevention and therapy. Curr Cancer Drug Targets. 2008;8(7):634–646. doi:10.2174/156800908786241050
267. Aziz N, Kim MY, Cho JY. Anti-inflammatory effects of luteolin: a review of in vitro, in vivo, and in silico studies. J Ethnopharmacol. 2018;225:342–358. doi:10.1016/j.jep.2018.05.019
268. Xiong J, Wang K, Yuan C, et al. Luteolin protects mice from severe acute pancreatitis by exerting HO-1-mediated anti-inflammatory and antioxidant effects. Int J Mol Med. 2017;39(1):113–125. doi:10.3892/ijmm.2016.2809
269. Liu X, Meng J. Luteolin alleviates LPS-induced bronchopneumonia injury in vitro and in vivo by down-regulating microRNA-132 expression. Biomed Pharmacother. 2018;106:1641–1649. doi:10.1016/j.biopha.2018.07.094
270. Chen CY, Peng WH, Wu LC, Wu CC, Hsu SL. Luteolin ameliorates experimental lung fibrosis both in vivo and in vitro: implications for therapy of lung fibrosis. J Agric Food Chem. 2010;58(22):11653–11661. doi:10.1021/jf1031668
271. Liu B, Yu H, Baiyun R, et al. Protective effects of dietary luteolin against mercuric chloride-induced lung injury in mice: involvement of AKT/Nrf2 and NF-κB pathways. Food Chem Toxicol. 2018;113:296–302. doi:10.1016/j.fct.2018.02.003
272. Hsuan CF, Hsu HF, Tseng WK, et al. Glossogyne tenuifolia extract inhibits TNF-α-induced expression of adhesion molecules in human umbilical vein endothelial cells via blocking the NF-kB signaling pathway. Molecules. 2015;20(9):16908–16923. doi:10.3390/molecules200916908
273. Bian D, Liu M, Li Y, Xia Y, Gong Z, Dai Y. Madecassoside, a triterpenoid saponin isolated from Centella asiatica herbs, protects endothelial cells against oxidative stress. J Biochem Mol Toxicol. 2012;26(10):399–406. doi:10.1002/jbt.21434
274. Luo Y, Yang YP, Liu J, et al. Neuroprotective effects of madecassoside against focal cerebral ischemia reperfusion injury in rats. Brain Res. 2014;1565:37–47. doi:10.1016/j.brainres.2014.04.008
275. Bian GX, Li GG, Yang Y, et al. Madecassoside reduces ischemia-reperfusion injury on regional ischemia induced heart infarction in rat. Biol Pharm Bull. 2008;31(3):458–463. doi:10.1248/bpb.31.458
276. Lu GX, Bian DF, Ji Y, et al. Madecassoside ameliorates bleomycin-induced pulmonary fibrosis in mice by downregulating collagen deposition. Phytother Res. 2014;28(8):1224–1231. doi:10.1002/ptr.5120
277. Pollier J, Goossens A. Oleanolic acid. Phytochemistry. 2012;77:10–15. doi:10.1016/j.phytochem.2011.12.022
278. Ayeleso TB, Matumba MG, Mukwevho E. Oleanolic acid and its derivatives: biological activities and therapeutic potential in chronic diseases. Molecules. 2017;22(11). doi:10.3390/molecules22111915
279. Zhang DH, Yang L, Cohn L, et al. Inhibition of allergic inflammation in a murine model of asthma by expression of a dominant-negative mutant of GATA-3. Immunity. 1999;11(4):473–482. doi:10.1016/S1074-7613(00)80122-3
280. Kim SH, Hong JH, Lee YC. Oleanolic acid suppresses ovalbumin-induced airway inflammation and Th2-mediated allergic asthma by modulating the transcription factors T-bet, GATA-3, RORγt and Foxp3 in asthmatic mice. Int Immunopharmacol. 2014;18(2):311–324. doi:10.1016/j.intimp.2013.12.009
281. Santos RS, Silva PL, Oliveira GP, et al. Effects of oleanolic acid on pulmonary morphofunctional and biochemical variables in experimental acute lung injury. Respir Physiol Neurobiol. 2011;179(2–3):129–136. doi:10.1016/j.resp.2011.07.008
282. Santos RS, Silva PL, de Oliveira GP, et al. Oleanolic acid improves pulmonary morphofunctional parameters in experimental sepsis by modulating oxidative and apoptotic processes. Respir Physiol Neurobiol. 2013;189(3):484–490. doi:10.1016/j.resp.2013.08.019
283. Kim W, Lim D, Kim J. p-Coumaric acid, a major active compound of bambusae caulis in taeniam, suppresses cigarette smoke-induced pulmonary inflammation. Am J Chin Med. 2018;46(2):407–421. doi:10.1142/S0192415X18500209
284. Lee CH, Wu SL, Chen JC, et al. Eriobotrya japonica leaf and its triterpenes inhibited lipopolysaccharide-induced cytokines and inducible enzyme production via the nuclear factor-κB signaling pathway in lung epithelial cells. Am J Chin Med. 2008;36(6):1185–1198. doi:10.1142/S0192415X0800651X
285. Peng XP, Li XH, Li Y, Huang XT, Luo ZQ. The protective effect of oleanolic acid on NMDA-induced MLE-12 cells apoptosis and lung injury in mice by activating SIRT1 and reducing NF-κB acetylation. Int Immunopharmacol. 2019;70:520–529. doi:10.1016/j.intimp.2019.03.018
286. Thimmulappa RK, Fuchs RJ, Malhotra D, et al. Preclinical evaluation of targeting the Nrf2 pathway by triterpenoids (CDDO-Im and CDDO-Me) for protection from LPS-induced inflammatory response and reactive oxygen species in human peripheral blood mononuclear cells and neutrophils. Antioxid Redox Signal. 2007;9(11):1963–1970. doi:10.1089/ars.2007.1745
287. To C, Ringelberg CS, Royce DB, et al. Dimethyl fumarate and the oleanane triterpenoids, CDDO-imidazolide and CDDO-methyl ester, both activate the Nrf2 pathway but have opposite effects in the A/J model of lung carcinogenesis. Carcinogenesis. 2015;36(7):769–781. doi:10.1093/carcin/bgv061
288. Chen T, Mou Y, Tan J, et al. The protective effect of CDDO-Me on lipopolysaccharide-induced acute lung injury in mice. Int Immunopharmacol. 2015;25(1):55–64. doi:10.1016/j.intimp.2015.01.011
289. Nichols DP, Ziady AG, Shank SL, Eastman JF, Davis PB. The triterpenoid CDDO limits inflammation in preclinical models of cystic fibrosis lung disease. Am J Physiol Lung Cell Mol Physiol. 2009;297(5):L828–836. doi:10.1152/ajplung.00171.2009
290. Kulkarni AA, Thatcher TH, Hsiao HM, et al. The triterpenoid CDDO-Me inhibits bleomycin-induced lung inflammation and fibrosis. PLoS One. 2013;8(5):e63798. doi:10.1371/journal.pone.0063798
291. Wang YY, Zhang CY, Ma YQ, He ZX, Zhe H, Zhou SF. Therapeutic effects of C-28 methyl ester of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid (CDDO-Me; bardoxolone methyl) on radiation-induced lung inflammation and fibrosis in mice. Drug Des Devel Ther. 2015;9:3163–3178. doi:10.2147/DDDT.S80958
292. Wu SJ, Tam KW, Tsai YH, Chang CC, Chao JC. Curcumin and saikosaponin a inhibit chemical-induced liver inflammation and fibrosis in rats. Am J Chin Med. 2010;38(1):99–111. doi:10.1142/S0192415X10007695
293. Brinker AM, Ma J, Lipsky PE, Raskin I. Medicinal chemistry and pharmacology of genus Tripterygium (Celastraceae). Phytochemistry. 2007;68(6):732–766. doi:10.1016/j.phytochem.2006.11.029
294. Carvalho PR, Silva DH, Bolzani VS, Furlan M. Antioxidant quinonemethide triterpenes from Salacia campestris. Chem Biodivers. 2005;2(3):367–372. doi:10.1002/cbdv.200590016
295. Li JJ, Yan YY, Sun HM, et al. Anti-cancer effects of pristimerin and the mechanisms: a critical review. Front Pharmacol. 2019;10:746. doi:10.3389/fphar.2019.00746
296. Zhao Q, Liu Y, Zhong J, et al. Pristimerin induces apoptosis and autophagy via activation of ROS/ASK1/JNK pathway in human breast cancer in vitro and in vivo. Cell Death Discov. 2019;5:125. doi:10.1038/s41420-019-0208-0
297. Yang CC, Hsiao LD, Tseng HC, Kuo CM, Yang CM. Pristimerin Inhibits MMP-9 expression and cell migration through attenuating NOX/ROS-dependent NF-κB activation in rat brain astrocytes challenged with LPS. J Inflamm Res. 2020;13:325–341. doi:10.2147/JIR.S252659
298. Shaaban AA, El-Kashef DH, Hamed MF, El-Agamy DS. Protective effect of pristimerin against LPS-induced acute lung injury in mice. Int Immunopharmacol. 2018;59:31–39. doi:10.1016/j.intimp.2018.03.033
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.