")
Back to Journals » OncoTargets and Therapy » Volume 12
CHFR promotes the migration of human gastric cancer cells by inducing epithelial-to-mesenchymal transition in a HDAC1-dependent manner
Authors Yang S, He F, Dai M, Pan J, Wang J, Ye B
Received 15 October 2018
Accepted for publication 4 January 2019
Published 7 February 2019 Volume 2019:12 Pages 1075—1084
DOI https://doi.org/10.2147/OTT.S191016
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr William C. Cho
Shangwen Yang,1 Feiyun He,2 Mugen Dai,1 Jundi Pan,1 Jianbo Wang,1 Bin Ye1
1Department of Gastroenterology, The Fifth Affiliated Hospital of Wenzhou Medical University and Lishui Municipal Central Hospital, Lishui 323000, Zhejiang Province, China; 2Department of Gastroenterology, Lishui Chinese Medicine Hospital, Lishui 323000, Zhejiang Province, China
Background: Previous studies have illustrated that checkpoint with forkhead-associated and ring finger domains (CHFR) was frequently silenced in several cancer types due to promoter hypermethylation and functions as a tumor suppressor gene. However, the data from the public dataset reveal that CHFR is highly expressed in human gastric cancer specimens, and the biological function of CHFR in gastric cancer is still not well understood.
Materials and methods: The clinical association between CHFR expression and the overall survival of gastric cancer patients as well as cancer metastasis was analyzed according to public datasets. The CHFR expression in clinical specimens and human gastric cancer cell lines was detected by immunohistochemistry and Western blotting, respectively. Gain (overexpression) and loss (silencing) of function experiments were used to elucidate the role of CHFR in gastric cancer. The migration ability of gastric cancer cells was determined by wound healing and transwell assays. Cell cycle distribution was analyzed using fluorescence-activated cell sorting experiment. The expression of the proteins in cancer cells was measured using Western blot analysis.
Results: According to the analysis from Kaplan–Meier plotter dataset, CHFR expression was negatively associated with overall survival of gastric cancer patients. Our data revealed that exogenous expression of CHFR not only arrested cell cycle but also led to dramatically enhanced cell migration, while silencing of CHFR significantly inhibited cell migration in gastric cancer cells. This result is consistent with the data from the Human Cancer Metastasis Dataset, in which CHFR level is found to significantly increase in metastatic gastric cancer. The overexpression of CHFR promoted epithelial–mesenchymal transition (EMT) in both SGC-7901 and AGS cells, while HDAC1 was inhibited. Interestingly, suberoylanilide hydroxamic acid, a HDAC1 antagonist, could effectively increase cell migration in both cell lines via enhancement of EMT.
Conclusion: Our data indicated that CHFR exerted positive effects on cell migration of human gastric cancer by promoting EMT via downregulating HDAC1.
Keywords: human gastric cancer, migration, CHFR, EMT, HDAC1
Introduction
Gastric cancer is the third most common cause of cancer-related death worldwide, with a high incidence rate in China.1,2 Although the incidence of gastric cancer has been significantly declining in the past few decades, approximated one million cases are diagnosed yearly.3 The gastric carcinogenesis is a complicated process that involves the abnormality of various genes.4 Due to the recurrence and metastasis of advanced gastric cancer, the prognosis remains unsatisfactory. Therefore, it is urgent to further elucidate the progression of gastric cancer at the molecular level.
The epithelial–mesenchymal transition (EMT) was originally recognized as a process during which epithelial cells lose their cell polarity and cell–cell adhesion and become mesenchymal stem cells.5 EMT plays an indispensable role in numerous biological processes, such as mesoderm and neural tube development, wound healing, organ fibrosis, and initiation of cancer cell metastasis by generating cells with stem-like properties.5 Various transcription factors, including Twist, Snail, Slug, and ZEB1, can induce EMT.6 The upregulation of N-cadherin and vimentin, and the downregulation of E-cadherin are the biomarkers of cells undergoing EMT.7 The abnormal activation of EMT also contributes to carcinogenesis and cancer progression.7 Because EMT could facilitate the migration and invasion of epithelial tumor cells, targeting EMT is a promising strategy for anticancer treatment, especially for overcoming cancer cell metastasis.
CHFR plays an important role in cell–cycle regulation by delaying entry into metaphase in response to microtubule stress.8,9 In multiple studies, CHFR gene was found to be significantly silenced by promoter methylation or mutated in a number of cancer types, including gastric cancer,10 human non-small-cell lung cancer,11 esophageal cancer,12 and colorectal cancer.13 Although increasing evidence supports that CHFR functions as a tumor-suppressor protein, its silencing could sensitize the endometrial cancer cells to paclitaxel.14 However, the biological function of CHFR in human gastric cancer remains poorly understood. In fact, the percentage of the methylation of CHFR promoter in human gastric cancer is only 34.3%.15 The CHFR protein expression level is relatively abundant in human gastric cancer specimens according to The Human Protein Atlas dataset. In the present study, the role of CHFR in cell migration of gastric cancer cells and its underlying mechanism was elucidated.
Materials and methods
Clinical specimens and cell culture
Forty-five gastric cancer biopsy specimens and corresponding paracancerous tissues were collected from patients of The Fifth Affiliated Hospital of Wenzhou Medical University at the time of surgery and immediately stored in liquid nitrogen until use. All patients provided written informed consent for this study, and the project was approved by the Institutional Ethics Committee of The Fifth Affiliated Hospital of Wenzhou Medical University and was conducted in accordance with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The tissues were fixed in buffered 10% formalin, transferred to 70% ethanol, embedded in paraffin, sectioned into 5 μm sections, and kept at −80°C.
The human gastric cancer cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in DMEM medium (Hyclone) supplemented with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA), 2 mM L-glutamine (Thermo Fisher Scientific), 1% penicillin (100 units/mL), and streptomycin (100 μg/mL) (Thermo Fisher Scientific). All cell lines used in this study were cultured in a humidified incubator in an atmosphere of 5% (v/v) CO2 at 37°C.
Immunohistochemistry
Tissues were deparaffinized and rehydrated. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide in methanol. Heat-induced antigen retrieval was carried out for all sections in 0.01 M citrate buffer, pH 6.0, using a steamer at 95°C. All primary antibodies were diluted with 5% BSA in PBS to a concentration of 1:50 and applied to the sections. The sections were incubated at room temperature for 45 minutes followed by incubation with a Dako EnVision+ System HRP Labeled Polymer for 30 minutes at room temperature. Diaminobenzidine was then applied for 10 minutes. The sections were counterstained with hematoxylin, dehydrated, coverslipped, and visualized.
Western blotting
Cells were lysed in RIPA buffer containing protease inhibitor cocktail (Sigma-Aldrich Co.). After electrophoresis on a 10% SDS–PAGE gel, proteins were transferred onto polyvinylidene difluoride membranes. The membranes were blocked with 5% BSA in PBS for at least 2 hours and incubated with primary antibodies at 4°C overnight. The corresponding horseradish peroxidase (HRP)-conjugated secondary antibody was added and incubated at room temperature for 1 hour. Signal was visualized after chemiluminescence reaction with HRP substrate.
The primary antibodies against CHFR, Snail, E-cadherin, N-cadherin, β-catenin, claudin-1, and HDAC1 were purchased from CST (Danvers, MA, USA). The antibodies against β-actin and HDAC1 inhibitor suberoylanilide hydroxamic acid (SAHA) were purchased from Sigma-Aldrich Co.
RNA interference of CHFR
Short interfering RNAs (siRNA) for CHFR and non-targeting siRNA negative control were obtained from Genepharma (Shanghai, China). The sequences for siRNA against CHFR were as follows: siRNA-1, 5′-GUGCAAAGUAUGGAUGCCATT-3′; siRNA-2, 5′-CACCACGCCAUGAAAUUCATT-3′. The sequence for negative control siRNA: 5′-UUCUCCGAACGUGUCACGU-3′. Cells were transfected with siRNA using Lipofectamine 2000 reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. Before any treatment, cells were incubated for 24 hours and the silencing efficiency of siRNA was determined by Western blotting assay.
Plasmid construction and transfection
The coding sequence (CDS) of human CHFR mRNA (CCDS53849.1) was synthesized and subcloned into pcDNA3.1 vector to construct the CHFR overexpression plasmid. The integrity of the plasmid construct was confirmed by DNA sequencing. The Flag-HDAC1 overexpression plasmid was purchased from Vigene Biosciences (Shandong, China). The plasmid was allowed to form complexes with Lipofectamine 2000 (Thermo Fisher Scientific) for 20 minutes at room temperature, and transfection was carried out at 37°C for 24 hours. The expression efficiency of the plasmid was determined by Western blotting assay.
In vitro migration assay
For the migration assay, a transwell system (8 μm pore size; Corning Life Science) was used according to the manufacturer’s instructions. The cells were seeded into the upper chamber and cultured in serum-free DMEM medium. The lower compartment was filled with DMEM with 10% FBS. After incubation for the indicated time, the cells remaining in the upper chamber were removed, and the cells at the bottom of the insert were fixed with iced methanol, stained in 0.5% crystal violet, and counted under a microscope (Olympus Corp., Tokyo, Japan). The results were averaged over three independent experiments.
Wound healing assay
AGS and MKN-1 cells were incubated in a six-well plate to 100% confluence in DMEM medium with 10% FBS. Then, the cells were allowed to grow in serum-free medium overnight. A 200 μL tip was used to produce a clean wound across the center of the well. The cell debris was washed away using PBS, and the cells were allowed to migrate in FBS-free DMEM medium for the indicated time. The images of the wound healing process were photographed and the gap distance normalized to control was calculated.
Cell cycle distribution assay
After serum starvation overnight, cells were treated as indicated and then washed with cold PBS twice and fixed in cold 70% ethanol at least overnight. Then, cells were centrifuged and washed with cold PBS twice again. Subsequently, cells were resuspended in PBS and stained with propidium iodide solution (10 μg/mL) for 30 minutes in dark at room temperature. Cells were then analyzed with flow cytometry (Beckman) to determine the cell cycle distribution.
Statistical analysis
All data were expressed as the mean ± SD from at least three independent experiments. All statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). Statistical significance was determined using two-sided Student’s t-test, and P<0.05 was considered significant.
Results
CHFR is upregulated in human gastric cancer specimens and negatively associated with the overall survival rate of gastric cancer patients
To determine the clinical relevance of CHFR in human gastric cancer, we first analyzed the relevant data in a public database (www.KMplot.com). The result indicated that CHFR levels were negatively associated with the overall survival rate of patients with gastric cancer significantly, which was inconsistent with its anticancer function reported previously (log-rank test, P<0.01, Figure 1A). Although numerous publications declared that CHFR was silenced in multiple cancer types including gastric cancer due to promoter hypermethylation as mentioned earlier, the data of The Human Protein Atlas reveals that the CHFR protein levels in human gastric cancer specimens are very high (data not shown). To further confirm this phenomenon, the CHFR protein expression in gastric cancer tissues and their paracancerous tissues was compared using immunohistochemistry. We found that the level of CHFR was obviously lower in paracancerous tissues than in gastric cancer tissues (representative pictures are shown, Figure 1B). Representative images of gastric cancer tissues with different levels of CHFR expression are shown in Figure 1C.
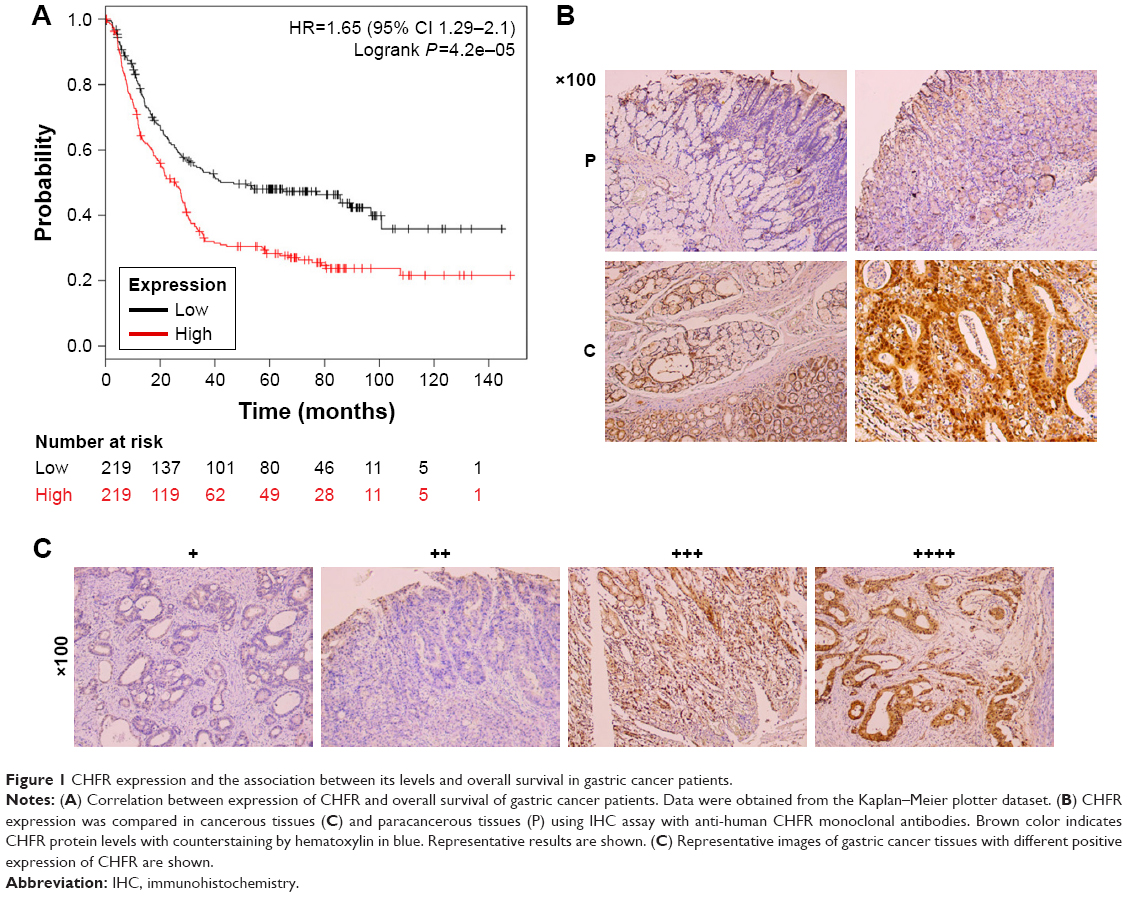 | Figure 1 CHFR expression and the association between its levels and overall survival in gastric cancer patients. |
CHFR is positively associated with migration ability of gastric cancer cell lines
To examine the role of CHFR in gastric cancer, the basal expression of CHFR was determined by Western blotting, and the migration ability was measured by wound healing and transwell assays in seven human gastric cancer cell lines. As shown in Figure 2A, the protein levels of CHFR were found to be different in the gastric cancer cell lines. CHFR expression was undetectable in AGS cells; low level in BGC823, MKN-1, MKN-45, and SGC-7901 cells; and high level in HGC-27 and MKN-28 cells. Then, we chose AGS and MKN-1 cells to compare their migration ability by using wound healing assay. As shown in Figure 2B, a significantly accelerated rate of cells migrating into the wounded area was observed in MKN-1 cells compared to AGS cells at 24 hours and 48 hours (AGS group: 12.5%±2.4% and 21.2%±2.5%; MKN-1 group: 22.5%±7.2% and 32.8%±4.0%, respectively). Thus, these data illustrated that MKN-1 cells, which expressed higher levels of CHFR, were more aggressive than AGS cells.
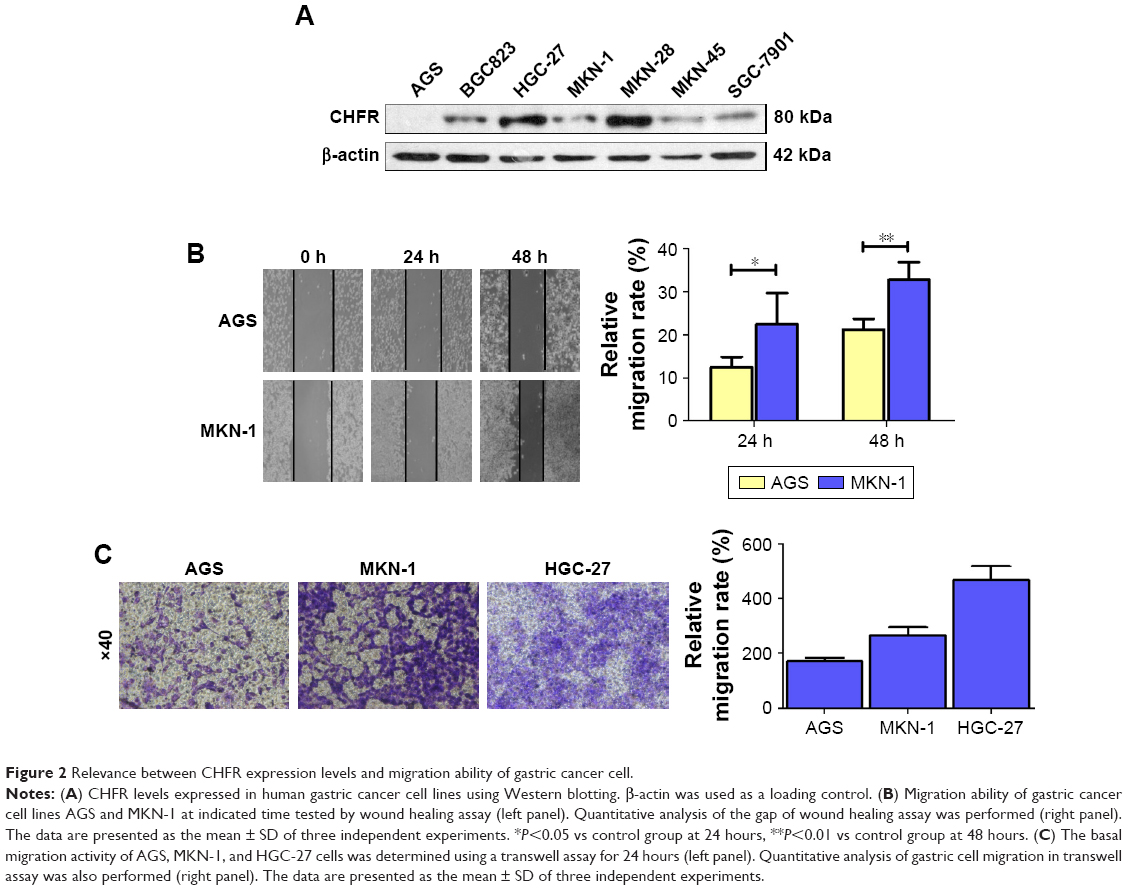 | Figure 2 Relevance between CHFR expression levels and migration ability of gastric cancer cell. |
To further verify these findings, we compared the migration ability of the AGS, MKN-1, and HGC-27 cells by using the transwell system assay. As shown in Figure 2C, the migration activity of HGC-27 cells was the strongest, while the migration activity of AGS cells was the weakest (HGC-27 group: 466±51 cells/view; MKN-1 group: 265±31 cells/view; AGS group: 171±13 cells/view). These data suggested that the gastric cancer cells expressing high levels of CHFR were more aggressive. Taken all together, these results indicated that upregulation of CHFR might be related to the onset of gastric cancer cell migration.
CHFR promotes cell migration in human gastric cancer cell
To study the biological functions of CHFR in gastric cancer, SGC-7901 and AGS cells with low level CHFR were transfected with its overexpression plasmid for further analysis. Firstly, we checked the transfection efficiency using Western blotting, and the results indicated that CHFR expression was effectively increased in both SGC-7901 and AGS cells after transfection (Figure 3A). To confirm how overexpression of CHFR influences cell cycle distribution, we performed fluorescence-activated cell sorting assay and found that the cell cycle in both SGC-7901 and AGS cell lines with ectopic expression of CHFR was significantly arrested at G1 (P<0.01, Figure 3B). In addition, as shown in Figure 3C, overexpression of CHFR significantly promoted the migration ability of both SGC-7901 and AGS cells compared to their control groups (P<0.01).
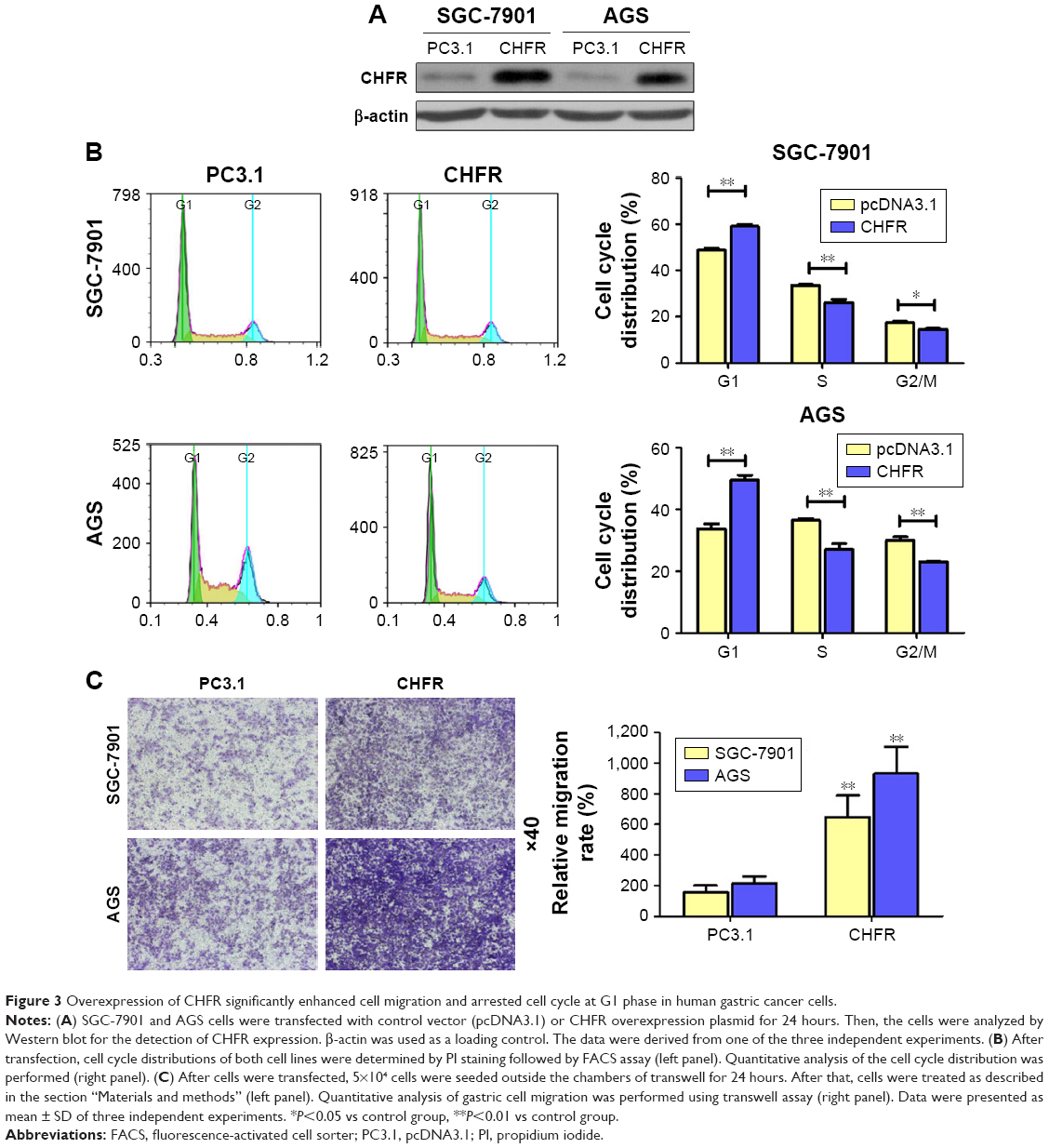 | Figure 3 Overexpression of CHFR significantly enhanced cell migration and arrested cell cycle at G1 phase in human gastric cancer cells. |
As there is no available publication focusing on the regulatory role of CHFR in the migration of gastric cancer cells, we used the Human Cancer Metastasis Datasets to analyze the association between CHFR level and gastric cancer metastasis.16 As shown in Figure 4A, CHFR mRNA was significantly higher in gastric cancer with metastasis than the ones without (P<0.01). To further validate the pro-mobility role of CHFR in gastric cancer cells, CHFR gene expression was silenced using siRNA in HGC-27 cells, and cell migration ability was then determined. As shown in Figure 4B, the silencing efficiency of two siRNAs against CHFR was detected by Western blotting, and siRNA-2 was chosen for further experiments, as it could inhibit the CHFR expression more effectively. Our data indicated that CHFR silencing significantly inhibited cell migration compared with the control at both 24 and 48 hours in HGC-27 cells (P<0.01, Figure 4C).
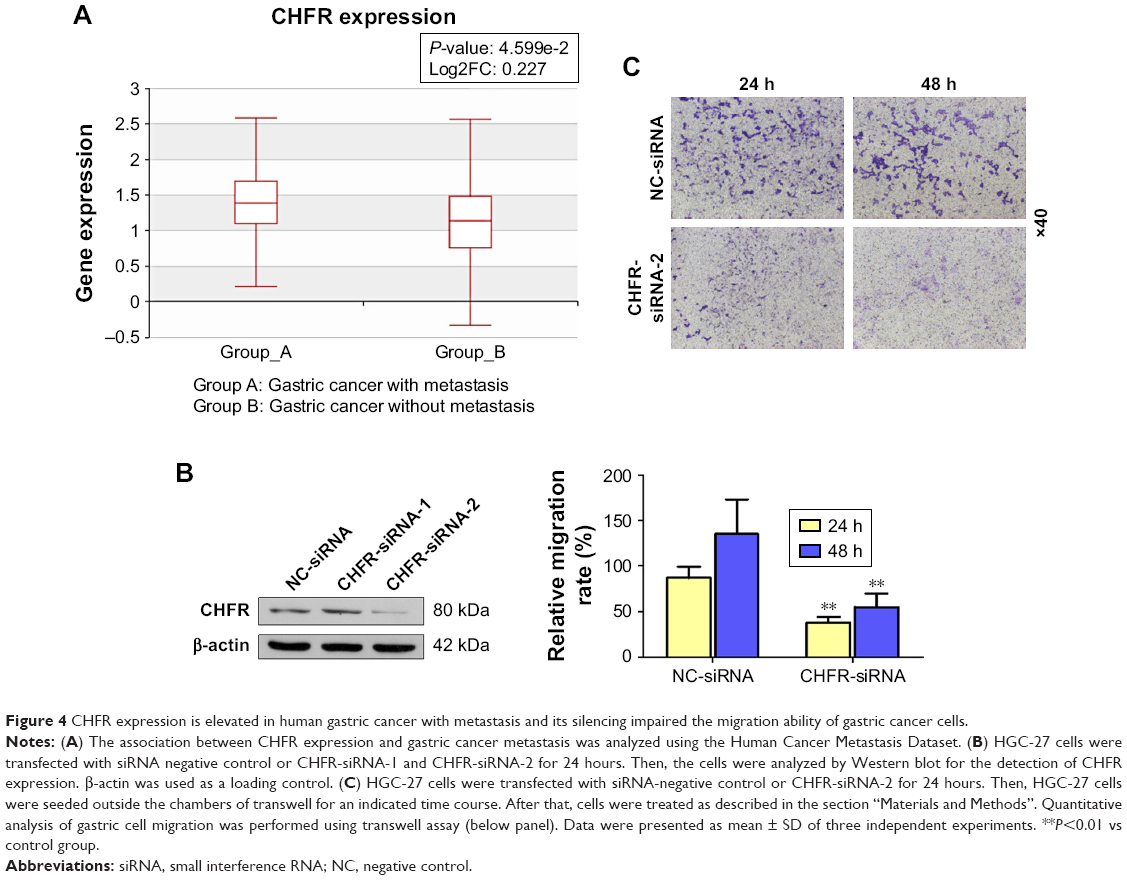 | Figure 4 CHFR expression is elevated in human gastric cancer with metastasis and its silencing impaired the migration ability of gastric cancer cells. |
Taken together, all these findings demonstrated that CHFR indeed induced cell cycle arrest and more importantly could enhance migration activity in gastric cancer cells.
CHFR augments the migration ability of gastric cancer cell by promoting EMT with HDAC1 is involved
Next, whether CHFR promoted the gastric cancer cell migration by affecting cell EMT was determined. The regulatory effects of CHFR on the expression of EMT-related transcription factors or biomarker proteins were detected using Western blotting assay. As shown in Figure 5A, overexpression of CHFR significantly upregulated the expression of mesenchymal markers N-cadherin and transcription factor Snail with no change in the expression of claudin-1 and β-catenin, while downregulated the expression of epithelial marker E-cadherin. Interestingly, we detected that CHFR effectively inhibited the expression of HDAC1 in both cell lines, which is also consistent with results from previous investigation17 (Figure 5A). Next, SGC-7901 cells were co-transfected with CHFR, flag-HDAC1 plasmid, and HA-Ub overexpression plasmid. The ubiquitination of HDAC1 was determined using immunoprecipitation–Western blot assay. As shown in Figure 5B, ectopic expression of CHFR obviously promoted the ubiquitination of HDAC1, which suggested that CHFR may decrease the HDAC1 expression by promoting its degradation.
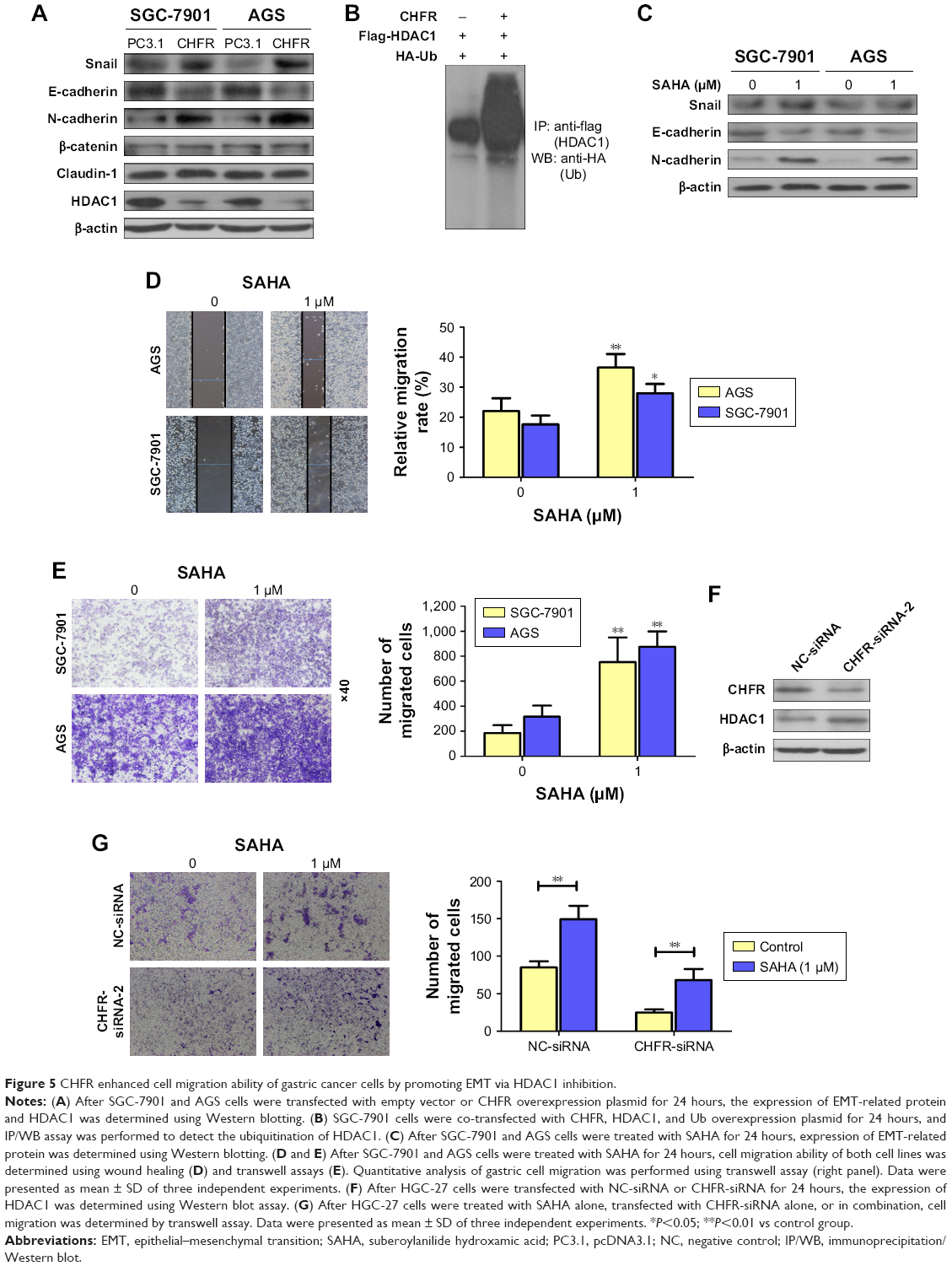 | Figure 5 CHFR enhanced cell migration ability of gastric cancer cells by promoting EMT via HDAC1 inhibition. |
To further confirm the role of HDAC1 on EMT in human gastric cancer, SAHA (Sigma-Aldrich Co.), an antagonist for HDAC1, was subjected to SGC-7901 and AGS cells. Our data revealed that inhibition of HDAC1 downregulated the expression of E-cadherin and upregulated the expression of Snail and N-cadherin, which means SAHA indeed promotes the EMT in both SGC-7901 and AGS cells (Figure 5C). Furthermore, SAHA also significantly promoted the migration ability of SGC-7901 and AGS cells (Figure 5D and E). Interestingly, CHFR silencing could enhance the HDAC1 expression in HGC-27 cells as expected (Figure 5F). Finally, transwell assay was performed to detect the cell migration of HGC-27 cells with CHFR silencing alone, SAHA treatment alone, or in combination. As shown in Figure 5G, SAHA could attenuate the inhibitory effect of CHFR silencing on cell migration of HGC-27 cells. All these findings suggest that CHFR may enhance the migration ability of gastric cancer cells by promoting EMT via the inhibition of HDAC1.
Discussion
Frequent hypermethylation of tumor-suppressor genes is one of the multiple pivotal factors for the carcinogenesis of gastric cancer.15 Furthermore, many methylated or hypermethylated genes participate in tumor initiation, progression, and metastasis and function as oncogenes or tumor suppressors under different conditions.18 Previous evidence reported that CHFR was inactivated in many tumors due to the hypermethylation in CpG island on its promoter.19 Additionally, downregulation of CHFR expression has proved to be a prognosticator of distasteful prognosis and chemosensitivity to taxane-based therapy.20 The hypermethylation in CHFR promoter is frequently observed in several cancer types, such as gastric cancer, colorectal cancer, and esophageal cancer, while it is much less frequent in non-small-cell lung cancer and independently associated with poor prognosis in acute myeloid leukemia.21–24 Although various studies have declared that CHFR is a tumor suppressor, the cognizance of how CHFR works in cancers, especially in human gastric cancer, is still extremely poor.
In the current study, we investigated the biological functions of CHFR and the underlying mechanism in human gastric cancer for the first time to our best knowledge. First of all, clinicopathological characteristics of CHFR expression were analyzed using online data which are available in public datasets. To our surprise, there was a negative correlation between CHFR expression and overall survival of gastric cancer patients according to the data of Kaplan–Meier plotter dataset (Figure 1A). This finding is inconsistent with the previous publication, in which CHFR was deemed to be a tumor suppressor.25,26 Although increasing publications have indicated that CHFR was frequently hypermethylated in gastric cancer, the percentage was only 34.3%.15 In fact, our immunohistochemistry data also indicated that the protein level of CHFR was much higher in gastric cancer tissues compared with the paracancerous tissues and that a large proportion of the tumor specimens express high level of CHFR (Figure 1B and C). Our data suggested that CHFR expression, but not its promoter hypermethylation, might be a useful biomarker for predicting a worse prognosis in patients with gastric cancer.
Next, we measured the background expression of CHFR in several human gastric cancer cell lines, and compared their basal migration activity using wound healing assays and transwell experiments. It seems that there is a positive association between CHFR levels and the migration ability of gastric cancer cells (Figure 2). Next, a CHFR overexpression plasmid was constructed and transfected into SGC-7901 and AGS cells, which express low levels of CHFR. Our results indicated that the ectopic expression of CHFR could not only arrest the cell cycle in G1 phase but could also significantly promote cell migration in both cell lines (Figure 3). This is a rather unexpected result, as CHFR was considered to be a tumor suppressor since it was discovered. On the other hand, this finding is reasonable, as CHFR level is negatively associated with the overall survival of gastric cancer patients and is significantly highly expressed in metastatic gastric cancer tissues (Figures 1A and 4A). To further confirm the regulatory effect of CHFR on the migration ability of gastric cancer cells, siRNA was used to silence the CHFR expression in HGC-27 cells. As expected, inhibition of CHFR significantly repressed the cell migration of HGC-27 cells (Figure 4C). All these findings indicate that CHFR promotes the cell migration in human gastric cancer.
The key initial step of sequential processes involved in the development of distant metastasis of cancer cells is EMT.5 At the molecular level, EMT causes an increased expression of mesenchymal markers N-cadherin and vimentin, and decreased expression of epithelial maker E-cadherin.27,28 As shown in Figure 5A, we found that overexpression of CHFR significantly upregulated the expression of mesenchymal marker N-cadherin and transcriptional factor Snail, while downregulated the expression of epithelial marker E-cadherin. Overall, these data clearly demonstrated that CHFR increased the migration of gastric cancer cells by promoting EMT. As previous research indicated that CHFR might be linked to cancer metastasis by promoting HDAC1 degradation, we also observed that CHFR indeed decreased HDAC1 expression, which was probably by promoting its ubiquitination (Figure 5A and B). Although it was reported that CHFR negatively regulates the cell migration in multiple cancer types17 and many papers also indicated that HDAC1 is reported to induce EMT in most cancers,29 HDAC1 inhibitor was reported to induce EMT in colon cancer cells, and more importantly, low dose of SAHA promoted cell migration in human gastric cancer.30,31 As expected, our data indeed revealed that SAHA not only promoted EMT in both SGC-7901 and AGS cells, but also enhanced the cell migration of both the cell lines (Figure 5C–E). Our data also indicated that CHFR silencing effectively upregulated the expression of HDAC1 (Figure 5F). Interestingly, low dose of SAHA significantly attenuated the inhibitory effect of CHFR silencing on cell migration of HGC-27 cells.
Conclusion
Our findings suggest that CHFR is a negative biomarker for overall survival in patients with gastric cancer, and functions as a novel oncogene to promote cell migration by inducing EMT via the inhibition of HDAC1 at least in gastric cancer cells. Therefore, the CHFR/HDAC1/EMT axis may be a novel molecular therapeutic target for the treatment of gastric cancer, especially the metastatic tumors.
Data availability
All data generated or analyzed during this study are included in this published article or available from public datasets.
Acknowledgments
This study was financially supported by the public project grant (2015ZA226) from the Administration of Traditional Chinese Medicine of Zhejiang Province, and the public project grant (2017RC22) from Science and Technology Bureau of Lishui, Zhejiang Province, China. Shangwen Yang and Feiyun He are co-first authors for this study.
Author contributions
Conceived and designed the experiments: Bin Ye and Shangwen Yang; performed the experiments: Shangwen Yang, Feiyun He, Mugen Dai, Jundi Pan, and Jianbo Wang; analyzed the data: Bin Ye and Shangwen Yang; wrote the manuscript: Bin Ye and Shangwen Yang. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global Cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global Cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
Zheng L, Wang L, Ajani J, Xie K. Molecular basis of gastric cancer development and progression. Gastric Cancer. 2004;7(2):61–77. | ||
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. | ||
Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nature Reviews Cancer. 2018;18(2):128–134. | ||
Heerboth S, Housman G, Leary M, et al. EMT and tumor metastasis. Clin Transl Med. 2015;4(1):6. | ||
Scolnick DM, Halazonetis TD. CHFR defines a mitotic stress checkpoint that delays entry into metaphase. Nature. 2000;406(6794):430–435. | ||
Kang D, Chen J, Wong J, Fang G. The checkpoint protein Chfr is a ligase that ubiquitinates Plk1 and inhibits cdc2 at the G2 to M transition. J Cell Biol. 2002;156(2):249–260. | ||
Ding Y, Lian HF, Du Y. Clinicopathological significance of CHFR promoter methylation in gastric cancer: a meta-analysis. Oncotarget. 2018;9(11):10083–10090. | ||
Mizuno K, Osada H, Konishi H, et al. Aberrant hypermethylation of the Chfr prophase checkpoint gene in human lung cancers. Oncogene. 2002;21(15):2328–2333. | ||
Shibata Y, Haruki N, Kuwabara Y, et al. CHFR expression is downregulated by CpG island hypermethylation in esophageal cancer. Carcinogenesis. 2002;23(10):1695–1700. | ||
Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest. 2005;115(6):1503–1521. | ||
Wang X, Yang Y, Xu C, et al. CHFR suppression by hypermethylation sensitizes endometrial cancer cells to paclitaxel. Int J Gynecol Cancer. 2011;21(6):996–1003. | ||
Li Y, Yang Y, Lu Y, et al. Predictive value of CHFR and MLH1 methylation in human gastric cancer. Gastric Cancer. 2015;18(2):280–287. | ||
Zheng G, Ma Y, Zou Y, Yin A, Li W, Dong D. HCMDB: the human cancer metastasis database. Nucleic Acids Res. 2018;46(D1):D950–D955. | ||
Oh YM, Kwon YE, Kim JM, et al. CHFR is linked to tumour metastasis through the downregulation of HDAC1. Nat Cell Biol. 2009;11(3):295–302. | ||
Shi H, Wang X, Wang J, Pan J, Liu J, Ye B. Association between CHFR gene hypermethylation and gastric cancer risk: a meta-analysis. Onco Targets Ther. 2016;9:7409–7414. | ||
Derks S, Cleven AH, Melotte V, et al. Emerging evidence for Chfr as a cancer biomarker: from tumor biology to precision medicine. Cancer Metastasis Rev. 2014;33(1):161–171. | ||
Sanbhnani S, Yeong FM. CHFR: a key checkpoint component implicated in a wide range of cancers. Cell Mol Life Sci. 2012;69(10):1669–1687. | ||
Kawasaki T, Ohnishi M, Nosho K, et al. CpG island methylator phenotype-low (CIMP-low) Colorectal cancer shows not only few methylated CIMP-high-specific CpG islands, but also low-level methylation at individual loci. Mod Pathol. 2008;21(3):245–255. | ||
Soutto M, Peng D, Razvi M, et al. Epigenetic and genetic silencing of CHFR in esophageal adenocarcinomas. Cancer. 2010;116(17):4033–4042. | ||
Pillai RN, Brodie SA, Sica GL, et al. Chfr protein expression predicts outcomes to taxane-based first line therapy in metastatic NSCLC. Clin Cancer Res. 2013;19(6):1603–1611. | ||
Gao L, Liu F, Zhang H, Sun J, Ma Y. CHFR hypermethylation, a frequent event in acute myeloid leukemia, is independently associated with an adverse outcome. Genes Chromosomes Cancer. 2016;55(2):158–168. | ||
Kashima L, Toyota M, Mita H, et al. CHFR, a potential tumor suppressor, downregulates interleukin-8 through the inhibition of NF-kappaB. Oncogene. 2009;28(29):2643–2653. | ||
Kwon YE, Bae SJ, Kim M, Seol JH. Sumoylation negatively regulates the stability of CHFR tumor suppressor. Biochem Biophys Res Commun. 2013;430(1):213–217. | ||
Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29(34):4741–4751. | ||
Vuoriluoto K, Haugen H, Kiviluoto S, et al. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene. 2011;30(12):1436–1448. | ||
Lei W, Zhang K, Pan X, et al. Histone deacetylase 1 is required for transforming growth factor-beta1-induced epithelial-mesenchymal transition. Int J Biochem Cell Biol. 2010;42(9):1489–1497. | ||
Ji M, Lee EJ, Kim KB, et al. HDAC inhibitors induce Epithelial-Mesenchymal transition in colon carcinoma cells. Oncol Rep. 2015;33(5):2299–2308. | ||
Lu H, Yang XF, Tian XQ, et al. The in vitro and vivo anti-tumor effects and molecular mechanisms of suberoylanilide hydroxamic acid (SAHA) and MG132 on the aggressive phenotypes of gastric cancer cells. Oncotarget. 2016;7(35):56508–56525. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.