")
Back to Journals » OncoTargets and Therapy » Volume 11
Chelerythrine induced cell death through ROS-dependent ER stress in human prostate cancer cells
Authors Wu S, Yang Y, Li F, Huang L, Han Z, Wang G, Yu H, Li H
Received 21 November 2017
Accepted for publication 31 January 2018
Published 8 May 2018 Volume 2018:11 Pages 2593—2601
DOI https://doi.org/10.2147/OTT.S157707
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yao Dai
Songjiang Wu, Yanying Yang, Feiping Li, Lifu Huang, Zihua Han, Guanfu Wang, Hongyuan Yu, Haiping Li
Department of Urology, Enze Hospital of Taizhou Enze Medical Center (Group), Taizhou, China
Introduction: Prostate cancer is the most common noncutaneous cancer and the second leading cause of cancer-related mortality worldwide and the third in USA in 2017. Chelerythrine (CHE), a naturalbenzo[c]phenanthridine alkaloid, formerly identified as a protein kinase C inhibitor, has also shown anticancer effect through a number of mechanisms. Herein, effect and mechanism of the CHE-induced apoptosis via reactive oxygen species (ROS)-mediated endoplasmic reticulum (ER) stress in prostate cancer cells were studied for the first time.
Methods: In our present study, we investigated whether CHE induced cell viability decrease, colony formation inhibition, and apoptosis in a dose-dependent manner in PC-3 cells. In addition, we showed that CHE increases intracellular ROS and leads to ROS-dependent ER stress and cell apoptosis.
Results: Pre-treatment with N-acetyl cysteine, an ROS scavenger, totally reversed the CHE-induced cancer cell apoptosis as well as ER stress activation, suggesting that the ROS generation was responsible for the anticancer effects of CHE.
Conclusion: Taken together, our findings support one of the anticancer mechanisms by which CHE increased ROS accumulation in prostate cancer cells, thereby leading to ER stress and caused intrinsic apoptotic signaling. The study reveals that CHE could be a potential candidate for application in the treatment of prostate cancer.
Keywords: chelerythrine, reactive oxygen species, endoplasmic reticulum stress, apoptosis, prostate cancer
Introduction
Prostate cancer is the most common noncutaneous cancer and the second leading cause of cancer-related mortality for adult men in the USA and Europe.1 The risk for prostate cancer increases exponentially after the age of 50.2 Despite advances in clinical treatment, by antihormonal therapy, radiotherapy, and chemotherapy, prostate cancer remains a major cause of cancer-related morbidity and mortality.3–5 Over 70% of cancer metastasis from prostate cancer develops bone metastases because of the insensitivity with the abovementioned clinical treatments. An increasing number of patients experience tumor progression to hormone-refractory prostate cancer, resulting in serious problems in the treatment of prostate cancer.6 In fact, most cancer-related deaths are due to drug resistance and eventual chemotherapy failure.7 Hence, finding effective agents for treating the advanced prostate cancer is of urgent need.
Chelerythrine (CHE), a natural benzo[c]phenanthridine alkaloid, is extracted from plant species, such as Chelidonium majus, Macleaya cordata, and Sanguinaria canadensis.8 CHE has been used as inhibitors of protein kinase C9 and the Bcl-2 family proteins. It shows a diversity of biological activities, including anticancer,10 antidiabetes,11 antifungus,12 and protecting lipopolysaccharide-induced endotoxic shock.13 CHE was reported to induce apoptosis and the G1-phase cell cycle arrest in human promyelocytic leukemia HL-60 cells.14 CHE also induced apoptosis through inhibiting Bcl-2 expression and activating the mitochondrial pathway in hepatocellular carcinoma cells.15 These data suggest that CHE may influence the malignant potential of tumor types other than hepatoma and leukemia.
Compared with normal cells, many types of cancer cells usually acquire higher levels of reactive oxygen species (ROS) or adaptive mechanisms to resist constant intrinsic oxidative stress.16,17 Oxidative stress plays a crucial role in the processes of cellular viability and function. Furthermore, targeting ROS and oxidative stress can also be seen as a good strategy to eliminate cancer cells.18 Indeed, this treatment concept was already exemplified by various ROS-enhancing drugs such as paclitaxel,19 trisenox,20 and cisplatin.21 Recently, CHE has been reported to enhance the production of ROS, which stimulates autophagy in non-small cell lung cancer cells.22 Previous studies have shown that CHE was a potentially useful antineoplastic agent against head and neck squamous cell carcinoma tumors.23 Furthermore, treatment with CHE resulted in minimal toxicity in nude mice.23 ROS are normal by-products of various cellular processes, such as signal transduction, mitochondrial metabolism, DNA repair, and protein folding.24,25 Therefore, the oxidative stress response plays a significant role in keeping the balance between prosurvival and proapoptotic signaling pathways.26 Varieties of proapoptotic signaling pathways can be stimulated in an uncontrolled high level of ROS status, such as mitochondrial dysfunction, endoplasmic reticulum (ER) stress, and DNA damage.27,28 Thus, the ROS-inducing agent CHE may be effective in killing cancer cells. However, the effect and mechanism of CHE on prostate cancer are unknown.
In the present study, we aimed to investigate the anticancer effects and the underlying mechanism of CHE, especially the ROS function in prostate cancer. Our study demonstrated that CHE showed excellent anticancer potential effects against PC-3 cells through triggering ROS accumulation, whereafter inducing ER stress-dependent apoptosis. Blockage of the ROS production by selective inhibitor NAC could totally reverse all the anticancer effects of CHE. Together, these data suggest that CHE could be a potential agent for prostate cancer therapy.
Materials and methods
Reagents
CHE was obtained from Aladdin (Shanghai, China). Antibodies against B-cell lymphoma 2 (Bcl2, sc-492), Bcl2-associated protein x (Bax, sc-493), GAPDH (sc-32233), and horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against cleaved form of poly (ADP-ribose) polymerase (cleaved-PARP, 5625S), phosphorylated eukaryotic initiation factor 2α (p-eIF2α, 3398S), eIF2α (9722S), and activating transcription factor-4 (ATF4, 11815S) were obtained from Cell Signaling Technology (Danvers, MA, USA). NAC, DMSO, and MTT were purchased from Sigma-Aldrich (St Louis, MO, USA). Fluorescein isothiocyanate (FITC) Annexin V Apoptosis Detection Kit I and propidium iodide (PI) were purchased from BD Pharmingen (Franklin Lakes, NJ, USA). ROS probe 2′7′-dichlorodihydrofluorescein diacetate (DCFH-DA) was purchased from Thermo Fisher Scientific (Carlsbad, CA, USA).
Prostate cancer cell lines
Human prostate cancer PC-3 cells and DU145 cells were purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). Cells were cultured in DMEM/F12 medium (Gibco, Eggenstein, Germany) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin (Mediatech Inc., Manassas, VA, USA) in a humidified atmosphere of 5% CO2 at 37°C.
Cell viability assay
To measure the prostate cancer cells viability after CHE treatment, cells were seeded on 96-well plates at a density of 6×103 per well and allowed to attach overnight in Roswell Park Memorial Institute (RPMI) 1640 containing 10% heat-inactivated FBS. CHE was dissolved in DMSO and diluted with 1640 medium to final concentrations of 0.5, 1, 2.5, 5, 7.5, 10, 15, and 20 μM. The prostate cancer cells were incubated with CHE for 24 or 48 h before the MTT assay.
Cell apoptosis analysis
PC-3 cells were plated on 60-mm dishes for 12 h and then treated with CHE (5, 7.5, or 10 μM) for 24 h in the presence or absence of NAC (5 mM). Cells were then harvested and washed thrice with ice-cold PBS. The washed cell samples were incubated with 3 μL Annexin-V for 10 min in the dark, then incubated with 2 μL PI for 5 min, and then evaluated for apoptosis using an FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA).
Colony formation assay
PC-3 cells were plated at 500 cells per well in 6-well plates and cultured complete growth media for 24 h. Cells were then exposed to CHE. Cells were allowed to grow for 7 days and colonies emerging were stained with crystal violet solution. A colony was defined as a cluster of at least 50 cells that can often only be determined microscopically.
Western blot analysis
Total proteins from cultured cells were prepared and protein levels were measured by using the Bradford assay (Bio-Rad, Hercules, CA, USA). Proteins were separated using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes. After blocking with tris-buffered saline containing 0.05% Tween 20 (TBST) and freshly prepared 5% nonfat milk for 1.5 h at room temperature, membranes were incubated with different primary antibodies overnight at 4°C. Membranes were then washed in TBST and incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. Immunoreactivity was visualized using an ECL substrate (Bio-Rad, Hercules, CA, USA). Densitometric measurements were performed using ImageJ (National Institute of Health, Bethesda, MD, USA).
Measurement of ROS generation
Intracellular ROS contents were measured by flow cytometry utilizing DCFH-DA. Briefly, cells were plated at 5×105 density in 6-well plates and allowed to attach for 12 h. Cells were then exposed to CHE for 1.5 h. NAC pretreatments were carried out at 5 mM for 1 h. Following treatments, cells were stained with 10 μM DCFH-DA at 37° for 30 min in the dark. Cells were collected and dihydrodichlorofluorescein (DCF) fluorescence was analyzed by FACS Calibur flow cytometer (BD Biosciences).
Statistical analysis
All experiments were performed in triplicate (n=3). Data are expressed as means ± SEM. Statistical analyses were performed using GraphPad Prism 5.0 (GraphPad, San Diego, CA, USA). Statistical differences between two groups were assessed by Student’s t-test. A p-value <0.05 was considered statistically significant.
Results
CHE reduced cell viability in human prostate cancer cells
To determine the cytotoxic effects of CHE (Figure 1A) in prostate cancer cell lines, an MTT assay was performed to evaluate the viability in human prostate cancer PC-3 and DU145 cells. As shown in Figure 1B, CHE treatment for 24 or 48 h significantly decreased the viability of PC-3 and DU145 cells in a dose-dependent manner. The IC50 value for CHE was 5.2 μM for 24 h or 3.8 μM for 48 h in PC-3 cells, and 6.1 μM for 24 h or 4.3 μM for 48 h in DU145 cells. We then utilized the colony formation assay which shows whether prostate cancer cells are able to form colonies when the cells were treated with CHE. As shown in Figure 1C and D, CHE prevented colony formation at 7.5 and 10 μM levels. Collectively, our findings show that CHE reduces the growth of prostate cancer cells and induces apoptotic cell death.
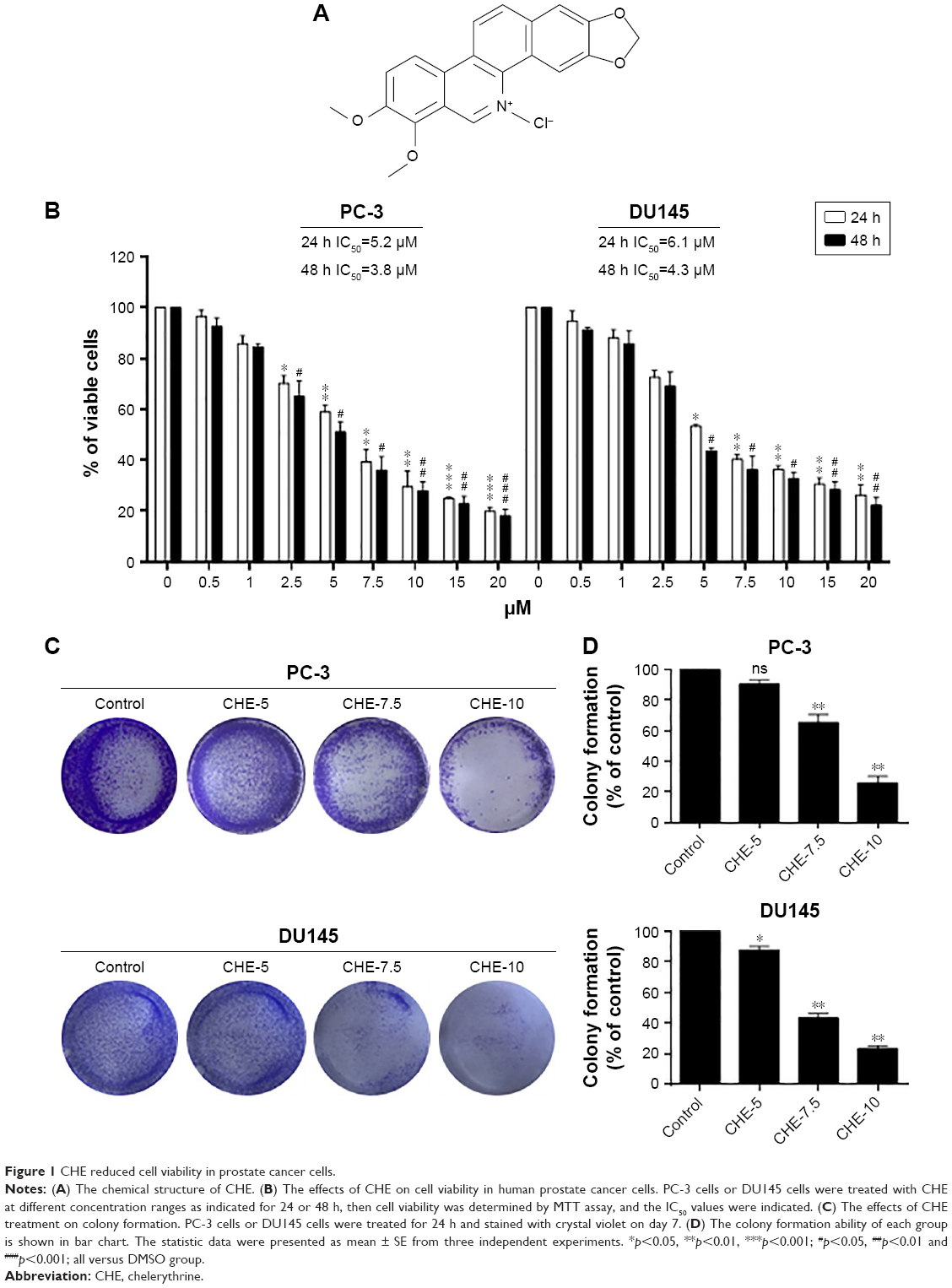 | Figure 1 CHE reduced cell viability in prostate cancer cells. |
CHE induced cell apoptosis in human prostate cancer cells
Next, we investigated the proapoptosis effects of CHE by flow cytometry analysis using Annexin V-FITC+PI staining. As shown in Figure 2A and B, flow cytometry analysis showed that after treatment with CHE for 24 h in PC-3 cells, the apoptosis cells were significantly increased up to about 32% in a dose-dependent way. We further examined the expression of apoptosis-associated proteins by western blot analysis in PC-3 cells. As shown in Figure 2C, CHE treatment decreased the protein level of Bcl-2 and increased the cleaved PARP in a dose-dependent manner. In addition, Bax protein did not show obvious changes after CHE treatment.
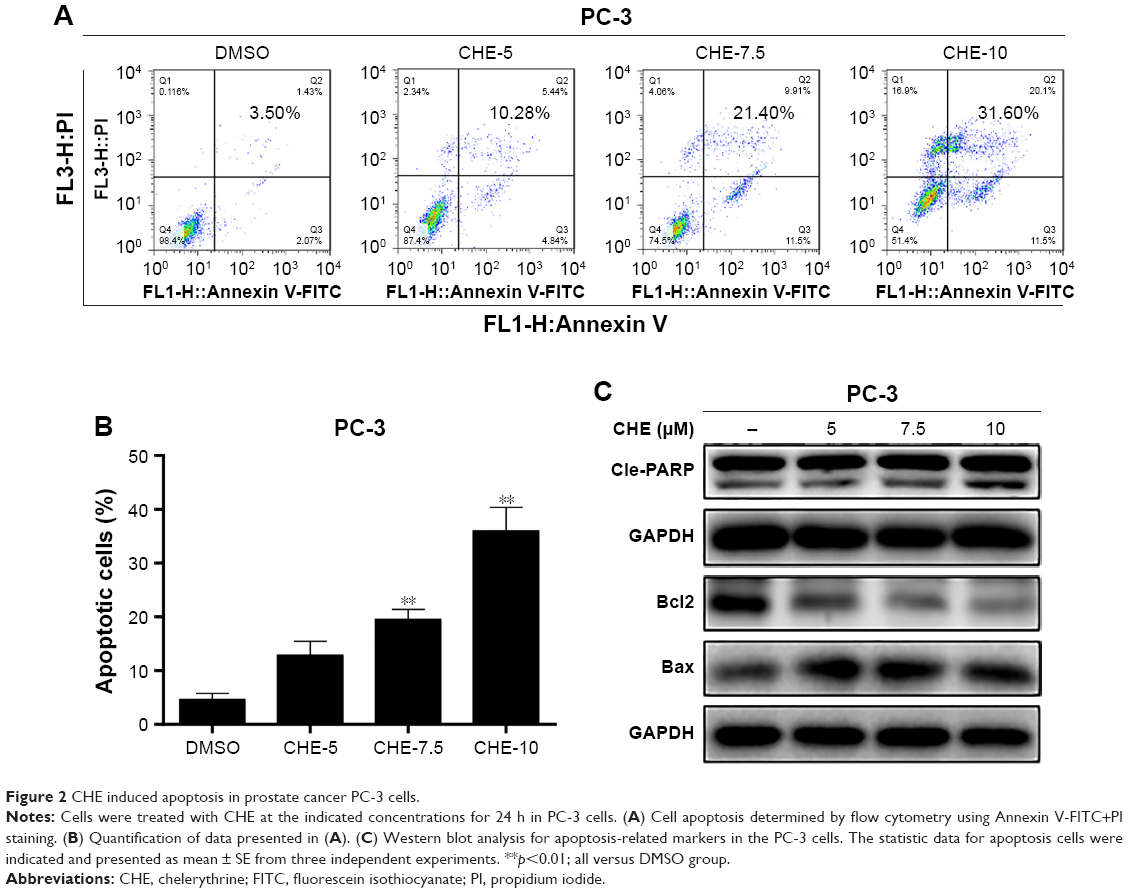 | Figure 2 CHE induced apoptosis in prostate cancer PC-3 cells. |
CHE increased ROS accumulation in PC-3 cells
A previous study has shown the induction of ROS-stimulated distinctive autophagy by CHE in non-small cell lung cancer cells.22 Here, we first investigated the effects of CHE on ROS production in PC-3 cells by flow cytometry analysis using DCFH-DA fluorescent dyes. As shown in Figure 3A and B, when treated with CHE at 10 μM for different time points in PC-3 cells, the results showed that the intracellular H2O2 levels were increased in a time-dependent manner. It indicated that CHE could induce ROS accumulation in PC-3 prostate cancer cells. We pretreated prostate cancer cells with NAC before exposing the cells to CHE and measured ROS levels. NAC is commonly used as a precursor of glutathione and can also interact directly with ROS.29 As expected, NAC pretreatment decreased the intracellular ROS levels (Figure 3C and D). Thus, these data strongly demonstrated that CHE can induce ROS accumulation in prostate cancer cells.
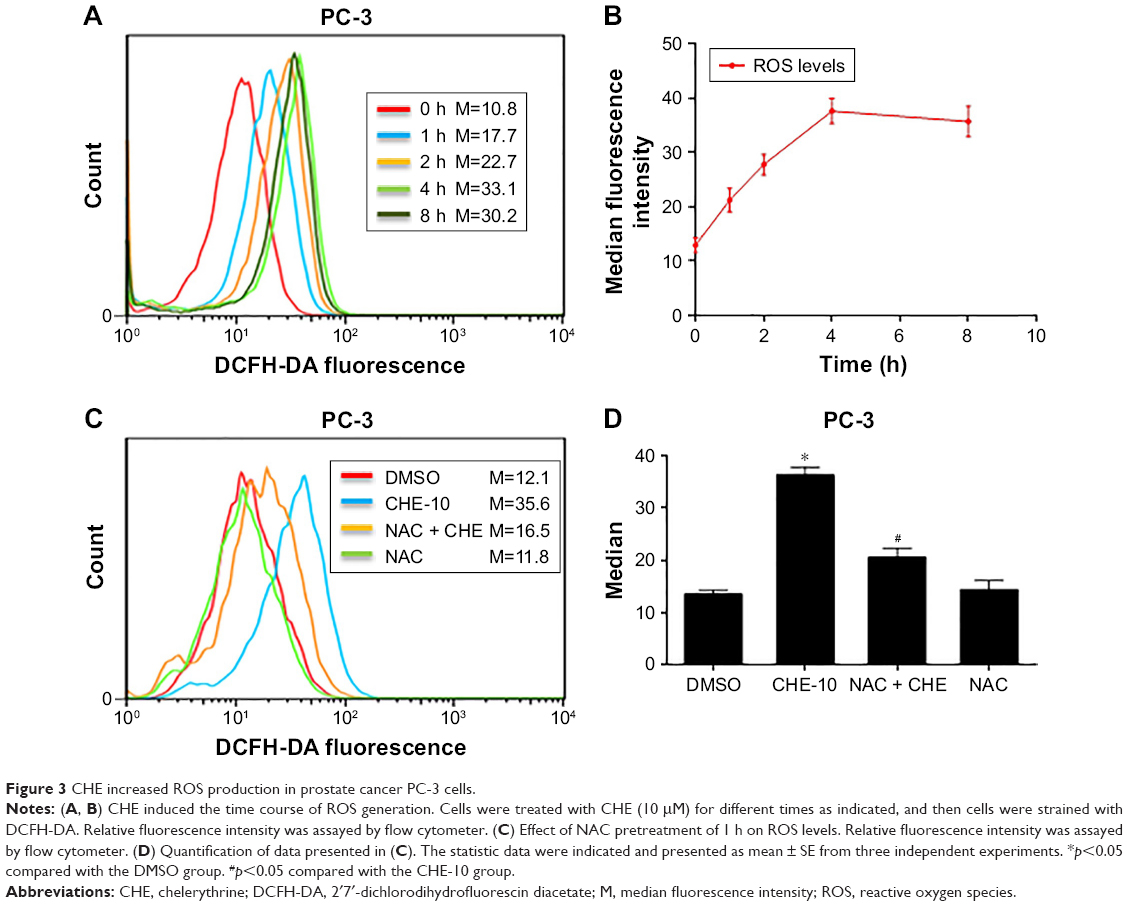 | Figure 3 CHE increased ROS production in prostate cancer PC-3 cells. |
Blockage of ROS generation reversed CHE-induced cell apoptosis in PC-3 cells
Next, we determined whether ROS generation was involved in the anticancer effects of CHE in prostate cancer cells. PC-3 cells were pretreated with 10 mM NAC for 1 h and then treated with 10 μM CHE for 24 h. As shown in Figure 4A and B, pretreatment of cells with NAC was able to normalize CHE-induced apoptosis in Annexin V-FITC+PI staining experiment. The effects of ROS blockage were further validated via western blot analysis. As shown in Figure 4C, NAC prevented CHE-induced alterations of apoptosis-related proteins. These findings vigorously demonstrated the vital role of ROS generation in mediating anticancer effects of CHE in prostate cancer cells.
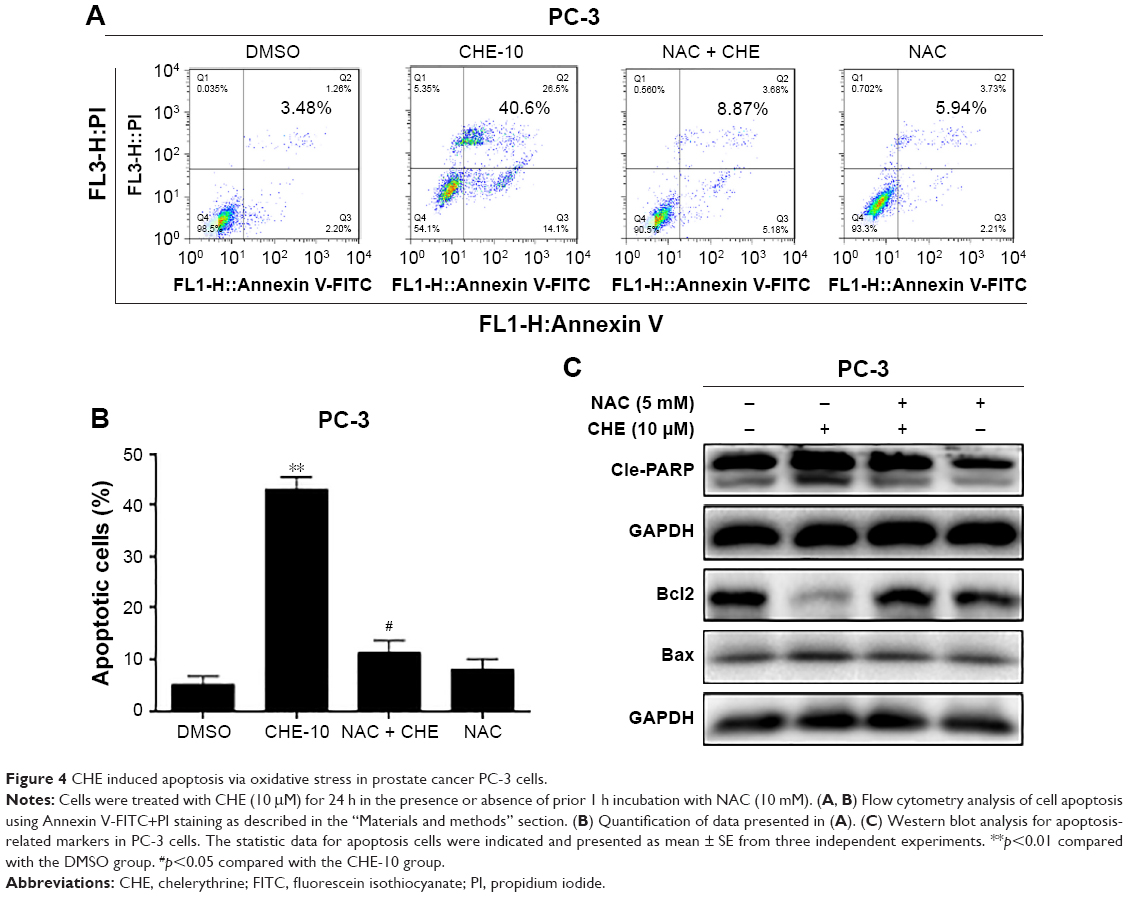 | Figure 4 CHE induced apoptosis via oxidative stress in prostate cancer PC-3 cells. |
CHE induced cell apoptosis through ROS-mediated ER stress in PC-3 cells
ROS generation was reported to activate various proapoptotic pathways, including the ER stress-induced cancer cell apoptosis pathway.30 Therefore, we tested whether CHE-mediated ROS activates the ER stress pathway in prostate cancer cells. When PC-3 cells were treated with CHE for different time intervals, we noticed a transient increase in the level of p-eIF2α 2–8 h after CHE treatment (Figure 5A and B). ATF4 expression also increased in a similar manner with p-eIF2α (Figure 5A and B). Moreover, CHE-mediated ROS was clearly inducing ER stress in cells as pretreatment with NAC normalized the induction of p-eIF2α and ATF4 (Figure 5C and D). Taken together, all these results indicate that CHE-induced cell apoptosis is, at least partly, mediated by the ROS-dependent ER stress pathway.
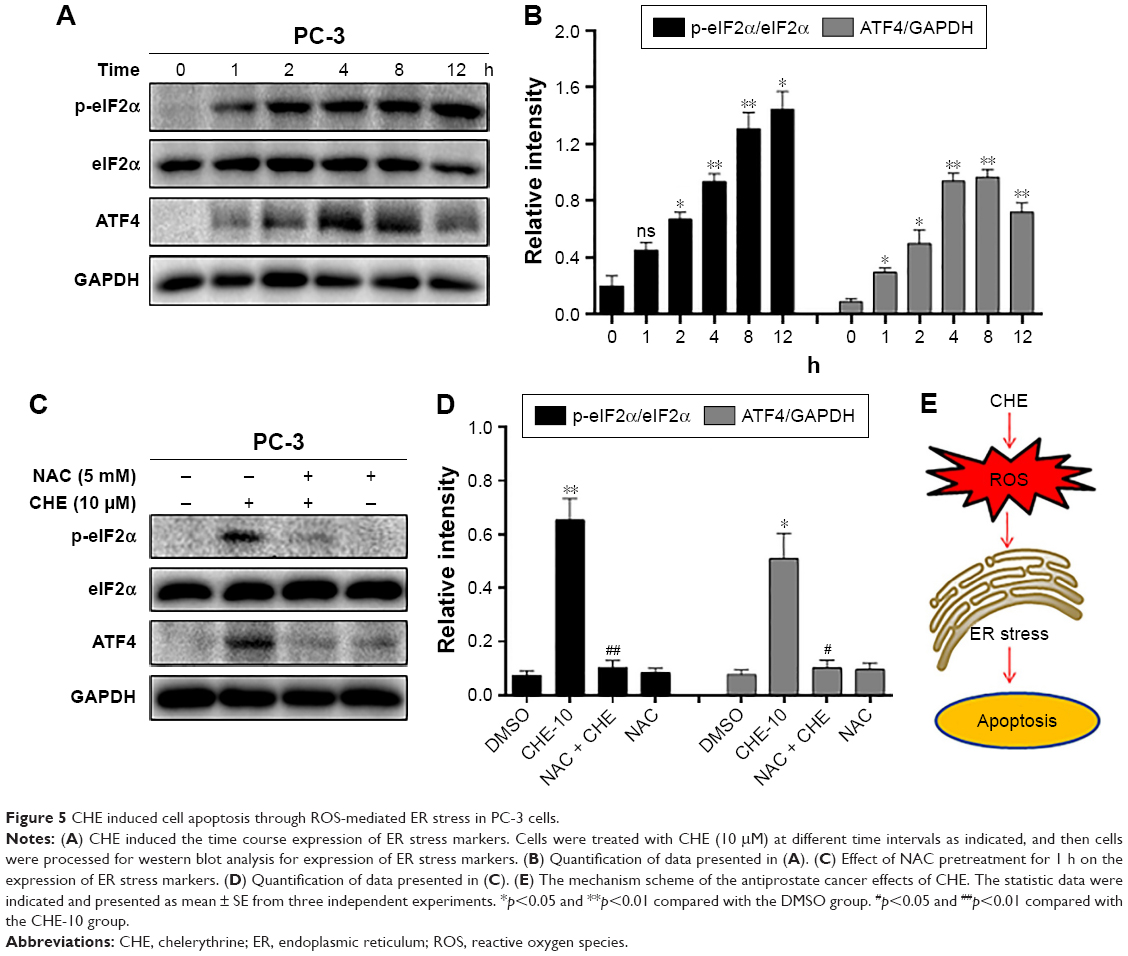 | Figure 5 CHE induced cell apoptosis through ROS-mediated ER stress in PC-3 cells. |
Discussion
In this study, we provide sufficient evidence to demonstrate that CHE shows excellent anticancer effects in prostate cancer cells via inducing ROS-dependent ER stress. Recently, accumulating evidence suggests that increasing oxidative stress might be a promising strategy to inhibit cancer cells.31 The inductions of cancer cell apoptosis by some anticancer agents such as cisplatin,21 farnesol,32 and arsenic trioxide20 have been reported to be mediated by ROS accumulation. Interestingly, increased accumulation of ROS by CHE was also observed in prostate cancer cells in Figure 3A. Importantly, ROS was directly involved in inducing apoptosis as decreasing the levels of ROS alleviated CHE-induced negative growth function. Furthermore, our study shows that ROS levels are correlated with the induction of apoptosis and ER stress pathway in prostate cancer. These salient findings are summarized in Figure 5E. In summary, our study presents a promising strategy for prostate cancer therapy that preferentially eradicates cancer cells by targeting ROS generation.
ER is usually well known to regulate cellular responses to stress.33 Numerous signals were reported to break the ER function and consequently induce ER stress. ER plays a significant role in cancer cell apoptosis signaling pathway and it also becomes a novel signaling target for candidate of cancer therapy drugs.34 Recently, agents inducing ER stress-mediated cancer cell apoptosis have become a novel signaling for the development of cancer therapy.35,36 Besides, it was reported that some agents show their proapoptotic effects through inducing ER stress in prostate cancer cells.37,38 Therefore, targeting ER stress can be a promising anticancer strategy. In consistent with these findings, we first proved that CHE treatment concomitantly induces ER stress response in prostate cancer PC-3 cells, which increases the ER stress-related proteins such as p-eIF2α and ATF4 in a time-dependent manner. As expected, NAC pretreatment fully reversed all these changes in ER induced by CHE. Collectively, all these results indicate that ROS production is the critical upstream regulator of CHE-induced ER stress in prostate cancer cells.
Under physiological conditions, the maintenance of moderate levels of intracellular ROS is important both in keeping redox balance and in cell proliferation.39 Recently, it has been reported that ROS generation is partly linked to ER stress activation.40 Enhanced ROS generation or oxidative stress might be associated with subsequent ER stress and ER stress-dependent cancer cell apoptosis. However, a moderate increase in ROS levels may usually trigger cell growth and proliferation.41 Indeed, excessive ROS and free radicals may lead to severe damage to mitochondria, ER, and DNA repair. Besides, overproduced ROS can promote the process involved in the activation of apoptotic signaling.42 Moreover, Vrablic et al has reported that ROS overproduction could reduce the mitochondrial membrane potential, which eventually caused an increase in the level of some other proapoptotic factors in the cytosol.43 We observed that CHE treatment induced ROS accumulation within 60 min at the apoptosis-inducing concentrations (Figure 3A and B). More importantly, blockage of ROS by NAC almost reversed the cytotoxicity and apoptosis induced by CHE in PC-3 cells. All these data validated that CHE induces cancer cell death via activating ROS production.
Conclusion
We demonstrated for the first time that CHE induced a distinctive apoptosis of ROS-dependent ER stress in prostate cancer cells, and the decreased level of ROS reversed the CHE-induced cell viability decrease, cell death, and apoptosis in prostate cancer cells. The discovery of the activation of ROS-mediated ER stress apoptotic pathway through CHE may provide a new strategy for prostate cancer treatment.
Acknowledgments
This work was supported by grants from Taizhou Science and Technology Bureau (No. 1501KY41) and Taizhou Enze Medical Center (No. 15EZB7).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. | ||
Vickers AJ, Sjoberg DD, Ulmert D, et al. Empirical estimates of prostate cancer overdiagnosis by age and prostate-specific antigen. BMC Med. 2014;12:26. | ||
Gravis G, Audenet F, Irani J, et al. Chemotherapy in hormone-sensitive metastatic prostate cancer: evidences and uncertainties from the literature. Cancer Treat Rev. 2017;55:211–217. | ||
Rydzewska LHM, Burdett S, Vale CL, et al. Adding abiraterone to androgen deprivation therapy in men with metastatic hormone-sensitive prostate cancer: a systematic review and meta-analysis. Eur J Cancer. 2017;84:88–101. | ||
Spratt DE, Dess RT, Feng FY. Reply to Pirus Ghadjar and Thomas Wiegel’s Letter to the Editor re: Daniel E. Spratt, Robert T. Dess, Zachary S. Zumsteg, et al. A systematic review and framework for the use of hormone therapy with salvage radiation therapy for recurrent prostate cancer. Eur Urol. In press. http://dx.doi.org/10.1016/j.eururo.2017.06.027: salvage radiotherapy and hormone therapy: change is coming, just not quite yet. Eur Urol. Epub. 2017 Oct 27. | ||
Shimada K, Nakamura M, Ishida E, Konishi N. Molecular roles of MAP kinases and FADD phosphorylation in prostate cancer. Histol Histopathol. 2006;21(4):415–422. | ||
Frame FM, Maitland NJ. Cancer stem cells, models of study and implications of therapy resistance mechanisms. Adv Exp Med Biol. 2011;720:105–118. | ||
Malikova J, Zdarilova A, Hlobilkova A. Effects of sanguinarine and chelerythrine on the cell cycle and apoptosis. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2006;150(1):5–12. | ||
Herbert JM, Augereau JM, Gleye J, Maffrand JP. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun. 1990;172(3):993–999. | ||
Vogler M, Weber K, Dinsdale D, et al. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ. 2009;16(7):1030–1039. | ||
Zheng W, Qiu L, Wang R, et al. Selective targeting of PPARgamma by the natural product chelerythrine with a unique binding mode and improved antidiabetic potency. Sci Rep. 2015;5:12222. | ||
Yang XJ, Miao F, Yao Y, et al. In vitro antifungal activity of sanguinarine and chelerythrine derivatives against phytopathogenic fungi. Molecules. 2012;17(11):13026–13035. | ||
Niu X, Mu Q, Li W, Huang H, Yao H, Li H. Protective effects of chelerythrine against lipopolysaccharide-induced endotoxic shock in mice. Inflammation. 2014;37(6):1968–1975. | ||
Vrba J, Dolezel P, Vicar J, Modriansky M, Ulrichova J. Chelerythrine and dihydrochelerythrine induce G1 phase arrest and bimodal cell death in human leukemia HL-60 cells. Toxicol In Vitro. 2008;22(4):1008–1017. | ||
Zhang ZF, Guo Y, Zhang JB, Wei XH. Induction of apoptosis by chelerythrine chloride through mitochondrial pathway and Bcl-2 family proteins in human hepatoma SMMC-7721 cell. Arch Pharm Res. 2011;34(5):791–800. | ||
Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11(2):85–95. | ||
Pervaiz S, Clement MV. Tumor intracellular redox status and drug resistance–serendipity or a causal relationship? Curr Pharm Des. 2004;10(16):1969–1977. | ||
Kardeh S, Ashkani-Esfahani S, Alizadeh AM. Paradoxical action of reactive oxygen species in creation and therapy of cancer. Eur J Pharmacol. 2014;735:150–168. | ||
Alexandre J, Hu Y, Lu W, Pelicano H, Huang P. Novel action of paclitaxel against cancer cells: bystander effect mediated by reactive oxygen species. Cancer Res. 2007;67(8):3512–3517. | ||
Jeanne M, Lallemand-Breitenbach V, Ferhi O, et al. PML/RARA oxidation and arsenic binding initiate the antileukemia response of As2O3. Cancer Cell. 2010;18(1):88–98. | ||
Bragado P, Armesilla A, Silva A, Porras A. Apoptosis by cisplatin requires p53 mediated p38alpha MAPK activation through ROS generation. Apoptosis. 2007;12(9):1733–1742. | ||
Tang ZH, Cao WX, Wang ZY, et al. Induction of reactive oxygen species-stimulated distinctive autophagy by chelerythrine in non-small cell lung cancer cells. Redox Biol. 2017;12:367–376. | ||
Chmura SJ, Dolan ME, Cha A, Mauceri HJ, Kufe DW, Weichselbaum RR. In vitro and in vivo activity of protein kinase C inhibitor chelerythrine chloride induces tumor cell toxicity and growth delay in vivo. Clin Cancer Res. 2000;6(2):737–742. | ||
Singh I. Mammalian peroxisomes: metabolism of oxygen and reactive oxygen species. Ann NY Acad Sci. 1996;804:612–627. | ||
Nordberg J, Arner ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med. 2001;31(11):1287–1312. | ||
Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48(6):749–762. | ||
He L, He T, Farrar S, Ji L, Liu T, Ma X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell Physiol Biochem. 2017;44(2):532–553. | ||
Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol. Epub 2017 Jun 3. | ||
Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci. 2003;60(1):6–20. | ||
Verfaillie T, Rubio N, Garg AD, et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012;19(11):1880–1891. | ||
Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10(3):175–176. | ||
Machida K, Tanaka T. Farnesol-induced generation of reactive oxygen species dependent on mitochondrial transmembrane potential hyperpolarization mediated by F(0)F(1)-ATPase in yeast. FEBS Lett. 1999;462(1–2):108–112. | ||
Jheng JR, Ho JY, Horng JT. ER stress, autophagy, and RNA viruses. Front Microbiol. 2014;5:388. | ||
Gajate C, Matos-da-Silva M, Dakir el H, Fonteriz RI, Alvarez J, Mollinedo F. Antitumor alkyl-lysophospholipid analog edelfosine induces apoptosis in pancreatic cancer by targeting endoplasmic reticulum. Oncogene. 2012;31(21):2627–2639. | ||
Hill DS, Martin S, Armstrong JL, et al. Combining the endoplasmic reticulum stress-inducing agents bortezomib and fenretinide as a novel therapeutic strategy for metastatic melanoma. Clin Cancer Res. 2009;15(4):1192–1198. | ||
Fiskus W, Saba N, Shen M, et al. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer Res. 2014;74(9):2520–2532. | ||
Lee WJ, Chien MH, Chow JM, et al. Nonautophagic cytoplasmic vacuolation death induction in human PC-3M prostate cancer by curcumin through reactive oxygen species-mediated endoplasmic reticulum stress. Sci Rep. 2015;5:10420. | ||
Ryu S, Lim W, Bazer FW, Song G. Chrysin induces death of prostate cancer cells by inducing ROS and ER stress. J Cell Physiol. 2017;232(12):3786–3797. | ||
Martin KR, Barrett JC. Reactive oxygen species as double-edged swords in cellular processes: low-dose cell signaling versus high-dose toxicity. Hum Exp Toxicol. 2002;21(2):71–75. | ||
Verfaillie T, Garg AD, Agostinis P. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett. 2013;332(2):249–264. | ||
Boonstra J, Post JA. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene. 2004;337:1–13. | ||
Liao YJ, Bai HY, Li ZH, et al. Longikaurin A, a natural ent-kaurane, induces G2/M phase arrest via downregulation of Skp2 and apoptosis induction through ROS/JNK/c-Jun pathway in hepatocellular carcinoma cells. Cell Death Dis. 2014;5:e1137. | ||
Vrablic AS, Albright CD, Craciunescu CN, Salganik RI, Zeisel SH. Altered mitochondrial function and overgeneration of reactive oxygen species precede the induction of apoptosis by 1-O-octadecyl-2-methyl-rac-glycero-3-phosphocholine in p53-defective hepatocytes. FASEB J. 2001;15(10):1739–1744. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.