")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Characterization of Stable and Reactive Metabolites of the Anticancer Drug, Ensartinib, in Human Liver Microsomes Using LC-MS/MS: An in silico and Practical Bioactivation Approach
Authors Abdelhameed AS , Attwa MW , Kadi AA
Received 31 July 2020
Accepted for publication 29 October 2020
Published 30 November 2020 Volume 2020:14 Pages 5259—5273
DOI https://doi.org/10.2147/DDDT.S274018
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Ali S Abdelhameed, Mohamed W Attwa, Adnan A Kadi
Department of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, Riyadh 11451, Kingdom of Saudi Arabia
Correspondence: Ali S Abdelhameed Tel +966 1146 98314
Fax +966 1146 76 220
, Email [email protected]
Background: Ensartinib (ESB) is a novel anaplastic lymphoma kinase inhibitor (ALK) with additional activity against Abelson murine leukemia (ABL), met proto-oncogene (MET), receptor tyrosine kinase (AXL), and v-ros UR2 sarcoma virus oncogene homolog 1 (ROS1) and is considered a safer alternative for other ALK inhibitors. ESB chemical structure contains a dichloro-fluorophenyl ring and cyclic tertiary amine rings (piperazine) that can be bioactivated generating reactive intermediates.
Methods: In vitro metabolic study of ESB with human liver microsomes (HLMs) was performed and the hypothesis of generating reactive intermediates during metabolism was tested utilizing trapping agents to capture and stabilize reactive intermediates to facilitate their LC-MS/MS detection. Reduced glutathione (GSH) and potassium cyanide (KCN) were utilized as trapping agents for quinone methide and iminium intermediates, respectively.
Results: Four in vitro ESB phase I metabolites were characterized. Three reactive intermediates including one epoxide and one iminium intermediates were characterized. ESB bioactivation is proposed to occur through unexpected metabolic pathways. The piperazine ring was bioactivated through iminium ions intermediates generation, while the dichloro-phenyl group was bioactivated through a special mechanism that was revealed by LC-MS/MS.
Conclusion: These findings lay the foundations for additional work on ESB toxicity. Substituents to the bioactive centers (piperazine ring), either for blocking or isosteric replacement, would likely block or interrupt hydroxylation reaction that will end the bioactivation sequence.
Keywords: in vitro phase-I metabolites, iminium intermediates, epoxide intermediates, human liver microsomes
Introduction
Cancer is the second leading cause of death worldwide and is accountable for about 9.6 million casualties in 2018, with almost one-sixth of global deaths.1 Earlier studies have shown that molecular-targeted therapeutic approaches were successful in treating cancer cases by targeting genes, oncogenes, and tumor suppressors involved in cancer progression.2 Lung cancer has the highest mortality rate globally among all cancer types. In 2012, 1.59 million deaths were attributed to lung cancer, which is 20% of all cancer deaths.3 Non-small-cell lung cancers (NSCLCs) account for 90% of all lung cancer cases and have various subtypes that are triggered by several activated oncogenes.4,5 The treatment of patients with NSCLC was greatly improved by applying active personalized therapies.6–8 Anaplastic lymphoma kinase (ALK) is a member of the receptor tyrosine kinase (RTK) family.9 ALK inhibitors are anticancer agents that act on tumors with ALK gene rearrangements (eg, the EML4-ALK fusion virus).10 Translocations of EML4-ALK were observed in 4–7% of patients with NSCLC.11
First-generation ALK inhibitors (eg, crizotinib) became less active due to NSCLC resistance mutations; however, new-generation ALK inhibitors (eg, brigatinib and ensartinib (ESB)) were designed to treat these mutations.12,13 ESB is a novel ALK inhibitor with additional activity against ABL, MET, ROS1, and Axl. It provided promising preliminary data in terms of safety and clinical activity.14,15 In phase I/II clinical trial, ESB was well tolerated and active in patients with ALK-positive NSCLC16 and showed intracranial responses in patients with ALK-positive NSCLC who developed CNS metastases.17
Drug detoxification occurs through metabolic pathways that improve the hydrophilicity of xenobiotics and endogenous compounds facilitating their elimination outside the human body. Additionally, compared with the precursor molecules, the produced metabolites demonstrated lower toxicity profiles. Nonetheless, in special cases, reactive metabolites (unstable intermediates) were produced exhibiting greater toxicity.18–20 These unstable intermediates are considered electron-deficient species, so covalent bonds can alter DNA and proteins, which is the preliminary step in organ toxicity.21,22 Characterization of the generation of reactive intermediates is crucial to understanding drug-induced toxicity. Reactive intermediates are often generated through metabolic pathways in phase I metabolism and cannot be identified by direct methodology as they are unstable species. Thus, nucleophile trapping can be utilized to seize the generated reactive species forming stable adducts that could be characterized using liquid chromatography-tandem mass spectrometry (LC-MS/MS).23–26
The IUPAC designation of ESB is 6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl) ethoxy]-N-(4-{[(3R,5S)-3,5-dimethyl-1-piperazinyl] carbonyl} phenyl)-3-pyridazinecarboxamide (Figure 1). ESB chemical structure contains a dichloro-fluorophenyl ring and cyclic tertiary amine rings (piperazine). Human liver microsomes (HLMs) can generate unstable iminium while metabolizing piperazine that can be trapped using potassium cyanide (KCN).27–30 The dichloro-fluorophenyl ring can be bioactivated to an unstable epoxide species by oxidative defluorination mechanism generating quinone methide intermediate that can be captured by reduced glutathione (GSH).31 The formed stable adducts, which were formed by nucleophilic-electrophilic reactions, can be separated by high-performance liquid chromatography (HPLC) and characterized by mass spectrometry (MS) analyzers.23,24,27,32,33 These reactive electrophiles may contribute to the ESB reported side-effects.
 |
Figure 1 Chemical structure of ensartinib. |
In patients receiving ESB treatment, common toxicities were as follows: vomiting (26%), fatigue (22%), nausea (36%), rash (56%), and pruritus (28%), with about 23% of the patients exhibiting a grade 3–4 treatment-related toxicity (including pruritus and rash).17,34 Here, LC-MS/MS was employed to characterize in vitro phase I metabolites of ESB generated in the incubation of HLMs. The generation of reactive electrophiles was also tested using capturing agents. Notably, ESB bioactivation was observed to take place via unexpected pathways.
Piperazine ring was bioactivated forming unstable iminium ion species. The dichloro-3-fluorophenyl ring was bioactivated by an unexpected metabolic reaction verified by LC-MS/MS. ESB generated three reactive metabolites through three ways of bioactivation that were detected via in silico testing and in vitro phase I metabolic practical experiments. This could be a new strategy for reducing the side-effects of the newly designed drugs without affecting and even improving its pharmacological activity. Targeted modifications for the characterized structural alerts and bioactive centers may improve the safety of ESB while maintaining its efficacy, which was confirmed by the DEREK module for StarDrop software. Four in vitro phase I metabolites, one GSH, and two cyano adducts of ESB were characterized. The proposed reaction involved oxidation, hydroxylation, and cyanide or GSH addition. In silico testing of the toxicity of ESB-related metabolites was performed using DEREK software. To the best of our knowledge, this is the first experiment of in vitro phase I metabolites of ESB, including structural characterization of novel ESB reactive intermediates.
Chemicals and Methods
Chemicals and Instruments
HLMs (product number: M0567), collected from various male human livers, were obtained from Sigma-Aldrich (St. Louis, Missouri, USA) and stored at −70°C until use. Organic solvents and reference powders were of HPLC grade and analytical grade (AR), respectively. HPLC grade water was prepared in-house by a Milli-Q Plus filtration instrument (Millipore, USA). ESB (98.51%) and NADPH (99.99%) were obtained from MedChemExpress (USA). Acetonitrile, formic acid, ammonium formate, GSH, and KCN were acquired from Sigma-Aldrich (USA). LC-MS/MS analysis was performed using Agilent 1200 HPLC connected to Agilent 6410 triple quadrupole. Electrospray ionization (ESI) was used as an interface. Incubation of ESB with HLMs was performed in the SW22 Shaking Water Bath (JULABO, Seelbach, Germany) adjusted at 37°C.
Chromatography Conditions
Both liquid chromatographic and mass optimized conditions for resolution and identification of the ESB-HLM incubation mixture are provided in Table 1.
|
Table 1 LC-MS/MS Optimized Chromatographic Parameters |
In silico Prediction of ESB Metabolites Using WhichP450™ Software
In this study, we aim to recognize the susceptibility of metabolism key sites, demonstrated by decreasing the site vulnerability, which is revealed by the composite site lability (CSL) scores, and examine the proposed regioselectivity of major isoforms responsible for metabolism. The outcomes from the WhichP450™ module of StarDrop software, shown in a pie chart, were used as an indication of the most likely CYP450 isoform that plays an important role in ESB metabolism.
In silico Prediction of ESB Reactive Metabolites Using XenoSite Reactivity Model and DEREK Software
XenoSite reactivity model in silico studies, freely available online at http://swami.wustl.edu/xenosite, were conducted to detect liable sites that can produce reactive intermediates forming stable adducts with GSH and cyanide.35 This prognostic model was built on neural networks of more than 680 molecules. It has a short processing time.36,37 The SMILES format of the ESB chemical structure was uploaded to the XenoSite reactivity model online website. DEREK software was employed to check for structural alerts, verify our proposed bioactivation pathway, and propose structural modification at the bioactive sites that may stop the toxicity sequence.
HLM Incubation
ESB was solubilized in dimethyl sulfoxide (DMSO). To avoid nonspecific protein binding, 1 mg/mL of HLMs was used. The characterization for ESB-related metabolites was performed by incubation of ESB (30 μM) with HLMs (1.0 mg/mL) in 1 mL of 50 mM phosphate buffer (pH, 7.4) plus 3.3 mM MgCl2 in a thermostatic shaking water bath (37°C) for 2 h. Incubation was initiated by adding NADPH to a final concentration of 1.0 mM after 5 min of preincubation and terminated by adding 2 mL of ice-cold ACN as a precipitating agent and extraction solvent. Excess precipitated protein was discarded by centrifugation at 9000 g for 15 min at 4°C. The purified supernatants were transferred to clean tubes; then, evaporation to dryness was carried out under the influence of a nitrogen stream. The evaporated samples were reconstituted in the mobile phase; then, they were transferred to HPLC vials for LC-MS/MS analysis (Agilent 6410 QqQ).38–41 The ESB-HLM incubation was repeated using the same procedure by adding GSH or KCN for capturing reactive metabolites. All metabolic procedures were conducted three times using controls (without NADPH or HLMs) to confirm that all identified stable and reactive metabolites were metabolically generated.
Identification of ESB Reactive Metabolites
After a full mass scan (MS) over a specific m/z range, extraction of molecular ion peaks (MIP) from the total ion chromatograms (TIC) was utilized to find the hypothesized metabolites. Chromatographic peaks (CP) of the predicted metabolites were exposed to fragmentation using high purity nitrogen under the collision-induced dissociation (CID) mode within the collision cell of the triple quadrupole, resulting in the generation of fragment ions required to reconstruct the chemical structures into the proposed chemical structure. Thus, the fragmentation pattern of ESB was used as a confirmatory and interpretation tool for the predicted chemical structures of the in vitro stable and reactive metabolites generated during ESB metabolism.
Results and Discussion
In silico Prediction of ESB Metabolites
The ESB metabolic landscape exhibits the vulnerability of each chemical site towards CYP3A4 metabolism in absolute terms, to guide the ESB metabolites prediction and the chemical structure optimization for enhancing metabolic stability. This reveals that dimethyl piperazine ring and dichlorophenyl group are labile which is consistent with experimental laboratory work. The CSL is shown in the metabolic landscape at the top-right corner and the P450 column of the dataset (Figure 2). CYP3A4 was predicted to play an important role in ESB metabolism. CSL value is 0.98 indicating high liability for ESB to metabolism. The metabolic liability of ESB is mainly due to N7, C1, C2, C6, and C8 of substituted methyl piperazine ring.
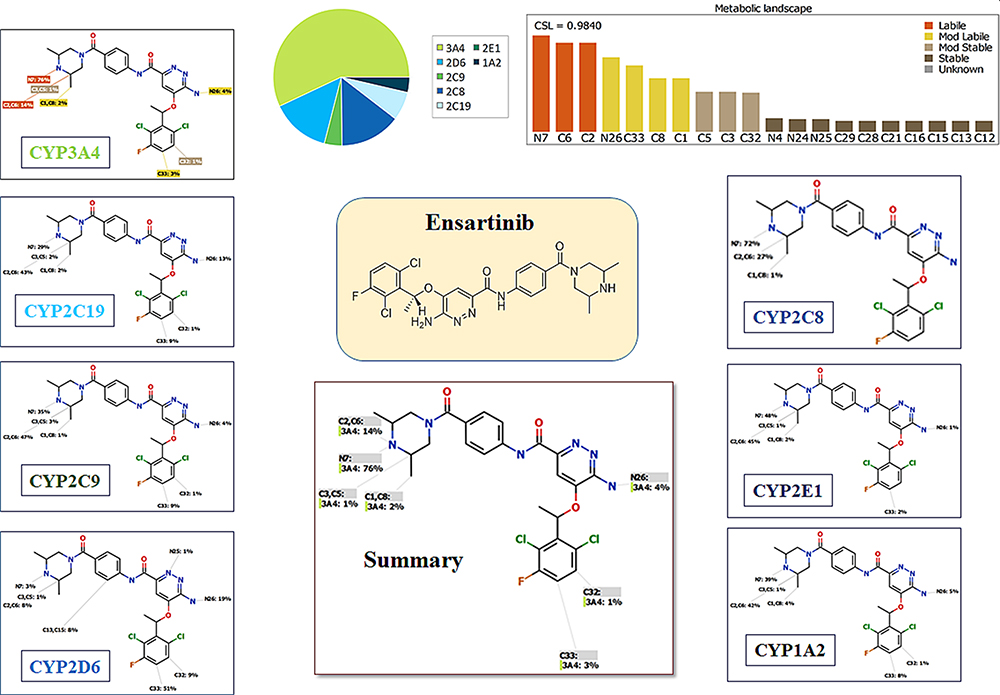 |
Figure 2 Suggested metabolic labile sites for ensartinib (ESB) by CYPP450 module of StarDrop software. |
In silico ESB Bioactivity and Toxicity Prediction
The vulnerable electrophilic atomic sites of ESB were proposed for cyanide and GSH attack and conjugation. The outcomes were represented by a color scale bar that has a color gradient as follows: the red color indicates the maximum possibility of reactive intermediate generation at the site, whereas the white color indicates no possibility of reactive intermediate formation (Figure 3A). The cyanide model indicates that the piperazine ring could be metabolized generating reactive iminium intermediates that could be attacked by cyanide ion forming cyano adduct. Furthermore, dichlorophenyl and pyridazine rings were predicted for GSH conjugation. The faint blue color means that forming reactive intermediates are less probable, which was confirmed by in vitro experiments. The outcomes of the predictions are shown in Figure 3A.
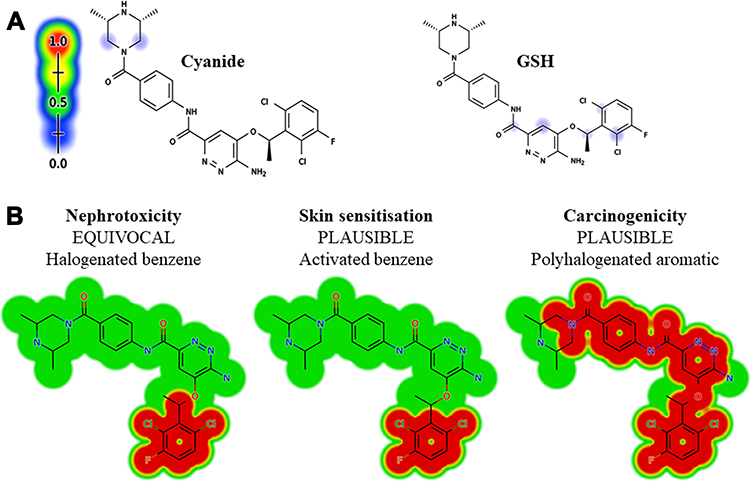 |
Figure 3 Outcomes of in silico reactivity tool expectation by the Xenosite including Cyanide and GSH (A). Structural alerts of ESB as proposed by DEREK toxicity predictor software (B). |
As proposed by DEREK software (in silico toxicity evaluation program), ESB and related metabolites were expected to exhibit skin sensitization, nephrotoxicity, and carcinogenicity due to activated benzene, halogenated benzene, and polyhalogenated aromatic, respectively, which were toxicity alerts by the software (Figure 3B). On the other hand, M1, M2, and M3 were expected to exhibit carcinogenicity due to alkyl aryl moiety. Table 2 lists the in vitro ESB stable metabolites with DEREK results for the predicted toxicity profile (Figure 3B).
|
Table 2 Qualitative Toxicity Prediction (DEREK) of the ESB and Its Metabolites |
ESB Dissociation Pattern
The ESB CP appeared at 24.0 min in the TIC (Figure 4A). CID of the ESB at m/z 561 resulted in the generation of three qualitative fragment ions at m/z 447, 257, and 138, showing weak chemical bonds in the ESB structure (Figure 4B, Scheme 1). The fragment ion at m/z 447 reveals the loss of the dimethyl piperazine ring, whereas the other two fragment ions at m/z 138 and m/z 257 have qualitative significance in the structural rebuilding of the related metabolites.
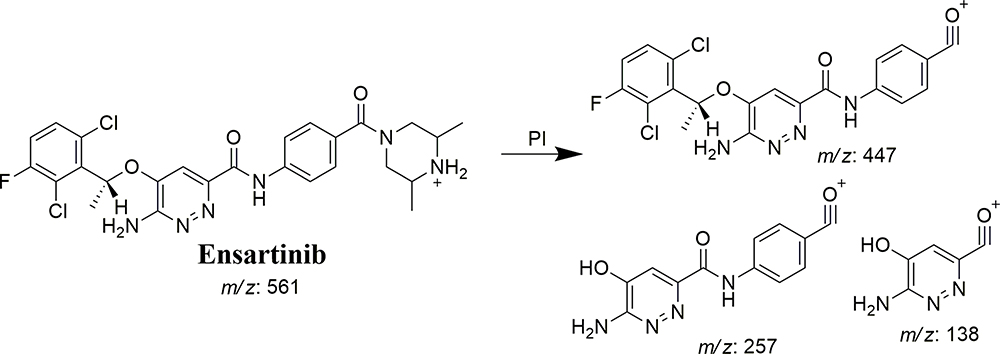 |
Scheme 1 ESB fragment ions. |
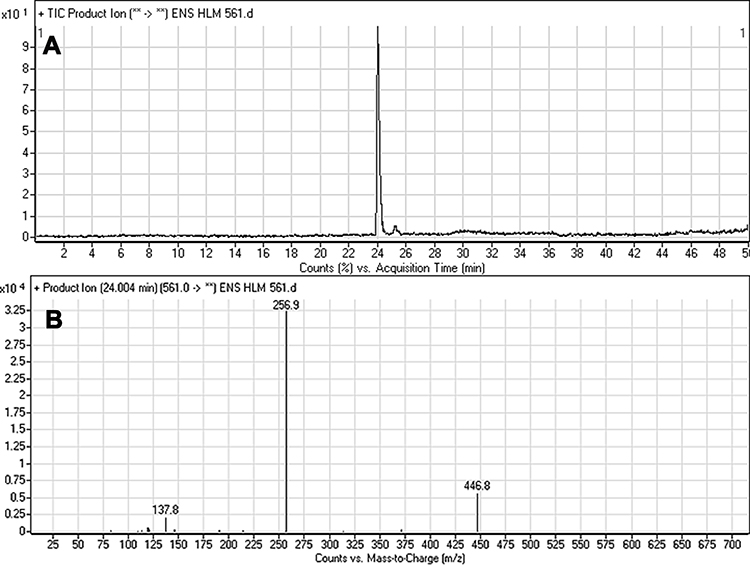 |
Figure 4 Fragment ion (m/z 561) chromatogram displaying the ESB peak at 24.0 min (A). Fragment ions of ESB (B). |
Identification of ESB Stable and Reactive Metabolites
Four ESB metabolites were formed by two phase I metabolic pathways (oxidation and hydroxylation). One GSH adduct and two cyano adducts were characterized (Table 3). One GSH conjugate and two cyano adducts were identified after HLM incubation of ESB in the presence of trapping agents (GSH or KCN). Compared to CID of ESB, neutral loss of HCN (27 m/z) or fragment ion with 25 m/z more was indicative of a cyano group addition.
|
Table 3 In vitro Metabolites and Reactive Intermediates of Ensartinib |
M1 and M2 Fragmentation Patterns
The ESB575 CPs appeared at two elution times (20.9 min and 23.3 min) in the TIC (Figure 5A). The CID of M1 and M2 ions at m/z 575 resulted in the generation of different fragment ions for the two peaks, whereas the CID of M1 and M2 ions at m/z 447 and 257 led to the generation of two qualitative fragment ions (Figure 5B and C). Compared to the CID of ESB, the fragment ions at m/z 447 and 257 were indicative of the oxidation metabolic pathway occurring at the piperazine moiety without knowing the exact place of hydroxylation, so a Markush structure was used to indicate the ambiguous structural assignment (Scheme 2). Oxidation may occur at α-carbon atoms to the tertiary nitrogen of piperazine moiety or the methyl group attached to the piperazine ring.
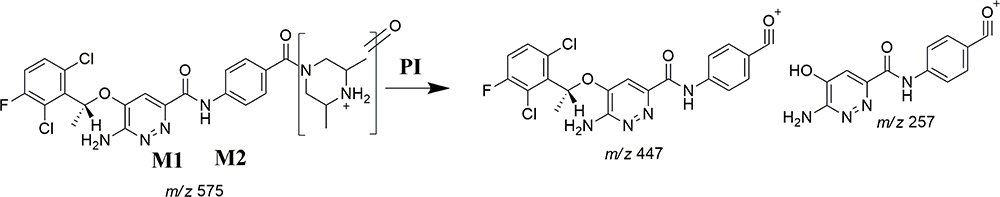 |
Scheme 2 M1 and M2 fragment ions. |
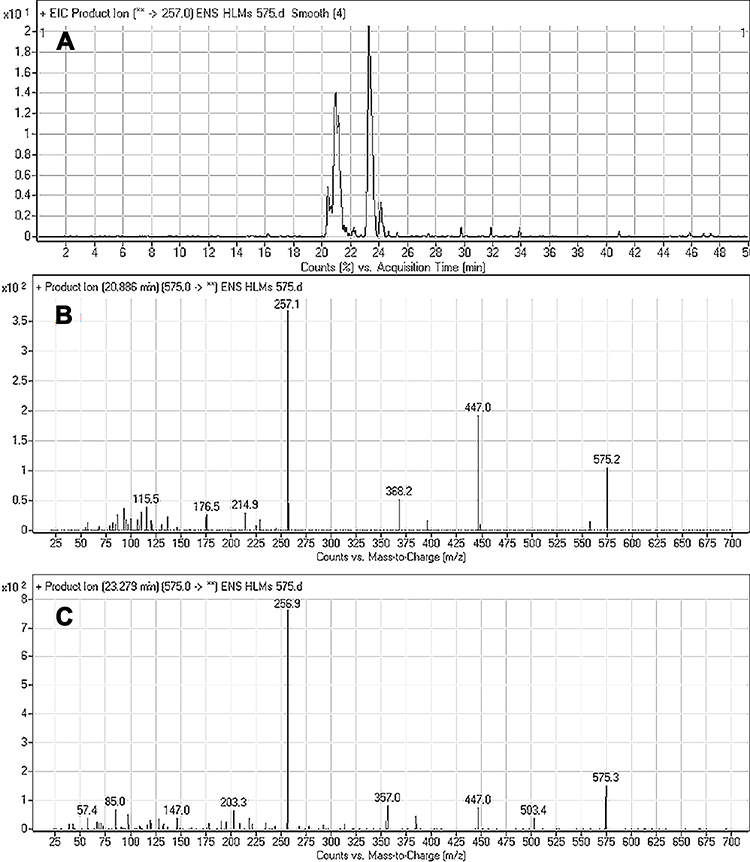 |
Figure 5 Fragment ion (m/z 575) chromatogram displaying the M1 and M2 peaks at 20.9 min and 23.3 min, respectively (A). Fragment ions of M1 (B). Fragment ions of M2 (C). |
M3 and M4 Fragmentation Patterns
M3 and M4 CPs appeared at two elution times (18.9 min and 21.5 min), respectively, in the TIC (Figure 6A). The CID of M3 and M4 ions at m/z 577 resulted in the generation of various fragment ions for the two peaks. The CID of M3 ion generated five fragment ions at m/z 463, 371, 327, 257, and 120 (Figure 6B). Compared to the CID of ESB, the fragment ions at m/z 463 (16 m/z more) were indicative of the hydroxylation metabolic pathway occurring at phenyl ring confirmed by the four fragment ions at m/z 371, 327, 257, and 120 (Scheme 3). The CID of M4 ion resulted in the generation of three fragment ions at m/z 447, 257, and 147. The exact place of hydroxylation at the phenyl ring is not known; thus, a Markush structure was used to detect the ambiguous structural assignment (Figure 6C). Compared to CID of ESB, the fragment ions at m/z 447 and 257 were indicative of the hydroxylation metabolic pathway occurring at the piperazine moiety without knowing the exact place of hydroxylation; therefore, a Markush structure was used to indicate the ambiguous structural assignment (Scheme 4). Hydroxylation may occur at α-carbon atoms to the tertiary nitrogen of piperazine moiety or the methyl group attached to the piperazine ring (Scheme 4).
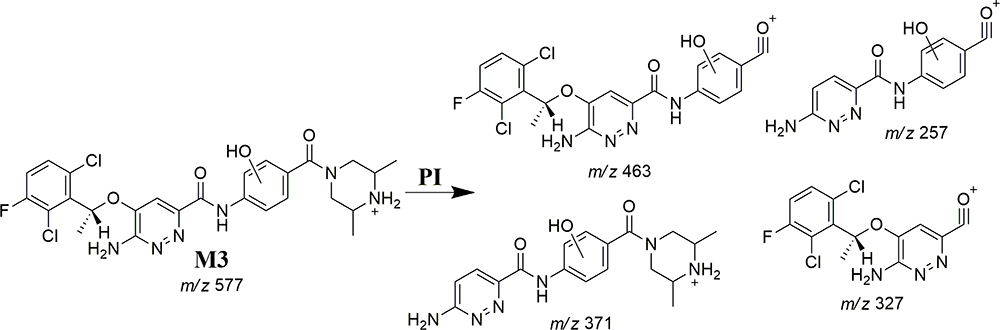 |
Scheme 3 M3 fragment ions. |
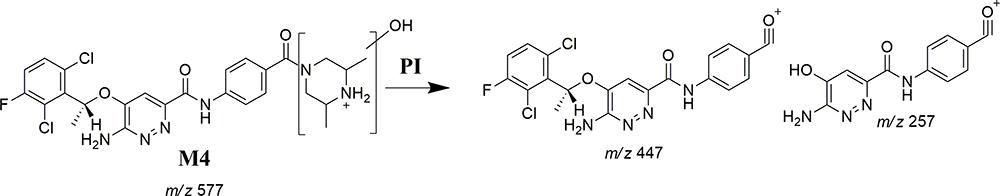 |
Scheme 4 M4 fragment ions. |
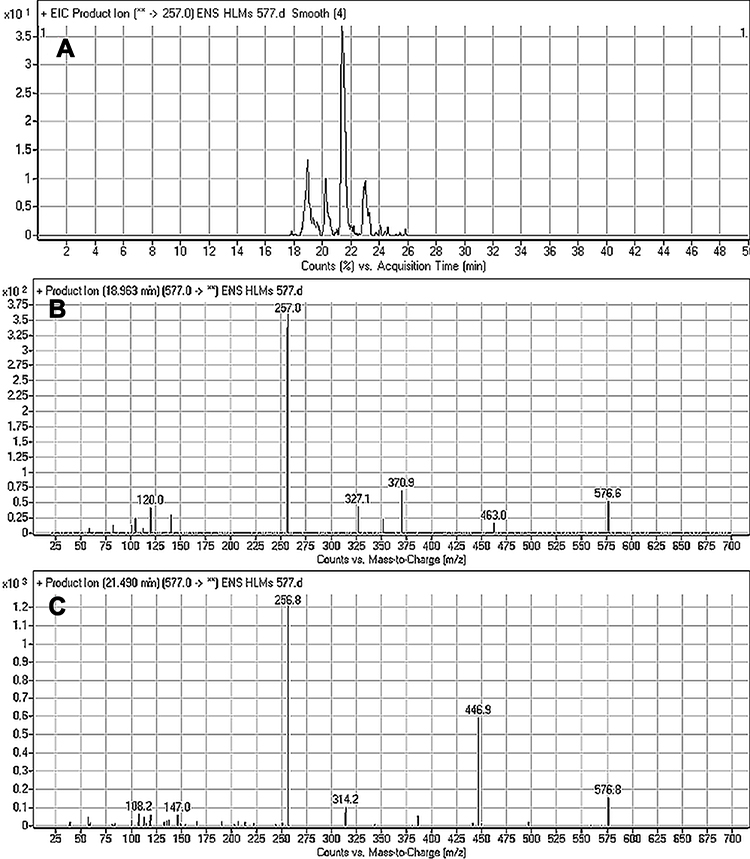 |
Figure 6 Fragment ion (m/z 575) chromatogram displaying the M3 and M4 peaks at 18.9 min and 21.5 min, respectively (A). Fragment ions of M3 (B). Fragment ions of M4 (C). |
ESB602a and ESB602b Fragmentation Patterns
The ESB602 CP appeared at the elution time (26.5 min) in the TIC (Figure 7A). ESB602 molecular ion fragmentation resulted in the formation of three qualitative fragment ions at m/z 557, 447, and 257 (Figure 7B). Compared to CID of ESB, the fragment ions at m/z 447 and 257 proposed that all metabolic pathways occurred at the dimethyl piperazine moiety, keeping the remaining structure unchanged. DI at 557 represented the neutral loss of one molecule of water and one molecule of HCN, revealing hydroxylation and cyano addition at the piperazine ring. The hydroxylation metabolic reaction and cyano addition occurred at the two α-carbon atoms to the tertiary nitrogen at the piperazine ring (Scheme 5).
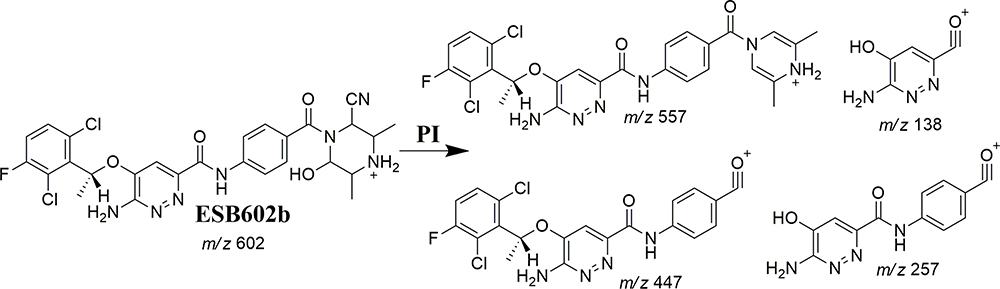 |
Scheme 5 ESB602b fragment ions. |
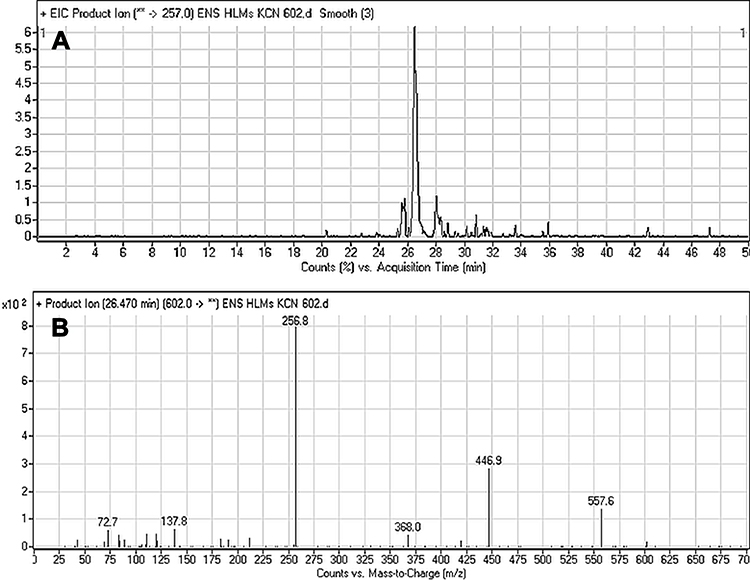 |
Figure 7 Fragment ion (m/z 602) chromatogram displaying the ESB602 peak at 26.5 min (A). Fragment ions of ESB602 (B). |
ESB866 Fragmentation Pattern
The ESB866 CP appeared at 16.8 min in the TIC chromatogram (Figure 8A). CID of ESB866 ion at m/z 866 resulted in the generation of five fragment ions at m/z 848, 790 496, 371, and 257 (Figure 8B). Compared to the CID of ESB, the fragment ions at m/z 371 and 257 indicated the metabolic bioactivation and then conjugation with GSH at the dichlorophenyl ring. We proposed that epoxide was formed by the direct oxidation of the dichlorophenyl ring generating reactive intermediate (quinone methide) attacked by the GSH-forming adduct (Scheme 6). This mechanism of metabolic bioactivation has been discussed for other compounds (eg, X-376 and diclofenac).31,42,43
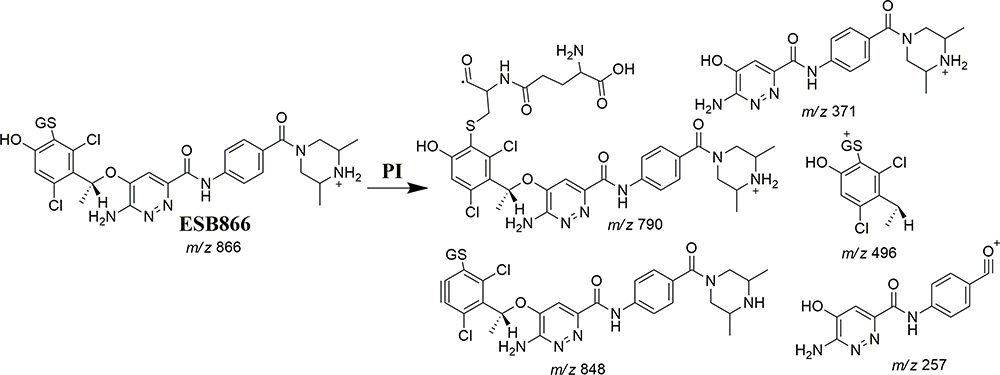 |
Scheme 6 ESB866 fragment ions. |
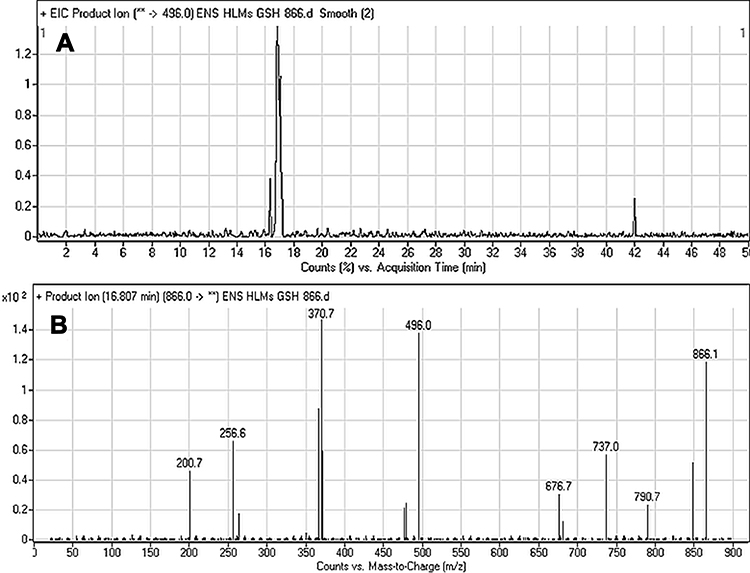 |
Figure 8 Fragment ion (m/z 866) chromatogram displaying the ESB866 chromatographic peak at 16.8 min (A). Fragment ions of ESB866 (B). |
ESB Bioactivation Sequence
ESB602 cyanide adduct characterization revealed the formation of unstable iminium metabolites in the in vitro metabolism of ESB and HLMs. The metabolic bioactivation process was initiated by hydroxylation metabolic reaction at the bioactivated piperazine ring in ESB followed by water loss (dehydration), which generated unstable metabolites. These unstable metabolites (reactive intermediates) could be stabilized by cyanide nucleophile forming cyano adducts (ESB602) (Scheme 7). The proposed pathway of the iminium intermediate formation in ESB in vitro metabolism has been previously discussed in the literature.44–47
ESB866 GSH conjugate revealed the formation of reactive intermediate through oxidative defluorination process in the ESB in vitro metabolism generating quinone methide intermediate that was captured by GSH (Scheme 8).31 We proposed that epoxide was formed by the direct oxidation of the dichlorophenyl ring generating reactive intermediate that was attacked by GSH-forming adducts. The formation of GSH conjugate was confirmed using negative precursor ion mode (272) and positive neutral loss (129) mode of triple quadrupole mass analyzer.
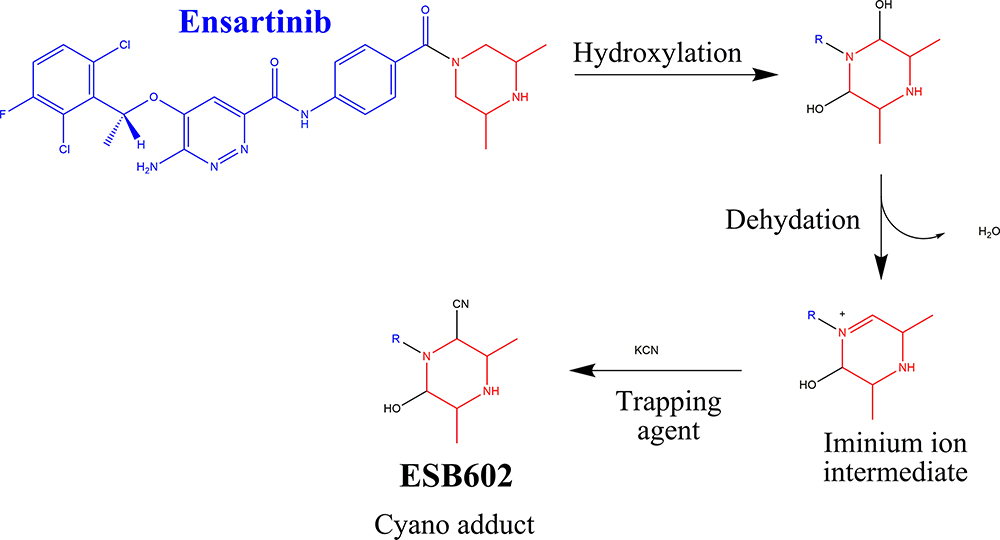 |
Scheme 7 Supposed metabolic sequence for reactive iminium intermediate formation at piperazine rings in ESB metabolism and a potential capturing strategy. |
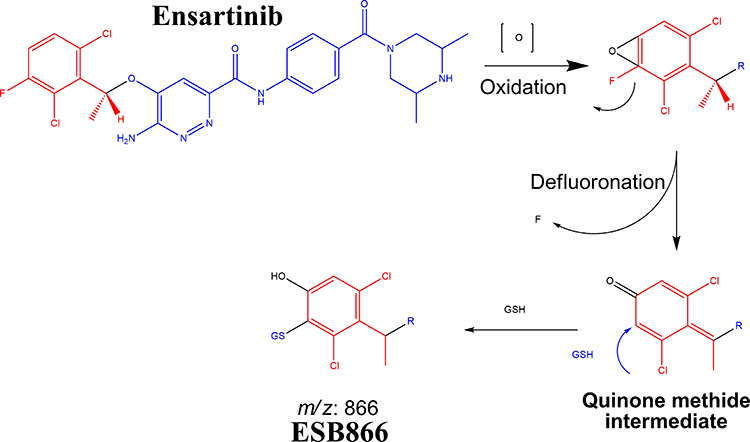 |
Scheme 8 Supposed pathways of GSH conjugation through oxidative defluorination mechanism generating quinone methide intermediate. |
Conclusions
Two metabolic reactions (oxidation and hydroxylation) generated four stable phase I ESB metabolites in the in vitro metabolism of ESB. The bioactivation sequence resulted in the generation of one GSH conjugate and one cyano adduct (Figure 9). The piperazine ring was metabolically bioactivated due to the generation of an unstable reactive intermediate that may be responsible for ESB side-effects. The dichlorophenyl ring was bioactivated by oxidative defluorination pathway generating quinone methide intermediate, which was verified using LC-MS/MS. These findings provide the basis for further studies on ESB toxicity. Substituents to the bioactive centers (piperazine ring) for either blocking or isosteric replacement would likely block or interrupt the hydroxylation reaction that will end the bioactivation sequence. Future drug discovery studies can be conducted following this concept, which allows designing novel drugs with more safety profile without influencing their pharmacological effects.
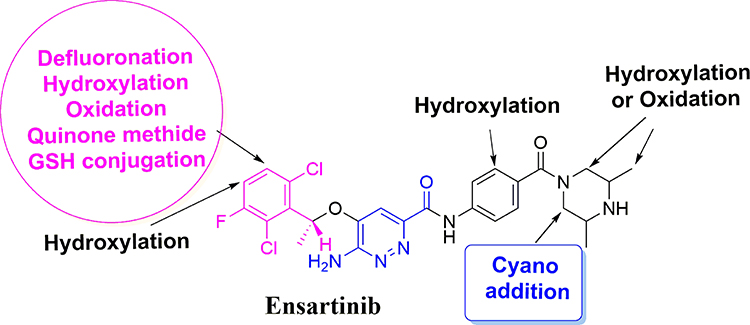 |
Figure 9 Chemical structure of ensartinib displaying various metabolic phase I pathways and the proposed bioactive sites. |
Abbreviations
ESB, ensartinib; ESI, electrospray ionization; HLMs, human liver microsomes; TKIs, tyrosine kinase inhibitors; LC-MS/MS, liquid chromatography tandem mass spectrometry; GSH, reduced glutathione; KCN, potassium cyanide; NSCLCs, Non-small cell lung cancers; ALK, Anaplastic lymphoma kinase; CSL, composite site lability; HPLC, high-performance liquid chromatography; ABL, Abelson murine leukemia; MET, met proto-oncogene; ROS1, v-ros UR2 sarcoma virus oncogene homolog 1; Axl, receptor tyrosine kinase.
Ethics
The study’s design (in vitro experiments using commercially available human liver microsomes) exempts it from the need of the Ethics Committees approval.
Acknowledgments
The authors extend their appreciation to the Deputyship for Research & Innovation, “Ministry of Education” in Saudi Arabia for funding this research work through the project number IFKSURG-1435-025
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest for this work.
References
1. World Health Organization. Department of information, evidence and research, WHO cancer fact sheet; 2018. https://www.who.int/news-room/fact-sheets/detail/cancer.
2. Barinaga M. Cancer research: from bench top to bedside. Science. 1997;278:1036–1039. doi:10.1126/science.278.5340.1036
3. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi:10.1002/ijc.29210
4. Ettinger DS, Akerley W, Bepler G, et al. Non–small cell lung cancer. J Natl Comprehen Cancer Netw. 2010;8:740–801. doi:10.6004/jnccn.2010.0056
5. Larsen JE, Cascone T, Gerber DE, Heymach JV, Minna JD. Targeted therapies for lung cancer. Cancer J (Sudbury, Mass). 2011;17:512. doi:10.1097/PPO.0b013e31823e701a
6. Cardarella S, Johnson BE. The impact of genomic changes on treatment of lung cancer. Am J Respir Crit Care Med. 2013;188:770–775. doi:10.1164/rccm.201305-0843PP
7. Li T, Kung H-J, Mack PC, Gandara DR. Genotyping and genomic profiling of non–small-cell lung cancer: implications for current and future therapies. J Clin Oncol. 2013;31:1039–1049.
8. Moreira AL, Thornton RH. Personalized medicine for non–small-cell lung cancer: implications of recent advances in tissue acquisition for molecular and histologic testing. Clin Lung Cancer. 2012;13:334–339. doi:10.1016/j.cllc.2012.01.004
9. Fisher E, Charbonneau H, Tonks N. Protein tyrosine phosphatases: a diverse family of intracellular and transmembrane enzymes. Science. 1991;253:401–406. doi:10.1126/science.1650499
10. Gridelli C, Peters S, Sgambato A, Casaluce F, Adjei AA, Ciardiello F. ALK inhibitors in the treatment of advanced NSCLC. Cancer Treat Rev. 2014;40:300–306. doi:10.1016/j.ctrv.2013.07.002
11. Goldstraw P, Ball D, Jett JR, et al. Non-small-cell lung cancer. Lancet. 2011;378:1727–1740. doi:10.1016/S0140-6736(10)62101-0
12. Sen Zhang F, Keats J, Ning Y, et al. AP26113, a potent ALK inhibitor, overcomes mutations in EML4-ALK that confer resistance to PF-02341066. AACR Poster Session. 2010.
13. Squillace RM, Anjum R, Miller D, et al. AP26113 possesses pan-inhibitory activity versus crizotinib-resistant ALK mutants and oncogenic ROS1 fusions. 2013.
14. Karachaliou N, Santarpia M, Gonzalez Cao M, et al. Osimertinib in the treatment of non-small-cell lung cancer: design, development and place in therapy. Expert Opin Investig Drugs. 2017;26:713–722.
15. Ziogas DC, Tsiara A, Tsironis G, et al. Treating ALK-positive non-small cell lung cancer. Ann Transl Med. 2018;6:141.
16. Horn L, Infante JR, Reckamp KL, et al. Ensartinib (X-396) in ALK-positive non–small cell lung cancer: results from a first-in-human phase I/II, multicenter study. Clin Cancer Res. 2018.
17. Horn L, Infante JR, Reckamp KL, et al. Ensartinib (X-396) in ALK-positive non–small cell lung cancer: results from a first-in-human phase I/II, multicenter study. Clin Cancer Res. 2018. doi:10.1158/1078-0432.ccr-17-2398
18. Evans DC, Watt AP, Nicoll-Griffith DA, Baillie TA. Drug−protein adducts: an industry perspective on minimizing the potential for drug bioactivation in drug discovery and development. Chem Res Toxicol. 2004;17:3–16. doi:10.1021/tx034170b
19. Kalgutkar AS, Dalvie DK, O’Donnell JP, Taylor TJ, Sahakian DC. On the diversity of oxidative bioactivation reactions on nitrogen- containing xenobiotics. Curr Drug Metab. 2002;3:379–424. doi:10.2174/1389200023337360
20. Boelsterli UA. Xenobiotic Acyl glucuronides and Acyl CoA thioesters as protein- reactive metabolites with the potential to cause idiosyncratic drug reactions. Curr Drug Metab. 2002;3:439–450. doi:10.2174/1389200023337315
21. Knowles SR, Uetrecht J, Shear NH. Idiosyncratic drug reactions: the reactive metabolite syndromes. Lancet. 2000;356:1587–1591. doi:10.1016/S0140-6736(00)03137-8
22. Ju C, Uetrecht J. Mechanism of idiosyncratic drug reactions: reactive metabolites formation, protein binding and the regulation of the immune system. Curr Drug Metab. 2002;3:367–377. doi:10.2174/1389200023337333
23. Ma S, Zhu M. Recent advances in applications of liquid chromatography–tandem mass spectrometry to the analysis of reactive drug metabolites. Chem Biol Interact. 2009;179:25–37. doi:10.1016/j.cbi.2008.09.014
24. Stepan AF, Walker DP, Bauman J, et al. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: a perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem Res Toxicol. 2011;24:1345–1410. doi:10.1021/tx200168d
25. AlRabiah H, Kadi AA, Attwa MW, Abdelhameed AS. A simple liquid chromatography-tandem mass spectrometry method to accurately determine the novel third-generation EGFR-TKI naquotinib with its applicability to metabolic stability assessment. RSC Adv. 2019;9:4862–4869. doi:10.1039/C8RA09812C
26. Attwa MW, Kadi AA, Abdelhameed AS, Alhazmi HA. Metabolic stability assessment of new parp inhibitor talazoparib using validated LC–MS/MS methodology: in silico metabolic vulnerability and toxicity studies. Drug Des Devel Ther. 2020;14:783–793. doi:10.2147/DDDT.S239458
27. Masic LP. Role of cyclic tertiary amine bioactivation to reactive iminium species: structure toxicity relationship. Curr Drug Metab. 2011;12:35–50. doi:10.2174/138920011794520044
28. Zhang Z, Chen Q, Li Y, et al. In vitro bioactivation of dihydrobenzoxathiin selective estrogen receptor modulators by cytochrome P450 3A4 in human liver microsomes: formation of reactive iminium and quinone type metabolites. Chem Res Toxicol. 2005;18:675–685. doi:10.1021/tx0496789
29. Park BK, Boobis A, Clarke S, et al. Managing the challenge of chemically reactive metabolites in drug development. Nat Rev Drug Discov. 2011;10:292–306. doi:10.1038/nrd3408
30. Attwa MW, Kadi AA, Darwish HW, Amer SM, Al-shakliah NS. Identification and characterization of in vivo, in vitro and reactive metabolites of vandetanib using LC–ESI–MS/MS. Chem Cent J. 2018;12:99. doi:10.1186/s13065-018-0467-5
31. Waldon DJ, Teffera Y, Colletti AE, et al. Identification of quinone imine containing glutathione conjugates of diclofenac in rat bile. Chem Res Toxicol. 2010;23:1947–1953. doi:10.1021/tx100296v
32. Ma S, Subramanian R. Detecting and characterizing reactive metabolites by liquid chromatography/tandem mass spectrometry. J Mass Spectrom. 2006;41:1121–1139. doi:10.1002/jms.1098
33. Tolonen A, Turpeinen M, Pelkonen O. Liquid chromatography–mass spectrometry in in vitro drug metabolite screening. Drug Discov Today. 2009;14:120–133. doi:10.1016/j.drudis.2008.11.002
34. Zweig J, Wakelee H. The evolution of frontline therapy in ALK-positive advanced NSCLC: which ALK TKI to use upfront?.Am J Hematol Oncol. 2017;13:32–36.
35. Hughes TB, Dang NL, Miller GP, Swamidass SJ. Modeling reactivity to biological macromolecules with a deep multitask network. ACS Cent Sci. 2016;2:529–537. doi:10.1021/acscentsci.6b00162
36. Matlock MK, Hughes TB, Swamidass SJ. XenoSite server: a web-available site of metabolism prediction tool. Bioinformatics. 2015;31:1136–1137. doi:10.1093/bioinformatics/btu761
37. Zaretzki J, Matlock M, Swamidass SJ. XenoSite: accurately predicting CYP-mediated sites of metabolism with neural networks. J Chem Inf Model. 2013;53:3373–3383. doi:10.1021/ci400518g
38. Abdelhameed AS, Kadi AA, Attwa MW, AlRabiah H, Ahmad A. Validated LC-MS/MS assay for quantification of the newly approved tyrosine kinase inhibitor, dacomitinib, and application to investigating its metabolic stability. PLoS One. 2019;14:e0214598. doi:10.1371/journal.pone.0214598
39. Abdelhameed AS, Attwa MW, Kadi AA. An LC-MS/MS method for rapid and sensitive high-throughput simultaneous determination of various protein kinase inhibitors in human plasma. Biomed Chromatogr. 2017;31:e3793. doi:10.1002/bmc.3793
40. Kadi AA, Abdelhameed AS, Darwish HW, Attwa MW, Al‐Shakliah NS. A highly efficient and sensitive LC-MS/MS method for the determination of afatinib in human plasma: application to a metabolic stability study. Biomed Chromatogr. 2016;30:1248–1255. doi:10.1002/bmc.3674
41. Abdelhameed AS, Attwa MW, Al-Shaklia NS, Kadi AA. A highly sensitive LC-MS/MS method to determine novel Bruton’s tyrosine kinase inhibitor spebrutinib: application to metabolic stability evaluation. Royal Soc Open Sci. 2019;6:190434. doi:10.1098/rsos.190434
42. Attwa MW, Kadi AA, Abdelhameed AS. Phase I metabolic profiling and unexpected reactive metabolites in human liver microsome incubations of X-376 using LC-MS/MS: bioactivation pathway elucidation and in silico toxicity studies of its metabolites. RSC Adv. 2020;10:5412–5427. doi:10.1039/C9RA09115G
43. Mandal M, Mitra K, Grotz D, et al. Overcoming Time-Dependent Inhibition (TDI) of cytochrome P450 3A4 (CYP3A4) resulting from bioactivation of a fluoropyrimidine moiety. J Med Chem. 2018;61:10700–10708. doi:10.1021/acs.jmedchem.8b01326
44. AlRabiah H, Kadi AA, Attwa MW, Abdelhameed AS, Mostafa GAE. Reactive intermediates in copanlisib metabolism identified by LC-MS/MS: phase I metabolic profiling. RSC Adv. 2019;9:6409–6418. doi:10.1039/C8RA10322D
45. Attwa MW, Kadi AA, Abdelhameed AS. Detection and characterization of olmutinib reactive metabolites by LC–MS/MS: elucidation of bioactivation pathways. J Sep Sci. 2020;43:708–718. doi:10.1002/jssc.201900818
46. Attwa MW, Kadi AA, Abdelhameed AS. Characterization of reactive intermediates formation in dacomitinib metabolism and bioactivation pathways elucidation by LC-MS/MS: in vitro phase I metabolic investigation. RSC Adv. 2018;8:38733–38744. doi:10.1039/C8RA06709K
47. Attwa MW, Kadi AA, Abdelhameed AS. Reactive intermediates and bioactivation pathways characterization of avitinib by LC–MS/MS: in vitro metabolic investigation. J Pharm Biomed Anal. 2019;164:659–667. doi:10.1016/j.jpba.2018.11.033
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.