")
Back to Journals » Infection and Drug Resistance » Volume 15
Characterization of Fluoroquinolone-Resistant and Multidrug-Resistant Mycobacterium tuberculosis Isolates Using Whole-Genome Sequencing in Tianjin, China
Authors Wang Z , Sun R, Mu C, Wang C, Zhao H, Jiang L, Ju H, Dai W, Zhang F
Received 11 February 2022
Accepted for publication 2 April 2022
Published 13 April 2022 Volume 2022:15 Pages 1793—1803
DOI https://doi.org/10.2147/IDR.S361635
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Zhirui Wang,* Rui Sun,* Cheng Mu, Chunhua Wang, Hui Zhao, Lina Jiang, Hanfang Ju, Wenxi Dai, Fan Zhang
Tuberculosis Reference Laboratory, Tianjin Center for Tuberculosis Control, Tianjin, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Fan Zhang, Tuberculosis Reference Laboratory, Tianjin Center for Tuberculosis Control, No. 124, Chifeng Road, Heping District, Tianjin, 300041, People’s Republic of China, Tel +86-22-27124491, Fax +86-22-27117595, Email [email protected]
Objective: Tuberculosis (TB) caused by Mycobacterium tuberculosis remains a global concern. This study aimed to determine the molecular characteristics of fluoroquinolone-resistant and multidrug-resistant M. tuberculosis strains using whole-genome sequencing to predict drug resistance in M. tuberculosis in Tianjin, China, which has not been established previously.
Methods: Twenty-one fluoroquinolone-resistant and multidrug-resistant M. tuberculosis strains were isolated from sputum samples. Phenotypic drug resistance against 12 anti-tuberculosis drugs was determined using drug susceptibility testing. Whole-genome sequencing was performed to predict drug resistance in M. tuberculosis based on genome regions associated with drug resistance. The sensitivity of whole-genome sequencing for predicting drug resistance was calculated based on phenotypic drug susceptibility testing information.
Results: Among the 21 isolates, mutations in 15 genome regions associated with drug resistance, including rpoB for rifampicin; katG and inhA promoter for isoniazid; gyrA and gyrB for ofloxacin and moxifloxacin; rpsL for streptomycin; rrs for streptomycin, amikacin, kanamycin and capreomycin; pncA and panD for pyrazinamide; embB, embC-embA, aftA, and ubiA for ethambutol; ethA for protionamide; and folC for para-aminosalicylic acid, were detected. Compared with traditional drug susceptibility testing results, the sensitivities for whole-genome sequencing of rifampin, isoniazid, ofloxacin, moxifloxacin, streptomycin, ethambutol, pyrazinamide, kanamycin, and amikacin resistance were 100%, 90.48%, 95.24%, 92.86%, 95.27%, 85.71%, 66.67%, 50%, and 50%, respectively. The sensitivities for whole-genome sequencing of capreomycin, protionamide, and para-aminosalicylic acid were not calculated because only one isolate showed phenotypic drug resistance. Mutations determined in drug susceptibility-associated genes can explain phenotypic drug resistance in most isolates. Notably, these mutations were absent in certain drug-resistant isolates, indicating other drug resistance mechanisms.
Conclusion: Whole-genome sequencing represents an effective diagnostic tool for fluoroquinolone-resistant and multidrug-resistant TB though it has some obstacles. Whole-genome sequencing should be used to predict drug resistance prior to performing traditional phenotypic drug susceptibility testing in Tianjin, China.
Keywords: Mycobacterium tuberculosis, tuberculosis, fluoroquinolone, mutation, drug resistance, whole-genome sequencing
Introduction
Tuberculosis (TB), which is caused by the slow-growing bacterium Mycobacterium tuberculosis, can spread via cough aerosols and remains a global concern. According to a recent report from the World Health Organization (WHO), the estimated number of new cases of active TB was 9.9 million and the number of deaths was 1.3 million in 2020 alone.1 The emergence of drug-resistant TB, especially multidrug-resistant (MDR) TB, which is resistant to both isoniazid and rifampicin, represents a challenge to global TB control.1 Second-line drugs are recommended for the treatment of MDR-TB, such as fluoroquinolones, including ofloxacin, levofloxacin, and moxifloxacin, which are broad-spectrum antibacterial agents and are considered as the most effective second-line drugs for treating MDR-TB as recommended by the WHO.1 However, the emergence of fluoroquinolone-resistant MDR M. tuberculosis strains has been increasingly reported worldwide,2–4 hindering TB management and complicating the therapy required for these patients. Treatment outcomes of fluoroquinolone-resistant MDR-TB remain poor,4 and an appropriate use of drugs for treating patients with fluoroquinolone-resistant MDR-TB is recommended by the WHO.1 However, prior to developing a treatment strategy, an accurate and rapid diagnosis of fluoroquinolone-resistant MDR-TB is necessary.
Traditional diagnosis of TB caused by drug-resistant M. tuberculosis relies on culture-based techniques that are time-consuming and labor-intensive. Recently, techniques for the rapid molecular diagnosis of TB have been increasingly used worldwide, such as the Xpert MTB/RIF assay, which can be used to detect the presence of M. tuberculosis and its resistance to rifampicin,5 and Xpert MTB/XDR (not for sale in China), which can detect the presence of M. tuberculosis showing resistance to isoniazid, fluoroquinolones, ethionamide, and second-line injectable drugs.3 However, these molecular techniques cannot simultaneously detect resistance to other antitubercular drugs, especially pyrazinamide and other second-line drugs. Recently, whole-genome sequencing of M. tuberculosis has been increasingly used to detect drug resistance genes in M. tuberculosis based on chromosomal mutations in existing genes.6–9 However, the diversity of fluoroquinolone-resistant MDR M. tuberculosis varies geographically, and its prevalence has not been determined in Tianjin, China. Therefore, we performed whole-genome sequencing of fluoroquinolone-resistant MDR M. tuberculosis isolates in the present study, which was compared with traditional drug susceptibility testing (DST) data to develop diagnostics and treatment strategies in Tianjin, China.
Materials and Methods
Ethics Board Approval
This study was approved by the ethics board of the Tianjin Centers for Disease Control and Prevention, Tianjin, China (approval no.: TJCDC191). In this study, informed consent was waived because all M. tuberculosis strains were selected from the Tianjin drug-resistance tuberculosis surveillance program based on the DST phenotype. In addition, it was difficult to obtain the informed consent of participants because the samples (sputum) used were residual samples and not fresh samples collected for this study. These strains were used only for scientific research and were not used for clinical diagnosis.
Strains
From July 2017 to March 2020, a total of 2257 M. tuberculosis strains isolated from sputum samples of patients with TB were consecutively collected at the Tianjin Center of Tuberculosis Control; 21 of these isolates were fluoroquinolone-resistant MDR M. tuberculosis. The time points for collection of 21 fluoroquinolone-resistant MDR M. tuberculosis isolates are shown in Table 1. The sputum specimens obtained from patients were inoculated into Bactec MGIT 960 culture tubes to verify positive isolates after standard NALC-NaOH decontamination. Positive isolates were identified by culturing on Lowenstein-Jensen (L-J) medium containing p-nitrobenzoic-acid and thiophene-2-carboxylic hydrazide. Prior to DST on L-J medium using the proportion method, enrichment of the positive isolates was performed on L-J medium. All colonies of each M. tuberculosis culture on L-J agar slants were harvested and dissolved in 7H9 broth containing 20% glycerol to reach a suspension turbidity higher than that of McFarland 4.93 (the maximum). The bacterial suspension was then aliquoted to volumes of 1mL and stored at −80°C.
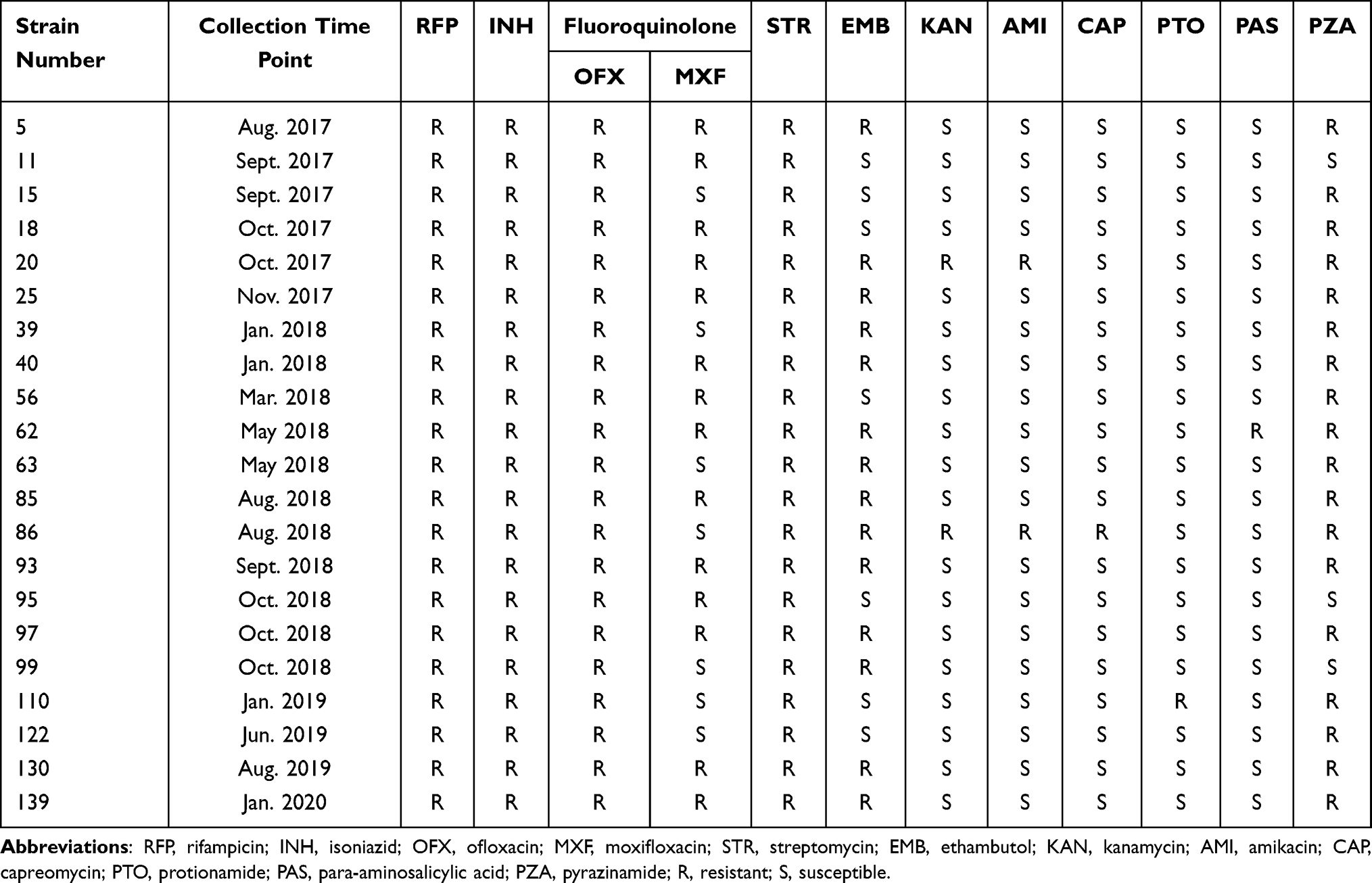 |
Table 1 Drug Resistance Phenotypic Pattern of 21 Fluoroquinolone-Resistant and Multidrug- Resistant Mycobacterium tuberculosis Strains |
DST
According to the Tianjin drug-resistance tuberculosis surveillance program, DST using the proportion method was performed for 11 anti-TB drugs on L-J medium, containing rifampicin (40 mg/L), isoniazid (0.2 mg/L), streptomycin (4 mg/L), ethambutol (2 mg/L), ofloxacin (4 mg/L), moxifloxacin (2 mg/L), amikacin (30 mg/L), kanamycin (30 mg/L), capreomycin (40 mg/L), protionamide (30 mg/L), and para-aminosalicylic acid (1 mg/L). DST for pyrazinamide susceptibility was performed using a Bactec MGIT 960 pyrazinamide kit, with a critical concentration of 100 mg/L for pyrazinamide. M. tuberculosis H37Rv was used as the reference isolate, which was susceptible to all drugs when each batch of 10 strains was tested. The results are reported as either susceptible or resistant. DST by L-J medium and Bactec MGIT 960 was performed three times to obtain consistent results.
DNA Extraction
A large inoculum containing colonies of the isolates was suspended in TE buffer containing lysozyme and proteinase K and incubated overnight at 37°C. Subsequently, CTAB/chloroform extraction and ethanol precipitation were performed to obtain DNA. DNA concentration was tested using a Qubit Fluorometer 3.0.
Whole-Genome Sequencing
Whole-genome sequencing was performed at Shanghai Gene-Optimal Science & Technology (Shanghai, China). DNA was sheared into approximately 200–300-bp fragments using a Covaris sonicator and prepared using the TIANSeq Whole-Genome Sequencing DNA Sample Preparation Kit (TianGen, Beijing, China). The samples were sequenced using an Illumina HiSeq 2500 sequencer. Base-calling was performed using PE 150 software. FastQC was used for quality control of SAM-TB. Cutadapt (version 1.15) was used to remove linkers and low-quality sequences. The retained high-quality sequences were aligned to the reference genome H37Rv (NC000962.3) using BWA (version 0.7.17). Picard (version 2.0.1) was used to mark and remove PCR repeat reads. SAM tools (version 1.6)/VarScan (version 2.3.6) were used to identify single-nucleotide polymorphisms and short indels. Delly (version 0.8.7) was used to identify larger deletions (≥50bp). By default, SAM-TB detects mutations that meet the following conditions: base matrix ≥30, mapping quality ≥30, ≥5 reads aligned to the site and no less than two reads that support the mutation, and mutation frequency ≥75%. The sequencing data were compared with that of M. tuberculosis isolate H37Rv to identify single-nucleotide polymorphisms. Known resistance loci of M. tuberculosis including the following 15 genes: rpoB, katG, inhA promoter, gyrA, gyrB, rpsL, rrs, pncA, panD, embB, embC-embA, aftA, ubiA, folC, and ethA2,6,10–21 were defined based on previously published studies. Data analysis was performed for isolates to identify single-nucleotide polymorphisms of the above loci and to predict drug resistance.
Lineage Identification
M. tuberculosis lineages and sub lineages were identified based on previously reported single-nucleotide polymorphisms via whole-genome sequencing.22
Data Analysis
The sensitivity, specificity, positive predictive value (PPV), and negative predictive value were calculated based on DST information, which is regarded as the gold standard.
Results
Phenotyping
Among 21 fluoroquinolone-resistant MDR M. tuberculosis strains, 100% (21/21) of the strains were resistant to rifampicin, isoniazid, ofloxacin, and streptomycin; 66.7% (14/21) of the strains were resistant to moxifloxacin; 85.7% (18/21) of the strains were resistant to pyrazinamide; 66.7% (14/21) of the strains were resistant to ethambutol; 9.5% (2/21) of the strains were resistant to kanamycin; 9.5% (2/21) of the strains were resistant to amikacin; 4.8% (1/21) of the strains were resistant to capreomycin; 4.8% (1/21) of the strains were resistant to protionamide; and 4.8% (1/21) of the strains were resistant to para-aminosalicylic acid (Table 1).
Genotyping
Among the 21 fluoroquinolone-resistant MDR M. tuberculosis strains, 95.2% (20/21) belonged to the East Asian lineage (lineage 2.2.1, 19 strains; lineage 2.2.2, 1 strain) and 4.8% (1/21) belonged to the Euro-American lineage (lineage 4.4.2, 1 strain) (Table 2). Among the 21 fluoroquinolone-resistant MDR strains, mutations in 15 genome regions associated with drug resistance were detected via whole-genome sequencing (Table 3), including rpoB for rifampicin; katG and inhA promoter for isoniazid; gyrA and gyrB for ofloxacin and moxifloxacin; rpsL for streptomycin; rrs for streptomycin, amikacin, kanamycin, and capreomycin; pncA and panD for pyrazinamide; embB, embC-embA, aftA, and ubiA for ethambutol; ethA for protionamide; and folC for para-aminosalicylic acid.
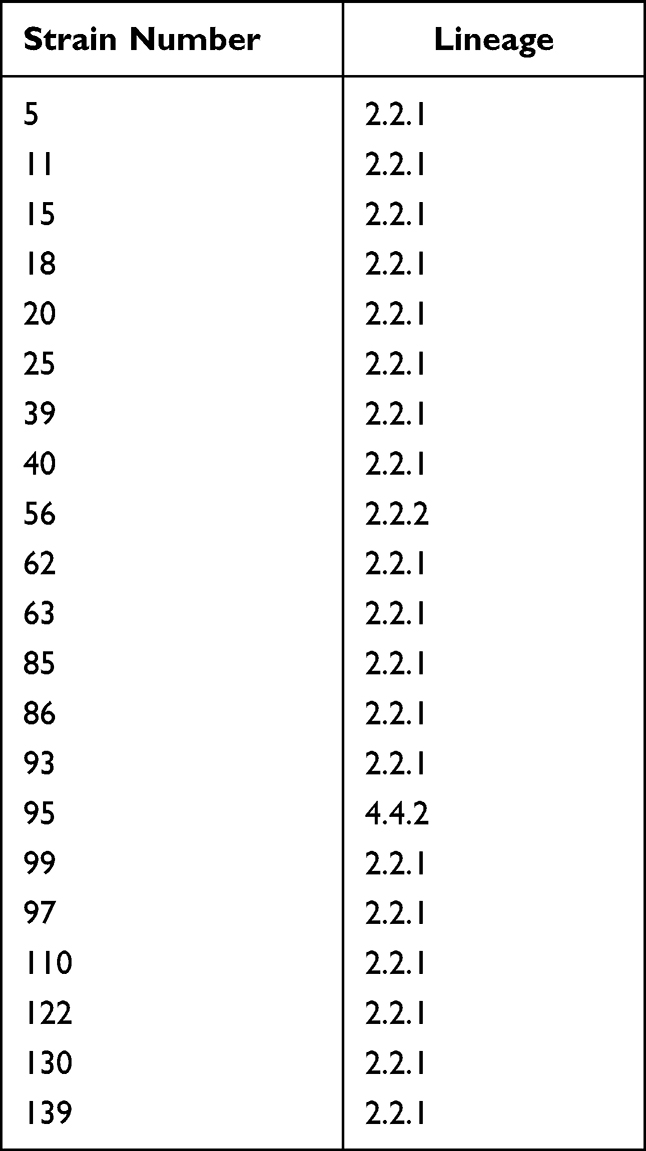 |
Table 2 Lineages of 21 Fluoroquinolone-Resistant and Multidrug-Resistant Mycobacterium tuberculosis Strains |
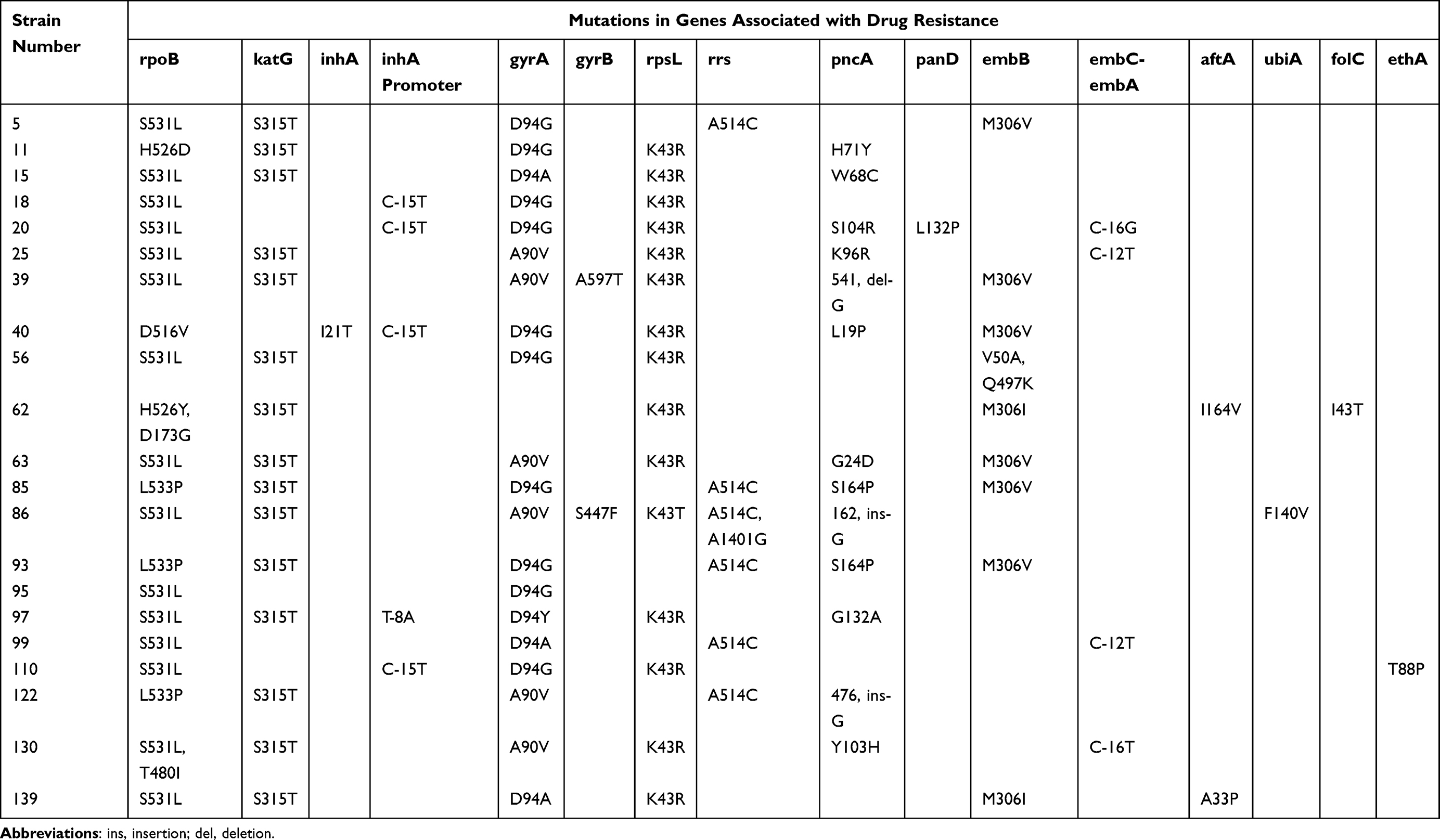 |
Table 3 Gene Mutation Pattern of 21 Fluoroquinolone-Resistant and Multidrug-Resistant Mycobacterium tuberculosis Strains |
rpoB Gene
All 21 fluoroquinolone-resistant MDR strains harbored rpoB gene mutations. Among these, 15 strains had an S531L mutation, 3 strains had an L533P mutation, 2 strains had an H526D mutation, and 1 strain had a D516V mutation (Table 3). These mutations in rpoB can confer rifampin resistance. Thus, 100% (21/21) of the strains were resistant to rifampin based on these genotypic mutations.
katG Gene and inhA Promoter
Among the 21 fluoroquinolone-resistant MDR strains, 14 strains had S315T mutations in katG, 3 strains had −15 C-T mutations in the inhA promoter, 1 strain had an −15 C-T mutation in the inhA promoter combined with an I21T mutation in inhA, 1 strain had an S315T mutation in the katG gene combined with an −8 T-A mutation in the inhA promoter, and 2 strains did not contain any mutations in these genes. The three gene mutations, S315T, −15 C-T, and −8 T-A, can confer isoniazid resistance, and the lack of mutations in these two genes cannot cause isoniazid resistance. Thus, 90.5% (19/21) of the strains were resistant to isoniazid based on these genotypic mutations.
gyrA and gyrB Genes
Among the 21 fluoroquinolone-resistant MDR strains, 14 strains had D94 mutations in gyrA (D94G, 10 strains; D94A, 3 strains; D94Y, 1 strain), 4 strains had A90V mutations in gyrA, 1 strain had an A90V mutation in gyrA and an A597T mutation in gyrB, 1 strain had an A90V mutation in gyrA and an S447F mutation in gyrB, and 1 strain did not have any mutation in gyrA or gyrB. The mutations D94G, D94A, D94Y, and A90V in gyrA were in the quinolone-resistance-determining region (QRDR) and can cause fluoroquinolone resistance. The A597T and S447F mutations in gyrB were outside of the QRDR and were considered to present fluoroquinolone genotypic sensitivity. The strains without any mutations in gyrA and/or gyrB were also considered to present fluoroquinolone genotypic sensitivity. Thus, 95.2% (20/21) of the strains were resistant to fluoroquinolones based on these genotypic mutations. Among these, 95.2% (20/21) and 61.9% (13/21) of MTB strains were resistant to ofloxacin and moxifloxacin, respectively.
rpsL and rrs
Among the 21 fluoroquinolone-resistant MDR strains, 14 strains had K43R mutations in rpsL, 5 strains had A514C mutations in rrs, 1 strain had a K43T mutation in rpsL combined with A514C and A1401G mutations in rrs gene, and 1 strain did not have any mutation in these two genes. The mutations K43R in rpsL and A514C in rrs can confer streptomycin resistance; strains without mutations in rpsL and/or rrs were considered susceptible to streptomycin. Thus, 95.2% (20/21) of the strains were resistant to streptomycin based on these genotypic mutations. In addition, the mutation A1401G in rrs can confer resistance to second-line injectable drugs (amikacin, kanamycin, and capreomycin), and the strains without mutation A1401G in rrs and without/with mutation A514C in rrs gene were designated as susceptible to second-line injectables. Thus, 4.8% (1/21) of the isolates were resistant to second-line injectables based on these genotypic mutations.
pncA and panD
Among the 21 fluoroquinolone-resistant MDR strains, 12 strains had mutations in pncA: L19P (n = 1), G24D (n = 1), W68C (n = 1), H71Y (n = 1), K96R (n = 1), Y103H (n = 1), G132A (n = 1), S164P (n = 2), 1 deletion (Del) pattern (Del G at position 541 [n = 1]), 2 insertion (Ins) patterns (G Ins at position 162 [n = 1], and G Ins at position 476 [n = 1]); and 1 strain had mutation S104R in pncA combined with mutation L132P in panD (n = 1). Among these mutations in pncA, the Y103H, 541 Del G, 476 Ins G, and 162 Ins G mutations have not been reported in the Genome-wide M. tuberculosis variation database or the WHO guidelines. Any nonsynonymous mutations in pncA detected in this study were considered to present genotypic resistance. Thus, 61.9% (13/21) of the isolates were resistant to pyrazinamide based on these genotypic mutations.
embB Gene, embC-embA Intergenic Region, and aftA and ubiA Genes
Among the 21 fluoroquinolone-resistant MDR strains, 9 strains had mutations in embB, M306V (n = 6), M306I (n = 2), Q497K combined with V50A (n = 1), and 12 strains did not have any mutations. In the embC-embA intergenic region, 4 out of 21 strains had mutations: C-T mutations at the −12 position (n = 2), C-G mutation at the −16 position (n = 1), and C-T mutation at the −16 position (n = 1). Among the 21 strains, two had A33P and I164V mutations in aftA combined with mutation M306I in embB. One ethambutol-resistant strain harbored a single ubiA mutation, F140V. The effects of mutation V50A in embB and mutation F140V in ubiA on ethambutol resistance were not investigated in this study, and were therefore considered to present ethambutol genotypic susceptibility. These nonsynonymous mutations in embB, excluding the mutation Q497K which was present in both phenotypically susceptible and resistant isolates, were considered to present ethambutol genotypic resistance. Thus, 57.1% (12/21) of the isolates were resistant to ethambutol based on these genotypic mutations.
folC Gene
Among the 21 fluoroquinolone-resistant MDR strains, one strain had an I43T mutation in folC, and this mutation was considered to represent para-aminosalicylic acid genotypic resistance. Thus, 4.8% (1/21) of the strains were resistant to para-aminosalicylic acid based on this genotypic mutation.
Comparisons Between Phenotype and Genotype
Compared to traditional DST results, the genomic resistance of the isolates to rifampin, isoniazid, ofloxacin, moxifloxacin, and streptomycin was at 100%, 90.48%, 95.24%, 92.86%, and 95.27%, respectively. Among these five drugs, the PPV of moxifloxacin resistance was 65%, while the PPV for the remaining drugs was 100%. The genomic resistance of the isolates to ethambutol, pyrazinamide, kanamycin, and amikacin was 85.71%, 66.67%, 50%, and 50%, respectively (Table 4). The sensitivities of capreomycin, protionamide, and para-aminosalicylic acid to whole-genome sequencing were not calculated because only one isolate showed phenotypic drug resistance. The resistance of the isolates to all drugs is shown in Table 4.
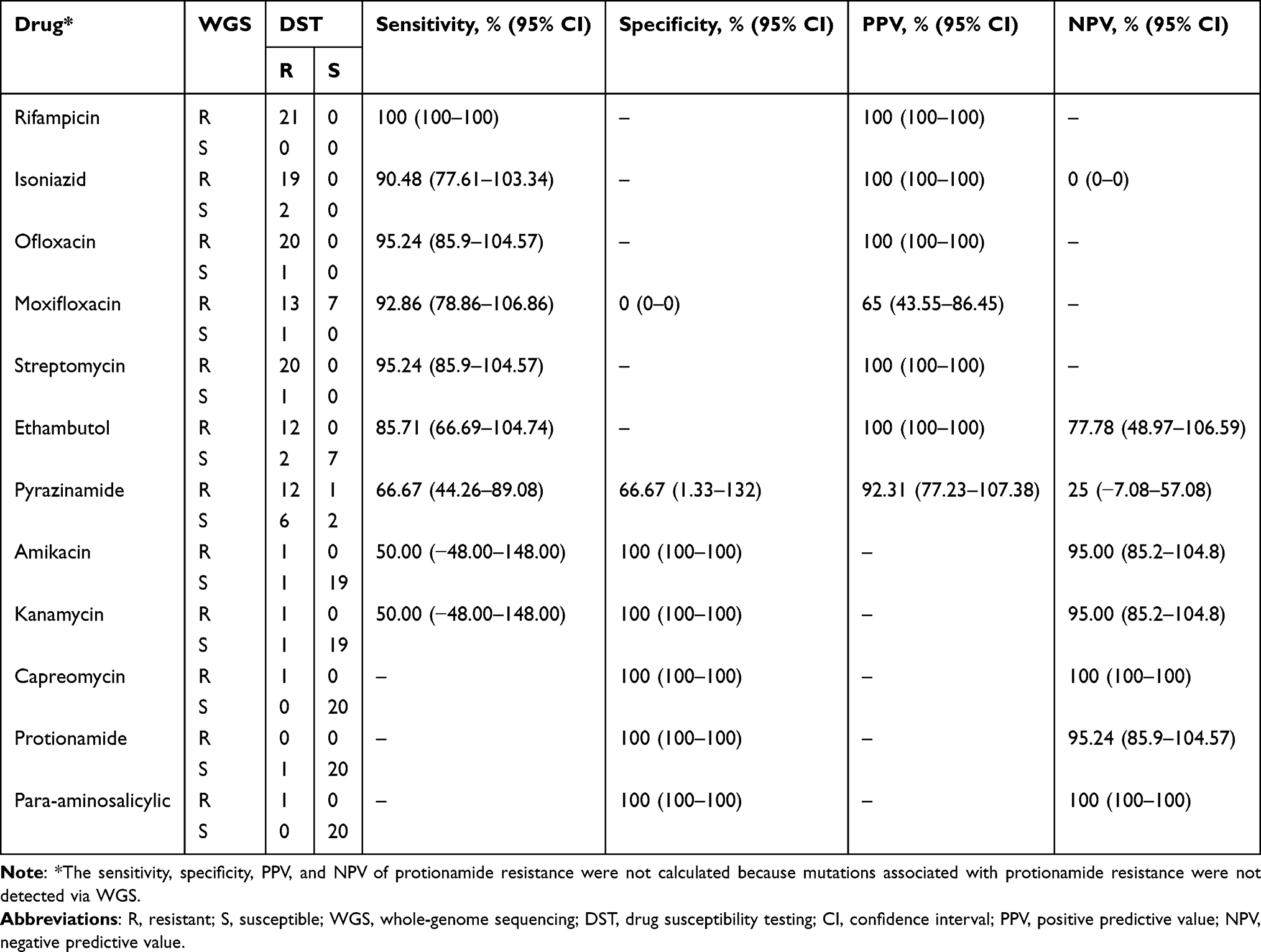 |
Table 4 Performance of WGS Compared with DST |
Discussion
Phenotypic rifampin resistance can be explained by mutations in the 81-bp rifampin resistance-determining region (between codons 507 and 533) in the rpoB gene; the rpoB gene mutation frequency has been reported as follows: rpoB531, rpoB526, and rpoB516.10 However, in the present study, the common mutation was S531L, followed by L533P, H526D, and D516V; these mutations accounted for 100% of the phenotypic rifampin resistance. This suggests that rpoB533 also plays an important role in the prediction of phenotypic rifampin resistance. These findings suggest that whole-genome sequencing can be performed to predict rifampin resistance, and that if the presence of M. tuberculosis and its resistance to rifampin is detected using Xpert MTB/RIF assays to test specimens of patients (such as sputum), then whole-genome sequencing should be performed to predict resistance to other antitubercular drugs to shorten the time of diagnosis of fluoroquinolone-resistant MDR-TB.
Phenotypic isoniazid resistance has been found to be mainly associated with katG315, inhA promoter, and ahpC-oxyR intergenic region mutations, of which katG315 and inhA-15 promoter mutations contribute to approximately 83% of phenotypic isoniazid resistance.11 In the present study, the common mutation was katG315, followed by inhA promoter mutations; these mutations explained 90.5% of phenotypic isoniazid resistance, approaching the cumulative frequency previously reported.11
Phenotypic fluoroquinolone resistance can be caused by mutations in the QRDR of gyrA and gyrB genes, which are located between gyrA codons 74 and 113 and between gyrB codons 500 and 540 in M. tuberculosis. The common gyrA mutations are D94 and A90, and the common gyrB mutations are N538 and D500.2 In the present study, we found that the common mutation in the QRDR of gyrA was D94, followed by A90, and no mutation was found in the QRDR of gyrB. This suggests that single mutations D94 and A90 in the QRDR of gyrA represented the major mutations in fluoroquinolone-resistant MDR M. tuberculosis. The proportion (95.2%) of fluoroquinolone-resistant MDR M. tuberculosis strains with mutations identified via whole-genome sequencing was higher than that reported previously.8 We found mutations in the QRDR that accounted for 95.2% of phenotypic fluoroquinolone resistance, 95.2% of phenotypic ofloxacin resistance, and 92.8% of phenotypic moxifloxacin resistance. However, seven out of 21 strains showed ofloxacin resistance and moxifloxacin susceptibility based on DST, although these strains had mutations in QRDR that accounted for the ofloxacin resistance. In addition, one of the strains showing ofloxacin resistance and moxifloxacin resistance did not have mutations in QRDR. This difference may be because the DST method (moxifloxacin concentration 2 mg/L) used in this study was different from other methods, such as MIC (moxifloxacin concentration 0.5 mg/L or 0.25 mg/L),3 and because other mechanisms for moxifloxacin resistance were not involved, such as heteroresistance.23 This finding suggests that if ofloxacin is considered as the only fluoroquinolone for detecting phenotypic fluoroquinolone resistance, then whole-genome sequencing can be effective in predicting the phenotypic resistance. However, if moxifloxacin is considered as the only fluoroquinolone for detecting phenotypic fluoroquinolone resistance, then whole-genome sequencing may be ineffective in predicting the phenotypic resistance.
Phenotypic streptomycin resistance is explained by mutations in rpsL, rrs, and gidB.13 In the present study, we found that the common mutations that conferred resistance to streptomycin were rpsL K43R and rrs A514C, excluding gidB, similar to that reported previously.13 Phenotypic resistance to second-line injectables (amikacin, kanamycin, and capreomycin) can be caused by mutations in the rrs gene, tlyA gene, and eis promoter.24 In the present study, among the two second-line injectable-resistant M. tuberculosis strains, only one strain (strain 86) had mutation A1401G in rrs, whereas the other did not, suggesting that this mutation may not be enough to explain phenotypic resistance to second-line injectables.
Phenotypic pyrazinamide resistance has been reported to be associated with mutations in pncA and its promoter, and in panD and rpsA genes, including nucleotide changes, deletions, and insertions that cause amino acid changes and frameshifts.14,25 In the present study, we found that mutations occurring in pncA were scattered throughout the pncA gene; mutation L132P in panD occurred in combination with mutation S104R in pncA in one isolate, and single mutations in panD and rpsA were not detected. These findings suggest that mutations in pncA are the primary cause of pyrazinamide resistance, which is similar to that reported previously.14,17 In addition, mutation H71Y in pncA, which has been found to confer pyrazinamide resistance in a previous study,26 was found in one pyrazinamide-susceptible isolate, and six pyrazinamide-resistant isolates did not have any mutations in pncA, panD, or rpsA. This suggests that predicting pyrazinamide resistance using whole-genome sequencing can be complex and that other mechanisms for pyrazinamide resistance, such as efflux proteins,27 should be investigated.
Phenotypic ethambutol resistance has been reported to be associated with mutations in the embCAB locus, mainly embB and the embC-embA intergenic region, and the ubiA and aftA genes.19 In the present study, 85.7% (12/14) ethambutol-resistant isolates harbored mutations embB M306 and embC-embA in the intergenic region at−12 and −16 position conferring ethambutol resistance; the number of resistant isolates was lower than that reported in a previous study.18 We also found mutations in ubiA and aftA, but they did not represent major causes of ethambutol resistance.19
Phenotypic para-aminosalicylic acid resistance has been reported to be associated with mutations in ribD, folC, and thyA.21 In the present study, only one para-aminosalicylic acid resistant isolate was identified among the 21 fluoroquinolone-resistant MDR strains, which harbored the mutation I43T in folC conferring para-aminosalicylic acid resistance,21 without any mutation in both ribD and thyA genes.
In the present study, only one isolate was resistant to protionamide and did not harbor any putative mutations in the ethA gene,28 which confers protionamide resistance; however, it harbored a novel mutation, T88P, in ethA. The effect of the mutation T88P on protionamide resistance was not investigated in this study.
In the present study, mutations that represent markers for drug resistance were determined in drug susceptibility-associated genes detected via whole-genome sequencing, which explained the phenotypic drug resistance in the majority of isolates. However, it is important to note the absence of these mutations in certain drug-resistant isolates, indicating the presence of other mechanisms of drug resistance, such as the strain genetic background,29 mixed infection,7 heteroresistance,23 and efflux pumps.30 In addition, even though the traditional phenotypic DST method is the current gold standard, it is not perfect in detecting drug resistance in M. tuberculosis, such as fluoroquinolone resistance and low-level drug resistance.31
Whole-genome sequencing, as a clinical tool, has been used to detect the presence of M. tuberculosis and to predict its resistance pattern based in drug resistance mutations. In the present study, the rates of rifampin resistance, isoniazid resistance, ofloxacin resistance, and moxifloxacin resistance, ranging from 90.48% to 100%, were higher than the diagnostic minimal sensitivity of sequencing (>90% and >95%) against the phenotypic reference standard recommended by the WHO;17 the rates were similar to those reported previously, ranging from 89.2–100%, 90–100%, 80–100%, and 60–90.6%, respectively.9 Notably, mutations in gyrA found in moxifloxacin-susceptible isolates, such as D94G (strain 110), which is regarded as a mutation associated with high minimum inhibitory concentration, may misclassify moxifloxacin susceptibility. The rates of streptomycin resistance (95.24%) and ethambutol resistance (85.71%) in this study were consistent with those reported previously, ranging from 57.1–100% and 71.4–100%, respectively.9 The rate of pyrazinamide resistance (66.67%) in this study was considerably lower than the diagnostic minimal sensitivity of sequencing (>85%) against the phenotypic reference standard recommended by the WHO;17 however, it was similar to that of a previous study, ranging from 43.2% to 100%.9 We did not compare the rates of second-line injectables (amikacin, kanamycin, and capreomycin), protionamide, and para-aminosalicylic acid resistance determined in this study with those reported previously because only one or two isolates showed phenotypic drug resistance. These findings suggest that whole-genome sequencing can be used to rapidly predict drug resistance in fluoroquinolone-resistant MDR isolates but cannot replace the traditional phenotypic DST method. Furthermore, whole-genome sequencing with a validated bioinformatics pipeline can predict drug susceptibility in M. tuberculosis. Therefore, phenotypic DST may not be needed for some predicted drug-susceptible strains, such as rifampicin-susceptible strains.31 The turn-around time of whole-genome sequencing (7 days) is much shorter than that of phenotypic DST (4 weeks), but the cost of whole-genome sequencing is higher than that of phenotypic DST.
The limitations of this study were as follows: only 21 fluoroquinolone-resistant MDR isolates were collected, and genome-wide association studies and other mechanisms of drug resistance were not considered to evaluate the genotypic resistance of fluoroquinolone-resistant MDR isolates.
Conclusion
In conclusion, whole-genome sequencing represents an effective diagnostic tool for MDR-TB showing fluoroquinolone resistance, and may help clinicians to develop accurate, individual, and clinical therapeutic strategies for such patients, although there have been obstacles in developing such diagnosis. Whole-genome sequencing should be applied to predict drug resistance prior to performing traditional phenotypic DST in Tianjin, China.
Acknowledgments
This work was supported by the Tianjin Municipal Health Commission (grant number: ZC20199).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. World Health Organization. Global Tuberculosis Report 2021. Licence: CC BY-NC-SA 3.0 IGO. Geneva: World Health Organization;2021.
2. Avalos E, Catanzaro D, Catanzaro A, et al. Frequency and geographic distribution of gyrA and gyrB mutations associated with fluoroquinolone resistance in clinical Mycobacterium tuberculosis isolates: a systematic review. PLoS One. 2015;10(3):e0120470. doi:10.1371/journal.pone.0120470
3. Cao Y, Parmar H, Gaur RL, et al. Xpert MTB/XDR: a 10-color reflex assay suitable for point-of-care settings to detect isoniazid, fluoroquinolone, and second-line-injectable-drug resistance directly from Mycobacterium tuberculosis-positive sputum. J ClinMicrobiol. 2021;59(3):e02314–20. doi:10.1128/JCM.02314-20
4. Lee EH, Yong SH, Leem AY, et al. Improved fluoroquinolone-resistant and extensively drug-resistant tuberculosis treatment outcomes. Open Forum Infect Dis. 2019;6(4):ofz118. doi:10.1093/ofid/ofz118
5. Zong K, Luo C, Zhou H, et al. Xpert MTB/RIF assay for the diagnosis of rifampicin resistance in different regions: a meta-analysis. BMC Microbiol. 2019;19(1):177. doi:10.1186/s12866-019-1516-5
6. Deelder W, Christakoudi S, Phelan J, et al. Machine learning predicts accurately Mycobacterium tuberculosis Drug resistance from whole genome sequencing data. Front Genet. 2019;10:922. doi:10.3389/fgene.2019.00922
7. Köser CU, Bryant JM, Becq J, et al. Whole-genome sequencing for rapid susceptibility testing of M. tuberculosis. N Engl J Med. 2013;369(3):290–292. doi:10.1056/NEJMc1215305
8. Maruri F, Guo Y, Blackman A, et al. Resistance-conferring mutations on whole-genome sequencing of fluoroquinolone-resistant and -susceptible Mycobacterium tuberculosis isolates: a proposed threshold for identifying resistance. Clin Infect Dis. 2021;72(11):1910–1918. doi:10.1093/cid/ciaa496
9. Papaventsis D, Casali N, Kontsevaya I, et al. Whole genome sequencing of Mycobacterium tuberculosis for detection of drug resistance: a systematic review. Clin MicrobiolInfect. 2017;23(2):61–68. doi:10.1016/j.cmi.2016.09.008
10. Zaw MT, Emran NA, Lin Z. Mutations inside rifampicin-resistance determining region of rpoB gene associated with rifampicin-resistance in Mycobacterium tuberculosis. J Infect Public Health. 2018;11(5):605–610. doi:10.1016/j.jiph.2018.04.005
11. Seifert M, Catanzaro D, Catanzaro A, et al. Genetic mutations associated with isoniazid resistance in Mycobacterium tuberculosis: a systematic review. PLoS One. 2015;10(3):e0119628. doi:10.1371/journal.pone.0119628
12. Sekiguchi J, Miyoshi-Akiyama T, Augustynowicz-Kopeć E, et al. Detection of multidrug resistance in Mycobacterium tuberculosis. J Clin Microbiol. 2007;45(1):179–192. doi:10.1128/JCM.00750-06
13. Bwalya P, Yamaguchi T, Solo ES, et al. Characterization of mutations associated with streptomycin resistance in multidrug-resistant Mycobacterium tuberculosis in Zambia. Antibiotics. 2021;10(10):1169. doi:10.3390/antibiotics10101169
14. Tam KK, Leung KS, Siu GK, et al. Direct detection of pyrazinamide resistance in Mycobacterium tuberculosis by use of pncA PCR sequencing. J ClinMicrobiol. 2019;57(8):e00145–19. doi:10.1128/JCM.00145-19
15. Charoenpak R, Santimaleeworagun W, Suwanpimolkul G, et al. Association between the phenotype and genotype of isoniazid resistance among Mycobacterium tuberculosis isolates in Thailand. Infect Drug Resist. 2020;13:627–634. doi:10.2147/IDR.S242261
16. Chernyaeva EN, Shulgina MV, Rotkevich MS, et al. Genome-wide Mycobacterium tuberculosis variation (GMTV) database: a new tool for integrating sequence variations and epidemiology. BMC Genomics. 2014;15:308. doi:10.1186/1471-2164-15-308.
17. World Health Organization.The Use of Next-Generation Sequencing Technologies for the Detection of Mutations Associated with Drug Resistance in Mycobacterium Tuberculosis Complex: TechnicalGuide. World Health Organization; 2018.
18. Brossier F, Sougakoff W, Bernard C, et al. Molecular analysis of the embCAB locus and embR gene involved in ethambutol resistance in clinical isolates of Mycobacterium tuberculosis in France. Antimicrob Agents Chemother. 2015;59(8):4800–4808. doi:10.1128/AAC.00150-15
19. Andres S, Gröschel MI, Hillemann D, et al. A diagnostic algorithm to investigate pyrazinamide and ethambutol resistance in rifampin-resistant Mycobacterium tuberculosis isolates in a low-incidence setting. Antimicrob Agents Chemother. 2019;63(2):e01798–18. doi:10.1128/AAC.01798-18
20. Ramaswamy SV, Amin AG, Göksel S, et al. Molecular genetic analysis of nucleotide polymorphisms associated with ethambutol resistance in human isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2000;44(2):326–336. doi:10.1128/AAC.44.2.326-336.2000
21. Luo M, Li K, Zhang H, et al. Molecular characterization of para-aminosalicylic acid resistant Mycobacterium tuberculosis clinical isolates in southwestern China. Infect Drug Resist. 2019;12:2269–2275. doi:10.2147/IDR.S207259
22. Barbier M, Wirth T. The evolutionary history, demography, and spread of the Mycobacterium tuberculosis complex. MicrobiolSpectr. 2016;4(4):53. doi:10.1128/microbiolspec.TBTB2-0008-2016
23. Rigouts L, Miotto P, Schats M, et al. Fluoroquinolone heteroresistance in Mycobacterium tuberculosis: detection by genotypic and phenotypic assays in experimentally mixed populations. Sci Rep. 2019;9(1):11760. doi:10.1038/s41598-019-48289-9
24. Georghiou SB, Magana M, Garfein RS, et al. Evaluation of genetic mutations associated with Mycobacterium tuberculosis resistance to amikacin, kanamycin and capreomycin: a systematic review. PLoS One. 2012;7(3):e33275. doi:10.1371/journal.pone.0033275
25. Rahman A, Ferdous SS, Ahmed S, et al. Pyrazinamide susceptibility and pncA mutation profiles of Mycobacterium tuberculosis among multidrug-resistant tuberculosis patients in Bangladesh. Antimicrob Agents Chemother. 2017;61(9):e00511–17. doi:10.1128/AAC.00511-17
26. Khan MT, Malik SI, Ali S, et al. Pyrazinamide resistance and mutations in pncA among isolates of Mycobacterium tuberculosis from Khyber Pakhtunkhwa, Pakistan. BMC Infect Dis. 2019;19(1):116. doi:10.1186/s12879-019-3764-2
27. Zhang Y, Zhang J, Cui P, et al. Identification of novel efflux proteins Rv0191, Rv3756c, Rv3008, and Rv1667c involved in pyrazinamide resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2017;61(8):e00940–17. doi:10.1128/AAC.00940-17
28. Tan Y, Su B, Zheng H, et al. Molecular characterization of prothionamide-resistant Mycobacterium tuberculosis isolates in Southern China. Front Microbiol. 2017;8:2358. doi:10.3389/fmicb.2017.02358
29. Fenner L, Egger M, Bodmer T, et al. Effect of mutation and genetic background on drug resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2012;56(6):3047–3053. doi:10.1128/AAC.06460-11
30. Goossens SN, Sampson SL, Van Rie A. Mechanisms of drug-induced tolerance in Mycobacterium tuberculosis. ClinMicrobiolRev. 2020;34(1):e00141-20. doi:10.1128/CMR.00141-20
31. Jajou R, van der Laan T, de Zwaan R, et al. WGS more accurately predicts susceptibility of Mycobacterium tuberculosis to first-line drugs than phenotypic testing. J AntimicrobChemother. 2019;74(9):2605–2616. doi:10.1093/jac/dkz215
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.