")
Back to Journals » International Journal of Nanomedicine » Volume 14
Characterization Of Blood–Brain Barrier Crossing And Tumor Homing Peptides By Molecular Dynamics Simulations
Authors Arcangeli C , Lico C , Baschieri S , Mancuso M
Received 7 August 2019
Accepted for publication 17 October 2019
Published 30 December 2019 Volume 2019:14 Pages 10123—10136
DOI https://doi.org/10.2147/IJN.S225793
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Video abstract presented by Caterina Arcangeli.
Views: 288
Caterina Arcangeli,1 Chiara Lico,2 Selene Baschieri,2 Mariateresa Mancuso3
1Laboratory of Health and Environment, Italian National Agency for New Technologies, Energy and Sustainable Economic Development, ENEA, Rome, Italy; 2Laboratory of Biotechnology, Italian National Agency for New Technologies, Energy and Sustainable Economic Development, ENEA, Rome, Italy; 3Laboratory of Biomedical Technologies, Italian National Agency for New Technologies, Energy and Sustainable Economic Development, ENEA, Rome, Italy
Correspondence: Caterina Arcangeli; Mariateresa Mancuso
ENEA, Italian National Agency for New Technologies, Energy and Sustainable Economic Development, Casaccia Research Centre, Via Anguillarese 301, Rome 00123, Italy
Tel +39 06 3048 6898/4993
Fax +39 06 3048 6559/3644
Email [email protected]; [email protected]
Introduction: The new frontier of tumor diagnosis and treatment relies on the development of delivery strategies capable of allowing the specific targeting of the diagnostic agents/chemotherapeutics, avoiding side effects. In the case of brain tumors, achieving this goal is made more difficult by the presence of the blood–brain barrier (BBB). Peptides have been revealed as excellent candidates for both BBB crossing and specific cancer homing. Nanoparticles (NPs), functionalized with BBB crossing and tumor homing (TH) peptides, are emerging as smart theranostic systems. However, there is still poor knowledge concerning the molecular structure and dynamical properties of these peptides, essential requirements for a suitable functionalization of the delivery systems themselves.
Methods: In this work, by means of molecular dynamics (MD) simulations, we have extensively characterized the structural and dynamical behavior of several peptides, known to be endowed of BBB crossing and TH properties.
Results: The simulations point out that, on the basis of their conformational dynamics, the peptides can be classified in two main groups: 1) peptides assuming a specific structural conformation, a feature that could be important for interacting with the molecular target but that may limit their use as functionalizing molecules and 2) highly flexible peptides whose interaction with the target may be independent of a particular structural conformation and that may represent good candidates for the functionalization of theranostic NP-based platforms.
Discussion: Such findings may be useful for the de novo designing of NP-based delivery systems.
Keywords: conformational flexibility, free energy landscapes, essential dynamics, peptides, brain, theranostic platform
Introduction
Brain tumor remains one of the cancers more difficult to treat. Even if improvements have been achieved in 5-year disease-free survival rates through a multimodal treatment, the aggressive nature of such therapy strongly affects the quality of life of the patients.1 A theranostic approach, defined as an integrated system that can diagnose, deliver targeting therapies and monitor the response, could represent a promising strategy for this kind of tumor. Recently, a number of theranostic approaches, including, for example, carbon nanotubes, liposomes, dendrimers, polymeric vesicles2,3 and, more recently, small-molecule multifunctional probes,4–6 have been proposed for the cancer diagnosis and therapies. However, for brain tumors, the BBB still represents the main obstacle for drug delivery. Peptides have been recently revealed as excellent candidates for both BBB crossing and specific homing to brain cancer targets. Peptides are small in size, easy to produce and highly specific. They possess remarkable sequence flexibility and can be genetically/chemically conjugated with other molecules, for example, with NPs, of various origins, which, in turn, can be used as containers of chemotherapeutic agents, for a specific drug targeting and delivery approach.5–9
In this work, we have extensively characterized the structural and dynamical behavior, by means of MD simulations, of nine peptides selected from the wide variety of BBB-penetrating peptides covering different brain-permeation mechanisms such as BBB-homing and cell-penetrating peptides (CPP). Some CPPs have indeed been shown as excellent BBB carriers with different degrees of brain-influx selectivity.8 In the hypothesis of designing a platform capable of delivering drugs to brain tumors, the BBB-crossing peptides considered in our study were derived from a class of peptides that internalize into the cells by receptor-mediated transcytosis, a process overcoming the limitation of non-specific uptake by peripheral tissues and blood vessels. Similarly, in order to distinguish abnormal from normal tissues, the TH peptides were selected from those reported in the literature capable of specifically recognizing the overexpressed brain tumor receptors. Table 1 summarizes the amino acid sequences and the molecular targets of the selected peptides investigated in our work.
 |
Table 1 Peptides Sequences Used For Modeling And Simulation |
Even if several attempts to functionalize NPs of various origins with the peptides of Table 1 are already reported in literature,10–17 the design of such systems did not take advantage of structure-based knowledge of the peptides, an essential requisite for a suitable functionalization of the delivery system itself and, up to now, there is still poor knowledge concerning the molecular structure and dynamical properties of these peptides.
To the best of our knowledge, this is the first study that reports a detailed characterization, at atomic and temporal resolution, of structural, dynamical and physicochemical features of several BBB and TH peptides. We have exploited the capabilities of MD simulation to determine the conformation adopted in the physiological aqueous solution of all the peptides selected, as well as their temporal and structural fluctuations at atomic resolution. The simulations have put into evidence two classes of peptides on the basis of their conformational dynamics. Some peptides seem to adopt specific structural conformations, which could be important for the correct interaction with their molecular targets. The use of these peptides for functionalizing delivery systems may be made difficult by the necessity of correctly maintaining their structural and functional conformations. Other peptides, on the contrary, seem to be characterized by a very high structural flexibility. This feature suggests that their interaction with the targets could be independent of a particular structural conformation, making them good candidates for an adequate functionalization of a theranostic platform. Such findings may be useful for the de novo designing of NP-based delivery systems, before beginning the complex time-consuming and cost-expensive experimental production of the theranostic platforms.
Materials And Methods
Molecular Modeling
The amino acid sequences of the peptides used for the simulations were obtained from the literature (Table 1). All the peptides were modeled in their fully extended conformation by using the open-source PepFold 2.0,18,19 with the exception of EETI 2.5F, whose structure was modeled by homology modeling. The three-dimensional structure used as a template for EETI 2.5F consists of the squash trypsin inhibitor EETI-II (2IT7.pdb), which showed 67% of sequence identity. An ensemble of thirty homology models of the EETI 2.5F peptide were generated by Modeller v9.120 and ranked by their molecular probability density functional (pdf) values after the highest optimization level. The best homology-derived model, ie the model with the lowest pdf value, was used as the starting structure for the MD simulations.
Molecular Dynamics Simulation
All the simulations were performed using the GROMACS 2016.3 package21 with all-atom AMBER 99sb-ildn and OPLS-AA force fields,22,23 which are both employed in the simulation of biomolecules and are able to reproduce experimentally observed conformational ensembles of small peptides.24 The AMBER 99sb-ildn force field, in particular, has been proven to not particularly favor certain secondary structures.25 TIP3P26 and SPC/E27 water models were used in combination with AMBER 99sb-ildn and OPLS-AA force field, respectively.
The modeled peptides were placed in a dodecahedron box in which water molecules and 100 mM of NaCl, including neutralizing counter-ions, were added. Periodic boundary conditions (PBC) were applied to better describe the condition of full hydration and avoid edge effects. The energy of all the systems was minimized by using 5000 steps of steepest descent (SD), with a tolerance of 100 kJ mol−1nm−1. Water molecules were then equilibrated by keeping the peptide atoms constrained. Systems were equilibrated under the NVT ensemble for 200 ps followed by further 200 ps under the NPT ensemble. Unrestrained MD simulations were carried out in the NPT ensemble for different simulation lengths, as indicated in Table 2. A global stochastic thermostat,28 with a time constant of 0.1 ps, was used to keep the temperature at 310 K. The average pressure was kept at 1 bar with a time constant of 2 ps by using the isotropic Parrinello–Rahman barostat.29,30 Newton’s equation of motion was integrated using a leapfrog31 algorithm with a 2-fs time step. The dielectric constant was set to 1.0. The particle mesh Ewald (PME) method32 was used to compute the long-range electrostatic forces. For the short-range electrostatics and Van der Waals interactions, a cut-off of 1 nm was used. Rotational and translational motions of the system were removed and all bonds were constrained with the LINCS algorithm.33 Initial velocities were assigned according to Maxwell–Boltzmann distribution.
 |
Table 2 Summary Of Simulations Performed |
Table 2 summarizes the MD simulations carried out in this study.
Analysis Of Trajectories
The trajectories were analyzed with tools included in GROMACS. The Gromos method34 was used to perform the cluster analysis. The matrix of atom positional RMSD between pairs of structures was calculated for the Cα atoms of the peptide. The criterion of similarity for the two structures was positional RMSD < 0.10 nm for the Cα atoms of the peptide.
Essential dynamics (ED) method35 is based on the principal component analysis (PCA) of the covariance matrix of the positional fluctuations of the Cα atoms. After removal of the overall rotational and translational motions, the diagonalization of the matrix yielded the principal directions of the large amplitude concerted motions (essential eigenvectors) that characterize the essential subspace of the peptide’s internal dynamics. The cosine content of the first eigenvector’s projections, an index of the sampling convergence,36 was calculated. The metastable conformational states of the systems (minima) and the barriers connecting these states are obtained from the free energy landscape plots, which were given by:
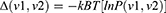
P(v1,v2) is the probability distribution of the molecular system along the first two eigenvectors.
Hydrogen bond (HB) and salt-bridge analyses were performed following the criteria adopted in Gromacs. An HB was assumed to exist during the simulation if the donor to acceptor distance was shorter than 0.35 nm and the hydrogen-donor-acceptor angle was lower than 30°. A salt-bridge was assumed to exist during the simulation if the distance between the side-chain oxygen (O) of Asp or Glu and the side-chain nitrogen (N) of Lys, Arg or His was less than 0.40 nm. The solvent-accessible surface area (SASA) was computed by following the algorithm adopted in Gromacs.37
The graphic representations of the peptide trajectory snapshots were made by means of VMD.38 The 3D free-energy landscape plots were obtained with homemade Python39 scripts.
Results And Discussion
The actual success of developing a theranostic platform for brain tumors is significantly determined by an effective knowledge of the physiochemical, structural and dynamical features of the molecules making up the system. In this respect, MD simulation has the great advantage of representing an inexpensive and powerful tool, providing the experimental designers with insights on both structural stability and dynamical flexibility of the systems, at temporal and spatial resolution.40 We have previously successfully employed MD simulations to provide insights for protein design,41 for structure-based NPs functionalization,42 and to evaluate dynamical processes at peptide–inorganic interfaces.43 Here, we report, for the first time, a detailed characterization, at spatial and temporal resolution, of long-scale MD-derived structural, dynamical and physicochemical features of several BBB-crossing and TH peptides.
The peptides investigated in our study were selected from the literature. Table 1 reports the names, the amino acid sequences and the molecular targets of the chosen peptides. Among the BBB-crossing peptides, we have focused our attention on Angiopep-2 (ang2) and on apolipoprotein E-derived peptide (ApoE) that specifically target the low-density lipoprotein receptor-related proteins (LRP1/LRP2), which are overexpressed in the brain as well as in tumors.44,45 Two additional BBB-crossing peptides, RVG29, a rabies virus-derived peptide which can enter into neurons by interaction with the nicotinic acetylcholine receptor (nAchR),46 and TGN, a phage-display derived peptide, whose sequence is actively transported across brain endothelial cells,13 were also included in our study. The TH peptides were selected from those reported in the literature to specifically recognize the overexpressed brain tumor receptors. CooP47 is a in vivo phage-display derived peptide that interacts with the mammary-derived growth inhibitor (MDGI), whose level of expression is higher in glioblastoma. Among the peptides that specifically bind α/β integrin receptors, usually overexpressed in glioblastoma and medulloblastoma, we selected RiGD, a small sequence with the presence of two copies of the RGD peptide, that recognizes αVβ3 receptor,48 and EETI 2.5F, an engineered mutant fragment of the Ecballium elaterium trypsin inhibitor (EETI-II), including the tripeptide RGD, that is known to recognize the α5β1, αVβ3, αVβ5 integrins.49 CLT-1, a phage display derived cyclic peptide, which recognizes fibrin–fibronectin complexes,50 and tLyp1, a truncated derivative of the tumor lymphatic targeting peptide that specifically binds neuropilin 1 receptor (NRP-1),51 were also considered in our investigation.
Only a sparse knowledge about the molecular structures of three of the considered peptides – namely of ApoE, EETI 2.5F and Lyp-1, the cyclic form of tLyp1 – is reported in literature.49,52–55 In order to obtain a full characterization of all nine peptides, we monitored their structural and dynamical behaviors in physiological solution by 0.95 and 1-μs-long MD simulations, as reported in Table 2.
A visual inspection of the molecular configurations sampled by the peptides in the simulations is shown in Figure 1. ApoE is the tandem dimer sequence of the receptor-binding domain (residues from 159 to 167) of the human ApoE (hApoE). In its native form and within the hApoE protein, a helix element represents the secondary structure of such domain.52 During our simulation, the isolated domain undergoes a misfolding process, and after 600 ns of the simulation, the peptide adopts a beta-hairpin conformation (Figure 1A and supplementary video 1A). In order to assess if this event was due to an inappropriate choice of the potential energy function,56 the peptide was simulated with a different force field (OPLSAA) and named ApoEo. A similar behavior was observed for ApoEo (Figure 1B and supplementary video 1B), indicating that this feature is independent of the force field used and is, presumably, the conformation adopted by this peptide in an aqueous system. EETI 2.5F is part of a family of peptides consisting of three interlocking disulfide bonds (Cys2-Cys24, Cys14-Cys26, Cys-20-Cys32) that form a structural motif known as a cysteine knot. This disulfide-bonded framework confers these peptides with high structural, thermal, chemical and proteolytic stability.49,53 The simulation snapshots show that the structural conformation adopted by the EETI 2.5F peptide in aqueous solution is maintained throughout the simulation with some fluctuations of the apical RGD-containing loop (Figure 1C and supplementary video 1C). CLT-1 peptide is characterized by a disulfide bond (Cys1-Cys10), which confers it with proteolytic stability. Such bond acts also as a structural constraint and the simulation snapshots put into evidence that after 300 ns of the simulation the circular conformation of the peptide undergoes deformations (Figure 1D and supplementary video 1D). The simulation snapshots of ang2, RVG29, TGN, CooP, tLyp1, and RiGD peptides capture different structural conformations indicating that they do not assume a specific and unique structure, exploring several conformational configurations (Figure 1E–J and supplementary videos 1E–1J).
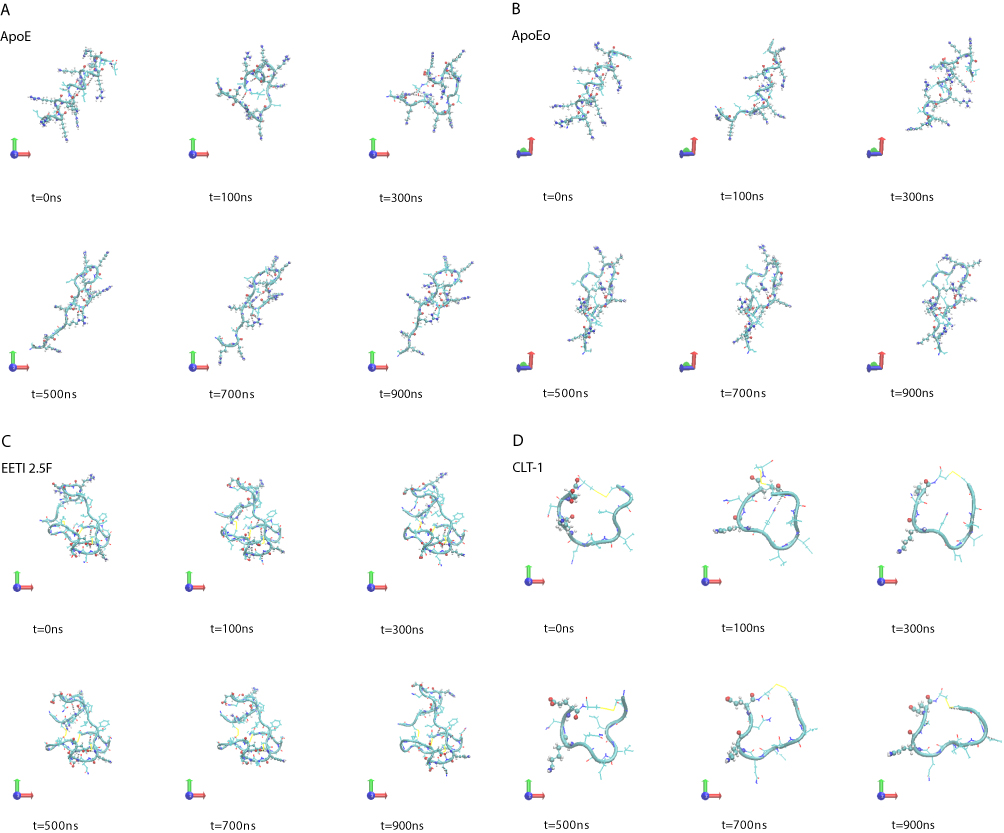 |
Figure 1 Simulation snapshots, taken at selected times, of (A) ApoE, (B) ApoEo, (C) EETI 2.5F, (D) CLT-1, (E) ang2, (F) RVG29, (G) TGN, (H) CooP, (I) tLyp1 and (J) RiGD of the MD trajectories. Notes: The peptide backbone is shown as a cyan ribbon. Charged residues (Arg, Lys, Glu, Asp) are represented by CPK model. The black dashed lines show hydrogen bonds. Color codes for the selected residues: carbon, cyan; hydrogen, white; nitrogen, blue; oxygen, red; sulfur, yellow. Movies of the peptide trajectories are available as supplementary data. |
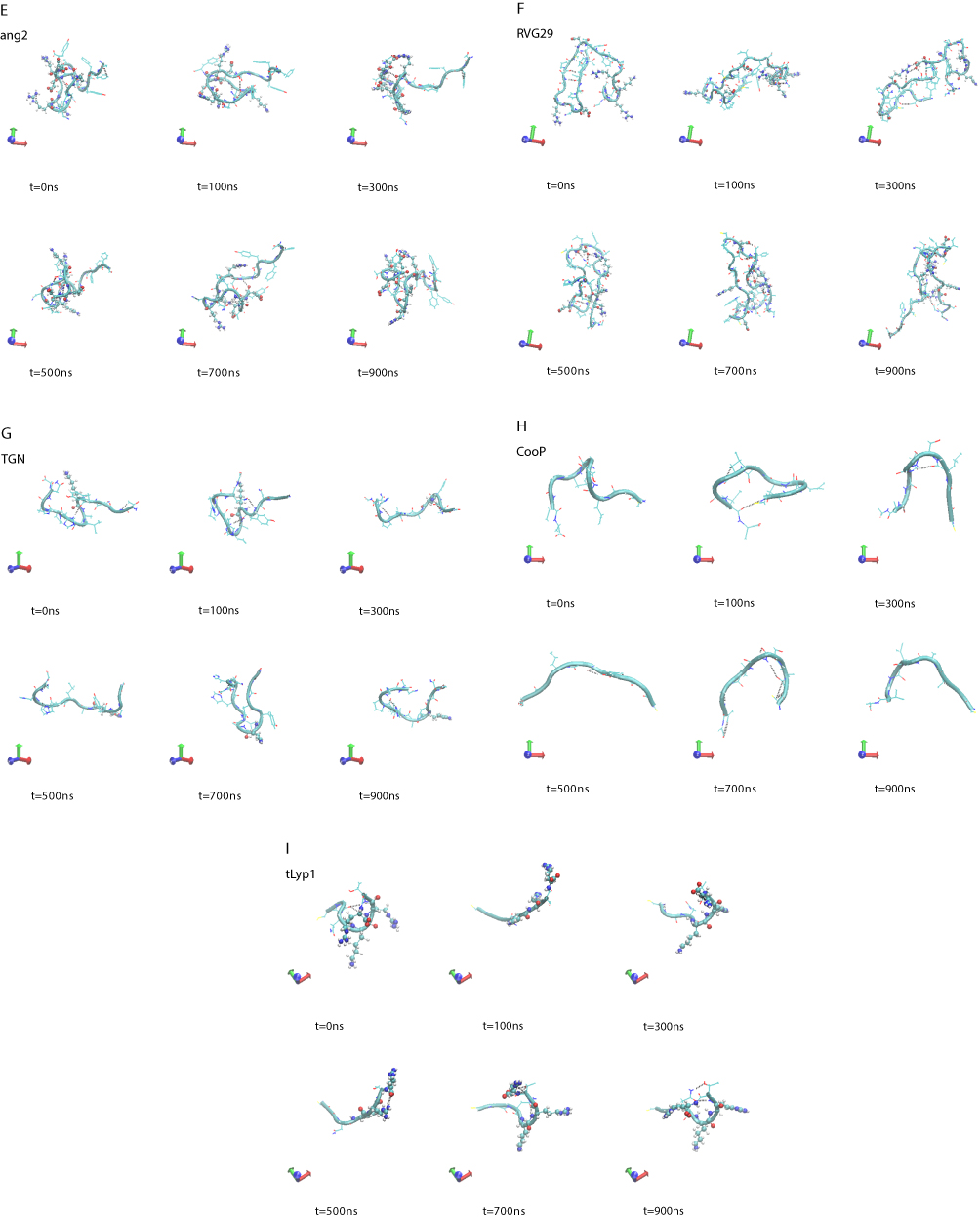 |
Figure 1 (Continued). |
In order to assess the overall stability of the MD simulations, we collected a number of structural and dynamical properties as a function of simulation time. An indicative measure of the stability was obtained by monitoring the RMSD of the Cα atoms coordinates from their initial values as a function of the simulation time (Figure 2). The RMSD values of both ApoE and ApoEo simulations reach a plateau within 500 ns of simulation, indicating that a conformational stability is always achieved also by using two different force fields. The RMSD values of the CLT-1 seem to reach a temporary stability within the first 300 ns, then the peptide starts to explore several conformational states. A different trend is observed for the remaining peptides. Indeed, the RMSD values of all the peptides show large fluctuations around 0.6 nm for ang2, 0.5 nm for TGN, 0.8 nm for RVG29, 0.4 nm for CooP, 0.2 nm for EETI 2.5F, 0.3 nm for tLyp1 and 0.3 nm for RiGD throughout all the simulation time.
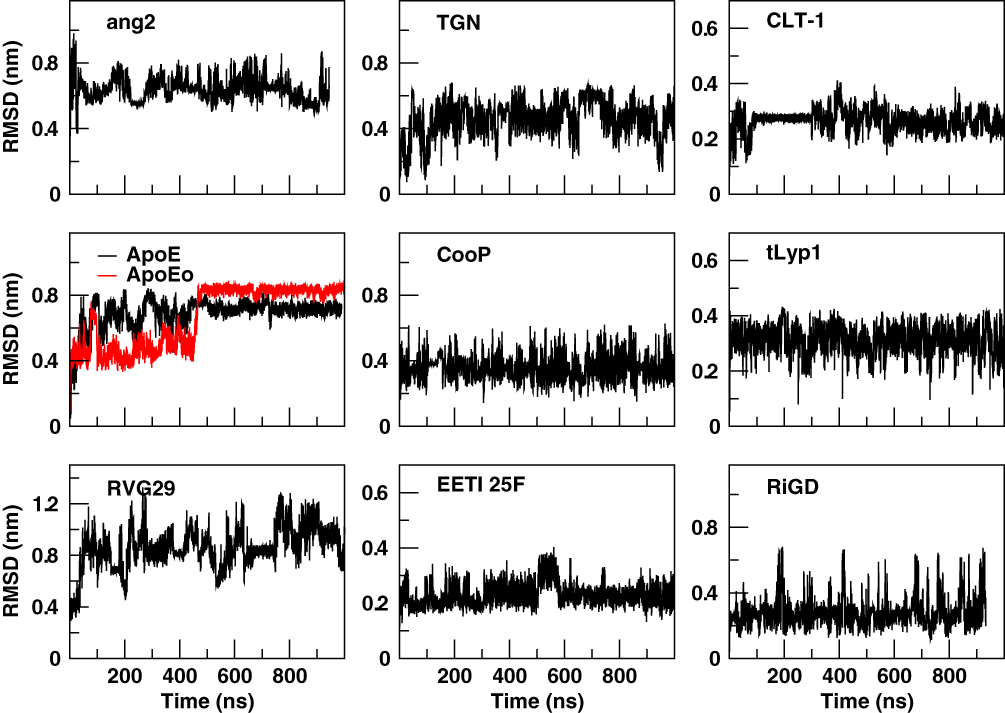 |
Figure 2 Cα RMSD of the peptides with respect to the equilibrated conformations as a function of simulation time. |
The cluster analysis of the trajectories supports these findings and reveals further structural features. The cumulative number of clusters (every 10 ns) throughout the trajectories of the peptides is reported in Figure 3. The number of clusters provides information about the level of population of the sampled conformational space during the simulation. A limited conformational sampling is observed for EETI 2.5F, whose total number of clusters is only about 25. The conformational sampling of ApoE and ApoEo reaches the stability after 500 ns of simulation, whereas CLT-1 shows a conformational stability only during the first 300 ns of simulation. The structural conformations of tLyp1 seem to converge, after 300 ns of simulation, into about 80 clusters. On the contrary, the time evolution of the number of clusters of ang2, RVG29, TGN, CooP, and RiGD shows an increasing trend. Generally, this behavior indicates that the peptides do not cluster into peculiar structures suggesting that the structures of these peptides may be intrinsically flexible.
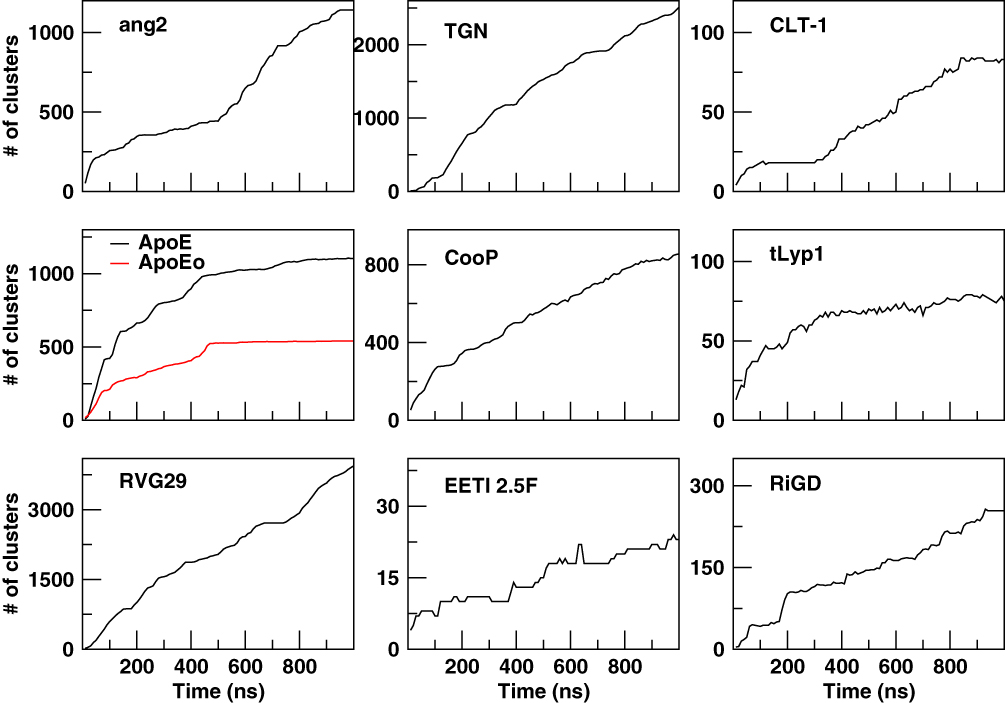 |
Figure 3 The cumulative number of clusters as a function of simulation time. |
To assess if the observed structural instability was due to an intrinsical flexibility of the peptides or to an insufficient sampling of the conformational states, the cosine content of the first eigenvector’s projections, an index of the sampling convergence,36 was calculated by means of ED analysis and reported in Table 3. Only RVG29 shows a value of cosine content of the first eigenvector more than 0.5, suggesting that longer simulation times are required to conclude that a structural convergence of this simulation was reached. This can occur when simulating the folding of a large peptide starting from a completely deployed structure, as is the case of the RVG29 peptide. The cosine content values of the first eigenvectors of ang2 and CLT-1 indicate that the structures of these peptides perform a semi-random conformational landscape sampling. On the contrary, the low values of cosine content registered for Apo, ApoEo, TGN, CooP, EETI 2.5F, tLyp1 and RiGD (less than 0.1) are strong indicators of a non-diffusive dynamics and clearly demonstrate good convergence for these simulations.
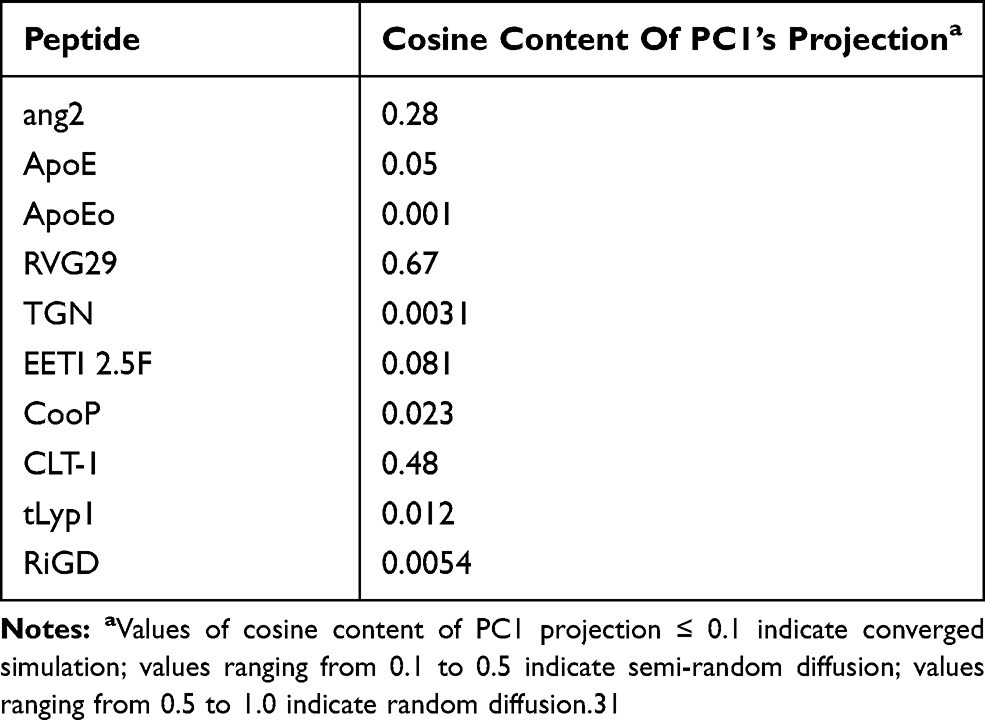 |
Table 3 Values Of Cosine Content Of First Eigenvector’s Projection Obtained By The PCA Analysis Of The Peptide Simulations |
In order to capture essential information on the peptide conformational sampling, we described the dynamical behavior of the peptide in terms of peptide’s free energy. The free energy landscape surfaces as a function of the first two eigenvectors (PC1 and PC2) of the peptides are shown in Figure 4. The free energy landscapes of ang2 and RVG29 show several narrow isoenergetic peaks (Figure 4A), whereas TGN, CooP, tLyp1 and RiGD (Figure 4B) show several and broad minima, suggesting that these peptides are highly flexible and characterized by several conformational states of similar energy. The free energy landscape of CLT-1 (Figure 4C) shows a single prominent minimum surrounded by few other minima. This is not surprising since CLT-1 is a cyclic peptide and the disulfide bond acts as a structural constraint. The free energy landscapes of ApoE, ApoEo and EETI 2.5F (Figure 4C) exhibit two prominent minima corresponding to two distinct conformational states. A pictorial view of the ApoE and of EETI 2.5F peptide motions along the first eigenvector is also shown in Figure 4D. ApoE shows a general collective motion involving almost all the residues. The consequence of this collective motion is a conformational change from the helix to the beta-hairpin element. Such a finding should not surprise, since it is already known that α-helix structures, usually stable in a hydrophobic environment, may undergo destabilization in hydrophilic environments, and structural transitions to a more stable β-hairpin conformation can occur.57 Large collective motions of the EETI 2.5F residues forming the RGD-containing loop above the knotted zone are observed. In particular, this loop seems to move away from the bottom. This behavior suggests that EETI 2.5F can assume two distinct conformational states during the simulation. This result seems to support a very recent finding,58 in which it was suggested that EETI 2.5F can bind αʋβ3 integrin by conformational selection from distinct conformational states adopted by the very flexible RGD-containing loop.
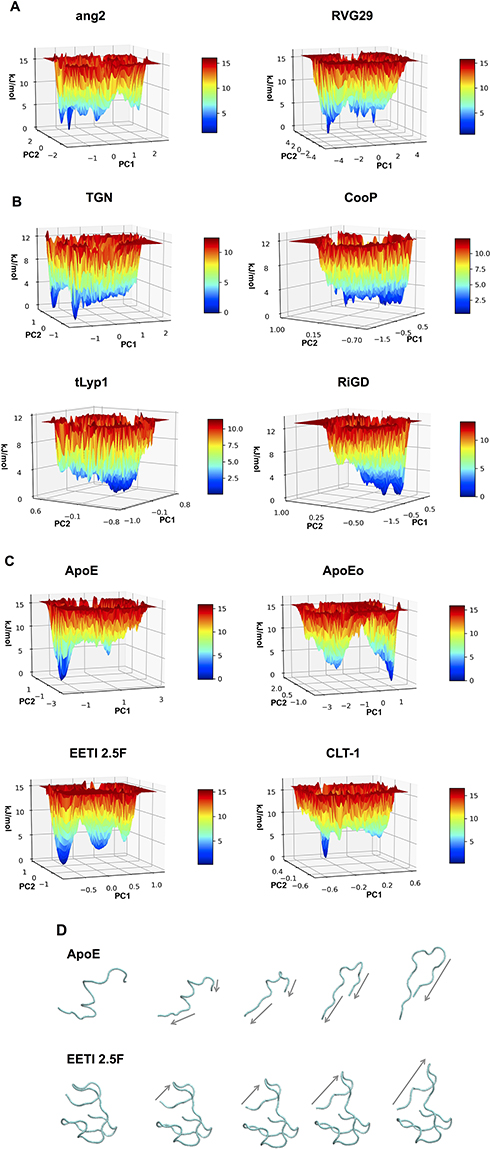 |
Figure 4 The 3D surfaces of free energy landscape of (A) ang2, RVG29, TGN, CooP, (B) tLyp1, RiGD, (C) ApoE, ApoEo, EETI 2.5F and CLT-1. (D) Selected configurations obtained by considering the Cα motions along the first eigenvector for ApoE and EETI 2.5f. Note: Arrows indicate the direction of the motion. |
The overall flexibility of the peptides was also determined to calculate the root mean square (RMSF) of Cα atoms after projecting the trajectories along their respective PC1 directions. This method has the advantage to show the flexibility only of the essential motions of the peptide, since PCA filters the global slow motions (large amplitude motions) from the fast motions,35 which usually are referred to as thermal fluctuations that can affect the measurement of the flexibility itself. Figure 5 reports the RMSF values averaged over atoms and normalized by the residue number. The overall flexibilities of ApoE, EETI 2.5F and CLT-1 are lower than those of TGN, RVG29, ang2, CooP, tLyp1 and RiGD. By this comparison and taking together all the results, peptides can be classified into two main groups: the first group characterized by a low structural flexibility and the second one by a high level of conformational flexibility. This finding may have functional implications, since conformational flexibility is often required for many peptide functions, eg for binding affinity and to improve the selectivity towards biological targets.59
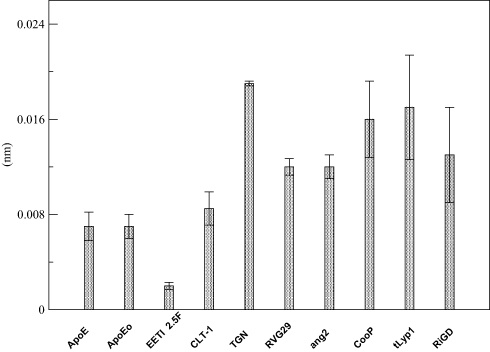 |
Figure 5 RMSF of the PC1’s projections. Note: The RMSF values are averaged over all Cα atoms and normalized by the number of residues. |
A further characterization of the structural and physicochemical properties of the peptides was obtained by performing an analysis of the hydrogen bonds (HBs), salt-bridges and of the solvent-accessible surface area (SASA), as a function of simulation time. As shown in Figure 6 and Table 4, the number of intra-peptide HBs of TGN, CLT-1, CooP, tLyp1, and RiGD show a quite constant trend, which oscillate around less than 5 hydrogen bonds (Table 3). The HB numbers of ApoE and ApoEo stabilize after 500 ns of simulation, suggesting that the hairpin conformation adopted by these peptides is stabilized by 6–7 hydrogen bonds. A constant number of HBs is also observed throughout the simulation of EETI 2.5F, indicating that the structure of this peptide is maintained stable by around 14 hydrogen bonds. The trend of the number of HBs of ang2 and RVG29, instead, follows an increasing course throughout the simulations.
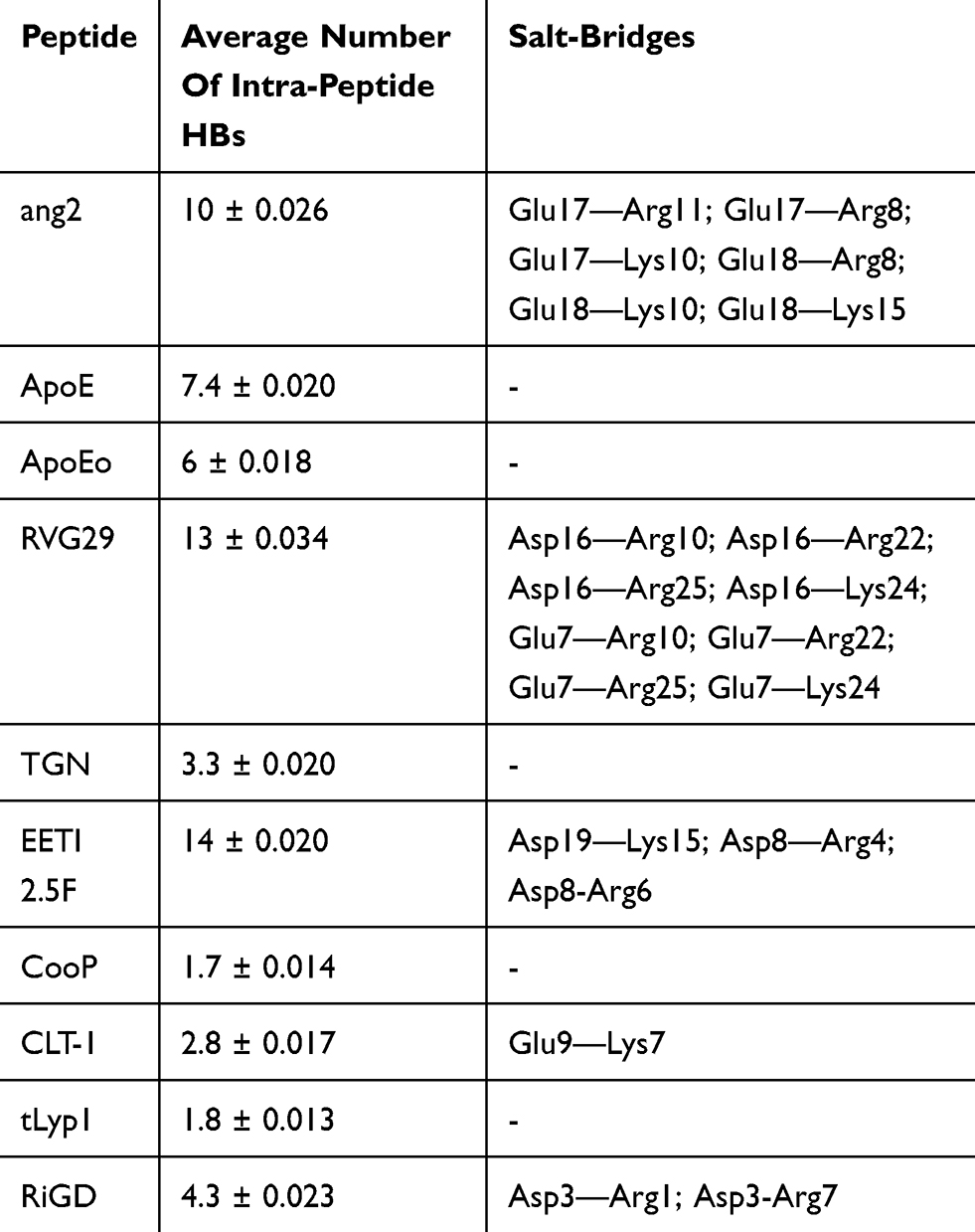 |
Table 4 Summary H-Bonding And Salt-Bridge Analyses |
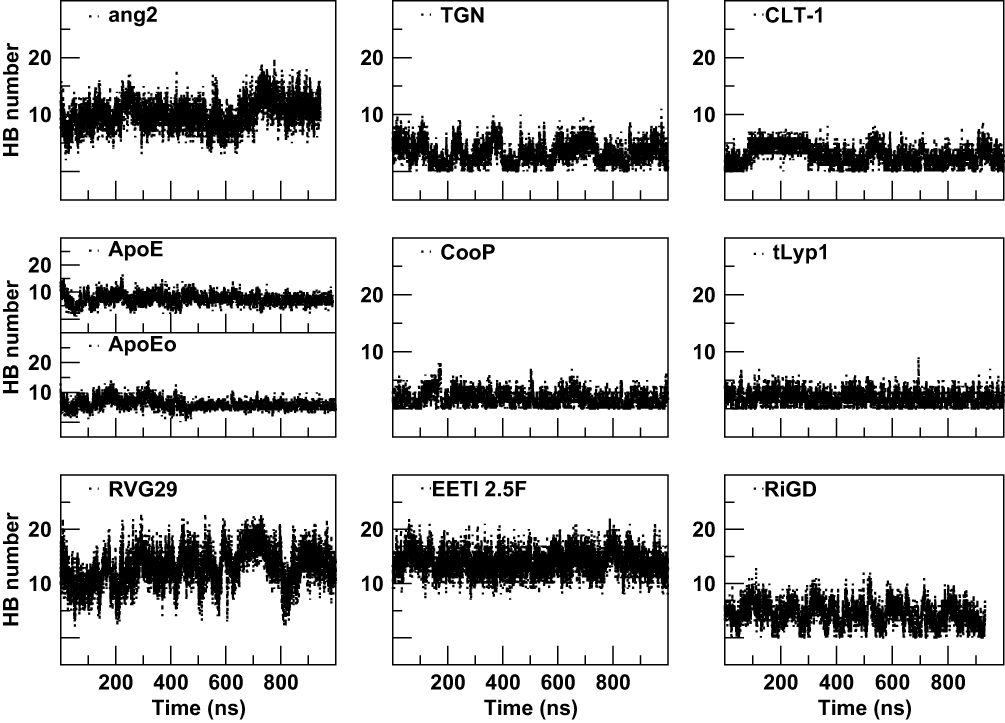 |
Figure 6 The intra-peptide HB number as a function of simulation time. |
The salt-bridges (ion pairs) were calculated by monitoring the distance between the side-chain O of Asp or Glu and the side-chain N of Arg, Lys or His. From the analysis were excluded ApoE, TGN, tLyp1, and CooP, since the Asp/Glu–Arg/Lys/His couples are not present in their sequences. All the salt-bridges calculated by using an O-N distance threshold of 4 Å are reported in Table 4. The table shows that ang2 and RVG29 peptides form 7 and 8 salt-bridges, respectively. The formation of 1, 2 and 3 salt-bridges is observed for CLT-1, RiGD and EETI 2.5F, respectively. However, analyzing the temporal evolution of these interactions during the simulations, only a few of them seem to actually contribute to the peptide structure stabilization. Consider in this respect Figure 7, in which only the salt-bridges present in the simulation with a time percentage greater than 60% are plotted. The Glu17—Arg11, Glu17—Arg8, and Asp3—Arg7 salt-bridges seem to confer the ang2 and RiGD peptides with strong stability. The continuous ruptures and formations, throughout the simulations, of the remaining salt-bridges reported in Table 4 (data not shown) indicate an intrinsic flexibility of these peptides despite their structural stability.
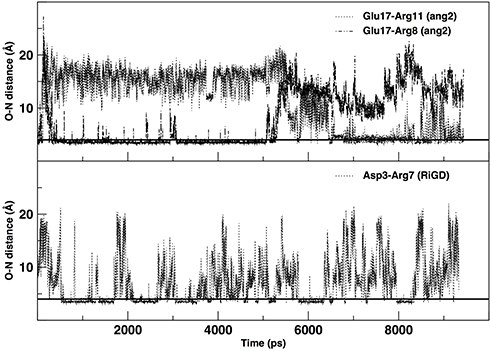 |
Figure 7 The temporal evolution of specific salt bridges of ang2 and RiGD peptides. Note: The black line, drawn at 4 Å, indicates the value of the O-N distance at which a salt bridge is considered inferred. |
The estimation of the solvent-accessible surface area gives information on the extent to which the residues of the peptide interact with the environment, in our simulation the solvent. As shown in Figure 8, quite constant trends of SASA, as a function of simulation time, are observed for CooP, CLT-1, EETI 2.5F, tLyp1 and RiGD peptides, indicating them as stable molecules. The SASA values of ang2, TGN and RVG29 peptides, instead, rapidly change during the simulation time, suggesting that these peptides undergo small conformational changes occurring around hydrophobic regions. As expected, the SASA values of ApoE and ApoEo vary only during the first 500 ns of simulation. After this time, they fluctuate around a constant value. This behavior suggests that the peptides adopt a stable conformation, with defined buried and solvent-exposed areas.
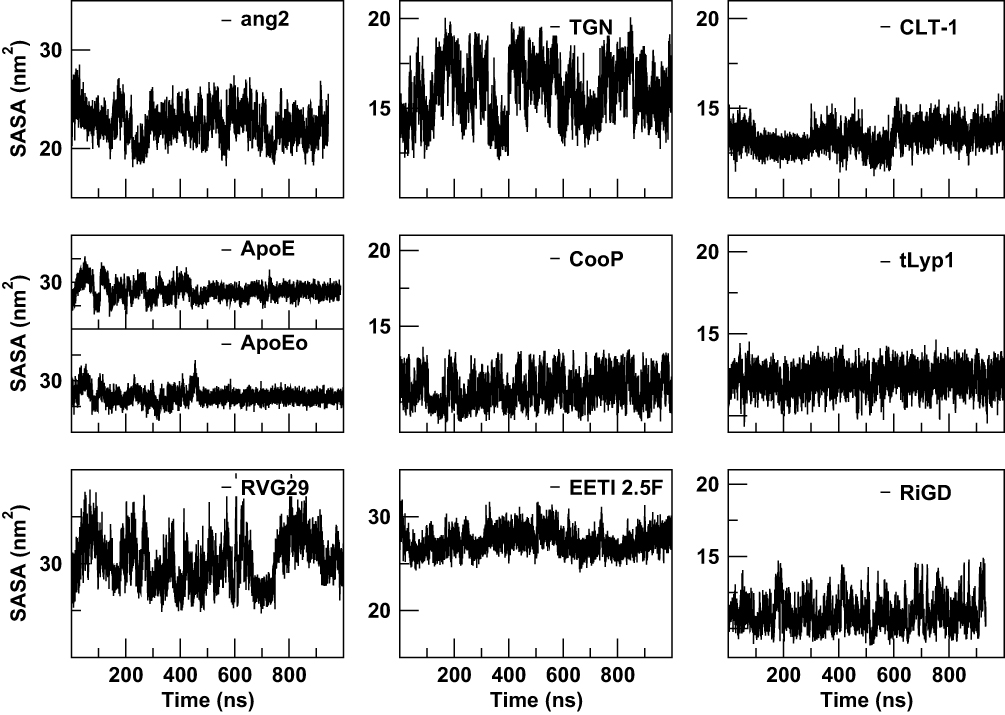 |
Figure 8 The solvent-accessible surface areas (SASA) of the peptides as a function of simulation time. |
On the overall, the high level of intramolecular interactions and the well-defined buried and solvent-exposed areas observed for ApoE, EETI 2.5F and CLT-1, result in a low conformational flexibility, suggesting that these peptides may interact with the target by preconfigured conformational states, as actually evidenced by PCA analysis. On the contrary, the small number of intramolecular interactions and the large number of isoenergetic conformational states, observed for ang2, TGN, CooP, tLyp-1 and RiGD, result in high conformational flexibility, which may allow these peptides to bind their molecular targets through an induced-fit like mechanism. Even if similar results were obtained for RVG29 peptide, the PCA-based convergence analysis evidenced that longer simulation times would be required to conclude that its conformational flexibility is actually an intrinsic feature of this peptide.
Conclusion
Designing and developing a NP-based platform, functionalized with BBB crossing and TH peptides, may represent a smart and promising approach to deliver drugs to brain tumors. Enormous benefits to such an approach may derive from the comprehension of the physicochemical, structural and dynamical features of the molecules making up the platform. Due to its capability to assess the stability, flexibility and dynamics of molecular systems, at both temporal and spatial resolutions, MD is considered a powerful tool.40 Here, we focused our attention on four BBB-crossing and five TH peptides and, to the best of our knowledge, for the first time we have characterized in detail their structural, dynamical and physicochemical features by exploiting the capabilities of MD simulation.
Our results demonstrate that the proposed computational approach could represent a valuable tool to evaluate the conformation adopted in the physiological aqueous solution of all the nine peptides as well as their temporal and structural fluctuations at atomic resolution.
The emerging picture is that the investigated peptides can be classified into two main groups on the basis of their conformational flexibility. Three peptides, ApoE, EETI 2.5F and CLT-1, assumed specific structural conformations during the simulations. The dynamical behavior of these peptides, captured by PCA-based free-energy landscapes, seems to be driven by large collective motions, which may be crucial for a suitable interaction with their molecular targets. Even if these peptides are characterized by an intrinsic stability, assessed by an overall low conformational flexibility and by a high number of intramolecular interactions, their use to functionalize delivery systems may be made difficult by the necessity of keeping unchanged their structural and functional conformations after the genetic/chemical functionalization of NPs. The remaining peptides (ang2, TGN, CooP, tLyp1 and RiGD), on the contrary, are clearly characterized by an intrinsical structural flexibility. This feature suggests that their interaction with the targets could be independent of a particular structural conformation (induced-fit binding), making them good candidates for a suitable functionalization of a theranostic platform. The simulation time for the RVG29 peptide resulted insufficient in order to conclude that all its conformational states were sampled. A comparative analysis of the structural and dynamical behavior of the free peptides, here described, with the same peptides anchored to a substrate mimicking the NPs would be also required for a full validation of the peptide classification here hypothesized. Current efforts are focusing on the set-up of a simulation procedure for this comparison.
Acknowledgments
This work was supported by the NANOCROSS project “Plant virus nanoparticles for blood-brain barrier crossing and medulloblastoma targeting” (IG 20314) granted by the Associazione Italiana per la Ricerca sul Cancro (AIRC) to M Mancuso. The computing resources used for this work were provided by CRESCO/ENEAGRID High Performance Computing infrastructure. The authors thank Dr. Massimo Pinto (ENEA) for his support in Python scripting and Dr. Andrea Quintiliani for English language editing and revision of the manuscript.
Disclosure
The authors report no conflicts of interest in this work
References
1. Strong MJ, Garces J, Vera JC, Mathkour M, Emerson N, Ware ML. Brain tumors: epidemiology and current trends in treatment. Brain Tumors Neurooncol. 2015;1(1):1–21. doi:10.4172/jbtn.1000102
2. Opoku-Damoah Y, Wang R, Zhou J, Ding Y. Versatile nanosystem-based cancer theranostics: design inspiration and predetermined routing. Theranostics. 2016;6(7):986–1003. doi:10.7150/thno.14860
3. Oliveira Silva C, Oliveira Pinho J, Margarida Lopes J, Almeida AJ, Gaspar MM, Reis C. Current trends in cancer nanotheranostics: metallic, polymeric, and lipid-based systems. Pharmaceutics. 2019;11(1):22. doi:10.3390/pharmaceutics11010022
4. Ding F, Chen Z, Kim WY, et al. A nano-cocktail of an NIR-II emissive fluorophore and organoplatinum(
5. Sun Y, Ding F, Zhou Z, et al. Rhomboidal Pt(II) metallacycle-based NIR-II theranostic nanoprobe for tumor diagnosis and image-guided therapy. PNAS. 2019;116(6):1968–1973. doi:10.1073/pnas.1817021116
6. Sun Y, Ding F, Chen Z, et al. Melanin-dot–mediated delivery of metallacycle for NIR-II/photoacoustic dual-modal imaging-guided chemo-photothermal synergistic therapy. PNAS. 2019;116(34):16729–16735. doi:10.1073/pnas.1908761116
7. Gao H. Progress and perspective on targeting nanoparticles for brain drug delivery. APBS. 2016;6(4):268–286. doi:10.1016/j.apsb.2016.05.013
8. Oller-Salvia B, Sànchez-Navarro M, Teixidò M. Blood-brain barrier: an emerging paradigm for brain delivery. Shuttle. Chem Soc Rev. 2016;45(17):4690–4707. doi:10.1039/C6CS00076B
9. Le Joncour V, Laakkonen P. Seek & destroy, use of targeting peptides for cancer detection and drug delivery. Bioorg Med Chem. 2018;26(10):2797–2806. doi:10.1016/j.bmc.2017.08.052
10. Gao H, Zhang S, Cao S, Yang Pang Z, Jiang X. Angiopep-2 and activable cell-penetrating peptide dual-functionalized nanoparticles for systemic glioma-targeting delivery. Mol Pharm. 2014;11(8):2755–2763. doi:10.1021/mp500113p
11. Wang S, Zhao C, Liu P, Wang Z, Ding J, Zhou W. Facile construction of dual-targeting delivery system by using lipid capped polymer nanoparticles for anti-glioma therapy. RSC Adv. 2018;8(1):444–453. doi:10.1039/C7RA12376K
12. Gao H, Qian J, Cao S, et al. Precise glioma targeting of and penetration by aptamer and peptide dual-functioned nanoparticles. Biomaterials. 2012;33(20):5115–5123. doi:10.1016/j.biomaterials.2012.03.058
13. Li J, Feng L, Fan L, et al. Targeting the brain with PEG-PLGA nanoparticles modified with phage-displayed peptides. Biomaterials. 2011;32(21):4943–4950. doi:10.1016/j.biomaterials.2011.03.031
14. Li J, Zhang C, Li J, et al. Brain delivery of NAP with PEG-PLGA nanoparticles modified with phage-display peptides. Pharm Res. 2013;30(7):1813–1823. doi:10.1007/s11095-013-1025-4
15. Feng X, Gao X, Kang T, et al. Mammary-derived growth inhibitor targeting peptide-modified PEG-PLA nanoparticles for enhanced targeted glioblastoma therapy. Bioconjug Chem. 2015;26(8):1850–1861. doi:10.1021/acs.bioconjchem.5b00379
16. Zhang B, Shen S, Liao Z. Targeting fibronectins of glioma extracellular matrix by CLT1 peptide-conjugated nanoparticles. Biomaterials. 2014;35(13):4088–4098. doi:10.1016/j.biomaterials.2014.01.046
17. Hu Q, Gu G, Liu Z, et al. F3 peptide-functionalized PEG-PLA nanoparticles co-administrated with tLyp-1 peptide for anti-glioma drug delivery. Biomaterials. 2013;34(4):1135–1145. doi:10.1016/j.biomaterials.2012.10.048
18. Shen Y, Maupetit J, Derreumaux P, Tufféry P. Improved PEP-FOLD approach for peptide and miniprotein structure predictor. J Chem Theor Comput. 2014;10(10):4745–4758. doi:10.1021/ct500592m
19. Thévenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tufféry P. PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acid Res. 2012;40(W1):W288–W293. doi:10.1093/nar/gks419
20. Fiser A, Sali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–491. doi:10.1016/S0076-6879(03)74020-8
21. Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. Gromacs: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. doi:10.1002/jcc.20291
22. Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS All-Atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118(45):11225–11236. doi:10.1021/ja9621760
23. Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparameterization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B. 2001;105(28):6474–6487. doi:10.1021/jp003919d
24. Tzanov A, Cuendet MA, Tuckerman ME. How accurately do current force fields predict experimental peptide conformations? An adiabatic free energy dynamics study. J Phys Chem B. 2014;118(24):6539−6552. doi:10.1021/jp500193w
25. Lindorff-Larsen K, Piana S, Palmo K, et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct Funct Bioinform. 2010;78(8):1950–1958. doi:10.1002/prot.22711
26. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. doi:10.1063/1.445869
27. Berendsen HJC, Grigera JR, Straatsma TP. The missing term in effective pair potentials. J Phys Chem. 1987;91(24):6269−6271. doi:10.1021/j100308a038
28. Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126(1):014101. doi:10.1063/1.2408420
29. Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys. 1981;52(12):7182–7190. doi:10.1063/1.328693
30. Nose S, Klein M. Constant pressure molecular dyncamics for molecular systems. Mol Phys. 1983;50(5):1055–1076. doi:10.1080/00268978300102851
31. Hockney RW, Eastwood JW. Computer Simulation Using Particles.
32. Essmann U, Perera L, Berkowitz ML, Darten T, Lee H, Pederson LGA. Smooth particle mesh ewald method. J Chem Phys. 1995;103(19):8577–8592. doi:10.1063/1.470117
33. Hess B, Bekker H, Berendsen HJC, Fraaije J. LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1997;18(12):1463–1472. doi:10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H
34. Daura X, Gademann K, Jaun B, Seebach D, van Gunsteren W, Mark A. Peptide folding: when simulation meets experiments. Angew Chem Int Ed. 1999;38(1–2):236−240. doi:10.1002/(ISSN)1521-3773
35. Amadei A, Linssen AB, Berendsen HJC. Essential dynamics of proteins. Proteins. 1993;17(4):412–425. doi:10.1002/prot.340170408
36. Hess B. Similarities between principal components of protein dynamics and random diffusion. Phys Rev E Stat Phys Plasmas Fluids Relat Interdiscip Topics. 2000;62(6Pt B):8438–8448. doi:10.1103/physreve.62.8438
37. Eisenhaber F, Lijnzaad P, Argos P, Sander C, Scharf M. The double cubic lattice method: efficient approaches to numerical integration of surface area and volume to dot surface contouring of molecular assemblies. J Comput Chem. 1995;16(3):273–284. doi:10.1002/(ISSN)1096-987X
38. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–38. doi:10.1016/0263-7855(96)00018-5
39. Van Rossum G, Drake JFL. Python Tutorial. The Netherlands: Centrum voor Wiskunde en Informatica Amsterdam; 1995.
40. Hollingsworth SA, Dror RO. Molecular dynamics simulations for all. Neuron. 2018;99(6):1129–1143. doi:10.1016/j.neuron.2018.08.011
41. Arcangeli C, Cantale C, Galeffi P, Rosato V. Structure and dynamics of the anti-AMCV scFv(F8): effects of selected mutations on the antigen combining site. J Struct Biol. 2008;164(1):119–133. doi:10.1016/j.jsb.2008.06.013
42. Arcangeli C, Circelli P, Donini M, et al. Structure-based design and experimental engineering of a plant virus nanoparticle for the presentation of immunogenic epitopes and as a drug carrier. J Biomol Struct Dyn. 2014;32(4):630–647. doi:10.1080/07391102.2013.785920
43. Polimeni M, Petridis L, Smith JC, Arcangeli C. Dynamics at a peptide-TiO2 anatase (101) interface. J Phys Chem B. 2017;121(38):8869–8877. doi:10.1021/acs.jpcb.7b04707
44. Demeule M, Règina A, Ché C, et al. Identification and design of peptides as a new drug delivery system for the brain. JPET. 2008;324(3):1064–1072. doi:10.1124/jpet.107.131318
45. Wang D, El-Amour SS, Dai M, et al. Engineering a lysosomal enzyme with a derivative of receptor binding domain of apoE enables delivery across the blood brain barrier. PNAS. 2013;110(8):2999–3004. doi:10.1073/pnas.1222742110
46. Oswald M, Geissler S, Goepferich A. Targeting the central nervous system (CNS): a review of rabies virus-targeting strategies. Mol Pharm. 2017;14(7):2177–2196. doi:10.1021/acs.molpharmaceut.7b00158
47. Hyvönen M, Enbäck J, Huhtala T. Novel target for peptide-based imaging and treatment of brain tumors. Mol Cancer Ther. 2014;13(4):996–1007. doi:10.1158/1535-7163.MCT-13-0684
48. Zhan C, Gu B, Xie C, Li J, Liu Y, Lu W. Cyclic RGD conjugated poly(ethylene glycol)-co-poly(lactic acid) micelle enhances paclitaxel anti-glioblastoma effect. J Control Release. 2010;143(1):136–142. doi:10.1016/j.jconrel.2009.12.020
49. Kimura RH, Levin AM, Cochran FV, Cochran JR. Engineered cysteine knot peptides that bind αʋβ3, αʋβ5 and α5β1 integrins with low-nanomolar affinity. Proteins. 2009;77(2):359–369. doi:10.1002/prot.22441
50. Pilch J, Brown DM, Komatsu M, et al. Peptides selected for binding to clotted plasma accumulate in tumor stroma and wounds. PNAS. 2006;103(8):2800–2804. doi:10.1073/pnas.0511219103
51. Roth L, Agemy L, Kotamraju VR, et al. Transtumoral targeting enabled by a novel neuropilin-binding peptide. Oncogene. 2012;31:3754–3763. doi:10.1038/onc.2011.537
52. Hsieh Y-H, Chou C-Y. Structural and functional characterization of human apoliprotein E 72-166 peptides in both aqueous and lipid environments. J Biomed Sci. 2011;18:4. doi:10.1186/1423-0127-18-4
53. Heitz A, Avrutina O, Le-Nguyen D, et al. Knottin cyclization: impact on structure and dynamics. BMC Struct Biol. 2008;8:54. doi:10.1186/1472-6807-8-54
54. Zhang X, Wang F, Shen Q, et al. Structure reconstruction of LyP-1: lc(LyP-1) coupling by amide bond inspires the brain metastatic tumor targeted drug delivery. Mol Pharm. 2018;15(2):430–436. doi:10.1021/acs.molpharmaceut.7b00801
55. Timur SS, Yalçın G, Çevik Ö, Andaç C, Gürsoy RN. Molecular dynamics, thermodynamic, and mutational binding studies for tumor-specific LyP-1 in complex with p32. J Biomol Struct Dyn. 2018;36(5):1134–1144. doi:10.1080/07391102.2017.1313779
56. Cino EA, Choy W-Y, Karttunen M. Comparison of secondary structure formation using 10 different force fields in microsecond molecular dynamics simulations. J Chem Theory Comput. 2012;8(8):2725–2740. doi:10.1021/ct300323g
57. Levy Y, Jortner J, Becker OM. Solvent effects on the energy landscapes and folding kinetics of polyarginine. PNAS. 2001;98(5):2188–2193. doi:10.1073/pnas.041611998
58. Van Agthoven JF, Shams H, Cochran FV, et al. Structural basis of the differential binding of engineered knottins 2.5F and 2.5D to integrins αʋβ3 and α5β1. Structure. 2019;27(9):1443–1451. doi:10.1016/j.str.2019.06.011
59. Vermeer LS, Lan Y, Abbate V, et al. Conformational flexibility determines selectivity and antibacterial, antiplasmodial, and anticancer potency of cationic α-helical peptides. J Biol Chem. 2012;287(41):120–133. doi:10.1074/jbc.M112.359067
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.