")
Back to Journals » International Journal of Nanomedicine » Volume 14
Characterization of apolipoprotein A-I peptide phospholipid interaction and its effect on HDL nanodisc assembly
Authors Patel H, Ding B, Ernst K, Shen L, Yuan W , Tang J , Drake LR , Kang J , Li Y, Chen Z, Schwendeman A
Received 12 July 2018
Accepted for publication 6 March 2019
Published 30 April 2019 Volume 2019:14 Pages 3069—3086
DOI https://doi.org/10.2147/IJN.S179837
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Hiren Patel,1 Bei Ding,2 Kelsey Ernst,3 Lei Shen,2 Wenmin Yuan,3 Jie Tang,3 Lindsey R Drake,1 Jukyung Kang,3 Yaoxin Li,2 Zhan Chen,2 Anna Schwendeman1,3,4
1Department of Medicinal Chemistry, College of Pharmacy, University of Michigan, Ann Arbor, MI, USA; 2Department of Chemistry, University of Michigan, Ann Arbor, MI, USA; 3Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, MI, USA; 4Biointerfaces Institute, University of Michigan, Ann Arbor, MI, USA
Background: Synthetic HDLs (sHDLs), small nanodiscs of apolipoprotein mimetic peptides surrounding lipid bilayers, were developed clinically for atheroma regression in cardiovascular patients. Formation of HDL involves interaction of apolipoprotein A-I (ApoA-I) with phospholipid bilayers and assembly into lipid-protein nanodiscs.
Purpose: The objective of this study is to improve understanding of physico-chemical aspects of HDL biogenesis such as the thermodynamics of ApoA-I-peptide membrane insertion, lipid binding, and HDL self-assembly to improve our ability to form homogeneous sHDL nanodiscs that are suitable for clinical administration.
Methods: The ApoA-I-mimetic peptide, 22A, was combined with either egg sphingomyelin (eSM) or 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) phospholipid vesicles to form sHDL. The sHDL assembly process was investigated through lipid vehicle solubilization assays and characterization of purity, size, and morphology of resulting nanoparticles via gel permeation chromatography (GPC), dynamic light scattering (DLS), and transmission electron microscopy (TEM). Peptide-lipid interactions involved were further probed by sum frequency generation (SFG) vibrational spectroscopy and attenuated total reflection-Fourier transform infrared spectroscopy (ATR-FTIR). The pharmacokinetics of eSM-sHDL and POPC-sHDL nanodiscs were investigated in Sprague Dawley rats.
Results: sHDL formation was temperature-dependent, with spontaneous formation of sHDL nanoparticles occurring only at temperatures exceeding lipid transition temperatures as evidenced by DLS, GPC, and TEM characterization. SFG and ATR-FTIR spectroscopy findings support a change in peptide-lipid bilayer interactions at temperatures above the lipid transition temperature. Lipid-22A interactions were stronger with eSM than with POPC, which resulted in the formation of more homogeneous sHDL nanoparticles with longer in vivo circulation time as evidenced the PK study.
Conclusion: Physico-chemical characteristics of sHDL are in part determined by phospholipid composition. Optimization of phospholipid composition may be utilized to improve the stability and homogeneity of sHDL.
Keywords: apoA-1 mimetic peptide, high-density lipoprotein (HDL), large unilamellar vesicle (LUV), sum frequency generation (SFG), dynamic light scattering (DLS), transmission electron microscopy (TEM)
Introduction
High levels of circulating high-density lipoprotein (HDL), commonly referred to as “good cholesterol” are reportedly associated with improved cardiovascular outcomes.1 Endogenous HDLs are 8–12 nm diameter nanoparticles composed of a mixture of lipids and amphipathic helix-containing proteins, primarily apolipoprotein A-I (ApoA-I).2 Endogenous HDL is formed by recruitment of phospholipids through ATP-binding cassette transporter (ABCA1) by ApoA-I leading to the formation of small discoidal HDL nanoparticles. These discoidal HDLs efflux excess cholesterol from macrophages in atherosclerotic plaques and deliver it to the liver for elimination in a process called reverse cholesterol transport.2 Thus, the idea of acute plaque reduction by direct infusion of cholesterol-free reconstituted or synthetic HDL (sHDL) nanodiscs, idiomatically referred to as “Drano® for the arteries”, has generated considerable interest as a method for reversing atherosclerosis.2,3
The reduction of atheroma burden following multiple infusions of reconstituted HDL (rHDL) products has been shown in several clinical trials.3,4 Clinically tested HDL products were based on either a full-length ApoA-I protein or small synthetic peptide mimetics of ApoA-I both combined with phospholipids to form nanodisc-shaped sHDL particles (Figure 1A).5–7 Following intravenous administration, sHDL nanoparticles uptake cholesterol from peripheral tissues and interact with endogenous lipoproteins in plasma by exchanging lipid and protein components (Figure 1B). The assembly of pharmaceutical grade sHDL nanodiscs and lipoprotein remodeling process in plasma all are driven by thermodynamics of ApoA-I peptide-phospholipid interactions. Thus, understanding physicochemical forces driving the initial insertion of ApoA-I peptide in lipid bilayers and sHDL particle assembly will offer practical insights into self-assembly of sHDL nanoparticles of high purity, size, homogeneity, and stability. It will guide efforts to produce clinical grade pure sHDL at the larger scale needed for patient dosing.
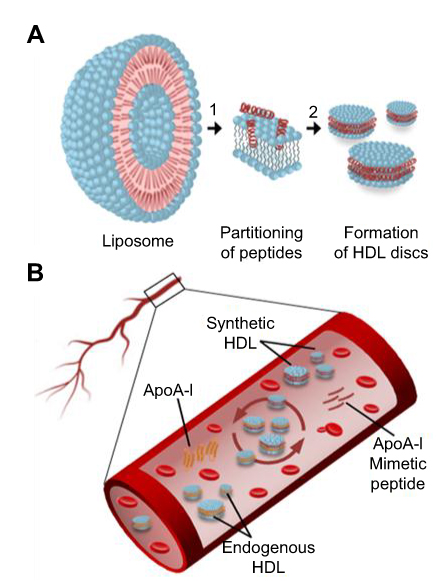 | Figure 1 Schematic presentation of ApoA-I peptide interaction with lipids. (A) Synthetic HDL (sHDL) particle assembly process: 1) ApoA-I peptides adsorb to the surface of liposomes and partition into the phospholipid bilayer; 2) Bilayer solubilization and assembly of sHDL occur after a critical peptide to lipid ratio is reached. (B) Lipoprotein remodeling in vivo. Upon administration, sHDL interacts with endogenous HDL in bloodstream by exchanging protein and phospholipid components. This interaction results in the formation of lipid-poor endogenous ApoA-I, lipid poor ApoA-I synthetic peptide, and HDL particles containing protein and peptide components. |
Current methods for sHDL formation include adding cholate surfactant to facilitate formation of ApoA-I-lipid-cholate micelles and subsequent surfactant removal by dialysis,6 disrupting liposomes by homogenization or sonication prior to the addition of ApoA-I to facilitate protein-lipid binding,3,7 co-lyophilization of ApoA-I peptide and lipid mixtures in organic solvent,8,9 and using microfluidics devices to facilitate mixing and binding of ApoA-I protein and phospholipid.10 Typically such methods generate HDL nanoparticles that require subsequent purification by processes that add to the complexity of the manufacturing process, including steps such as size-exclusion chromatography, ultracentrifugation, and dialysis.6,7,11 Additionally, the heterogeneity of HDL preparations and the presence of residual surfactants have led to dose-limiting toxicity in clinical trials.5,12 This toxicity, in turn, necessitated costly process development and additional animal and clinical safety studies.5,12,13 These problems may be avoided by using a production method that yields high-purity HDL.
The process of sHDL nanoparticle assembly involves a sequence of steps (Figure 1A), starting with the interaction of ApoA-I peptide monomers with lipid layers in which individual peptides begin to adsorb, partition, and insert into lipid bilayers. Later stages involve solubilization of the membrane, stripping of a lipid layer, and assembly of sHDL nanoparticles. Detailed understanding of the effects of sHDL composition, process temperatures, buffers, and component concentrations used during the assembly process on nanodisc purity, size, and morphology is critical to sHDL safety and efficacy. In this study, we examine the influence of phospholipid type on the initial adsorption/insertion of ApoA-I-mimetic peptide into bilayers, the kinetics of sHDL assembly, and the quality of the resulting sHDL particles.
The ApoA-I-mimetic peptide used in this study, ESP24218 (22A), has been previously tested in clinical trials in dyslipidemic patients.12,14–16 The 22A peptide was optimized for phospholipid binding properties, helix stability, and ability to facilitate cholesterol mobilization and esterification in patients.14,16 The two phospholipids tested here, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and egg sphingomyelin (eSM), have both been used in clinical sHDL formulations. POPC, used in the formulation of ETC-216 rHDL,12 is a phosphatidylcholine with unsaturated acyl fatty acid and a transition temperature (Tm) of −2°C.17 eSM, used to prepare CER-001 rHDL12 and ETC-642 sHDL,14 is a naturally occurring lipid with mostly saturated fatty acids and an effective Tm near 40°C.18 We chose to test these phospholipids because of their distinct physicochemical properties, most notably their transition temperatures (Tm). The Tm of a lipid is the temperature at which a lipid converts from the rigid gel ordered phase to the more fluid crystalline or liquid disordered phase. The fluidity of the lipid bilayer influences the ease with which proteins and peptides can insert into the bilayer, as in the formation of sHDL. The differences between these lipids have the potential to cause differences in peptide-lipid interactions in sHDL formation, ultimately dictating the characteristics and purity of the nanoparticles produced.
In this study, we examined the kinetics of solubilization of large unilamellar vehicles (LUVs) of POPC and eSM through spectrophotometric monitoring of turbidity as well as the particle size, purity, and morphology of the resulting sHDL particles via dynamic light scattering (DLS), gel permeation chromatography (GPC), and transmission electron microscopy (TEM). We also for the first time employed sum frequency generation (SFG) vibrational spectroscopy, attenuated total reflection-Fourier transform infrared spectroscopy (ATR-FTIR) to elucidate the mechanisms of the initial insertion of peptide into a lipid bilayer and the temperature dependence of this process on the molecular level. We believe this approach will be widely applicable to a variety of nanodisc systems to gain insights into nanoscale self-assembly.
Experimental section
Materials
ESP-24218 peptide (22A, PVLDLFRELLNELLEALKQKLK) and 5A peptide (DWLKAFYDKVAEKLKEAFPDWAKAAYDKAAEKAKEAA) were purchased from American Peptide Company (Sunnyvale, CA) and Bachem Americas Inc (Torrance, CA), respectively. The phospholipids, POPC and eSM, were purchased from Avanti Polar Lipids (Alabaster, AL) and Nippon Oil and Fats Corporation (Osaka, Japan). All other reagents were purchased from Sigma Aldrich, USA.
Lipid solubilization assay
LUVs were prepared by extrusion. Phospholipid stock solution in chloroform was dried by nitrogen gas and hydrated with 10 mM sodium phosphate buffer (pH 7.4). The suspension was subjected to 5–7 freeze-thaw cycles. The resulting multilamellar vesicles (MLVs) were extruded 10 times through 100 nm pore size Nuclepore membranes in a Lipex pressure extruder (Transferra Nanosciences Inc, Burnaby, BC) at 25°C for POPC and 45°C for eSM. Initial concentrations of 5.0 mg/mL in 10 mM sodium phosphate buffer (pH 7.4) were used for both lipid and peptide solutions. The solutions were mixed at a 1:1 wt/wt (~3.75:1 molar) lipid to peptide ratio and incubated at the designated temperature (25°C, 37°C, or 50°C). The kinetics of POPC/eSM LUV solubilization due to interaction with 22A and remodeling to form sHDL was indicated by the decrease in turbidity, measured by UV absorbance at 600 nm every 2 mins for 1 hr on BioTek’s Synergy II microplate spectrophotometer (Winooski, VT). After the first hour, measurements were taken at 2, 3, 4, 5, 6, 20 and 24 hrs time points.
Dynamic light scattering
Size distributions of liposomes and sHDL particles were measured by DLS. DLS experiments were done using a Malvern Zetasizer Nano ZS (Worcestershire, UK). sHDL solutions were diluted to 1.0 mg/mL peptide concentration prior to the measurements, and volume and intensity weighted size distributions were obtained at 25°C.
Transmission electron microscopy
For TEM, 3.0 µL of sHDL or liposome was deposited at room temperature on glow-discharged Quantifoil R2/2 200 mesh grids followed by blotting and vitrification with a Vitrobot (FEI, Hillsboro, OH). The samples were negatively stained with uranyl formate solution. All specimens were imaged on a Tecnai F20 transmission electron microscope (FEI) equipped with a field emission gun operated at 120 kV. Images were recorded at a magnification of ×60,000–150,000 and defocus value of approximately −1 µm on a Gatan US4000 CCD camera.
Gel permeation chromatography
The purity of sHDL was analyzed by GPC with UV detection at 220 nm using a Tosoh TSK gel G3000SWx 7.8 mm×30 cm column (Tosoh Bioscience, King of Prussia, PA) on a Waters Breeze Dual Pump system (Milford, MA). All sHDL samples were from the lipid solubilization assay with 1:1 wt/wt (~3.75:1 molar) lipid to peptide ratio. Samples were diluted to a final concentration of 5.0 mg/mL, 10.0 μL was injected, and injections were eluted with PBS at a flow rate of 1.0 mL/min. The analysis was performed at ambient temperature.
SFG spectroscopy
Solid-supported eSM/eSM and POPC/POPC lipid bilayers were deposited on a right-angle CaF2 prism by the Langmuir–Blodgett/Langmuir–Schaefer procedure as described.19–21 The first lipid monolayer was deposited on one square face of the prism with a KSV2000 LB deposition trough (Biolin Scientific, Finland). After plasma cleaning, the prism was immersed in the water subphase of the LB trough. Then, ~5 drops of 10.0 mg/mL lipid-chloroform solution were spread on the water surface. The two barrier arms were moved closer until the surface tension reached 34.0 mN/m. The prism was lifted at a velocity of 1.0 mm/min from the subphase while the positions of the barrier arms were controlled to maintain the surface tension at 34.0 mN/m. The first layer of lipid was deposited on the prism via this Langmuir–Blodgett process.21 For the second layer, lipid drops were added to the water surface in a 2.0 mL reservoir to reach the surface tension of 34.0 mN/m. The reservoir was then elevated so that the lipid molecules on the water surface contacted with the lipid monolayer deposited on the prism. This is the Langmuir–Schaefer process.22
Our SFG experimental setup and data analysis methodology were reported previously.19,20 The SFG spectra were collected from the interface between the lipid bilayer (deposited on CaF2 as presented above) and peptide solution. The concentration of the 22A peptide solution was 20.0 μg/mL. For each temperature condition, we recorded SFG spectra in the C-H stretching (2700–4000 cm−1) and amide I (1500–1800 cm−1) frequency regions. In order to deduce peptide orientation information, two polarization combinations, namely ssp (s-polarized SFG, s-polarized visible, p-polarized IR) and ppp (p-polarized SFG, p-polarized visible, and p-polarized IR), were adopted in these measurements. SFG experiments were carried out at three temperatures of 20°C, 37°C, and 50°C.
Attenuated total reflection-Fourier transform infrared spectroscopy
ATR-FTIR spectroscopy is a method of vibrational spectroscopy that can be used to assess the interactions between peptides and lipid bilayers and changes in peptide secondary structure and to supplement SFG results due to these interactions. The lipid bilayer was deposited on an ATR ZnSe crystal and the surface was washed with D2O to avoid the overlap of the O-H signal from water and the C-H stretch signal from the lipid bilayer.23 22A peptide was also dissolved in D2O at 20.0 μg/mL. ATR-FTIR spectra were recorded in the C-H stretching frequency range (2,800–3,000 cm−1) at three different temperatures of 25°C, 37°C, and 50°C.
Pharmacokinetic (PK) evaluation
Male Sprague-Dawley rats (7 weeks old) were purchased from Charles River Breeding Laboratories (Portage, MI). 22A-eSM and 22A-POPC sHDL particles were prepared following incubation of eSM and POPC liposomes with 22A peptide at 1:1 w/w ratio at 50°C for 30 mins. The resulting solutions were sterile filtered and the particle sizes were determined by DLS and were confirmed to be in the sHDL size range. To determine plasma stability and PK, 22A-eSM and 22A-POPC were dosed intravenously via tail vein at 100 mg/kg based on the peptide amount. Blood samples were collected pre-dose and at 1, 15, and 30 mins and 1, 3, 8, and 24 hrs post-infusion. Serum was separated, aliquoted, and stored at −80°C prior to analysis. Animal protocol was reviewed and approved by the University of Michigan Institutional Animal Care and Use Committee to assure compliance with the federal policies and guidelines specified by the Association for Assessment and Accreditation of Laboratory Animal Care.
The serum concentration of 22A peptide was measured by liquid chromatography combined with mass spectrometry detection (LC-MS/MS).9 Briefly, samples were spiked with the internal standard (alternative ApoA-I mimetic peptide, 5A) and peptides were extracted with methanol. The peptide concentrations were analyzed using an Agilent 6520 Accurate-Mass Q-TOF LC/MS equipped with a dual electrospray ionization source (Dual-ESI) (Agilent Technologies, CA). The HPLC separation was performed on an Agilent 300SB-C18 column (2.1×50 mm, 3.5 μm). Mass spectra were acquired in negative ion mode with the mass range set at m/z 100–3200. The conditions used for the ESI source included a capillary voltage of 3,500 V, a drying gas temperature of 332°C, a drying gas flow of 5 L/min, a nebulizer pressure of 45 psi, and a fragmentor voltage of 225 V. The extracted ion chromatograms of 22A, 22A metabolite, and 5A internal standard were exported from the total ion chromatogram at m/z 656.6, 832.5, and 844.4, respectively. The sum of peak areas of 22A and 22A metabolite was used for PK analysis.
Results
Kinetics of the solubilization of LUVs by 22A peptide
The temperature dependencies of LUV solubilization and sHDL assembly were examined for eSM and POPC at 25°C, 37°C, and 50°C. The 5.0 mg/mL solutions of LUV particles of eSM (133 nm in diameter) and POPC (120 nm in diameter) were turbid and had UV 600 nm absorbances of 0.78 and 0.26, respectively. Upon addition of 22A peptide, the turbidity decreased as 10 nm sHDL nanoparticles began to assemble. Thus, the kinetics of the decrease in turbidity was indicative of the sHDL assembly rates. sHDL assembly temperature dependencies and rates were markedly different for the two phospholipids (Figure 2). The formation of sHDL was observed for eSM LUVs at 37°C and 50°C, but not at 25°C (Figure 2A and C) whereas the formation of sHDL occurred at all three temperatures for POPC (Figure 2B and D). To evaluate the contribution of some fatty acid heterogeneity of animal source purified eSM to LUV solubilization by 22A peptide, we compared sHDL formation rates for eSM with fully synthetic sphingomyelin sSM (Figure S1). The sSM behaved similarly to eSM, with sHDL formation observed at 37°C (Figure S1B) and 50°C (Figure S1C) and no sHDL formation at 25°C (Figure S1A). It is well known that the process of peptide insertion into lipid bilayers is more favorable at temperatures above a lipid’s Tm thus, sHDL formation did not occur with eSM and sSM at 25°C, which is significantly below the lipid’s Tm of approximately 40°C. The Tm for POPC is −2°C, allowing for sHDL formation to occur at all three temperatures. At 50°C, the solubilization of eSM occurred much faster than for POPC, reaching near complete sHDL formation in 10 mins for eSM compared to 2.5 hrs for POPC, indicating stronger 22A-eSM interactions at this temperature. Similar faster solubilization for eSM compared to POPC occurred at 37°C as well. The rate of lipid solubilization also depended on the concentration of the components at the same lipid to peptide ratio, with sHDL formation occurring faster at peptide concentration of 10 and 5 mg/mL relative to 1 mg/mL at both 37°C (Figure S2A) and 50°C (Figure S2B).
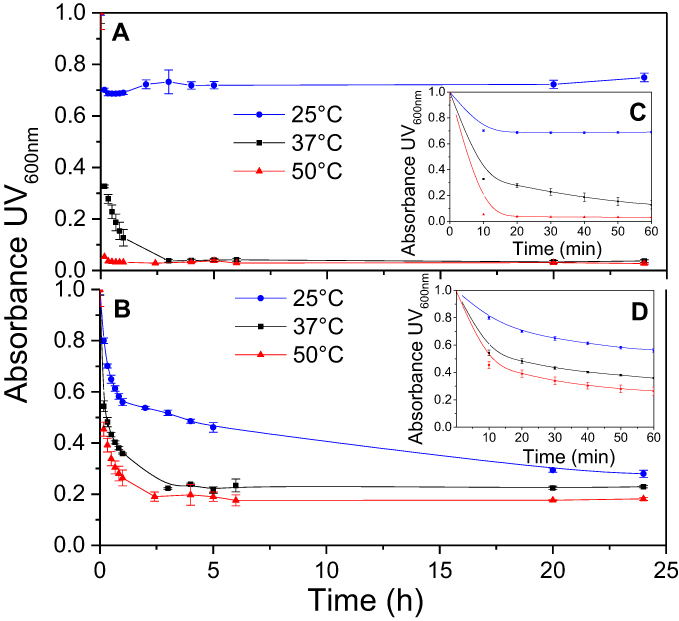 | Figure 2 Kinetics of synthetic HDL (sHDL) assembly examined by the decrease in UV600nm turbidity of large unilamellar vesicle (LUV) solutions during incubation with 22A peptide. Liposomes at 5 mg/mL concentration of egg sphingomyelin (eSM) (A/C) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) (B/D) were incubated with 5 mg/mL 22A peptide for 24 hrs at three different temperatures. sHDL assembly during the first hour of incubation is shown in the figure inserts (C and D). |
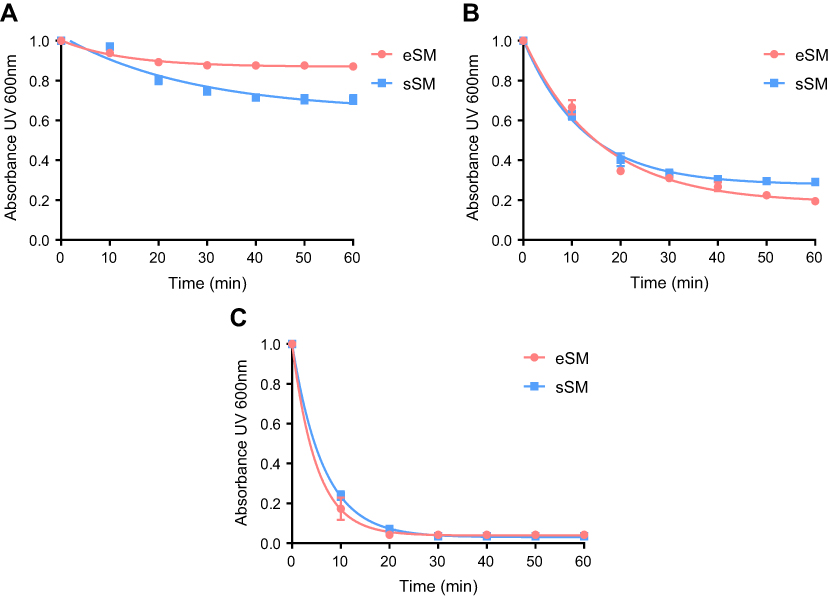 | Figure S1 Lipid solubilization assay for egg sphingomyelin (eSM, red) and synthetic sphingomyelin (sSM, blue). Kinetics of sHDL assembly examined by the decrease in UV600 nm turbidity of LUV solutions during incubation with 22A peptide. Liposomes at a concentration of 5 mg/mL were incubated with 5 mg/mL 22A peptide for 1 hr at 25°C (A), 37°C (B), and 50°C (C). |
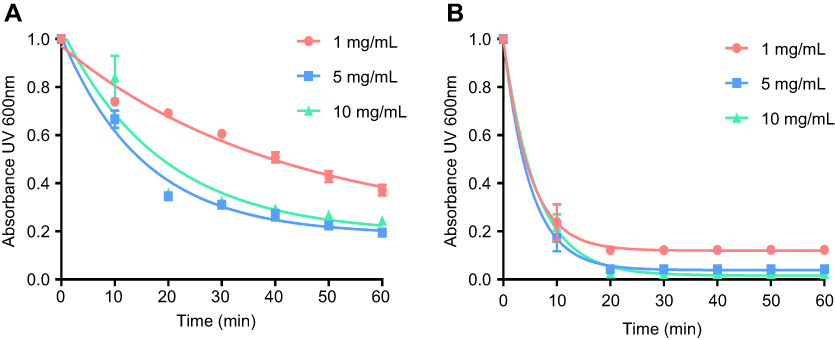 | Figure S2 Lipid solubilization assay at different concentrations. Kinetics of sHDL assembly examined by the decrease in UV600 nm turbidity of egg sphingomyelin (eSM) large unilamellar vesicles (LUVs) incubated with 22A peptide at 1:1 weight ratio and 1, 5, and 10 mg/mL peptide concentrations. Liposomes were incubated with 22A peptide for 1 hr at 37°C (A) and 50°C (B), respectively. Data are represented as mean±SD, and were analyzed via nonlinear fit-one phase decay by Graphpad Prism. |
Characterization of formed sHDL nanoparticles
To confirm the formation of sHDL following eSM and POPC vehicle solubilization, the resulting solutions were analyzed by DLS and TEM. In addition, the quality of sHDL was assessed by GPC following the removal of unreacted liposomes by filtration to avoid column fouling. At the end of a 24-hr solubilization experiment, the lipids were distributed among three phases: assembled sHDL (8–10 nm in diameter), remaining liposomes (LUVs, 100–130 nm in diameter), and aggregated liposomes or MLVs (approximately 1–2 µm in diameter). Because larger particles scatter light more strongly than smaller particles, the intensity averaged DLS fittings exaggerate the signal from liposomes and lipid aggregates and mask the presence of small sHDL. When heterogeneous solutions post incubation are analyzed, the relative abundance of particles is better described by the volume-averaged measurements (Figure 3A and C); however, the average particle sizes and the presence of larger liposomes and lipid aggregates are best described by intensity averaged values (Figure S3).24 The difference is most pronounced for POPC sHDL at 25°C, where a second peak occurs in the intensity-averaged plot (Figure S3B) absent from the volume-averaged plot (Figure 3C), indicative of some remaining POPC LUVs along with formed sHDL. Note that such difference is not evident for the intensity averaged plot for eSM at 25°C (Figure S3A) as sHDL is unable to form below lipid transition temperature. Similar to the solubilization results, complete formation of sHDL was observed by DLS volume measurement for eSM at 37°C and 50°C (Figure 3A) and for POPC at all temperatures (Figure 3C). Failure of eSM-sHDL to form at 25°C is evidenced by the large peak representative of remaining eSM LUVs via DLS (Figure 3A) and the absence of an sHDL peak via GPC (Figure 3B). The average sizes of all sHDL formulations were around 9 nm (Table S1). For eSM incubations at 25°C, no sHDL formation was observed and only LUVs (137.3±1.4 nm in diameter) were present. The representative GPC and DLS profiles of 22A peptide incubated in buffer without phospholipid present are shown in Supplemental Figures S4 and S5, respectively. Without lipid 22A appears to be initially self-associated at all concentrations of 1, 5, and 10 mg/mL. Dissociation of peptide to a monomeric form is observed only a 50°C in 1 mg/mL as obvious from the shift of GPC retention time from 9.6 mins (corresponding to a molecular weight of ~145,000 Da) to 14.9 mins (~2500 Da). Thus, the presence of lipids drives dissociation of peptide, bilayer insertion, and sHDL formation. It is possible that the dissociation of peptide aggregates and insertion of peptide in the lipid bilayers occurs faster based on the rapid sHDL formation observed at 5 and 10 mg/mL peptide concentrations at 50°C, Only 1 mg/mL dilute solutions.
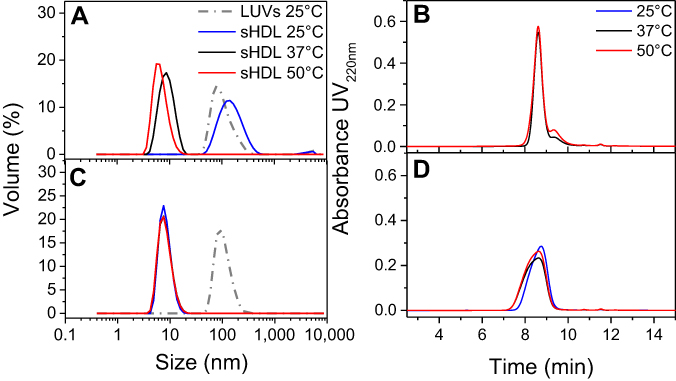 | Figure 3 Dynamic light scattering (DLS) volume distributions of particle sizes for egg sphingomyelin (eSM)-based (A) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC)-based (C) sHDL formed after 24 hr incubation of lipid large unilamellar vesicles (LUVs) and 22A peptide at different temperatures. The homogeneity of particle size assessed by gel permeation chromotography (GPC) following 24-hr incubations of 22A peptide with eSM (B) and POPC (D) LUVs at different temperatures. |
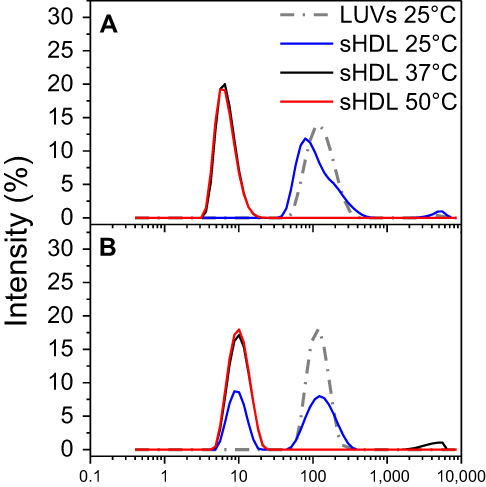 | Figure S3 Dynamic light scattering (DLS) intensity distributions of particle sizes for egg sphingomyelin (eSM)- (A) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC)-based (B) sHDL following 24-h incubation of lipid large unilamellar vesicles (LUVs) and 22A peptide at different temperatures. |
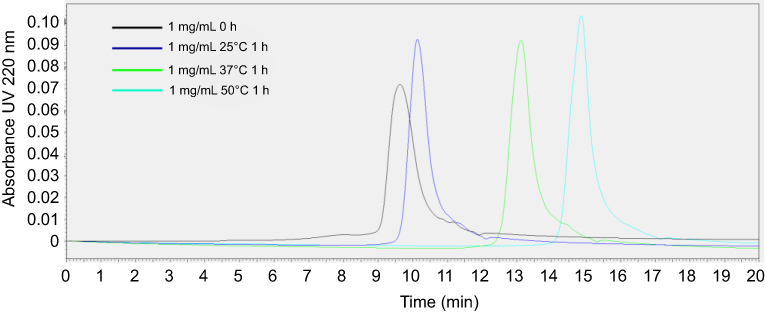 | Figure S4 Gel permeation chromotography (GPC) profile of 1 mg/mL 22A peptide solutions immediately after preparation and following 1-hr incubation at 25°C, 37°C, and 50°C. |
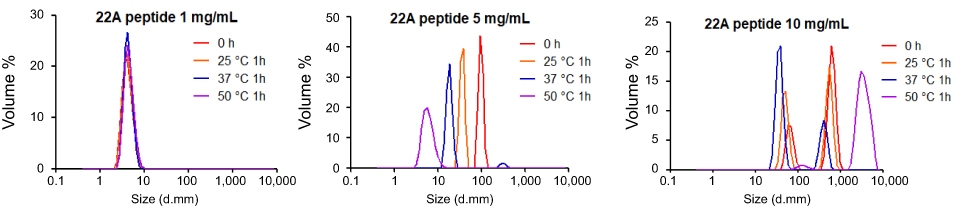 | Figure S5 Dynamic light scattering (DLS) profiles of 1, 5, and 10 mg/mL solutions of 22A peptide before and after 1-hr incubation at 25°C, 37°C, and 50°C. |
The TEM imaging experiments showed the formation of sHDL following peptide incubation with eSM at 37°C and 50°C and with POPC at all temperatures. Images were first obtained of pure eSM (Figure 4A) and POPC (Figure 4D) LUVs for comparison. The TEM images of sHDL show elliptical and round structures corresponding to edge-on and face-on projections of the discoidal particles (Figure 4). Similar TEM images of HDL particles have been reported elsewhere.6,25 The eSM-sHDL at 37°C (Figure 4B) and 50°C (Figure 4C) appeared to be more rounded and tightly packed compared to the somewhat heterogeneous shapes of POPC-sHDL at 37°C (Figure 4E) and 50°C (Figure 4F). sHDL particles were analyzed by GPC to assess size homogeneity (Figure 3B and D; Table S1). The sHDL formed by eSM-22A incubations had a sharper and narrower GPC peak than the POPC-sHDL, indicating higher homogeneity of eSM-sHDL, which is well correlated to the TEM results.
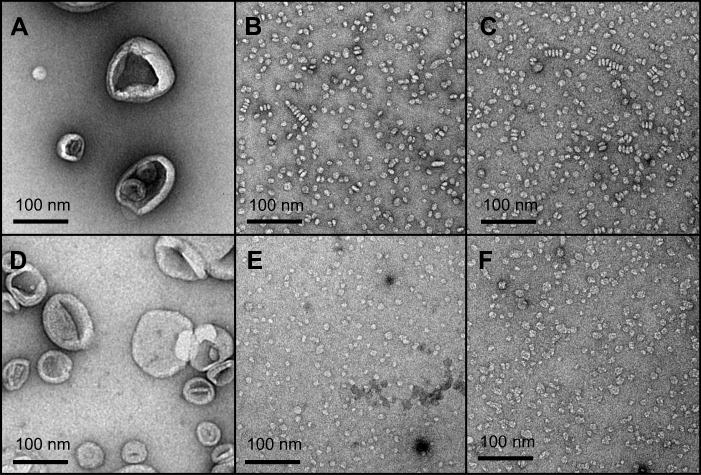 | Figure 4 Transmission electron microscopy (TEM) images of large unilamellar vesicles (LUVs) of egg sphingomyelin (eSM) (A) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) (D); sHDL nanoparticles formed following 1-hr incubations of 22A peptide with eSM (B) and POPC (E) LUVs at 37°C and with eSM (C) and POPC (F) LUVs at 50°C. |
SFG vibrational spectroscopy
SFG spectroscopy is a method of nonlinear vibrational spectroscopy that can be used to investigate molecular interactions on a surface or at an interface.26–29 (See Figure 5A for a schematic diagram of the experimental setup for SFG spectroscopy.) In a typical SFG bilayer experiment, a bilayer is deposited onto one face of a right-angle prism, then immersed in water or a peptide solution for the duration of the experiment.19,21 An infrared (IR) beam with a tunable frequency and a visible beam with a fixed frequency are overlapped on the surface or interface of interest, generating the SFG signal, which is a sum of the overlapping IR and visible beams.20,21 Unlike other vibrational spectroscopies, this technique is surface/interface specific, as only media lacking inversion symmetry generate SFG signals.21 This allows for analysis of the specific interactions and structures at an interface or surface without the need to subtract for effects from bulk media or other sources of interference. SFG signal intensity is recorded as a function of IR wavenumber and yields information concerning peptide-lipid interactions, lipid bilayer structure and flip-flop, and peptide secondary structure and orientation.19–22
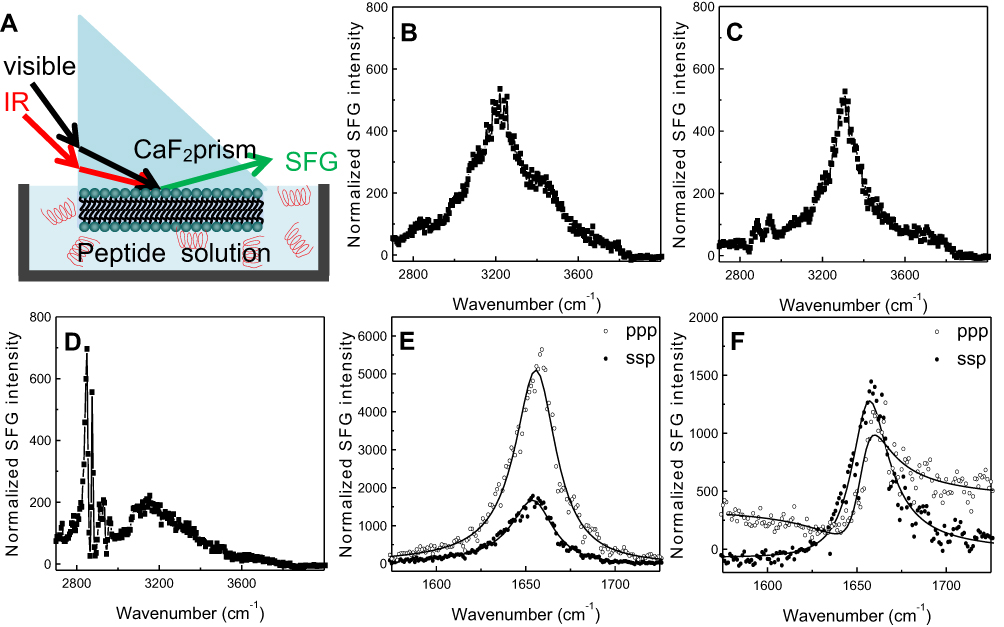 | Figure 5 Schematic diagram (A) of the experimental setup for sum frequency generation (SFG) spectroscopy analysis of lipid bilayer-peptide interactions; SFG O-H/N-H stretching signals collected from 22A peptide associated with egg sphingomyelin (eSM) at 20°C (B), 37°C (C), and 50°C (D); SFG amide range stretching signals collected from 22A peptide associated with eSM at 37°C (E) and 50°C (F); ppp refers to polarization combination of p-polarized SFG, p-polarized visible, p-polarized IR and ssp refers to s-polarized SFG, s-polarized visible, p-polarized IR. |
SFG spectra from the C-H stretching frequency range were collected at 20°C, 37°C, and 50°C to investigate the interactions of both lipids with 22A peptide. Two broad peaks, centered at 3,200 and 3,400 cm−1 dominated the SFG spectrum for eSM and 22A at 20°C; they result from O-H stretching in ordered water structures near the lipid bilayer surface (Figures 5B and S6A).22,30,31 The SFG spectrum for POPC at 20°C differed greatly, with a single peak at 3,300 cm−1 (Figure S6D). This peak represents the N-H stretch of ordered peptide secondary structure and suggests a strong interaction occurred between 22A peptide and the lipid bilayer.21 At the higher temperature of 37°C the 3,300 cm−1 N-H stretch peak appeared in the eSM spectrum (Figures 5C and S6B). Previously, the N-H stretching peak at 3,300 cm−1 has been reported on biomolecules with α-helical or β-sheet structures in SFG studies.32,33 The disappearance of the broad O-H peaks resulted from the disordering or the elimination of water molecules along the lipid bilayer surface due to increased peptide-lipid interactions.22,34 Two small C-H stretch peaks appeared at 2,875 and 2,940 cm−1 in the eSM spectrum at 37°C which may result from peptide side chains, lipid hydrocarbon chains, disruption of the lipid bilayer, or a combination of these factors (Figure 5C).21,22,26,35 The 3,300 cm−1 peak remained for POPC at 37°C (Figure S6E). Finally, at the temperature of ~50°C, the peaks at 2875 cm−1 and 2940 cm−1 grew significantly for eSM and became evident for POPC while the 3,300 cm−1 N-H peak disappeared for both lipids (Figures 5D, S6C, and S6F). The disappearance of the 3,300 cm−1 peak suggested the absence of ordered secondary structure at the high temperature and thus the increases in the 2,875 and 2,940 cm−1 peaks were from the disruption of the bilayer rather than peptide side chains or lipid hydrocarbon chains. The symmetry of the bilayer was broken, which can be explained by the removal of the first layer of the bilayer system by peptides. Thus, at 50°C, 22A peptide molecules begin to associate with lipids in the bilayer (especially the outer leaflet of the bilayer) and that the formed lipid-peptides complexes leave the bilayer. Once peptide-lipid complexes leave the bilayer, SFG N-H stretching of the peptide is no longer observed, though strong SFG signals can be observed from disruption of the bilayer.
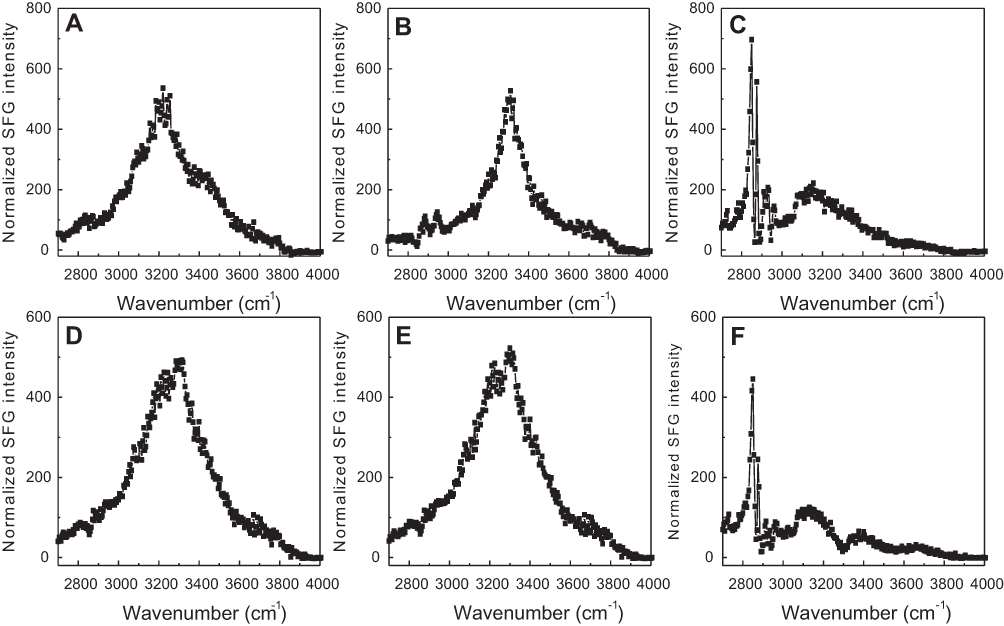 | Figure S6 Sum frequency generation (SFG) O-H/N-H stretching signals collected from 22A peptide associated with model lipid membranes of egg sphingomyelin (eSM) at 25°C (A), 37°C (B), and 50°C (C) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) at 20°C (D), 37°C (E), and 50°C (F). |
The SFG signal in the amide I frequency range is from the peptide backbone (mainly contributed by backbone C=O stretching), providing information on peptide secondary structure. Each secondary structure has a unique peak position with α-helix peaks appearing around 1,655 cm−1.21 By using different polarization combinations, the orientation of the peptide relative to the bilayer can be found.34 For the peptide-eSM interactions, at the low temperature, 20°C, weak signals centered around 1,652 cm−1 were detected for both lipids at both polarizations (Figures S7A and S7D). At the moderately elevated temperature, 37°C, the amide I signals significantly increased with the same peak-centers (Figures 5E, S7B, and S7E). The increase in the intensity at this temperature was likely due to the change in the peptide orientation and/or the increase in peptide adsorption numbers. Interestingly, at the high temperature, 50°C, the amide I signal intensities dropped for both lipids (Figures 5F, S7C, and S7F). We hypothesize that this occurs because the lipid bilayer is disrupted as lipid and peptide combine to form sHDL nanoparticles, also yielding lower signal intensity. At the high temperature, 50°C, the SFG intensity in the ppp spectra decreased with a slight peak-center shift to 1,658 cm−1 (Figure S7F). The minimal shift in the peak center suggests a change in the peptide environment occurred around 50°C. This environmental change may occur as the lipid bilayer is disrupted by sHDL nanoparticle formation. The similar amide I signal peak center observed at different temperatures remained suggesting that the α-helical secondary structure remained throughout the temperature changes. Therefore, SFG data matched the LUV solubilization and DLS results very well.
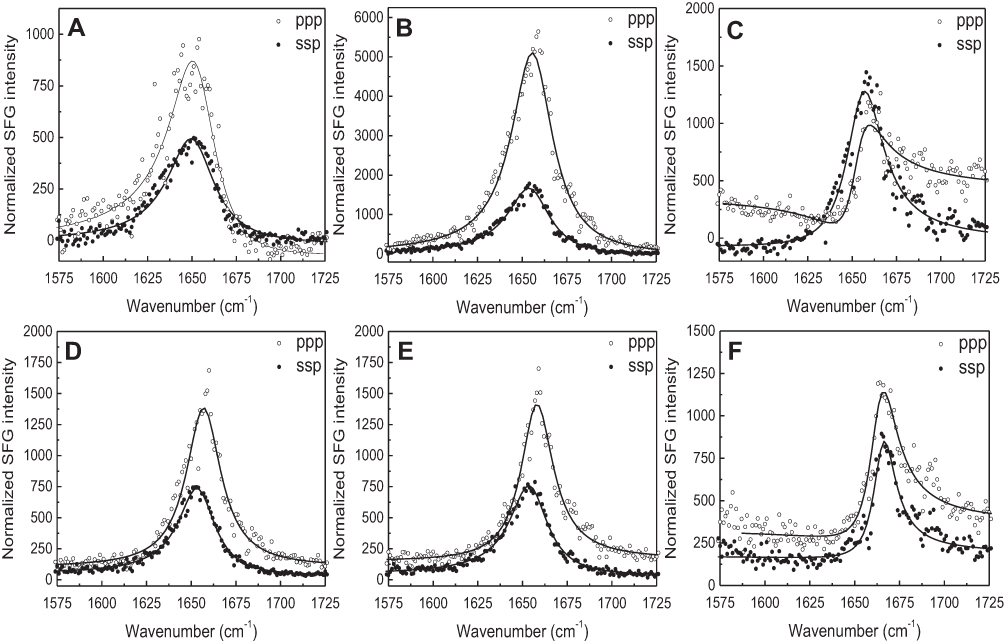 | Figure S7 The ssp and ppp sum frequency generation (SFG) amide I spectra collected from the 22A peptide associated with model lipid membranes of egg sphingomyelin (eSM) at 25°C (A), 37°C (B), and 50°C (C) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) at 25°C (D), 37°C (E), and 50°C (F). ppp refers to polarization combination of p-polarized SFG, p-polarized visible, p-polarized IR and ssp refers to s-polarized SFG, s-polarized visible, p-polarized IR. |
Attenuated total reflection-Fourier transform infrared spectroscopy
The ATR-FTIR spectra of peptide-free eSM and POPC bilayers in contact with D2O water showed three main peaks: 2,850 cm−1, 2,920 cm−1, and a small shoulder 2,955 cm−1 (Figures S8A and S8B). These are C-H stretching modes from the lipid chain.23 The peaks at 2,850 and 2,920 cm−1 represent C-H symmetric and asymmetric stretching of CH2 groups, respectively.35 Addition of 22A to the subphase in contact with the bilayer at 25°C, 37°C, and 50°C led to decreases in the C-H peaks at both 2,850 and 2,920 cm−1. Both peaks disappeared for eSM at 25°C, while the shoulder at 2,955 cm−1 increased and its center shifted toward 2,960 cm−1. At 37°C and 50°C for 22A-eSM and all temperatures for 22A-POPC dips started to appear at 2,850 and 2,920 cm−1, indicating that there were fewer C-H vibrations under these conditions. Decreases in these C-H peaks indicated disruption of the lipid bilayer.23 The delay in the appearance of dips in the eSM spectra versus POPC arises again from their different Tm values. At room temperature, eSM is in the ordered gel phase while POPC is in the disordered liquid crystalline phase, thus 22A peptide does not interact with eSM but interacts with POPC at 25°C. As proposed in the SFG results section, 22A peptide started to disrupt the bilayer structure and form sHDL nanoparticles at higher temperatures. The ATR-FTIR data reported here correlate well with the above results from the solubilization, DLS, and SFG experiments.
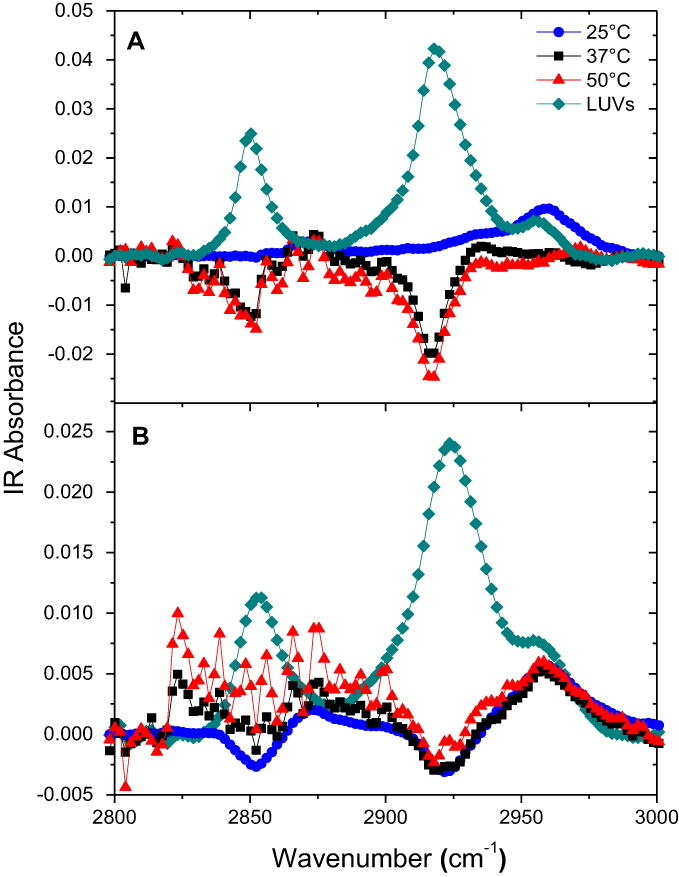 | Figure S8 Attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectra of egg sphingomyelin (eSM) bilayer only and 22A peptide with eSM difference spectra at three different temperatures. (A) ATR-FTIR spectra of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer only and 22A peptide with POPC difference spectra at three different temperatures (B). |
PK evaluation of sHDL nanodiscs
The PK of eSM-sHDL and POPC-sHDL sHDLs nanodiscs were investigated in Sprague-Dawley rats by quantifying serum 22A levels. The sHDL nanodiscs were prepared by incubation of eSM and POPC liposomes with 22A peptide at 50°C for 1 hr to obtain homogeneous particles with ~10 nm diameter for both lipids. Rats were dosed intravenously via tail vein at 100 mg/kg dose of 22A-sHDL and serum samples were collected pre-dose and at 0, 1, 15, and 30 mins and 1, 3, 8, and 24 hrs post-dose. To assess how the strength of 22A binding to phospholipid affects sHDL PK behavior, the serum concentrations of peptide were measured by LC-MS after administration. We have previously shown that 22A peptide has twice the circulation time in vivo when it is administered in a lipid-bound form of sHDL rather than as a free peptide.9 We hypothesized that stronger binding of 22A to eSM will result in slower kinetics of sHDL particle disassembly in blood, thus longer 22A residence time in vivo for eSM-sHDL relative to POPC-sHDL. The higher stability of eSM-sHDL in blood will also result in higher maximum plasma levels of 22A for this formulation relative to POPC-sHDL. The measurements from three rats per treatment were averaged to generate a PK profile (Figure 6). As predicted, serum levels of 22A in rats treated by the POPC-sHDL formulation were considerably lower than those of rats treated with the eSM-sHDL formulation, with the highest measured concentrations being 655.2 μg/mL for eSM-sHDL at 8 hrs and 340.7 μg/mL for POPC-sHDL at 3 hrs. The serum concentration of 22A for POPC-sHDL also peaked at an earlier time than that was observed for eSM-sHDL. The area under the curve (AUC) for 22A administered in POPC-based sHDL was approximately twofold lower relative to the AUC for the eSM-based sHDL with the values of 5193±322 and 10,247±61 μg*h/mL, respectively. These results indicate that stronger in vitro binding of 22A to eSM translates to higher blood stability of eSM-sHDL relative to POPC-sHDL and more favorable PK of 22A for eSM-based formulation.
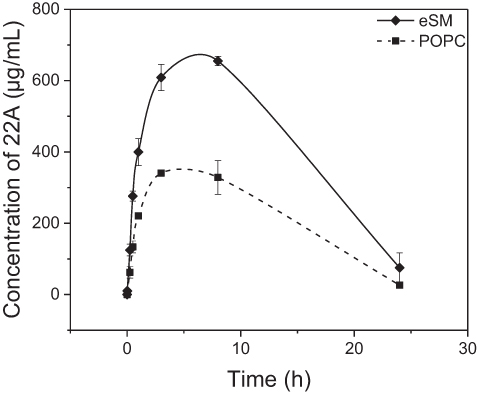 | Figure 6 Pharmacokinetics of 22A peptide following administration of assembled egg sphingomyelin (eSM) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC)-based sHDL to Sprague-Dawley rats at 100 mg/kg by intravenous injection. Peptide serum levels were determined by liquid chromotography-mass spectroscopy (LC-MS) (N=3 animals/group, mean±SEM). |
Discussion
A clear relationship between lipid composition and the temperature dependence of sHDL formation was observed in this series of experiments as well as in the work of other researchers.36,37 Spontaneous sHDL formation occurs at the temperatures near or above phospholipid Tm when the membrane fluidity is high enough for initial peptide insertion to occur and solubilization and assembly to begin. In the case of apolipoproteins, it is generally accepted that the protein determines the overall structure of the sHDL particle; however, in the case of the much shorter peptide, it is the lipid that dictates the kinetics and homogeneity of the overall formation of sHDL.
To explain this process further, we hypothesize that a nucleation-and-growth process accounts for spontaneous sHDL formation. According to this concept, peptide distributes equally on the vesicles and partitions between vesicles and aqueous phase. The initial partitioning is defined in part by the fluidity of lipid bilayer determined by heterogeneity of lipid composition (lipid type, fatty acid chain length, and saturation) and the effective Tm. Both bilayers are stressed by a high peptide content that would much more favorably be embedded in a locally curved topology such as a pore/breakage within the vesicles. However, such breakage requires large activation energy and the cooperative action of peptide addition. Energy fluctuations and random diffusion of peptide form a spontaneous cluster with atypically high local lipid density. This enables the vesicle to overcome the activation barrier and nucleate a pore, eventually leading toward nanoassembly of sHDL like structures.
Once nucleated, the area with peptide insertion will therefore attract more peptides from the aqueous phase and the accessibility of the inner lipid leaflet should increase as well. It will then follow the bilayer-to-edge micellization or solubilization scenario established by Almgren and co-workers.38,39 Accordingly, kinetically controlled solubilization of lipid by 22A will produce stable sHDL until it reaches thermodynamic equilibrium. We observed faster formation of sHDL at 22A concentration of 5 and 10 mg/mL rather than at 1 mg/mL. Interestingly, 22A appears to be aggregated at all temperatures; thus, initial insertion of peptide in lipid bilayers drives its disaggregation and sHDL formation. Isothermal titration calorimetry (ITC) has been previously used by other investigators to study the interaction between ApoA-I and phospholipids.40,41 Apoliprotein-lipid interaction and subsequent nanodisk formation have been shown to be a temperature-dependent process.37,42 As lipids must be in liquid-disordered state to fully interact with the peptides, stronger interactions would be expected at temperatures above the lipid transition temperature. Strengthened ApoA-I binding to lipids with increasing temperatures as evidenced by increasing ΔH in ITC has been observed elsewhere and attributed to increase bond formation and penetration of nonpolar peptide/protein regions into lipid bilayers.41
We implemented a combination of analytical techniques to elucidate the process of sHDL assembly through the solubilization of phospholipids eSM and POPC by the ApoA-I mimetic peptide 22A. The process was determined to be both lipid- and temperature-dependent, characteristics that are in part reliant upon each other, as the highly fluid state of lipid bilayers above their respective transition temperatures is most favorable for peptide insertion and sHDL formation. sHDL nanoparticles formed more rapidly with eSM than with POPC. DLS analysis confirmed that sHDL formed only at temperatures exceeding the Tm of each lipid, and GPC and TEM revealed that eSM formed nanoparticles of a more homogenous nature. Both the characterization and in vivo evaluation suggested that the phospholipid composition of sHDL could influence its structure, blood stability, and PK function. The results of this study along with subsequent experiments will enhance the understanding of the relationship between sHDL composition, assembly, and function. This knowledge will allow for the formulation of high-purity sHDL nanoparticles with life-saving therapeutic potential.
Acknowledgments
The authors acknowledge the editorial help of Dr Beth Schachter. This project was supported in part by AHA 13SDG17230049 (AS), Upjohn award (AS), R01 GM113832 (AS), R01 HL122416, AHA 16UFEL29850003 (KE), AHA 16POST27760002 (WY), Barbour Fellowship (BD), and T32 GM07767 (LD).
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. Am J Med. 1977;62(5):707–714. doi:10.1016/0002-9343(77)90874-9
2. Kingwell BA, Chapman MJ, Kontush A, Miller NE. HDL-targeted therapies: progress, failures and future. Nat Rev Drug Discov. 2014;13(6):445–464. doi:10.1038/nrd4279
3. Parolini C, Marchesi M, Lorenzon P, et al. Dose-related effects of repeated ETC-216 (recombinant apolipoprotein A-IMilano/1-Palmitoyl-2-Oleoyl Phosphatidylcholine complexes) administrations on rabbit lipid-rich soft plaques: in vivo assessment by intravascular ultrasound and magnetic resonance ima. J Am Coll Cardiol. 2008;51(11):1098–1103. doi:10.1016/j.jacc.2007.12.010
4. Shaw JA, Bobik A, Murphy A, et al. Infusion of reconstituted high-density lipoprotein leads to acute changes in human atherosclerotic plaque. Circ Res. 2008;103(10):1084–1091. doi:10.1161/CIRCRESAHA.108.182063
5. Degoma EM, Rader DJ. Novel HDL-directed pharmacotherapeutic strategies. Nat Rev Cardiol. 2011;8(5):266–277. doi:10.1038/nrcardio.2010.200
6. Ng KK, Lovell JF, Vedadi A, Hajian T, Zheng G. Self-assembled porphyrin nanodiscs with structure-dependent activation for phototherapy and photodiagnostic applications. ACS Nano. 2013;7(4):3484–3490. doi:10.1021/nn400418y
7. Song Q, Huang M, Yao L, et al. Lipoprotein-based nanoparticles rescue the memory loss of mice with Alzheimer’s disease by accelerating the clearance of amyloid-beta. ACS Nano. 2014;8(3):2345–2359. doi:10.1021/nn4058215
8. Schwendeman A, Sviridov DO, Yuan W, et al. The effect of phospholipid composition of reconstituted HDL on its cholesterol efflux and anti-inflammatory properties. J Lipid Res. 2015;56(9):1727–1737. doi:10.1194/jlr.M060285
9. Tang J, Li D, Drake L, et al. Influence of route of administration and lipidation of apolipoprotein A-I peptide on pharmacokinetics and cholesterol mobilization. J Lipid Res. 2016. doi:10.1194/jlr.M071043
10. Kim Y, Fay F, Cormode DP, et al. Single step reconstitution of multifunctional high-density lipoprotein-derived nanomaterials using microfluidics. ACS Nano. 2013;7(11):9975–9983. doi:10.1021/nn4039063
11. Zhao Y, Imura T, Leman LJ, Curtiss LK, Maryanoff BE, Ghadiri MR. Mimicry of high-density lipoprotein: functional peptide-lipid nanoparticles based on multivalent peptide constructs. J Am Chem Soc. 2013;135(36):13414–13424. doi:10.1021/ja404714a
12. Krause BR, Remaley AT. Reconstituted HDL for the acute treatment of acute coronary syndrome. Curr Opin Lipidol. 2013;24(6):480–486. doi:10.1097/MOL.0000000000000020
13. Easton R, Gille A, D’Andrea D, Davis R, Wright SD, Shear C. A multiple ascending dose study of CSL112, an infused formulation of ApoA-I. J Clin Pharmacol. 2014;54(3):301–310. doi:10.1002/jcph.194
14. Khan M, Lalwani N, Drake S, Crockatt J, Dasseux J-L. Single-dose intravenous infusion of ETC-642, a 22-Mer ApoA-I analogue and phospholipids complex, elevates HDL-C in atherosclerosis patients. Circulation. 2003;108:563–564.
15. Li D, Gordon S, Schwendeman A, Remaley AT. Apolipoprotein mimetics in the management of human disease. In: Anantharamaiah GM, Goldberg D, editors. Cham: Springer International Publishing; 2015:29–42. doi:10.1007/978-3-319-17350-4_3
16. Dasseux J-L, Sekul R, Buttner K, Cornut I, Metz G, Dufourcq J, inventors; Apolipoprotein A-I agonists and their use to treat dyslipidemic disorders. United States patent US 6630450B1. 2003 Oct 7.
17. Silvius JR. Thermotropic phase transitions of pure lipids in model membranes and their modifications by membrane proteins. Lipid-Protein Interact. 19822:239–281.
18. Mannock DA, McIntosh TJ, Jiang X, Covey DF, McElhaney RN. Effects of natural and enantiomeric cholesterol on the thermotropic phase behavior and structure of egg sphingomyelin bilayer membranes. Biophys J. 2003;84(2 Pt 1):1038–1046. doi:10.1016/S0006-3495(03)74920-0
19. Nguyen KT, Le Clair SV, Ye S, Chen Z. Molecular interactions between magainin 2 and model membranes in situ. J Phys Chem B. 2009;113(36):12358–12363. doi:10.1021/jp904154w
20. Ye S, Nguyen KT, Clair SV, Le Chen Z. In situ molecular level studies on membrane related peptides and proteins in real time using sum frequency generation vibrational spectroscopy. J Struct Biol. 2009;168(1):61–77. doi:10.1016/j.jsb.2009.03.006
21. Chen X, Chen Z. SFG studies on interactions between antimicrobial peptides and supported lipid bilayers. Biochim Biophys Acta. 2006;1758(9):1257–1273. doi:10.1016/j.bbamem.2006.01.017
22. Ding B, Glukhova A, Sobczyk-Kojiro K, Mosberg HI, Tesmer JJG, Chen Z. Unveiling the membrane-binding properties of N - Terminal and C - Terminal regions of g protein-coupled receptor kinase 5 by combined optical spectroscopies. Langmuir. 2014;30(3):823–831. doi:10.1021/la404055a
23. Zhang C, Wu FG, Hu P, Chen Z. Interaction of polyethylenimine with model cell membranes studied by linear and nonlinear spectroscopic techniques. J Phys Chem C. 2014;118(23):12195–12205. doi:10.1021/jp502383u
24. Allen T. Particle Size Measurement.
25. Antwerpen R, Van Chen GC, Pullinger CR, et al. Cryo-electron microscopy of low density lipoprotein and reconstituted discoidal high density lipoprotein : imaging of the apolipoprotein moiety. J Lipid Res. 1997;38(4):659–669.
26. Mermut O, Phillips DC, York RL, McCrea KR, Ward RS, Somorjai GA. In situ adsorption studies of a 14-Amino acid leucine-lysine peptide onto hydrophobic polystyrene and hydrophilic silica surfaces using quartz crystal microbalance, atomic force microscopy, and sum frequency generation vibrational spectroscopy. J Am Chem Soc. 2006;128(11):3598–3607. doi:10.1021/ja056031h
27. Yang Z, Li Q, Chou KC. Structures of water molecules at the interfaces of aqueous salt solutions and silica: cation effects. J Phys Chem. 2009;113(19)8201–8205.
28. Lu X, Spanninga SA, Kristalyn CB, Chen Z. Surface orientation of phenyl groups in poly(sodium 4-styrenesulfonate) and in poly(sodium 4-styrenesulfonate): poly(3,4-ethylenedioxythiophene) mixture examined by sum frequency generation vibrational spectroscopy. Langmuir. 2010;26(17):14231–14235. doi:10.1021/la101866v
29. Engel MFM, Vandenakker CC, Schleeger M, Velikov KP, Koenderink GH, Bonn M. The polyphenol EGCG inhibits amyloid formation less efficiently at phospholipid interfaces than in bulk solution. J Am Chem Soc. 2012;134(36):14781–14788. doi:10.1021/ja3031664
30. Jena KC, Hore DK. Variation of ionic strength reveals the interfacial water structure at a charged mineral surface. J Phys Chem C. 2009;113(34):15364–15372. doi:10.1021/jp905475m
31. Okur HI, Kherb J, Cremer PS. Cations bind only weakly to amides in aqueous solutions. J Am Chem Soc. 2013;135(13):5062–5067. doi:10.1021/ja3119256
32. Weidner T, Breen NF, Drobny GP, Castner DG. Amide or amine: determining the origin of the 3300 cm−1 NH mode in protein SFG spectra using 15N isotope labels. J Phys Chem B. 2009;113(47):15423–15426. doi:10.1021/jp908773c
33. Fu L, Wang Z, Yan ECY. Chiral vibrational structures of proteins at interfaces probed by sum frequency generation spectroscopy. Int J Mol Sci. 2011;12(12):9404–9425. doi:10.3390/ijms12129404
34. Ding B, Soblosky L, Nguyen K, et al. Physiologically-relevant modes of membrane interactions by the human antimicrobial peptide, LL-37, revealed by SFG experiments. Sci Rep. 2013;3:1854. doi:10.1038/srep01854
35. Ge A, Wu H, Darwish TA, James M, Osawa M, Ye S. Structure and lateral interaction in mixed monolayers of Dioctadecyldimethylammonium Chloride (DOAC) and stearyl alcohol. Langmuir. 2013;29(18):5407–5417. doi:10.1021/la400143k
36. Fukuda M, Nakano M, Sriwongsitanont S, Ueno M, Kuroda Y, Handa T. Spontaneous reconstitution of discoidal HDL from sphingomyelin-containing model membranes by apolipoprotein A-I Electron microscopy. J Lipid Res. 2007;48(4):882–889. doi:10.1194/jlr.M600495-JLR200
37. Wan CPL, Chiu MH, Wu X, Lee SK, Prenner EJ, Weers PMM. Apolipoprotein-induced conversion of phosphatidylcholine bilayer vesicles into nanodisks. Biochim Biophys Acta Biomembr. 2011;1808(3):606–613. doi:10.1016/j.bbamem.2010.11.020
38. Gustafsson J, Ora G, Almgren M, Disintegration of the lecithin lamellar phase by cationic surfactants. Langmuir. 1997;(13):6956–6963. doi:10.1021/la9705874
39. Almgren M. Vesicle transformations resulting from curvature tuning in systems with micellar, lamellar, and bicontinuous cubic phases. J Dispers Sci Technol. 2007;28:43–54. doi:10.1080/01932690600992613
40. Arnulphi C, Jin L, Tricerri MA, Jonas A. Enthalpy-driven apolipoprotein A-I and lipid bilayer interaction indicating protein penetration upon lipid binding. Biochemistry. 2004;43(38):12258–12264. doi:10.1021/bi036118k
41. Arnulphi C, Sánchez SA, Tricerri MA, Gratton E, Jonas A. Interaction of human apolipoprotein A-I with model membranes exhibiting lipid domains. Biophys J. 2005;89(1):285–295. doi:10.1529/biophysj.104.047480
42. Surewicz WK, Epand RM, Pownall HJ, Hui SW. Human apolipoprotein A-I forms thermally stable complexes with anionic but not with zwitterionic phospholipids. J Biol Chem. 1986;261(34):16191–16197.
Supplementary Materials
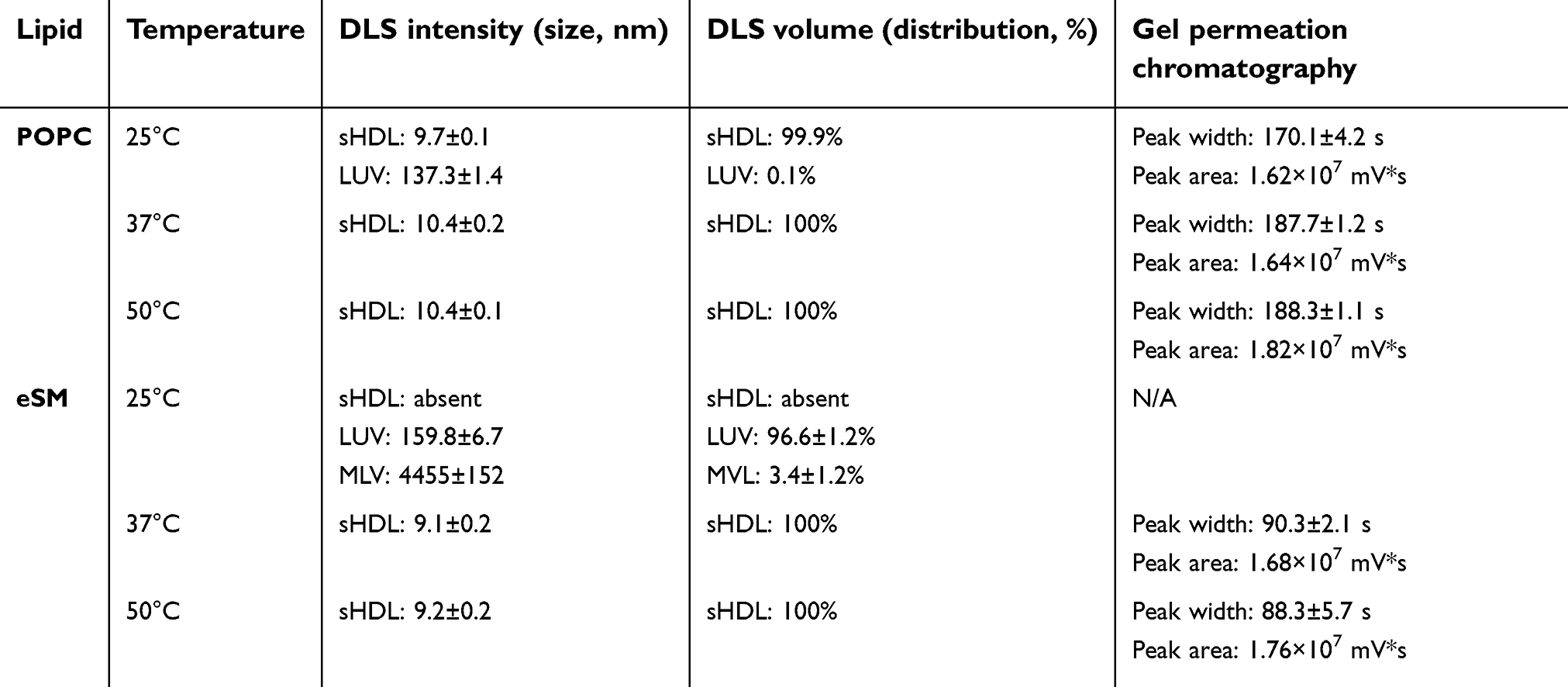 | Table S1 Size and purity characterization of POPC- and eSM-based sHDL assembled by 22A peptide and LUV incubations |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.