")
Back to Journals » Cancer Management and Research » Volume 11
Characterization of acquired receptor tyrosine–kinase fusions as mechanisms of resistance to EGFR tyrosine–kinase inhibitors
Authors Xu H, Shen J, Xiang J, Li H, Li B, Zhang T, Zhang L , Mao X, Jian H, Shu Y
Received 6 December 2018
Accepted for publication 15 April 2019
Published 9 July 2019 Volume 2019:11 Pages 6343—6351
DOI https://doi.org/10.2147/CMAR.S197337
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kenan Onel
Haiyuan Xu,1,2,* Jinge Shen,3,* Jianxing Xiang,4 Haiyan Li,4 Bing Li,4 Tengfei Zhang,4 Lu Zhang,4 Xinru Mao,4 Hong Jian,5 Yongqian Shu1
1Department of Oncology, First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, People’s Republic of China; 2Department of Oncology, Kunshan First People‘s Hospital affiliated to Jiangsu University, Kunshan 215300, People’s Republic of China; 3Department of Emergency, Shanghai Chest Hospital, Shanghai Jiaotong University, Shanghai 200000, People’s Republic of China; 4Burning Rock Biotech, Guangzhou 510000, People’s Republic of China; 5Department of Oncology, Shanghai Chest Hospital, Shanghai Jiaotong University, Shanghai 200000, People’s Republic of China
*These authors contributed equally to this work
Purpose: Responses to EGFR-targeted therapy are generally temporary, due to inevitable drug resistance. The prevalence and characteristics of receptortyrosine–kinase (RTK) fusion as acquired resistance to EGFR tyrosine–kinase inhibitors (TKIs) are rarely investigated.
Methods: We retrospectively reviewed genomic profiling data of 3873 EGFR (exons 18–21)-mutant lung cancer patients with more than once next-generation sequencing detection. A total of 16 patients who acquired RTK fusions during EGFR-TKI treatment with paired pre- and post-EGFR-TKI samples were identified. Their treatment history was collected.
Results: Newly acquired RTK fusions during EGFR-TKI treatment included RET (n=6, 37.5%), ALK (n=5, 31.3%), NTRK1 (n=4, 25.0%), ROS1 (n=1, 6.3%), and FGFR3 (n=1, 6.3%). All RET and EML4–ALK fusions were uncommon variants of KIF5B-RET and E2:A20 (V5), respectively. Interestingly, RET fusion occurred only after osimertinib treatment, and contributed to drug resistance in 50% (6 of 12) of patients treated with osimertinib, indicating that fusions had different prevalence when functioning as resistance mechanisms to EGFR TKIs. Moreover, we found that in all patients developing drug resistance to EGFR TKIs due to fusion emergence (n=16), those that had a treatment history of third-generation EGFR TKIs accounted for 75% (n=12).
Conclusion: We have extended the current knowledge of resistance mechanisms to EGFR TKIs in non-small-celllung cancer. Detection of RTK fusions should be included in genomic profiling panels to uncover potential resistance mechanisms of EGFR TKIs, which might inform therapeutic strategies, such as combination-therapy approaches, to circumvent tumorigenesis.
Keywords: receptor tyrosine kinase fusions, acquired resistance, EGFR tyrosine kinase inhibitors, lung cancer
Introduction
Improved understanding of the molecular changes that drive tumorigenesis has revolutionized the clinical management of non-small-cell lung cancer (NSCLC).1,2 Targeted therapy improves outcomes in EGFR-mutant NSCLC patients, but clinical responses to these drugs are generally incomplete and temporary, due to inevitable drug resistance.3,4 Acquired resistance will be the major limitation preventing EGFR-targeted therapy from having greater impact. Although several resistance mechanisms to EGFR tyrosine-kinase inhibitors (TKIs) have been identified, there are still many left to be discovered.5
Acquired resistance, which can be classified as “on‑target” and “off-target”, likely arises from the acquisition of new alterations under the selective pressure imposed by therapy. The emergence of EGFRT790M and EGFRC797S during treatment with EGFR-TKIs belongs to on‑target resistance, which limits the drug’s ability to inhibit EGFR through alterations of EGFR itself.6–8 Alterations activating signaling pathways downstream or in parallel with EGFR lead to off-target resistance, while also sustaining EGFR oncogenic signaling and tumor-cell proliferation, despite the inhibition of original EGFR. Such resistance mechanisms include amplifications in MET, HER2, BRAF, and fusions in receptor tyrosine kinases (RTKs).9–13 Fusions of such RTKs as RET, ALK, and ROS1, occurring in 1%–2%, 5%, and 1%–2% of NSCLC patients, respectively, have become potential therapeutic targets. Offin et al reported two EGFR-mutant lung cancer patients acquiring ALK rearrangements after osimertinib and responding to a combination of osimertinib and an ALK TKI.12 However, limited cases of acquired RTK fusions as mechanisms in EGFR TKIs have been reported. Meanwhile, the prevalence and characteristics of RTK fusions in acquired-resistance settings are rarely investigated.
We performed a retrospective study of 3,873 EGFR (exons 18–21)-mutant NSCLC patients with more than once detection by next-generation sequencing (NGS) and identified 16 patients with newly acquired RTK fusions. We aimed to interrogate the potential characteristics of RTK fusion as a resistance mechanism during EGFR-TKI treatment.
Methods
Patient selection
We retrospectively reviewed genomic profiling data of 3,873 EGFR (exons 18–21)-mutated NSCLC patients with more than once NGS detection. Their samples were profiled in a Clinical Laboratory Improvement Amendments–certified clinical molecular diagnostic laboratory (Burning Rock Biotech, Guangzhou, China) from September 2015 to October 2018, with the intent of investigating acquired fusion-related EGFR-TKI resistance. Sequencing panels used in this study were commercial panels provided by Burning Rock Biotech. The study was approved by the institutional review board of the First Affiliated Hospital of Nanjing Medical University. All other centers were covered by this protocol. All patients whose tissues or plasma samples were used in this research had provided written informed consent, in accordance with the Declaration of Helsinki.
DNA isolation
Tissue DNA was extracted using a QIAamp DNA formalin-fixed,paraffin-embedded tissue kit (Qiagen, Venlo, Netherlands) according to the manufacturer’s instructions. Circulating cell-free DNA was recovered from 4–5 mL plasma using a QIAamp circulating nucleic acid kit (Qiagen). To prepare cfDNA from pleural fluid samples, we first removed cells from pleural fluid by low-speed centrifugation, followed by high-speed centrifugation to remove any debris. The resultant supernatant was then subjected to cfDNA extraction using the circulating nucleic acid kit. DNA concentration was quantified using the fluorometry (Qubit 2.0; Thermo Fisher Scientific, Waltham, MA, US). A minimum of 50 ng DNA from plasma samples and 200 ng DNA from formalin-fixed, paraffin-embedded/pleural fluid samples was required for construction of the NGS library.
NGS-library preparation and capture-based targeted DNA sequencing
DNA shearing was performed using Covaris M220. End repair and A tailing were followed by adaptor ligation. Ligated fragments of 200–400 bp were selected by beads (Agencourt AMPure XP kit; Beckman Coulter, Brea, CA, US), hybridized with probe baits, selected by magnetic beads, and amplified by PCR. Indexed samples were sequenced on a NextSeq500 (Illumina, San Diego, CA, US) with pair-end reads.
NGS data-analysis pipeline
All reads were trimmed with Trimmomatic14 for adapters and then mapped to the human genome (hg19) with BWA software.15 Local alignment optimization, mark duplication, and variant calling were performed using the Genome Analysis Toolkit 3.2,16 Picard, and VarScan.17 Gene rearrangements were called with FACTERA18 and copy-number variants analyzed with an inhouse algorithm based on sequencing depth. Variants were filtered using the VarScan filter pipeline, with loci at depth <100 filtered out. At least two and five supporting reads were needed for insertions/deletions in plasma and tissue samples, respectively, while eight supporting reads were needed for single-nucleotide variants to be called in both plasma and tissue samples. According to the ExAC, 1000 Genomes, dbSNP, and ESP6500SI-V2 databases, variants with population frequency >0.1% were grouped as single-nucleotide polymorphisms and excluded from further analysis. Remaining variants were annotated with ANNOVAR19 and SnpEff v3.6.20
Results
Patient characteristics
Genomic profiles of 3,873 EGFR-mutated (exons 18-21) lung cancer patients who had received at least one NGS detection were reviewed retrospectively. A total of 9,012 samples were included in this study. Overall, 653 samples were sequenced using a 520-gene pan-cancer-related panel, 234 samples using a 295-gene pan cancer-related panel, 6,926 samples profiled using a panel of 168 lung cancer-related genes, 300 samples assessed using a 56-gene panel, and the remaining 899 samples were sequenced using a panel consisting of seven well-known lung cancer–driver genes (EGFR, ERBB2, BRAF, ROS1, RET, ALK, and MET) plus KRAS, a well-established prognostic factor. ALK, ROS1, and RET fusions can be detected by all panels, while NTRK and FGFR fusions can be detected by 168-, 295-, and 520-gene panels.
A total of 93 cases (2.4%) were identified with concomitant EGFR alterations and RTK fusions. Among them, 16 patients had positive RTK fusions after EGFR-TKI resistance but negative RTK fusions in pretreatment samples. The 16-patient cohort with paired pre- and post- EGFR TKI–treatment samples was used for following analyses, which investigated kinase fusions as a resistance mechanism to EGFR TKIs (Table 1). The overall workflow for patient selection is demonstrated in Figure 1.
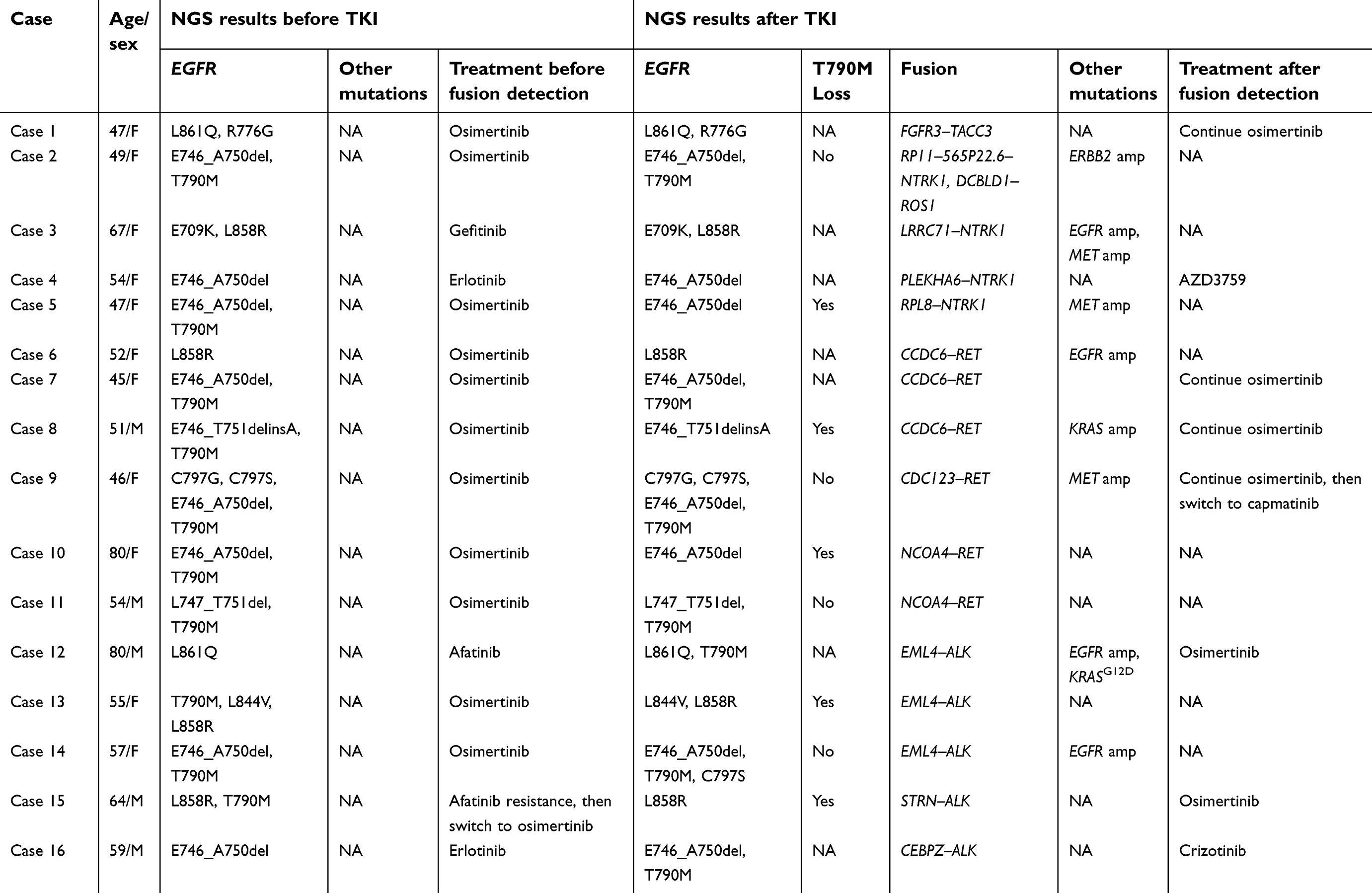 |
Table 1 Genomic profiles of paired pre- and post-EGFR-TKI treatment samples |
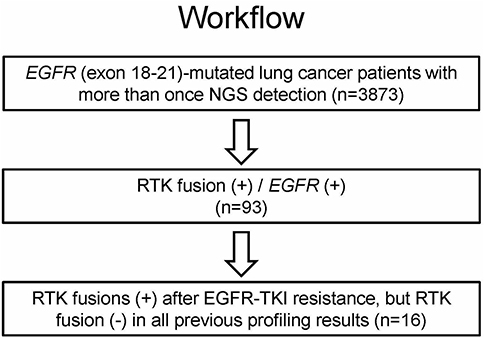 |
Figure 1 Workflow of patient selection. Abbreviations: RTK, receptor tyrosine kinase; TKI, tyrosine-kinase inhibitor. |
Molecular characteristics of pretreatment samples
In this cohort consisting of 16 EGFR and RTK coaltered patients, the median age was 54 years, ranging 45–80 years. Five (31.3%) were males and eleven (68.7%) females. All patients had been diagnosed with lung adenocarcinoma.
Samples profiled at baseline (before EGFR-TKI treatment proceeding to the detection of RTK fusion) consisted of 15 plasma samples and 1 FFPE (case 14). Genomic profiling results revealed that ten (62.5%) cases were positive for EGFR exon 19 deletion, four (25.0%) had L858R, and two (12.5%) L861Q. In addition, T790M was identified in ten (62.5%) cases and C797S/G were found in one (6.3%, case 9; Figure 2A).
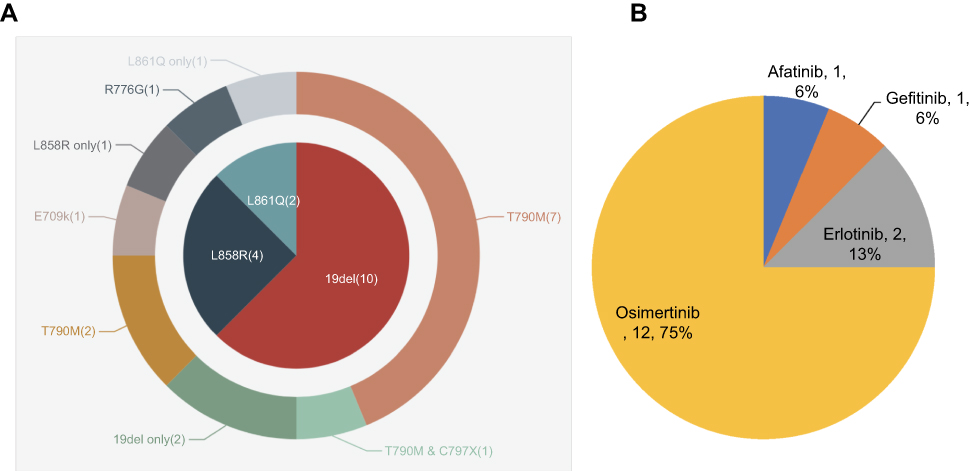 |
Figure 2 Molecular characteristics in 16 patients who acquired RTK fusions as resistance mechanisms to EGFR TKIs with paired pre- and posttreatment samples. Notes: (A) Distribution of EGFR alterations in pretreatment samples; (B) treatment history before emergence of fusions. Abbreviations: RTK, receptor tyrosine kinase; TKIs, tyrosine-kinase inhibitors. |
The 16 patients had different TKI-treatment history after positive EGFR-alteration detection. Before the emergence of RTK fusions, three patients (18.8%) received first-generation EGFR-TKI treatment (gefitinib, n=1, 6.3%; erlotinib, n=2, 12.5%), one case (6.3%) received second-generation TKI (afatinib) treatment, and 12 patients (75.0%) received third-generation TKI (osimertinib) treatment (Figure 2B). Of note, we classified case 15 into the osimertinib group, since we thought ALK fusion was likely induced by osimertinib.
Molecular characteristics of acquired fusions in posttreatment samples
After EGFR-TKI therapy, all 16 patients acquired RTK fusions. Posttreatment samples contained 15 plasma samples and one pleural effusion specimen (case 10). Among these, the RTK fusions involved consisted of RET (n=6, 37.5%), ALK (n=5, 31.3%), NTRK1 (n=4, 25.0%), ROS1 (n=1, 6.3%), and FGFR3 (n=1, 6.3%; Figure 3). Of note, one patient had concomitant ROS1 and NTRK1 fusions.
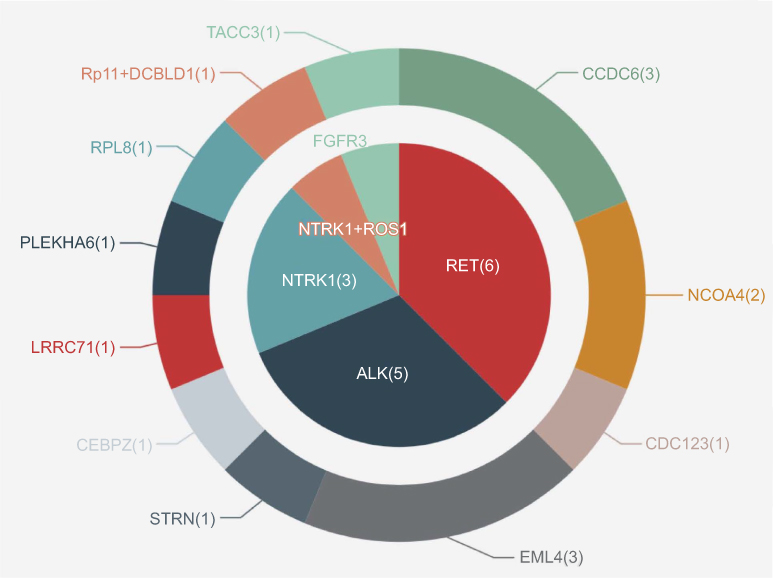 |
Figure 3 Variant distribution of acquired RTK fusions in EGFR-TKI posttreatment samples. Abbreviations: RTK, receptor tyrosine kinase; TKI, tyrosine-kinase inhibitor. |
Three RET-fusion partners were detected in six patients, including CCDC6 (n=3), NCOA4 (n=2), and CDC123 (n=1). The most common RET-fusion variant KIF5B–RET was not detected in this EGFR TKI–resistant cohort, coinciding with findings from other reports and one of our previous studies.13,21 Interestingly, we observed that all RET fusions occurred after osimertinib treatment, constituting 50% (six of 12) of patients with acquired RTK fusions following osimertinib treatment.
Partners of ALK identified in the five ALK+ patients included EML4 (n=3), STRN (n=1), and CEBPZ (n=1). All three EML4–ALK fusions emerging after EGFR-TKI resistance were the rare variant E2:A20 (V5). Four fusion partners of NTRK1 detected in this cohort contained LRRC71 (n=1), PLEKHA6 (n=1), RPL8 (n=1), and RP11-565P22.6 (n=1). The ROS1 partner identified here was DCBLD1 (n=1) and the FGFR3 partner TACC3 (n=1). Schematic structures of rearrangements of all RTK fusions are demonstrated in Figure 4.
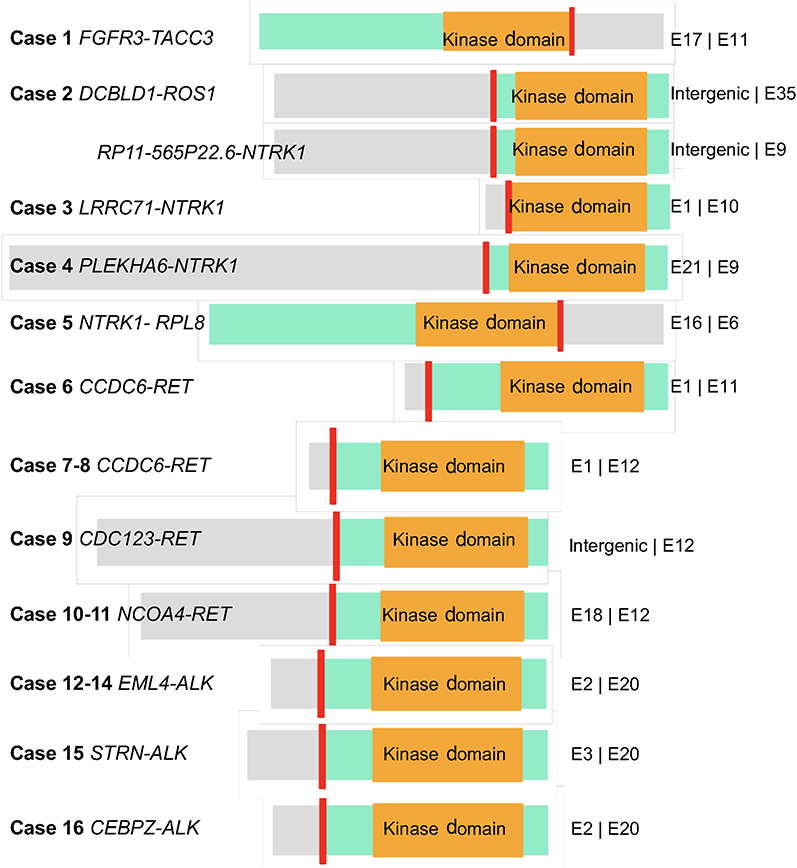 |
Figure 4 Schematic structure of all RTK fusions. Notes: The short red line indicates the breakpoint of each fusion. The green box represents the gene thatcontains kinase domain. The gray box represents the fusion-partner gene. The orange box represents the kinase domain. The length of the left and right fragments represents the exon number of two genes in fusion. Abbreviation: RTK, receptor tyrosine kinase. |
All the ten cases who had T790M in pretreatment samples received osimertinib treatment. We found five patients with acquired RTK fusions and disappearance of T790M after osimertinib treatment. Five patients still harbored T790M during the emergence of fusions. In addition, two cases (12 and 16) acquired T790M along with RTK fusions after first- or second-generation EGFR-TKI treatment. Besides T790M, several known resistance mechanisms occurred concomitantly with RTK fusion during EGFR-TKI treatment in eight patients, including EGFR amplification (amp; n=4), MET amp (n=3), ERBB2 amp (n=1), KRAS amp (n=1), KRASG12D (n=1), and EGFRC797S (n=1).
Discussion
In this study, we retrospectively investigated acquired RTK fusions as resistance mechanisms to EGFR TKIs in the largest NSCLC cohort with paired pre- and posttreatment samples to the best of our knowledge. It is conventionally considered that oncogenic driver mutations, such as ALK or ROS1 rearrangements, are mutually exclusive with EGFR mutations in NSCLC.22–25 Here, we report that RTK fusions as a result of ALK, ROS1, RET, FGFR3, and NTRK1 gene rearrangements emerged as resistance mechanisms to EGFR TKIs, providing a basis for the hypothesis that an actionable driver mutation could function as an acquired resistance mechanism to drugs targeting another actionable driver alteration.
We found that all six RET fusions emerged after third-generation TKI treatment, and half the patients developing osimertinib resistance in this selected cohort of ours had this caused by RET fusions (six of 12, 50.0%). This result corroborates a recent study stating that no ALK or RET fusions were found in tumor tissue from 174 patients who developed resistance to erlotinib or afatinib, but two ALK fusions and one RET fusion were found from 14 patients after progression developed during osimertinib treatment.12 It suggested that the prevalence of fusion events developed during different EGFR-TKI treatments might be distinct.
In addition, for the six patients with acquired RET fusions, no fusion variants were KIF5B–RET, the most common RET fusion. Other groups have also found non-KIF5B–RET fusions in EGFR-mutant patients with TKI resistance, which coincides with our observations. Reckamp et al studied nearly 33,000 samples and found that CCDC6– and NCOA4–RET fusions may contribute to anti-EGFR therapy resistance in NSCLC.21 Schrock et al assessed >3,500 EGFR-mutant patients and found that the two NSCLC RET fusions emerging after EGFR-TKI treatment were both CCDC6–RET.13 Our results also revealed that all three EML4–ALK fusions presented after resistance were the rare variant V5. The absence of common partners/variants in EGFR TKI–resistant patients suggested that common partners/variants themselves were strong enough to drive primary oncogenesis and rare partners/variants more likely functioned as potential acquired resistance mechanisms to EGFR TKIs. Therefore, it is potentially important to use NGS to identify precise fusion partners/variants and further guide treatment strategies, since heterogeneity of clinical responses exists among different fusion partners/variants.26–29 Owing to the limited number of paired pre- and post-EGFR TKI–treated samples, further studies are needed to validate our new observations.
We observed in our cohort that 75% of patients developed drug resistance after third-generation EGFR-TKI treatment, due to fusion emergence, compared with 18.8 and 6.3% after first- and second-generation EGFR TKIs, respectively. This observation supported the notion that newly acquired RTK fusions were not seen or seen rarely with earlier-generation EGFR TKIs, but could be found at a higher rate with osimertinib resistance.12 This underscores the necessity to detect fusion status after osimertinib resistance in clinical settings.
In vitro study revealed that RET fusions can lead to resistance to osimertinib in EGFR-mutant cells and combination treatment with cabozantinib can restore the response to osimertinib.12 The combination of EGFR TKIs and fusion-based targeted therapy has been reported to be successful in overcoming resistance to EGFR TKIs in clinical cases. Combination treatment with full-dose osimertinib and crizotinib in an EGFR-mutant tumor harboring both T790M and MET-mediated mechanisms of acquired resistance showed tolerable and effective clinical benefits.30 Furthermore, an NSCLC patient harboring EGFRL858R and RET fusion had an additional 7 months of stable disease after initiation of combinatorial treatment with afatinib and cabozantinib.13 The potential of such combinations to overcome multiple mechanisms of acquired resistance in EGFR-mutant NSCLC should be studied further.
Along with the emergence of RTK fusions during EGFR-TKI treatment, we identified polyclonal resistance alterations in eight cases. This suggests that accurate assessment of polyclonal drug resistance is limited by the number of resistance mechanisms assessed, the depth of sequencing, and the quantity of tumor DNA assessed in resistant samples. This points to the necessity to apply appropriate NGS panels to detect and monitor all classes of genomic alterations that may guide precise targeted therapies.
There were several limitations of this study. First, although we had a large EGFR-mutated NSCLC population at screening, due to the rare occurrence of RTK fusions, the cohort of postEGFR-TKI–treated RTK fusion–positive samples was still too small fully to reflect the underlying pattern of the fusion resistance mechanism. Studies in larger cohorts are needed to address this phenomenon further. Second, clinical information on patients who received combination therapies with EGFR TKIs and fusion-targeted drugs was unavailable, and thus could not provide clinical evidence for efficacy of such a combination strategy in our cohort.
Taken together, to the best of our knowledge, in the largest NSCLC cohort with paired pre- and post-treatment samples, these results highlighted the unique features of EGFR TKI–resistance mechanisms in EGFR-positive NSCLC and provide the rationale for combinatorial therapeutics tailored to the precise resistance mechanisms identified in patients who relapse on EGFR-TKI treatment.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Li T, Kung HJ, Mack PC, Gandara DR. Genotyping and genomic profiling of non-small-cell lung cancer: implications for current and future therapies. J Clin Oncol. 2013;31(8):1039–1049. doi:10.1200/JCO.2012.45.3753
2. Reck M, Rabe KF. Precision diagnosis and treatment for advanced non-small-cell lung cancer. N Engl J Med. 2017;377(9):849–861. doi:10.1056/NEJMra1703413
3. Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792. doi:10.1056/NEJMoa044238
4. Yang Z, Yang N, Ou Q, et al. Investigating novel resistance mechanisms to third-generation EGFR tyrosine kinase inhibitor osimertinib in non-small cell lung cancer patients. Clin Cancer Res. 2018;24(13):3097–3107. doi:10.1158/1078-0432.CCR-17-2310
5. Tang ZH, Lu JJ. Osimertinib resistance in non-small cell lung cancer: mechanisms and therapeutic strategies. Cancer Lett. 2018;420:242–246. doi:10.1016/j.canlet.2018.02.004
6. Kosaka T, Yatabe Y, Endoh H, et al. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res. 2006;12(19):5764–5769. doi:10.1158/1078-0432.CCR-06-0714
7. Thress KS, Paweletz CP, Felip E, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21(6):560–562. doi:10.1038/nm.3854
8. Ou SI, Cui J, Schrock AB, et al. Emergence of novel and dominant acquired EGFR solvent-front mutations at Gly796 (G796S/R) together with C797S/R and L792F/H mutations in one EGFR (L858R/T790M) NSCLC patient who progressed on osimertinib. Lung Cancer. 2017;108:228–231. doi:10.1016/j.lungcan.2017.04.003
9. Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104(52):20932–20937. doi:10.1073/pnas.0710370104
10. Takezawa K, Pirazzoli V, Arcila ME, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012;2(10):922–933. doi:10.1158/2159-8290.CD-12-0108
11. Ho CC, Liao WY, Lin CA, Shih JY, Yu CJ, Chih-Hsin Yang J. Acquired BRAF V600E mutation as resistant mechanism after treatment with osimertinib. J Thorac Oncol. 2017;12(3):567–572. doi:10.1016/j.jtho.2016.11.2231
12. Offin M, Somwar R, Rekhtman N, et al.Acquired ALK and RET gene fusions as mechanisms of resistance to osimertinib in EGFR-mutant lung cancers. JCO Precis Oncol. 2018;2:1–12. doi:10.1200/PO.18.00126
13. Schrock AB, Zhu VW, Hsieh WS, et al. Receptor tyrosine kinase fusions and BRAF kinase fusions are rare but actionable resistance mechanisms to EGFR tyrosine kinase inhibitors. J Thorac Oncol. 2018;13(9):1312–1323. doi:10.1016/j.jtho.2018.05.027
14. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi:10.1093/bioinformatics/btu170
15. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi:10.1093/bioinformatics/btp324
16. McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi:10.1101/gr.107524.110
17. Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–576. doi:10.1101/gr.129684.111
18. Newman AM, Bratman SV, Stehr H, et al. FACTERA: a practical method for the discovery of genomic rearrangements at breakpoint resolution. Bioinformatics. 2014;30(23):3390–3393. doi:10.1093/bioinformatics/btu549
19. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi:10.1093/nar/gkq603
20. Cingolani P, Platts A, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: sNPs in the genome of drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6(2):80–92. doi:10.4161/fly.19695
21. Reckamp KL, Rich TA, Chae YK, et al. Abstract 936: analysis of cell-free DNA from 32,991 advanced cancers reveals novel co-occurring activating RET alterations and oncogenic signaling pathway aberrations. Cancer Res. 2018;78(13 Supplement):936–936.
22. Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27(26):4247–4253. doi:10.1200/JCO.2009.22.6993
23. Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. doi:10.1038/nature05945
24. Horn L, Pao W. EML4-ALK: honing in on a new target in non-small-cell lung cancer. J Clin Oncol. 2009;27(26):4232–4235. doi:10.1200/JCO.2009.23.6661
25. Lin JJ, Ritterhouse LL, Ali SM, et al. ROS1 fusions rarely overlap with other oncogenic drivers in non-small cell lung cancer. J Thorac Oncol. 2017;12(5):872–877. doi:10.1016/j.jtho.2017.01.004
26. Yoshida T, Oya Y, Tanaka K, et al. Differential crizotinib response duration among ALK fusion variants in ALK-positive non-small-cell lung cancer. J Clin Oncol. 2016;34(28):3383–3389. doi:10.1200/JCO.2015.65.8732
27. Li Y, Zhang T, Zhang J, et al. Response to crizotinib in advanced ALK-rearranged non-small cell lung cancers with different ALK-fusion variants. Lung Cancer. 2018;118:128–133. doi:10.1016/j.lungcan.2018.01.026
28. Woo CG, Seo S, Kim SW, et al. Differential protein stability and clinical responses of EML4-ALK fusion variants to various ALK inhibitors in advanced ALK-rearranged non-small cell lung cancer. Ann Oncol. 2017;28(4):791–797. doi:10.1093/annonc/mdw693
29. Lin JJ, Zhu VW, Yoda S, et al. Impact of EML4-ALK variant on resistance mechanisms and clinical outcomes in ALK-positive lung cancer. J Clin Oncol. 2018;36(12):1199–1206. doi:10.1200/JCO.2017.76.2294
30. York ER, Varella-Garcia M, Bang TJ, Aisner DL, Camidge DR. Tolerable and effective combination of full-dose crizotinib and osimertinib targeting MET amplification sequentially emerging after T790M positivity in EGFR-mutant non-small cell lung cancer. J Thorac Oncol. 2017;12(7):e85–e88. doi:10.1016/j.jtho.2017.02.020
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.