")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Changing the face of hepatitis C management – the design and development of sofosbuvir
Authors Noell B, Besur S, deLemos A
Received 3 January 2015
Accepted for publication 12 March 2015
Published 24 April 2015 Volume 2015:9 Pages 2367—2374
DOI https://doi.org/10.2147/DDDT.S65255
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Bennett C Noell,* Siddesh V Besur,* Andrew S deLemos
Department of Medicine, Center for Liver Diseases and Transplantation, Carolinas Medical Center, Charlotte, NC, USA
*These authors contributed equally to this work
Abstract: The availability of direct-acting antiviral (DAA) therapy has launched a new era in the management of chronic hepatitis C. Sofosbuvir, a uridine nucleotide analog that inhibits the hepatitis C RNA-dependent RNA polymerase, is the backbone of chronic hepatitis C therapy. Acting at the catalytic site of the polymerase, sofosbuvir is highly potent in suppressing viral replication and has a high genetic barrier to resistance. Sofosbuvir is effective across all hepatitis C genotypes, and is a mainstay of interferon-free combination therapy. In Phase II and III studies, genotype 1 patients who took sofosbuvir in combination with another DAA such as the NS3-4A protease inhibitor, simeprevir, or the NS5A replication complex inhibitors, ledipasvir or daclatasvir, achieved a sustained virologic response rate of over 90%. Harvoni®, a combination tablet of sofosbuvir and ledipasvir, dosed once daily is recommended for 24 weeks for treatment-experienced genotype 1 patients with cirrhosis, but 12 weeks of therapy is sufficient for all other populations. While genotype 2 (12 weeks or 16 weeks) and treatment-naïve genotype 3 patients (24 weeks) have excellent response rates with sofosbuvir and ribavirin, treatment-experienced cirrhotic genotype 3 patients may need the addition of another DAA such as daclatasvir. Sofosbuvir is efficacious in special populations such as HIV–hepatitis C virus-coinfected patients and liver transplant recipients and has already made a profound impact in these groups. Since it is renally eliminated, patients with advanced kidney disease or on dialysis must await dosing recommendations. Sofosbuvir-based regimens appear to be well tolerated with headache and fatigue being the most common side effects. The opportunity to cure patients with hepatitis C with sofosbuvir combination therapy is likely to change the future for our patients, particularly if the emphasis shifts to identifying those patients unaware that they are infected and providing affordable access to treatment.
Keywords: NS5B polymerase inhibitor, ledipasvir, chronic hepatitis C, sustained virologic response
Introduction
Chronic hepatitis C (CHC) infection is a worldwide health concern affecting approximately 185 million people, about 3.5–4.4 million of whom reside in the US.1,2 Eighty percent of patients infected with hepatitis C develop a chronic infection, which will progress to cirrhosis in 20% of patients. End-stage liver disease due to CHC is currently the leading indication for liver transplantation in the US. Approximately 350,000 people across the world died from complications related to hepatitis C cirrhosis in 2010, and in the US, one million hepatitis C virus (HCV)-infected patients will have cirrhosis by 2020.3
The tremendous human cost from sequelae of CHC infection is now beginning to be framed in a new light due to the availability of highly effective all-oral therapies to cure hepatitis C infection. Patients no longer require treatment with pegylated interferon (Peg-IFN), which caused innumerable side effects resulting in poor adherence to therapy. Moreover, the patients most in need of treatment, namely cirrhotics and post-liver transplant patients, had dismal sustained virologic response (SVR) rates with Peg-IFN and ribavirin (RBV). With all-oral direct-acting antiviral (DAA) therapy, these patients now have actual life-saving therapies available with outstanding SVR rates. At the moment, it is not an exaggeration to say that sofosbuvir serves as the fulcrum, providing the foundation on which the change in caring for CHC patients is possible.
DAA therapy refers to pharmacological targets that specifically inhibit hepatitis C viral proteins. Identification of the four structural and six nonstructural proteins of HCV was possible only after the HCV RNA genome was sequenced (Figure 1). An intense effort by industry to develop attractive drug targets ensued but was initially complicated by the nature of the virus itself. The RNA polymerase NS5B does not have proofreading capability, and therefore, sequence diversity in the HCV genome exists at all times, even within individual patients. As a result, the first-generation DAAs, the NS3-4A protease inhibitors, telaprevir and boceprevir, were ineffective as monotherapy due to naturally occurring drug resistance mutations and required coadministration with Peg-IFN and RBV. Another example, simeprevir, a once-daily NS3-4A inhibitor, approved only a year ago in combination with Peg-IFN and RBV, is not recommended for genotype 1a patients who harbor the baseline Q80K resistance mutation. Sofosbuvir circumvents this problem by targeting the catalytic site of the NS5B viral polymerase, thereby profoundly diminishing viral replication directly.4 Furthermore, resistance mutations in the active site would be predicted to confer a lack of viral fitness (see Resistance section). These two factors, in combination with sequence conservation in the NS5B active site across all six hepatitis C genotypes, are responsible for the exceptional efficacy data of sofosbuvir.
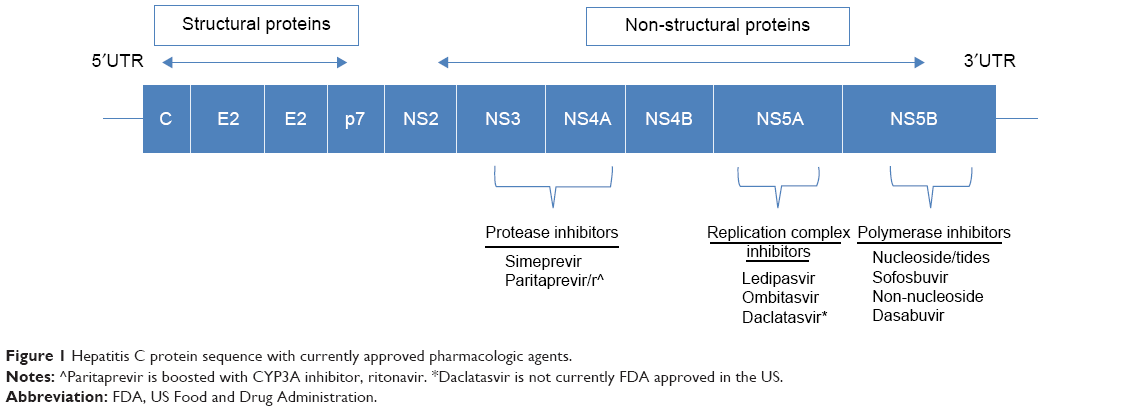 | Figure 1 Hepatitis C protein sequence with currently approved pharmacologic agents. |
Mechanism of action
NS5B is one of six nonstructural proteins encoded on the HCV genome. It is an RNA-dependent RNA polymerase responsible for replicating the HCV RNA genome which is a vital step in the HCV life cycle. The RNA-dependent RNA polymerase exhibits a classic fingers, palm, and thumb structure where interactions between the finger and thumb subdomains create the catalytic site that ensures synthesis of positive- and negative-strand HCV RNA.5 There are currently two types of NS5B inhibitors, nucleos(t)ide inhibitors and non-nucleoside inhibitors. Nucleoside inhibitors bind to the catalytic site of the RNA polymerase causing chain termination. Non-nucleoside inhibitors bind to a less conserved site resulting in a conformational change that distorts the positioning of residues binding RNA, thus inhibiting polymerization. Sofosbuvir belongs to the class of β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methylribose nucleoside inhibitors (Figure 2).
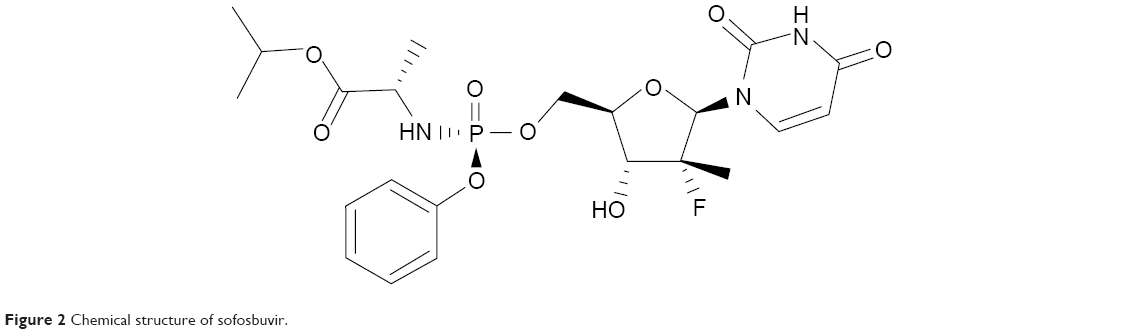 | Figure 2 Chemical structure of sofosbuvir. |
A variety of β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methylribose nucleosides have been shown to be potent inhibitors of HCV NS5B polymerase in clinical studies.6–8 Ultimately, second-generation compounds of these agents were selected due to improved potency and enhanced pharmacokinetics with hopes of enabling once-daily dosing.9 Unfortunately, many of the second-generation preliminary agents were not phosphorylated effectively in vivo due to being poor substrates to cellular kinases. In order to circumvent this rate-limiting step, a phosphoramidate prodrug strategy was used to synthesize compounds with improved bioavailability and transport into hepatocytes, thus enhancing intracellular concentrations of the active nucleoside. Sofosbuvir is a phosphoramidate prodrug that is metabolized in the liver to β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methyluridine-5′-monophosphate.8 The conversion of the monophosphate form to the active triphosphate involves four enzymatic steps and one nonenzymatic chemical step.10 The first step involves hydrolysis of the carboxyl ester moiety by serine protease, cathepsin A (CatA), and serine esterase, carboxylesterase 1 (CES1). The second step involves a nonenzymatic rapid chemical activation resulting in an alanyl phosphate intermediate. The third step of metabolism involves deamination by the cellular enzyme histidine triad nucleotide-binding protein 1 (Hint1), and the final two steps leading to the active triphosphate form are phosphorylation events catalyzed by cellular kinases UMP-CMP kinase and nucleoside diphosphate kinase (NDPK). Once converted to the triphosphate form, the uridine nucleotide acts as a defunct substrate for the NS5B polymerase causing chain termination, therefore ceasing RNA replication.
The active site moiety of NS5B is found in all HCV genotypes, which accounts for sofosbuvir’s pangenotypic activity. A study utilizing a molecular modeling approach used several quantitative structure–activity relationship (QSAR) properties to evaluate the performance of sofosbuvir on genotypes 1a, 2b, 3b, and 4a. Genotypes 1a and 3b were found to have the best QSAR values compared to genotypes 2b and 4a, suggesting that sofosbuvir would have better activity against HCV genotypes 1a and 3b.4 Lam et al demonstrated that sofosbuvir inhibited enzymatic activity of NS5B polymerase in genotypes 1–4 with similar 50% inhibitory concentrations between all the groups.11
Pharmacokinetics/drug–drug interactions
Sofosbuvir reaches peak plasma concentrations within 0.5–2 hours following oral administration and is approximately 60% bound to human plasma proteins. Administration with a standardized high-fat meal did not substantially affect its peak or area under the curve (AUC); therefore, it can be administered with or without food. Sofosbuvir undergoes extensive metabolism by the liver as described above to form the active nucleoside triphosphate, GS-461203. Dephosphorylation of GS-461203 results in the formation of the inactive metabolite GS-331007. Following a single 400 mg dose, the terminal half-life was 0.4 hours for sofosbuvir and 27 hours for the GS-331007 metabolite. Approximately 78% of the inactive metabolite is renally eliminated with the rest eliminated through the feces or as unchanged drug.
Population pharmacokinetic analyses in HCV-infected subjects indicated that race, sex, or age (19–75 years) had no clinically relevant effect on the exposure of sofosbuvir and GS-331007.12 Pharmacokinetics of sofosbuvir in pediatric patients has not been established. The AUC of the inactive metabolite, GS-331007, was increased 55%, 88%, and 451% in a pharmacokinetic study of HCV-negative subjects with mild, moderate, and severe renal impairment, respectively. Grade 4 laboratory abnormalities were limited to those with severe renal impairment.13 Dose adjustment for patients with mild-to-moderate renal impairment is currently not necessary. The safety and efficacy of sofosbuvir are currently being studied in patients with severe renal impairment including end-stage renal disease patients requiring dialysis. Currently, use in these patients is not recommended. The AUC of sofosbuvir increased by 126% and 143% in patients with hepatic impairment with Child-Pugh classes B and C, respectively. There was no difference in tolerability or effect in these patients, and the presence of cirrhosis had no clinically relevant effect on exposure.14 Dosage adjustment is not necessary for patients with mild, moderate, or severe hepatic impairment.
Sofosbuvir is a substrate of P-glycoprotein (P-gp); therefore, inhibitors or inducers of P-gp may alter serum concentrations of sofosbuvir. For example, the potent intestinal P-gp inducers (rifampin and St John’s wort) should be avoided, since they can decrease the sofosbuvir plasma concentration and reduce its efficacy. Coadministration with anticonvulsants is also not recommended. There was no effect on pharmacokinetic parameters when cyclosporine, darunavir/ritonavir, efavirenz, emtricitabine, methadone, or rilpivirine was coadministered with sofosbuvir. Sofosbuvir is not affected by cytochrome P450 metabolism, thus reducing the number of potential drug–drug interactions.15,16
Resistance
In vitro studies using HCV replicons to characterize resistance found S282T to be the only mutation across various genotypes and subtypes resistant to sofosbuvir. The S282T mutation conferred a 9.5-fold increase in the 50% effective concentration (EC50) of sofosbuvir against HCV replicons.17 Sofosbuvir remained susceptible to HCV replicons with resistant mutations to RBV, protease inhibitors, and NS5A inhibitors.17,18 Svarovskaia et al performed an extensive resistance analysis of 1,645 patients from Phase II and III sofosbuvir clinical trials using both deep sequencing and phenotypic analysis.19 There was no S282T mutation or polymorphisms present at baseline that were associated with failure to achieve SVR. Out of the 282 patients who did not achieve SVR, no resistant variants were detected during treatment in patients receiving dual and triple therapy; however, one patient receiving sofosbuvir monotherapy in the ELECTRON trial developed the S282T mutation.20 The S282T variant was present in greater than 99% of the viral population at 4 weeks posttreatment. This decreased to 27% at 8 weeks posttreatment and ultimately became undetectable at weeks 12 and 24 posttreatment. Analysis of all NS5B sequences identified L159F and V321A as sofosbuvir-treatment emergent substitutions. However, neither variant was associated with resistance to sofosbuvir in the replicon system.19 Sofosbuvir has a high genetic barrier to resistance. This may be due to the limited replicative fitness of the S282T strain of HCV compared to the wild type as Svarovskaia et al found, or could be due to sofosbuvir’s ability to rapidly reduce viral replication, thereby limiting the time resistant strains have to develop.19 The high barrier to resistance in addition to lack of cross-resistance between sofosbuvir and other classes of HCV inhibitors makes it an ideal candidate for combination therapy.
Sofosbuvir/ledipasvir
One combination recently approved by the US Food and Drug Administration (FDA) is sofosbuvir plus the NS5A replication complex inhibitor ledipasvir (Figure 3). Pharmacokinetic properties of combined ledipasvir, sofosbuvir, and circulating metabolite GS-331007 were studied in healthy adult subjects and subjects with CHC. While sofosbuvir and GS-331007 AUC and maximum concentration (Cmax) were similar in healthy subjects and those with CHC, ledipasvir AUC and Cmax were 24% lower and 32% lower, respectively, in HCV-infected subjects. Ledipasvir undergoes a slow oxidative metabolism through an unknown mechanism and is eliminated as an unchanged drug through biliary excretion. There was no detectable metabolism of ledipasvir by Cytochrome P450 enzymes. Ledipasvir solubility decreases as pH increases; thus, medications that increase gastric pH may result in a decreased concentration of ledipasvir. Omeprazole at doses of 20 mg once daily given 2 hours prior to ledipasvir resulted in lower AUC and Cmax. Only small decreases in ledipasvir AUC and Cmax were observed when given simultaneously with ledipasvir/sofosbuvir combination; therefore, proton-pump inhibitors at doses comparable to omeprazole 20 mg or lower can be administered simultaneously under fasted conditions.21 Amino acid substitutions Y93H and Q30E have conferred high levels of reduced susceptibility to ledipasvir in cell culture for genotypes 1a and 1b. Ledipasvir is fully active against the sofosbuvir resistance-associated substitution S282T in NS5B, and ledipasvir resistance-associated substitutions in NS5A are fully susceptible to sofosbuvir.
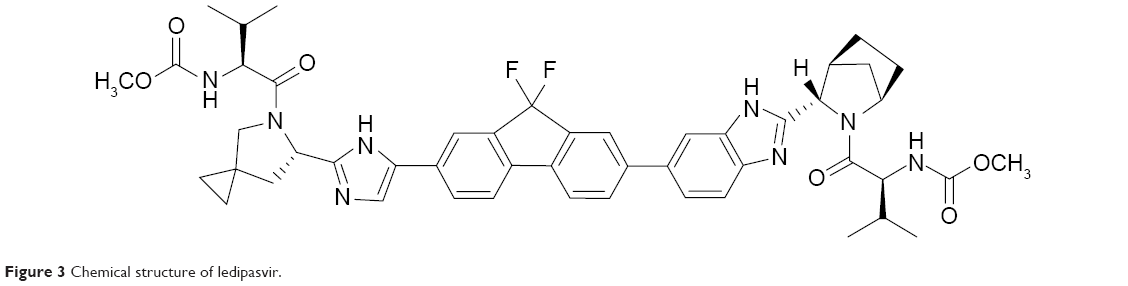 | Figure 3 Chemical structure of ledipasvir. |
Comparative effectiveness of sofosbuvir and current use in clinical practice
Genotype 1
Sofosbuvir (Sovaldi®) originally gained FDA approval on December 6, 2013, in combination with Peg-IFN and RBV for genotypes 1 and 4, and in combination with RBV for genotypes 2 and 3. This label was based upon five Phase III clinical trials in a total of 1,724 HCV-mono-infected subjects with genotypes 1–6 CHC (see package insert). NEUTRINO was a single-arm, open-label multicenter trial in treatment-naïve patients (n=327) with genotypes 1 and 4–6.22 Patients received 12 weeks of sofosbuvir (400 mg daily) with weight-based RBV (1,000 mg in patients <75 kg, 1,200 mg if >75 kg daily) plus weekly Peg-IFN alpha (180 μg). The majority of patients were Caucasian (79%), genotype 1 (89%), and non-cirrhotic (83%). The SVR rate at 12 weeks (SVR12) following treatment was 92% (genotype 1a) vs 82% (genotype 1b). The SVR12 in patients with cirrhosis was 80%, and 71% in patients with multiple predictors of poor response to Peg-IFN (genotype 1, advanced fibrosis [F3/F4], IL28B non-C/C, and baseline viral load >800,000 IU/ mL) (Table 1).
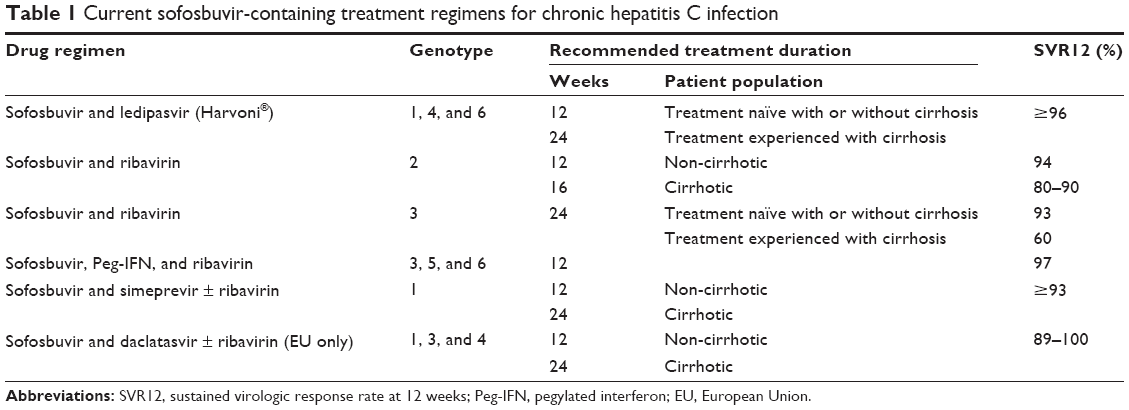 | Table 1 Current sofosbuvir-containing treatment regimens for chronic hepatitis C infection |
Shortly after the approval of Sovaldi® with Peg-IFN and RBV for genotype 1 CHC patients, Phase III clinical trials with the fixed-dose combination of sofosbuvir (400 mg) with the NS5A inhibitor, ledipasvir (90 mg), were completed. The anticipation for this combination was pronounced, particularly for genotype 1 CHC patients with cirrhosis, who were in need of a safe and effective all-oral DAA combination. A total of 1,518 subjects with genotype 1 CHC received sofosbuvir/ledipasvir (Harvoni®) in the ION trials. ION-323 enrolled treatment-naïve patients with early-stage or no fibrosis (Metavir F0–F2) to either 8 weeks or 12 weeks of Harvoni®. The SVR12 in the 8-week treatment group with or without RBV was 94%, and 96% in the 12-week treatment group. ION-124 compared 12 weeks vs 24 weeks in treatment-naïve patients with and without cirrhosis. The SVR12 in the 12-week arm was 99% (176/177) in patients without cirrhosis and 94% (32/34) in patients with cirrhosis, with again no difference with the addition of RBV. Lastly, ION-225 investigated the response rate of 12 weeks vs 24 weeks of Harvoni® in treatment-experienced patients with or without cirrhosis and included patients who had failed prior protease inhibitor therapy. These historically difficult-to-treat patients had outstanding response rates, and 24 weeks of therapy was superior to 12 weeks of therapy (99% vs 94% SVR). Based upon this compelling data, Harvoni® was approved by the FDA on October 10, 2014, for 12 weeks in treatment-naïve genotype 1 CHC patients with or without cirrhosis and 24 weeks or 12 weeks in treatment-experienced patients with or without cirrhosis, respectively.
A current alternative which was recommended by the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America (http://www.hcvguideline.org/) in 2014 prior to approval of Harvoni® for genotype 1 patients intolerant of or ineligible for Peg-IFN was sofosbuvir and the NS3-4A inhibitor, simeprevir (150 mg daily).26 While originally an off-label use, this combination was just recently added to simeprevir’s package insert based upon data from COSMOS.27 Ninety-five percent of non-cirrhotic patients attained an SVR following 12 weeks with this combination. The SVR12 was 96% after 24 weeks of simeprevir and sofosbuvir in patients with cirrhosis (n=23), and extending treatment is advised in this population.27 In Europe, the NS5A inhibitor, daclatasvir (60 mg daily, Daklinza®), is an additional DAA, which can be combined with sofosbuvir for genotype 1 patients with (24 weeks) or without (12 weeks) cirrhosis with SVR rates comparable to Harvoni® (see also daclatasvir package insert).28
Genotypes 2–4
Sofosbuvir is the backbone for genotypes 2–4 treatment as well. It should be given for 12–16 weeks in combination with weight-based RBV for genotype 2 patients with or without cirrhosis. Treatment-naïve patients do slightly better than treatment-experienced patients (SVR >90% vs >80%).29 Genotype 3 patients, however, require 24 weeks of therapy with sofosbuvir and RBV. Like genotype 2, treatment-experienced patients do not fare as well as treatment-naïve patients (SVR 77% vs 93%).30 In Europe, the addition of daclatasvir to sofosbuvir and RBV is an attractive option for genotype 3 and would be a combination to choose, particularly in treatment-experienced patients with cirrhosis (see also daclatasvir package insert).28 Lastly, like genotype 1, sofosbuvir was originally approved in combination with Peg-IFN and RBV for 12 weeks for genotype 4 patients. While this combination remains a viable option for interferon-eligible patients, 24 weeks of sofosbuvir and RBV or 12-weeks of Harvoni® is probably a better alternative (http://www.hcvguideline.org) (Table 1).
Hepatitis C treatment in special populations
Patients with HIV coinfection represent an important subset of CHC patients to target therapy. They are at high risk for fibrosis progression compared to mono-infected patients. Thus, sofosbuvir, which can be safely administered concomitantly with antiretroviral therapy, has been a blessing to coinfected patients. Twenty-four weeks of sofosbuvir and RBV are effective in genotype 1–3 patients.31 For genotype 1 coinfected patients, 12 weeks of Harvoni® therapy resulted in an SVR rate of 98%32 and appears to be preferable to 24 weeks of sofosbuvir/RBV. However, ledipasvir increases tenofovir levels and should be avoided in patients with a creatinine clearance below 60 mL/min (http://www.hcvguideline.org). Historically, post-liver transplant outcomes in patients transplanted for CHC were inferior to other indications for liver transplant due to allograft reinfection and recurrent hepatitis C infection.33 Moreover, an aggressive form of allograft reinfection, fibrosing cholestatic hepatitis C (FCH), contributed to early graft failure within the 1st year after liver transplant. The availability of sofosbuvir is poised to change the natural history of hepatitis C after transplant. In fact, a compassionate use program with sofosbuvir for patients with FCH and allograft cirrhosis was started prior to its FDA approval, and 59% (54/92) of patients achieved an SVR12.34 While exact guidelines on when to initiate therapy post-transplant in the DAA era are not clear, patients can now be treated before transplant35 to prevent allograft reinfection or after transplant36 with truly remarkable response rates and without the risk of immune graft dysfunction associated with Peg-IFN therapy.37
Safety profile of sofosbuvir
Sofosbuvir is used in combination with other DAAs, Peg-IFN, or RBV. Thus, attributing side effects directly to sofosbuvir should be considered with caution. Anemia due to Peg-IFN/RBV therapy is common and should be monitored closely in regimens containing these agents. In a pooled analysis of Phase III trials of patients who received a sofosbuvir/RBV regimen, 8% of patients had a Hgb <10 g/dL, and <1% had a Hgb <8.5 g/dL. The corresponding numbers from the FISSION study in which patients also received Peg-IFN were 14% and 2%, respectively.22 Not surprisingly, patients treated with interferon-sparing, sofosbuvir-based regimens had less treatment discontinuation rates and reported better health-related quality of life.
It is important to make note of adverse effects of sofosbuvir in combination with other DAAs. In the COSMOS trial, fatigue and headache were the most common adverse effects; however, simeprevir and sofosbuvir were also associated with pruritus (17%), rash (11%), and hyperbilirubinemia (7%). Sofosbuvir plus daclatasvir combination therapy was also associated with fatigue (11%) and headache (7.8%), as well as nausea. Only two patients required treatment discontinuation, which was likely unrelated to the study drug.28 Pooled data from Phase III trials using sofosbuvir in combination with ledipasvir revealed headache (11%–17%) and fatigue (13%–18%) as the most common side effects with very few patients (<1%) requiring treatment discontinuation due to side effects. Nausea, diarrhea, and insomnia were reported in 5%–10% of patients. Laboratory abnormalities with elevation in bilirubin and lipase levels were noted in ≤3% of patients, and may be a unique adverse effect of ledipasvir.
Conclusion and future challenges associated with the effective treatment of hepatitis C
The challenges in managing patients with CHC are in many ways different today in the era of highly effective DAA therapy. With the arrival of sofosbuvir ($1,000 per pill) and now Harvoni® ($1,125 per pill), concerns about cost have arisen. The cost of therapy must be further contemplated in the context of a strained US health care system where many patients with CHC are either uninsured or insured through government-sponsored plans.38 Insurance companies may decide to prioritize treating certain patients first over others (eg, those with more advanced fibrosis). Another factor both in the US and across the world involves identifying infected patients. In 2012, the US Centers for Disease Control and Prevention recommended one-time screening for CHC infection in persons born between 1945 and 1965.39 The implementation of this guideline is expected to identify a large number of previously undiagnosed patients who will require treatment. The public health mandate to find such patients is justified by our ability to now provide safe and effective therapy. While the cost of sofosbuvir or Harvoni® treatment today remains an issue, many providers feel that the real question is actually the relative cost of treating against the price paid by not treating – namely more patients at risk for complications from cirrhosis. In developing countries, where access and affordability are paramount, generic pharmaceutical companies, through licensing agreements with Gilead Sciences Inc., expect to be able to provide the drug at a fraction of the US cost. As a result, the main obstacle may become ensuring adherence to therapy.
Future challenges still exist for special patient populations, and guidelines for treating HIV–HCV-coinfected patients with Harvoni® will require updating once ongoing clinical trials are complete. The same can be said regarding the optimal timing and duration of therapy for post-liver transplant patients. Chronic kidney disease patients with CHC infection, including those on dialysis, must await dosing recommendations for sofosbuvir and Harvoni® or seek alternative DAA combinations that are not renal cleared. Finally, a small percentage of patients will not achieve an SVR with DAA therapy containing sofosbuvir, reflecting real-world outcomes where adherence is suboptimal. Retreatment with sofosbuvir should be possible, but the decision to change to a different combination of DAA and/or adding RBV therapy will need to be individualized. While these concerns are real, when placed in perspective, the chance to prevent patients from developing cirrhosis, liver cancer, and even in some cases the need for liver transplant is truly remarkable to envision.
Disclosure
Dr deLemos has received support for clinical trials from Vital Therapies. The remaining authors have no disclosures.
References
Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013;57(4):1333–1342. | ||
Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med. 2006;144(10):705–714. | ||
Davis GL, Alter MJ, El-Serag H, Poynard T, Jennings LW. Aging of hepatitis C virus (HCV)-infected persons in the United States: a multiple cohort model of HCV prevalence and disease progression. Gastroenterology. 2010;138(2):e511–e516. | ||
Elfiky AA, Elshemey WM, Gawad WA, Desoky OS. Molecular modeling comparison of the performance of NS5b polymerase inhibitor (PSI-7977) on prevalent HCV genotypes. Protein J. 2013;32(1):75–80. | ||
Chevaliez S, Pawlotsky JM. HCV genome and life cycle. In: Tan SL, editor. Hepatitis C Viruses: Genomes and Molecular Biology. Norfolk: Horizon Bioscience; 2006:5–47. | ||
Clark JL, Hollecker L, Mason JC, et al. Design, synthesis, and antiviral activity of 2′-deoxy-2′-fluoro-2′-C-methylcytidine, a potent inhibitor of hepatitis C virus replication. J Med Chem. 2005;48(17):5504–5508. | ||
Smith DB, Kalayanov G, Sund C, et al. The design, synthesis, and antiviral activity of monofluoro and difluoro analogues of 4′-azidocytidine against hepatitis C virus replication: the discovery of 4′-azido-2′-deoxy-2′-fluorocytidine and 4′-azido-2′-dideoxy-2′,2′-difluorocytidine. J Med Chem. 2009;52(9):2971–2978. | ||
Sofia MJ, Bao D, Chang W, et al. Discovery of a beta-d-2′-deoxy-2′-alpha-fluoro-2′-beta-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem. 2010;53(19):7202–7218. | ||
Murakami E, Niu C, Bao H, et al. The mechanism of action of beta-D-2′-deoxy-2′-fluoro-2′-C-methylcytidine involves a second metabolic pathway leading to beta-D-2′-deoxy-2′-fluoro-2′-C-methyluridine 5′-triphosphate, a potent inhibitor of the hepatitis C virus RNA-dependent RNA polymerase. Antimicrob Agents Chemother. 2008;52(2):458–464. | ||
Murakami E, Tolstykh T, Bao H, et al. Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J Biol Chem. 2010;285(45):34337–34347. | ||
Lam AM, Murakami E, Espiritu C, et al. PSI-7851, a pronucleotide of beta-D-2′-deoxy-2′-fluoro-2′-C-methyluridine monophosphate, is a potent and pan-genotype inhibitor of hepatitis C virus replication. Antimicrob Agents Chemother. 2010;54(8):3187–3196. | ||
Kirby B, Gordi T, Symonds W, Kearney BP, Mathias A. Population pharmacokinetics of sofosbuvir and its major metabolite (GS-331007) in healthy and HCV infected adult subjects [abstract]. Hepatology. 2013;58(S1):746A. | ||
Cornpropst M, Denning J, Clemons D, et al. The effect of renal impairment and end stage renal disease on the single-dose pharmacokinetics of PSI-7977 [abstract]. J Hepatol. 2012;56(S2):S433. | ||
Lawitz E, Rodriguez-Torres M, Cornpropst M, et al. The effect of hepatic impairment on the pharmacokinetics and antiviral activity of PSI-7977 in hepatitis C infected subjects treated for seven days [abstract]. J Hepatol. 2012;56(S2):S445–S446. | ||
Denning J, Cornpropst M, Flach SD, Berrey MM, Symonds WT. Pharmacokinetics, safety, and tolerability of GS-9851, a nucleotide analog polymerase inhibitor for hepatitis C virus, following single ascending doses in healthy subjects. Antimicrob Agents Chemother. 2013;57(3):1201–1208. | ||
Lawitz E, Rodriguez-Torres M, Denning JM, et al. Pharmacokinetics, pharmacodynamics, and tolerability of GS-9851, a nucleotide analog polymerase inhibitor, following multiple ascending doses in patients with chronic hepatitis C infection. Antimicrob Agents Chemother. 2013;57(3):1209–1217. | ||
Lam AM, Espiritu C, Bansal S, et al. Genotype and subtype profiling of PSI-7977 as a nucleotide inhibitor of hepatitis C virus. Antimicrob Agents Chemother. 2012;56(6):3359–3368. | ||
Rajyaguru S, Xu S, Hebner C, et al. Sofosbuvir selects the NS5B S282T mutation in vitro in genotype 1–6 replicons and is not cross-resistant to resistance associated variants Selected by other classes of antiviral inhibitors [abstract]. Hepatology. 2013;58(S1):739A. | ||
Svarovskaia ES, Dvory-Sobol H, Parkin N, et al. Infrequent development of resistance in genotype 1–6 hepatitis C virus-infected subjects treated with sofosbuvir in phase 2 and 3 clinical trials. Clin Infect Dis. 2014;59(12):1666–1674. | ||
Gane EJ, Stedman CA, Hyland RH, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med. 2013;368(1):34–44. | ||
German P, Yang J, West S, Han L, Sajwani K, Mathias A. Effect of food and acid reducing agents on the relative bioavailability and pharmacokinetics of ledipasvir/sofosbuvir fixed dose combination tablet [abstract 15]. Paper presented at: 15th International Workshop on Clinical Pharmacology of HIV and Hepatitis Therapy; 2014; Washington, D.C. | ||
Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med. 2013;368(20):1878–1887. | ||
Kowdley KV, Gordon SC, Reddy KR, ION-3 Investigators, et al. Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N Engl J Med. 2014;370(20):1879–1888. | ||
Afdhal N, Zeuzem S, Kwo P, ION-1 Investigators, et al. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med. 2014;370(20):1889–1898. | ||
Afdhal N, Reddy KR, Nelson DR, ION-2 Investigators, et al. Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med. 2014;370(16):1483–1493. | ||
AASLD/IDSA/IAS–USA [homepage on the Internet]. Recommendations for testing, managing, and treating hepatitis C. Available from: http://www.hcvguidelines.org. Accessed April 24, 2014. | ||
Lawitz E, Sulkowski MS, Ghalib R, et al. Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non-responders to pegylated interferon and ribavirin and treatment-naive patients: the COSMOS randomised study. Lancet. 2014;384(9956):1756–1765. | ||
Sulkowski MS, Gardiner DF, Rodriguez-Torres M, et al; AI444040 Study Group. Daclatasvir plus sofosbuvir for previously treated or untreated chronic HCV infection. N Engl J Med. 2014;370(3):211–221. | ||
Jacobson IM, Gordon SC, Kowdley KV, et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med. 2013;368(20):1867–1877. | ||
Zeuzem S, Dusheiko GM, Salupere R, et al. Sofosbuvir and ribavirin in HCV genotypes 2 and 3. N Engl J Med. 2014;370(21):1993–2001. | ||
Sulkowski MS, Naggie S, Lalezari J, et al; PHOTON-1 Investigators. Sofosbuvir and ribavirin for hepatitis C in patients with HIV coinfection. JAMA. 2014;312(4):353–361. | ||
Townsend KS, Osinusi A, Nelson AK, et al. High efficacy of sofosbuvir/ledipasvir for the treatment of HCV genotype 1 in patients coinfected with HIV on or off antiretroviral therapy: results from The NIAID ERADICATE Trial [abstract]. Hepatology. 2014;S1:59A. | ||
deLemos AS, Schmeltzer PA, Russo MW. Recurrent hepatitis C after liver transplant. World J Gastroenterol. 2014;20(31):10668–10681. | ||
Forns X, Charlton M, Denning J, et al. Sofosbuvir compassionate use program for patients with severe recurrent hepatitis C following liver transplantation. Hepatology. In press 2014. | ||
Curry MP, Forns X, Chung RT, et al. Sofosbuvir and ribavirin prevent recurrence of HCV infection after liver transplantation: an open-label study. Gastroenterology. 2014;148(1):100.e–107.e. | ||
Charlton M, Gane E, Manns MP, et al. Sofosbuvir and ribavirin for treatment of compensated recurrent hepatitis C virus infection after liver transplantation. Gastroenterology. 2014;148(1):108–117. | ||
Levitsky J, Fiel MI, Norvell JP, et al. Risk for immune-mediated graft dysfunction in liver transplant recipients with recurrent HCV infection treated with pegylated interferon. Gastroenterology. 2012;142(5):1132–1139e1131. | ||
Stepanova M, Kanwal F, El-Serag HB, Younossi ZM. Insurance status and treatment candidacy of hepatitis C patients: analysis of population-based data from the United States. Hepatology. 2011;53(3):737–745. | ||
Smith BD, Morgan RL, Beckett GA, Falck-Ytter Y, Holtzman D, Ward JW. Hepatitis C virus testing of persons born during 1945–1965: recommendations from the Centers for Disease Control and Prevention. Ann Intern Med. 2012;157(11):817–822. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.