")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
Changes of serum uric acid and total bilirubin in elderly patients with major postischemic stroke depression
Authors Gao J, Xu W, Han K, Zhu L, Gao L, Shang XL
Received 25 August 2017
Accepted for publication 9 November 2017
Published 27 December 2017 Volume 2018:14 Pages 83—93
DOI https://doi.org/10.2147/NDT.S149712
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Wai Kwong Tang
Jie Gao, Wei Xu, Kun Han, Lu Zhu, Lili Gao, Xiuli Shang
Department of Neurology, The First Affiliated Hospital, China Medical University, Shenyang, China
Background: This was a longitudinal study which investigated the relationship between serum uric acid (SUA) and total bilirubin (Tbil) upon admission in elderly stroke patients and the occurrence of postischemic stroke depression (IPSD) at 3, 6, and 9 months of post-stroke follow-up.
Subjects and methods: Data were analyzed for 525 acute ischemic stroke patients. Beck Depression Inventory (BDI) scores >17 and Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) were used separately to screen and diagnose IPSD at 3, 6, and 9 months post-stroke. Once IPSD was diagnosed, follow-up activities were terminated.
Results: High levels of SUA (odds ratio [OR]=2.08, P<0.01) and Tbil (OR=2.31, P<0.01) in the first 3 months post-stroke and low levels of SUA (OR=2.05, P=0.03) and Tbil (OR=2.79, P<0.01) from 3 to 6 months post-stroke were identified as risk factors for major IPSD. At 3 months, patients with SUA levels ≥406.5 µmol/L (males with SUA levels of ≥409.5 µmol/L and females with SUA levels ≥385.5 µmol/L) and Tbil levels ≥23.65 µmol/L were more likely to develop major IPSD. At 6 months, both SUA (area under curve [AUC]=0.625, P=0.005, cutoff =194.0 µmol/L) and Tbil (AUC=0.681, P=0.004, cutoff =6.75 µmol/L) had minor diagnostic values (AUC<0.700), although SUA levels ≤214.5 µmol/L (AUC=0.756, P=0.001) in female patients had a good diagnostic value (AUC=0.722, P=0.006) for major IPSD. At 9 months, major IPSD showed no statistical relationship with either SUA (χ2=2.33, P=0.13) or Tbil (χ2=0.41, P=0.84).
Conclusion: Higher levels of SUA and Tbil on admission were closely related to the occurrence of major IPSD within 3 months of stroke. Lower levels of these two biomarkers on admission were characteristic for the occurrence of major IPSD between 3 and 6 months post-stroke, while 6 months after stroke, there was no relationship between major IPSD and these two biomarkers.
Keywords: depression after stroke, stroke, total bilirubin, uric acid, longitudinal study
Introduction
Postischemic stroke depression (IPSD) is an important post-stroke complication and a chronic disorder that seriously affects the prognosis of patients.1,2 The biological processes and pathways of depression generally include inflammatory, oxidative, and nitrosative stress pathways; neurotransmitter systems; neurotrophins; and the regulation of neurogenesis and modulation of the hypothalamic–pituitary–adrenal (HPA) axis.3,4 Post-stroke depression (PSD) may have a similar pathological process to depression. Previous research has shown that the levels of some molecular markers are changed in the peripheral system of PSD patients, including C-reactive protein (CRP) and homocysteine,5–7 brain-derived neurotrophic factor (BDNF),8 and triiodothyronine.9 Of these pathophysiological factors, oxidative and nitrosative stress pathways are believed to be the most crucial pathophysiological factors,10 since the brain has high metabolic rates and low antioxidant levels.11
Serum uric acid (SUA) is the ultimate product of purine metabolism and undertakes 60% of the body’s antioxidant reactions.12 Uric acid (UA) can inhibit inflammatory cascades, reduce the permeability of the blood–brain barrier, and protect central nervous tissue.13 Similar to the situation reported for cardiovascular disease,14 hyperuricemia appears to be common in post-stroke patients and is suggestive of a better prognosis.15 However, low SUA levels after ischemic stroke have also been reported in previous experimental data and were indicative of poor outcomes.16 UA is also a known scavenger of peroxynitrite, which is the main oxidizing congener of nitric oxide and is involved in the biochemical pathogenesis of depression.17 However, both high18 and low19,20 SUA levels have been previously shown to be associated with depression.
Bilirubin is a powerful antioxidant and is the final catabolite of the heme fragmentation pathway.21 Some studies have shown that decreased serum bilirubin level could be an independent predictor of stroke incidence,22,23 and a high serum bilirubin level is related to the severity of stroke and a poor prognosis.24 However, other studies have shown that a high level of serum bilirubin is used as a mechanism which just only reflects the intensity of initial oxidative stress rather than outcome of stroke in acute ischemic stroke.25 The clear inconsistency between these earlier studies is interesting and has led to confusion with regard to our understanding of the specific relationship between bilirubin and stroke. Similar effects have been observed in studies which have evaluated the relationship between bilirubin and depression. For example, Miyaoka et al showed that high levels of biopyrrin (bilirubin oxidative metabolite) in the urine of psychiatric patients were correlated with depressive symptoms.26 In contrast, there is also strong evidence suggesting that low bilirubin levels represent a risk factor for depression.27
The precise relationship between IPSD and levels of SUA and Tbil has yet to be studied in depth, particularly in terms of the occurrence of IPSD at different phases after stroke. To our knowledge, only a limited number of studies have attempted to explain why IPSD may occur over different time periods following stroke.28,29 We hypothesized that levels of SUA and Tbil were implicated in IPSD. Consequently, the key purpose of the present study was to investigate the specific relationship between levels of SUA and Tbil at admission and the occurrence of IPSD.
Subjects and methods
Baseline participants and assessment
This was a prospective cohort study undertaken at China Medical University Affiliated Hospital between October 2014 and March 2016 involving 525 patients with ischemic stroke. Data regarding the incidence of postischemic stroke depression and physical outcomes were collected from stroke survivors every 3 months following their initial hospital admission.
Demographic and clinical data
For all subjects, a trained research assistant collected the demographic data (gender, age, marital status, and educational level) and evaluated stroke severity using the National Institute of Health Stroke Scale (NIHSS)30 within 48 hours of admission. The following morning, fasting blood was collected and levels of SUA and Tbil were determined.
Our specific inclusion criteria were as follows: 1) a first occurrence of an ischemic stroke that met the standards of the World Health Organization diagnostic criteria (not exceeding 2 weeks post-stroke); 2) a duration of clinical neurological function deficit lasting over 24 hours; 3) radiological magnetic resonance imaging confirmation31; and 4) over 55 years of age. Patients were excluded from the study if: 1) they refused to participate in the study; 2) there was apparent nonvascular etiologies (primary or metastatic neoplasms); 3) they were suffering from pre-stroke disability or depression, or antidepressant history; 4) they had a history of liver disease, hemolytic disease (such as Rh hemolytic disease and thalassemia), serious renal disease, or gout; 5) they were suffering from severe aphasia (language score ≥2 using the NIHSS), total blindness, deafness, or disturbance of consciousness; 6) they had a Barthel Index score <10; 7) they had an NIHSS score >20; or 8) their blood indices were insufficient upon admission, such as liver function, renal function, SUA, or Tbil.
SUA and Tbil assay
Levels of SUA and Tbil were determined by Roche kit (Hoffman-La Roche Ltd., Basel, Switzerland) with the use of a Hitachi 7170 automatic biochemical analyzer test (Hitachi High-Technologies, Tokyo, Japan). In our hospital department, normal serum SUA level is known to range from 208 to 428 μmol/L in male patients and from 155 to 357 μmol/L in females, while normal Tbil levels range from 3.0 to 22.0 μmol/L. As raw SUA and Tbil data were skewed, we recorded and divided these data into three tertiles: (tertile 1a ≤245.0 μmol/L, 245.0< tertile 2b <330.0 μmol/L, tertile 3c ≥330.0 μmol/L) and (tertile 1d ≤8.0 μmol/L, 8.0 tertile 2e <12.8 μmol/L, tertile 3f ≥12.8 μmol/L), respectively.
Follow-up
Patients meeting our specific inclusion criteria were invited to participate in the study and were followed up until 9 months post-stroke. Follow-up measurements were taken at 3, 6, and 9 months post-stroke. All assessments were performed by trained research assistants.
Ethical statement
The study was approved by the Regional Medical Research Ethical Committee of China Medical University. All subjects, or their legal representatives, provided informed written consent.
Assessment of IPSD
Subjects were invited to complete the BDI32 by telephone interview at 3, 6, and 9 months of follow-up as the primary screening standard. Patients with scores >17 were recruited in order to consider a final diagnosis of major IPSD in accordance with the DSM-IV33 by two professional clinicians who were blinded to laboratory results.
Statistical analysis
The SPSS 19.0 statistical package (SPSS Inc., Chicago, IL, USA) was used to analyze all data arising from the study. A χ2 test was used to assess differences in categorical variables data. For continuous, normally distributed data, the Student’s t-test was used. Risk factors (P≤0.10) were further analyzed by univariate and multivariate logistic regression. Relationships between gender and the levels of SUA and Tbil were analyzed using Spearman correlation analysis. Receiver operating characteristic curves were drawn to depict the diagnostic performance of SUA and Tbil levels in the occurrence of IPSD. All P-values were calculated as two-tailed, and significance level was set at P<0.05.
Results
Patients
A total of 1,006 ischemic stroke patients were screened and 760 met the specific entry criteria. During the follow-up process, 235 patients withdrew; consequently, the final data analysis included 525 patients. The follow-up procedure is depicted in Figure 1.
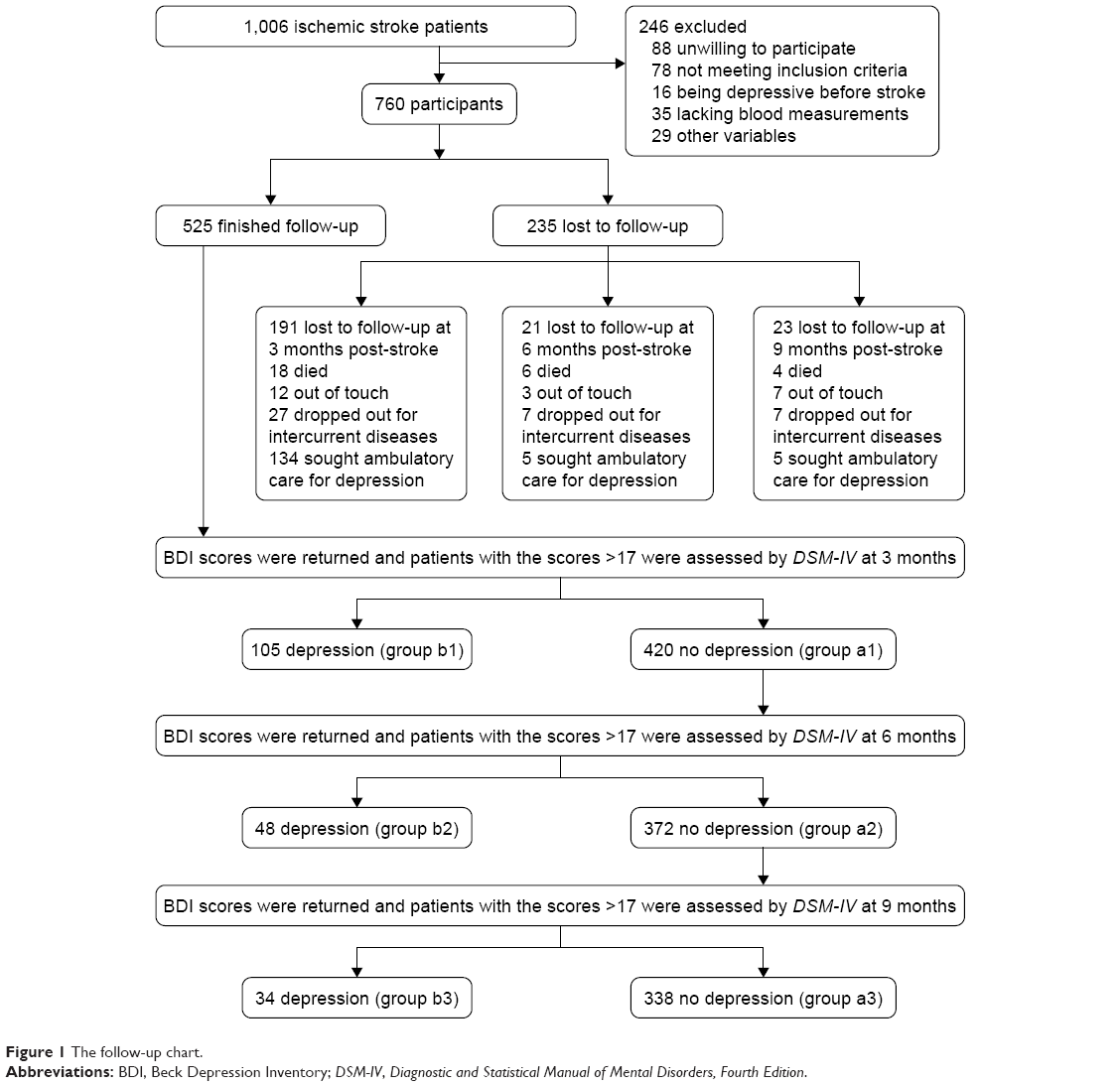 | Figure 1 The follow-up chart. |
Patients who withdrew from the study had a significantly higher mean age (mean ± SD: 68.26±7.48 years vs 64.00±13.19 years; t=5.28, P<0.01), and were more likely to have less than 9 years of formal education (46.6% vs 37.3%, P=0.03). Baseline characteristics of patients who completed the follow-up period are shown in Table 1.
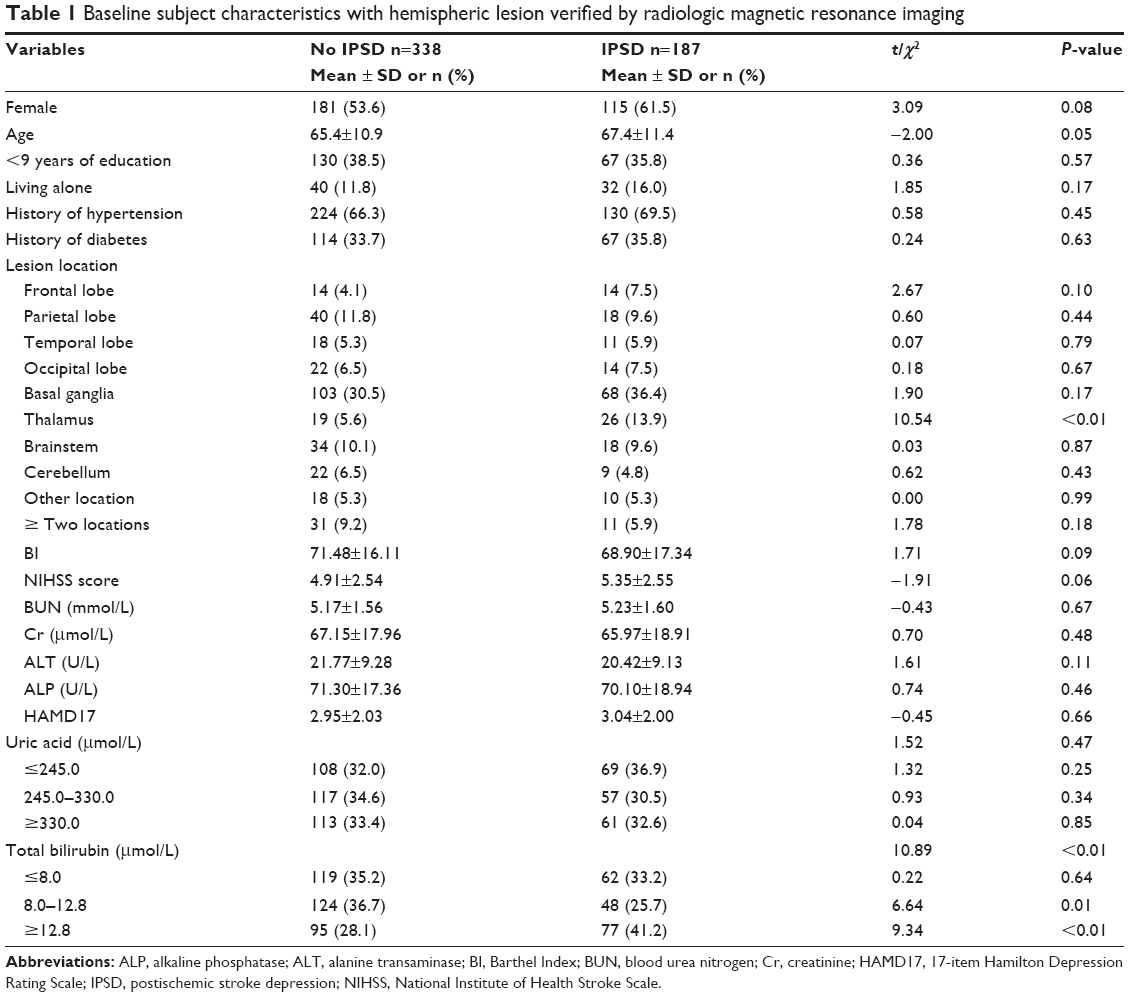 | Table 1 Baseline subject characteristics with hemispheric lesion verified by radiologic magnetic resonance imaging |
Correlation analysis between the levels of SUA and Tbil upon admission and the occurrence of IPSD
When no confounding factors were considered, a significant difference was observed in terms of Tbil levels when compared between patients with major IPSD and non-major IPSD (χ2=10.89, P<0.01; Table 1). Next, we entered all risk factors with P≤0.10 (Table 1) into a multivariate logistic regression model (ie, female gender, age, Barthel Index [BI], NIHSS, frontal lobe, thalamus, and Tbil level [tertile 3c]). Of these risk factors, multivariate logistic regression confirmed that Tbil levels (tertile 3c) were independently related to the occurrence of IPSD with an odds ratio (OR) of 1.93 (P<0.01) (Table 2).
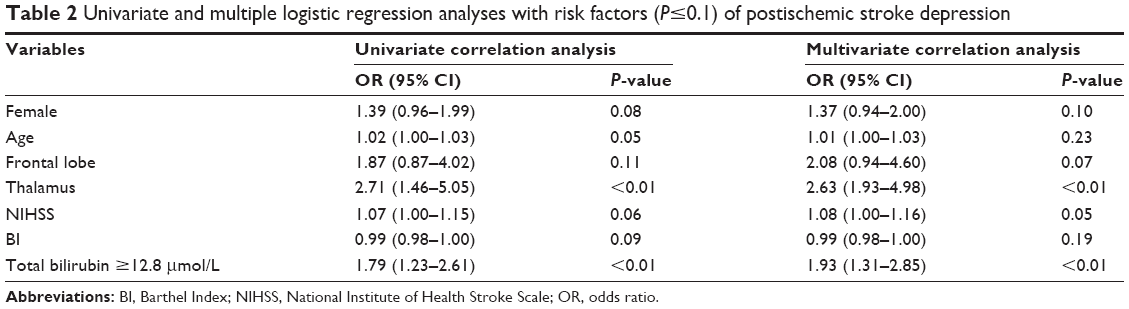 | Table 2 Univariate and multiple logistic regression analyses with risk factors (P≤0.1) of postischemic stroke depression |
The relationship between the occurrence of major IPSD and the levels of serum SUA and Tbil after adjustment for other risk factors
There were 105 patients with major IPSD within 3 months of stroke, 48 patients from 3 to 6 months post-stroke, and 34 patients between 6 and 9 months post-stroke. The incidence of major IPSD was significantly different when compared between these different time periods (χ2=24.96, P<0.01). Therefore, the analysis of relationships between different levels of SUA and Tbil and major IPSD was stratified according to different time periods following stroke (3 months, 3– 6 months, and 6–9 months post-stroke, as shown in Table 3).
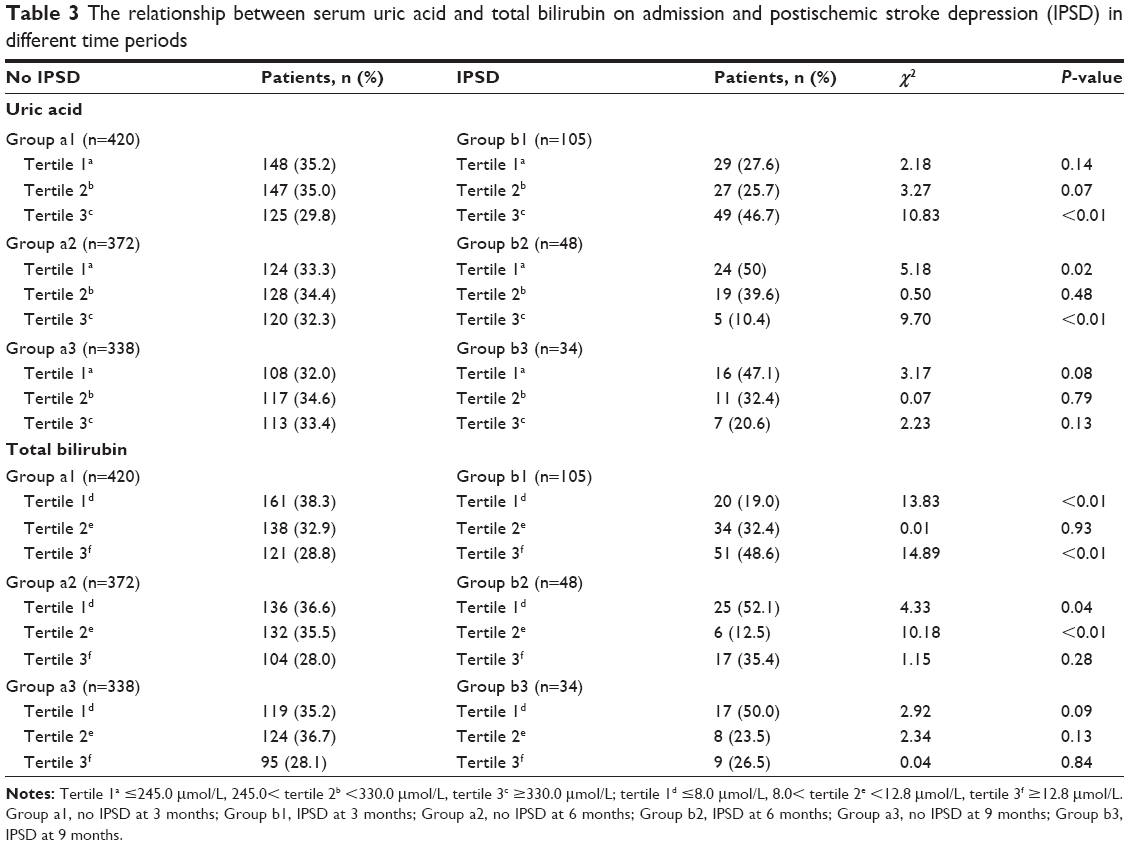 | Table 3 The relationship between serum uric acid and total bilirubin on admission and postischemic stroke depression (IPSD) in different time periods |
When we compared patients with major IPSD and non-major IPSD across the different time periods in terms of gender, age, education, hypertension, diabetes, NIHSS, BI, location, 17-item Hamilton Depression Rating Scale, blood urea nitrogen, creatinine, alkaline phosphatase, and alanine aminotransferase, we identified several risk factors where P≤0.1: age (t=−1.79, P=0.07), thalamus (χ2=5.47, P=0.02), frontal lobe (χ2=4.57, P=0.03), and NIHSS (t=−1.86, P=0.07) in the first 3 months post-stroke; female gender (χ2=5.81, P=0.02), age (t=−1.68, P=0.10), and NIHSS score (t=−2.78, P=0.01) from 3 to 6 months post-stroke; female gender (χ2=4.53, P=0.03), age (t=−2.43, P=0.02), and living alone (χ2=6.13, P=0.01) from 6 to 9 months post-stroke. Next, we used univariate and multivariate analyses to investigate major IPSD, SUA, and Tbil levels and the significant risk factors identified earlier (Table 2). The results of these analyses are shown in Table 4.
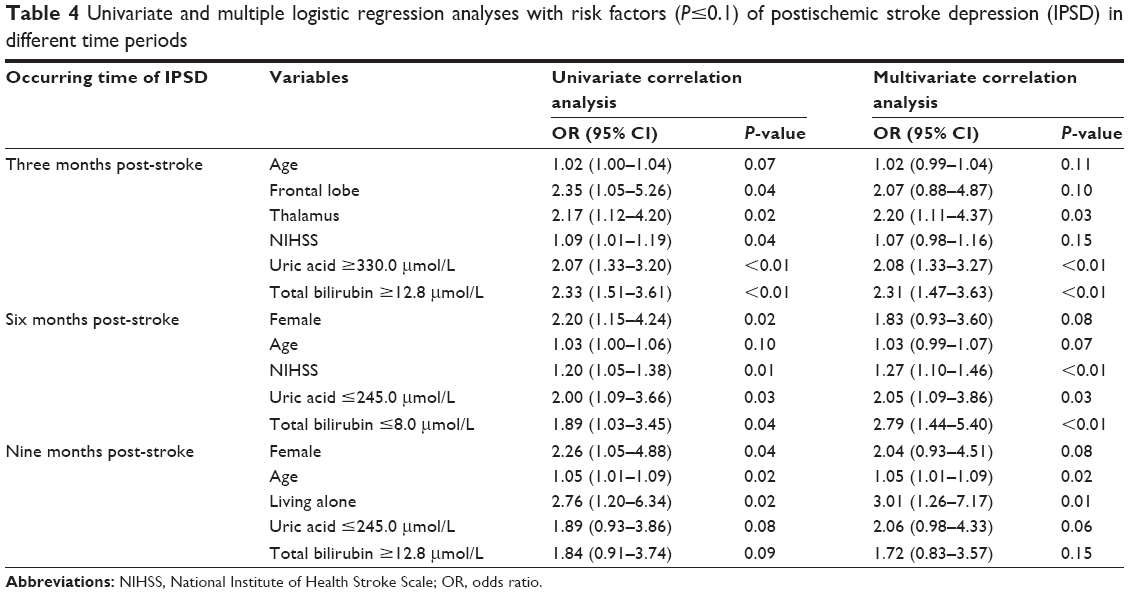 | Table 4 Univariate and multiple logistic regression analyses with risk factors (P≤0.1) of postischemic stroke depression (IPSD) in different time periods |
The sensitivity and specificity of SUA and Tbil levels to predict major IPSD in the first 6 months post-stroke
As shown in Table 5, a negative correlation was found between female gender and SUA level (r=–0.30, P<0.01). The predictive values of SUA and Tbil levels are shown in Table 6. In the first 3 months post-stroke, SUA levels ≥330.0 μmol/L (area under curve [AUC]=0.834, P<0.001, Figure 2A) and Tbil levels ≥12.8 μmol/L (AUC=0.782, P<0.001, Figure 2B) showed significant diagnostic value. Furthermore, patients with SUA levels ≥406.5 μmol/L, or Tbil levels ≥23.65 μmol/L, were more likely to be depressive. Meanwhile, male patients with SUA levels ≥409.5 μmol/L (AUC=0.920, P<0.001, Figure 2C) and females with SUA levels ≥385.5 μmol/L (AUC=0.756, P=0.001, Figure 2D) had a greater risk of suffering from major IPSD.
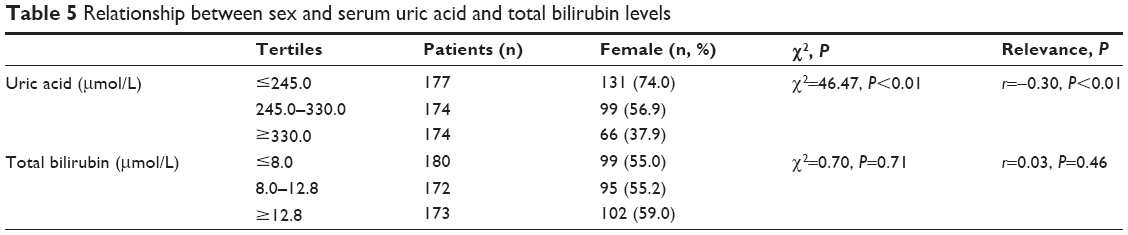 | Table 5 Relationship between sex and serum uric acid and total bilirubin levels |
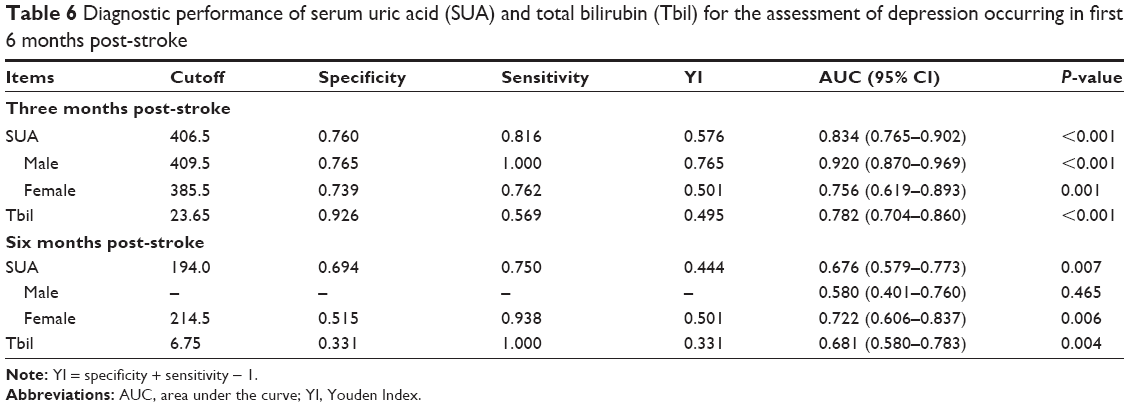 | Table 6 Diagnostic performance of serum uric acid (SUA) and total bilirubin (Tbil) for the assessment of depression occurring in first 6 months post-stroke |
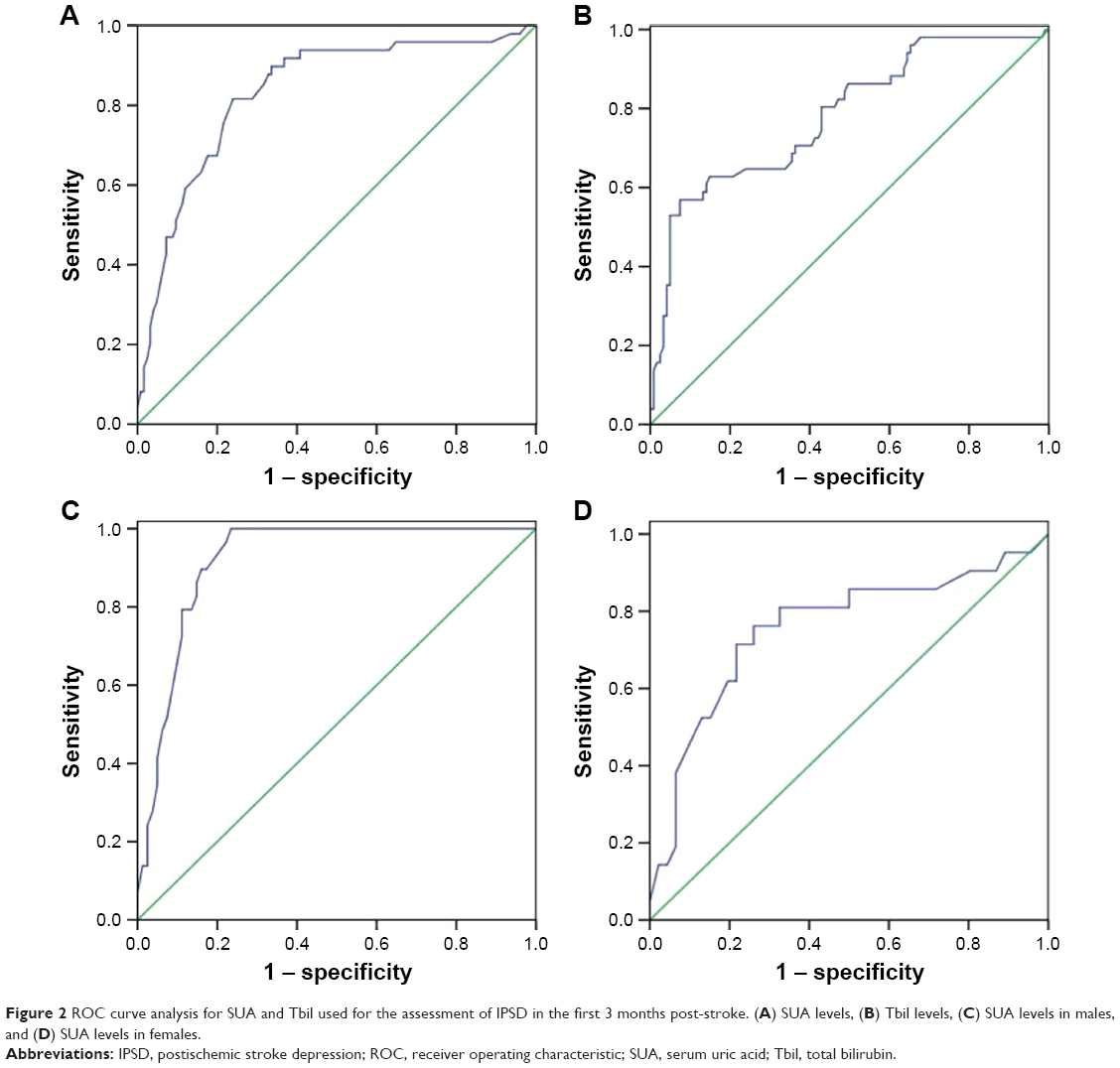 | Figure 2 ROC curve analysis for SUA and Tbil used for the assessment of IPSD in the first 3 months post-stroke. (A) SUA levels, (B) Tbil levels, (C) SUA levels in males, and (D) SUA levels in females. |
From 3 to 6 months post-stroke, the diagnostic performance of SUA levels ≤245 μmol/L (AUC=0.676, P=0.007, Figure 3A) and Tbil levels ≤8.0 μmol/L (AUC=0.681, P=0.004, Figure 3B) was statistically significant, although the diagnostic value was not very high. However, compared with male patients (AUC=0.580, P=0.465, Figure 3C), female patients with SUA levels ≤214.5 μmol/L (AUC=0.722, P=0.006, Figure 3D) were more likely to develop major IPSD.
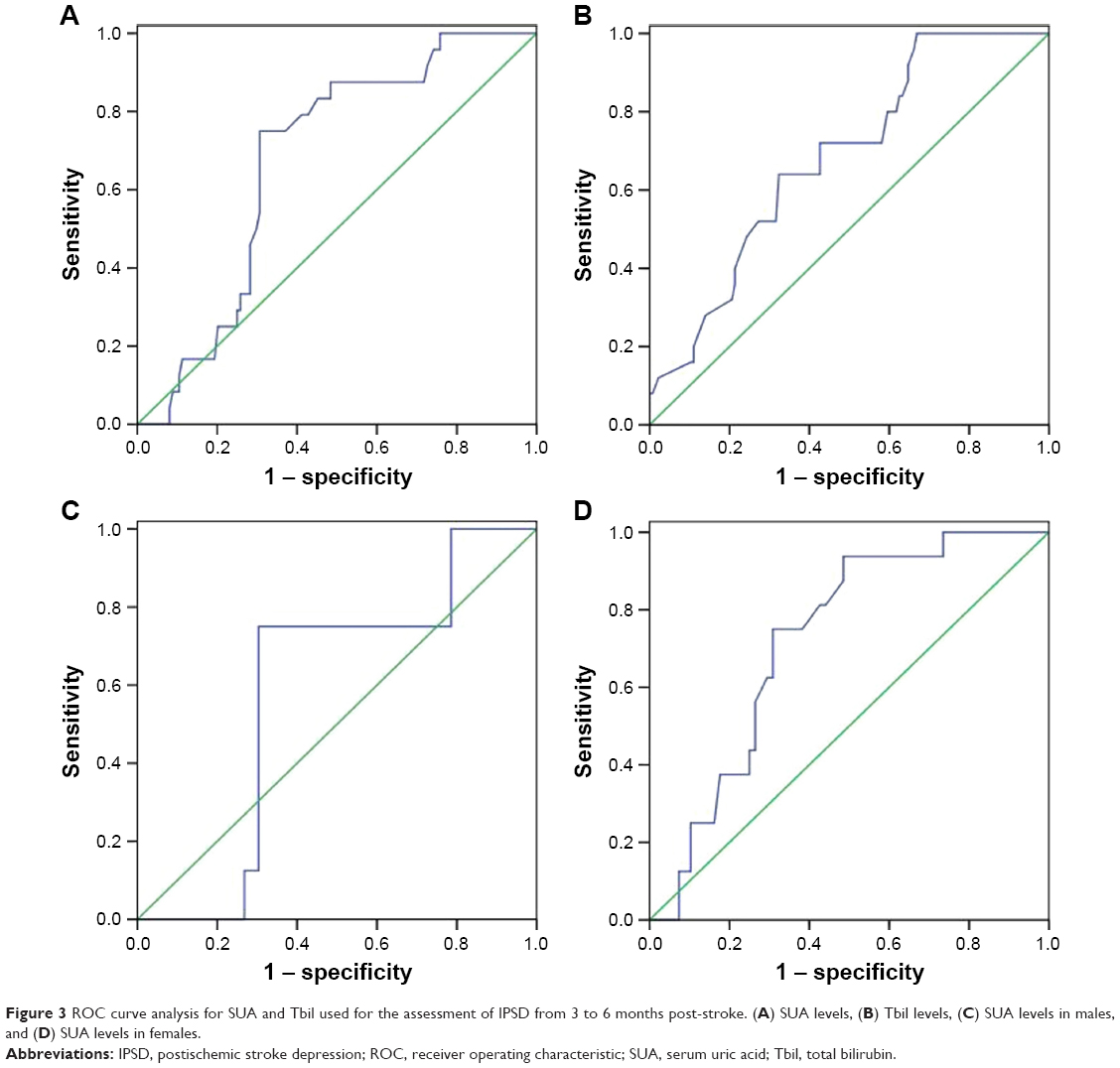 | Figure 3 ROC curve analysis for SUA and Tbil used for the assessment of IPSD from 3 to 6 months post-stroke. (A) SUA levels, (B) Tbil levels, (C) SUA levels in males, and (D) SUA levels in females. |
Discussion
Unlike previous studies, the present investigation focused on the occurrence of major IPSD across different time periods and attempted to link this information with levels of Tbil and SUA. The common characteristic of SUA and Tbil is that both these factors exhibit antioxidant reactions within the human body. Our study also confirmed that the changes observed on the levels of these two biomarkers showed excellent consistency in major IPSD patients. While the precise mechanism underlying IPSD has yet to be established, our present study provided significant evidence of the important pathological roles of both SUA and Tbil in IPSD.
Patients developing major IPSD in the first 3 months after stroke also showed higher levels of SUA and Tbil upon admission. In a previous study, Nanetti et al showed that acute ischemic stroke could cause the generation of free radicals and strong oxidative stress, and that this antioxidant capacity may limit the progression of ischemic injury.34 As powerful antioxidants, high concentrations of SUA and Tbil could reflect the level of oxidative stress in the first 3 months after stroke. Meanwhile, two previous meta-analyses reported that depression was strongly correlated to high oxidative stress by demonstrating a clear imbalance between antioxidants and oxidant activity.35 Previous research also implied that the pathophysiology of IPSD is probably involved in increased levels of oxidative stress in cerebral tissues.36 Firstly, oxidative stress, which involves enzymatic and nonenzymatic antioxidant factors expressed in the brain and periphery, could cause damage to proteins, DNA molecules, and lipids.37,38 Both SUA and Tbil are nonenzymatic antioxidants, much like glutathione and vitamins A, C, and E.12,37 Secondly, oxidative stress has been implicated in IPSD via the regulation of inflammation.35 A series of studies also provided support for the role of inflammatory factors in the development of IPSD.39–41 Under the influence of repeated inflammatory stimuli, microglia in the central nervous system can evolve into a source of inflammatory mediators, which may then influence brain neurotransmitter systems and neuronal integrity.42 Moreover, elevated levels of inflammatory cytokines can also affect the activity of 2,3-dioxygenase, which degrades tryptophan via the kynurenine pathway and ultimately leads to a reduction in the synthesis and availability of 5-hydroxytryptamine.43,44 It has also been reported that interaction between oxidative stress pathways and inflammatory pathways may lead to an increased level of neuronal apoptosis and neurodegeneration and a decline in the neurogenesis and neuroplasticity of depression.4,45,46 Thus, we can conclude that close associations between major IPSD and higher levels of SUA and Tbil in the first 3 months after stroke may be associated with high levels of oxidative stress. It has been reported that 60% of patients with depression during this particular time period showed recovery by 12 months post-stroke.47 It is possible that the symptoms of depression disappear at the same time as the disappearance of oxidative stress response.
However, this does not explain why patients with lower levels of SUA and Tbil at admission are more vulnerable to depression between 3 and 6 months post-stroke. One previous meta-analysis suggested that high levels of SUA at the onset of ischemic stroke could reduce levels of neurological damage and herald a better prognosis.15 Similar results were also reported by Perlstein et al for higher levels of Tbil.48 Some studies have also reported reduced levels of antioxidants in some patients with acute ischemic stroke.49 Thus, we can infer that lower levels of SUA and Tbil are related to more serious neurological damage than higher levels in acute stroke. It has also been reported that the more significant the stroke severity and functional impairment are, the greater the probability of suffering from depression.50 This is consistent with the higher NIHSS score in major IPSD patients observed between 3 and 6 months post-stroke. However, it is still unclear as to why patients with low levels of SUA and Tbil in our study developed depression after a 3-month delay instead of acutely. There may be several possible explanations for this phenomenon. Firstly, once the acute phase is over, patients with more severe ischemic stroke may gradually realize the seriousness of problems related to stroke,51 such as the decline in self-care and work ability, which would make such patients feel incompetent. Secondly, in patients with significant functional impairment such as post-stroke paresthesia, early paresthesia may lead to sleep disorders, and long-term sleep disorders inevitably lead to the occurrence of depression.52
From 6 to 9 months, we did not observe any significant association between major IPSD and the levels of SUA and Tbil. As we described in our previous publication, since biological changes and reconstruction in the brain tissue cease during this time frame, social psychology factors, rather than stroke severity, begin to play a more important role.53
In summary, our current study provides evidence that different levels of SUA and Tbil in the blood may be helpful in the early prevention of IPSD. This information may also be useful in selecting treatments for depression considering the different mechanisms of depression which occur over different time periods. In the first 3 months after stroke, depression may not need excessive intervention because acute oxidative stress could disappear by itself. However, from 3 to 6 months, early post-stroke rehabilitation treatment has a more significant effect upon the prevention of depression and recovery because of the relationship between depression and stroke severity. Six months after stroke, antidepressive therapies, such as antidepressant drugs or psychotherapy, may begin to exert effects upon the treatments used for major IPSD. Compared with other biomarkers, there are several superiorities of the two biomarkes (SUA and Tbil) for IPSD. First, SUA and Tbil represent affordable routine clinical tools which do not incur additional expense as compared with BDNF, which is limited to precise laboratory tests and not suitable for extensive clinical studies and follow-up. Second, as compared with CRP, SUA and Tbil are less affected by the following factors, such as fever, infection, immune system diseases, and so on. Third, as compared with triiodothyronine and other biomarkers, SUA and Tbil may be used to monitor the development of IPSD, since a previous report showed that the level of SUA changes as depression improves.11
Acknowledgment
This work was supported by grants from the Natural Science Foundation of China (81100243 and 81502181).
Disclosure
The authors report no conflicts of interest in this work.
References
Taylor-Piliae RE, Hepworth JT, Coull BM. Predictors of depressive symptoms among community-dwelling stroke survivors. J Cardiovasc Nurs. 2013;28(5):460–467. | ||
Babkair LA. Risk factors for poststroke depression: an integrative review. J Cardiovasc Nurs. 2017;49(2):73–84. | ||
Moylan S, Maes M, Wray NR, Berk M. The neuroprogressive nature of major depressive disorder: pathways to disease evolution and resistance, and therapeutic implications. Mol Psychiatry. May;18(5):595–606. | ||
Leonard B, Maes M. Mechanistic explanations how cell-mediated immune activation, inflammation and oxidative and nitrosative stress pathways and their sequels and concomitants play a role in the pathophysiology of unipolar depression. Neurosci Biobehav Rev. 2012;36(2):764–785. | ||
Tang CZ, Zhang YL, Wang WS, Li WG, Shi JP. Serum levels of high-sensitivity C-reactive protein at admission are more strongly associated with poststroke depression in acute ischemic stroke than homocysteine levels. Mol Neurobiol. 2016;53(4):2152–2160. | ||
Qian L, Ding L, Cheng L, et al. Early biomarkers for post-stroke cognitive impairment. J Neurol. Oct;259(10):2111–2118. | ||
Narasimhalu K, Lee J, Leong YL, et al. Inflammatory markers and their association with post stroke cognitive decline. Int J Stroke. 2015;10(4):513–518. | ||
Fang J, Cheng Q. Etiological mechanisms of post-stroke depression: a review. Neurol Res. 2009;31(9):904–909. | ||
Zhao J, Ren W, Lv D, Zhu Z, Wang Q, He J. Low triiodothyronine syndrome is a predictor of post-stroke depression. Int J Geriatr Psychiatry. 2017;32(3):352–353. | ||
Maes M. The cytokine hypothesis of depression: inflammation, oxidative & nitrosative stress (IO&NS) and leaky gut as new targets for adjunctive treatments in depression. Neuro Endocrinol Lett. 2008;29(3):287–291. | ||
Wen S, Cheng M, Wang H, et al. Serum uric acid levels and the clinical characteristics of depression. Clin Biochem. 2012;45(1):49–53. | ||
Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci U S A. 1981;78(11):6858–6862. | ||
Lucca G, Comim CM, Valvassori SS, et al. Increased oxidative stress in submitochondrial particles into the brain of rats submitted to the chronic mild stress paradigm. J Psychiatr Res. 2009;43(9):864–869. | ||
Wu CY, Hu HY, Chou YJ, et al. High serum uric acid levels are associated with all-cause and cardiovascular, but not cancer, mortality in elderly adults. J Am Geriatr Soc. 2015;63(9):1829–1836. | ||
Wang Z, Lin Y, Liu Y, et al. Serum uric acid levels and outcomes after acute ischemic stroke. Mol Neurobiol. 2016;53(3):1753–1759. | ||
Cherubini A, Polidori MC, Bregnocchi M, et al. Antioxidant profile and early outcome in stroke patients. Stroke. 2000;31(10):2295–2300. | ||
Kaster MP, Rosa AO, Santos AR, Rodrigues AL. Involvement of nitric oxide-cGMP pathway in the antidepressant-like effects of adenosine in the forced swimming test. Int J Neuropsychopharmacol. 2005;8(4):601–606. | ||
Tao R, Li H. High serum uric acid level in adolescent depressive patients. J Affect Disord. 2015;174:464–466. | ||
Wen S, Cheng M, Wang H, et al. Serum uric acid levels and the clinical characteristics of depression. Clin Biochem. 2012;45(1–2):49–53. | ||
Chaudhari K, Khanzode S, Khanzode S, Dakhale G, Saoji A, Sarode S. Clinical correlation of alteration of endogenous antioxidant-uric acid level in major depressive disorder. Indian J Clin Biochem. 2010;25(1):77–81. | ||
Stocker R, Yamamoto Y, McDonagh A, Glazer A, Ames B. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235(4792):1043–1046. | ||
Kimm H, Yun JE, Jo J, Jee SH. Low serum bilirubin level as an independent predictor of stroke incidence a prospective study in Korean men and women. Stroke. 2009;40(11):3422–3427. | ||
Targher G. Risk of ischemic stroke and decreased serum bilirubin levels: is there a causal link? Arterioscler Thromb Vasc Biol. 2014;34(4):702–704. | ||
Luo Y, Li J, Zhang J, Xu Y. Elevated bilirubin after acute ischemic stroke linked to the stroke severity. Int J Dev Neurosci. 2013;31(7):634–638. | ||
Pineda S, Bang OY, Saver JL, et al. Association of serum bilirubin with ischemic stroke outcomes. J Stroke Cerebrovasc Dis. 2008;17(3):147–152. | ||
Miyaoka T, Yasukawa R, Yasuda H, et al. Urinary excretion of biopyrrins, oxidative metabolites of bilirubin, increases in patients with psychiatric disorders. Eur Neuropsychopharmacol. 2005;15(3):249–252. | ||
Gordon WA, Hibbard MR. Poststroke depression: an examination of the literature. Arch Phys Med Rehabil. 1997;78(6):658–663. | ||
Robinson RG, Lipsey JR, Rao K, Price TR. Two-year longitudinal study of post-stroke mood disorders: comparison of acute-onset with delayed-onset depression. Am J Psychiatry. 1986;143(10):1238–1244. | ||
Kim JT, Park MS, Yoon GJ, et al. White matter hyperintensity as a factor associated with delayed mood disorders in patients with acute ischemic stroke. Eur Neurol. 2011;66(6):343–349. | ||
Brott T, Adams HP Jr, Olinger CP, et al. Measurements of acute cerebral infarction: a clinical examination scale. Stroke. 1989;20(7):864–870. | ||
Kunitz S, Gross C, Heyman A, et al. The pilot Stroke Data Bank: definition, design, and data. Stroke. 1984;15(4):740–746. | ||
Beck AT, Steer R, Brown G. Beck Depression Inventory Manual [M]. Psychological Corporation: San Antonio, TX; 1996. | ||
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington, DC: American Psychiatric Association; 1994. | ||
Nanetti L, Raffaelli F, Vignini A, et al. Oxidative stress in ischaemic stroke. Eur J Clin Invest. 2011;41(12):1318–1322. | ||
Palta P, Samuel LJ, Miller ER 3rd, Szanton SL. Depression and oxidative stress: results from a meta-analysis of observational studies. Psychosom Med. 2014;76(1):12–19. | ||
Nabavi SF, Dean OM, Turner A, Sureda A, Daglia M, Nabavi SM. Oxidative stress and post-stroke depression: possible therapeutic role of polyphenols? Curr Med Chem. 2015;22(3):343–351. | ||
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39(1):44–84. | ||
Girotti AW. Lipid hydroperoxide generation, turnover, and effector action in biological systems. J Lipid Res. 1998;39(8):1529–1542. | ||
Rawdin BJ, Mellon SH, Dhabhar FS, et al. Dysregulated relationship of inflammation and oxidative stress in major depression. Brain Behav Immun. 2013;31:143–152. | ||
Galecki P. Peripheral markers of inflammation, oxidative and nitrosative stress pathways and memory functions as a new target of pharmacotherapy in depression. Prog Neuropsychopharmacol Biol Psychiatry. 2018;80(Pt C):167. | ||
Doolin K, Farrell C, Tozzi L, Harkin A, Frodl T, O’Keane V. Diurnal hypothalamic-pituitary-adrenal axis measures and inflammatory marker correlates in major depressive disorder. Int J Mol Sci. 2017;18(10): pii: E2226. | ||
Chen Z, Jalabi W, Shpargel KB, et al. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci. 2012;32(34):11706–11715. | ||
Li W, Ling S, Yang Y, Hu Z, Davies H, Fang M. Systematic hypothesis for post-stroke depression caused inflammation and neurotransmission and resultant on possible treatments. Neuro Endocrinol Lett. 2014;35(2):104–109. | ||
Fujigaki H, Saito K, Fujigaki S, et al. The signal transducer and activator of transcription 1alpha and interferon regulatory factor 1 are not essential for the induction of indoleamine 2,3-dioxygenase by lipopolysaccharide: involvement of p38 mitogen-activated protein kinase and nuclear factor-kappaB pathways, and synergistic effect of several proinflammatory cytokines. J Biochem. 2006;139(4):655–662. | ||
Maes M, Mihaylova I, Kubera M, Uytterhoeven M, Vrydags N, Bosmans E. Lower whole blood glutathione peroxidase (GPX) activity in depression, but not in myalgic encephalomyelitis/chronic fatigue syndrome: another pathway that may be associated with coronary artery disease and neuroprogression in depression. Neuro Endocrinol Lett. 2011;32(2):133–140. | ||
Maes M, Galecki P, Chang YS, Berk M. A review on the oxidative and nitrosative stress (O&NS) pathways in major depression and their possible contribution to the (neuro)degenerative processes in that illness. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(3):676–692. | ||
Astrom M, Adolfsson R, Asplund K. Major depression in stroke patients: a 3-year longitudinal study. Stroke. 1993;24(7):976–982. | ||
Perlstein TS, Pande RL, Creager MA, Weuve J, Beckman JA. Serum total bilirubin level, prevalent stroke, and stroke outcomes: NHANES 1999–2004. Am J Med. 2008;121(9):781–788 e781. | ||
Cojocaru IM, Cojocaru M, Sapira V, Ionescu A. Evaluation of oxidative stress in patients with acute ischemic stroke. Rom J Intern Med. 2013;51(2):97–106. | ||
Ferro JM, Caeiro L, Figueira ML. Neuropsychiatric sequelae of stroke. Nat Rev Neurol. 2016;12(5):269–280. | ||
Berg A, Palomaki H, Lehtihalmes M, Lonnqvist J, Kaste M. Poststroke depression: an 18-month follow-up. Stroke. 2003;34(1):138–143. | ||
Vandeputte M, de Weerd A. Sleep disorders and depressive feelings: a global survey with the Beck Depression Scale. Sleep Med. 2003;4(4):343–345. | ||
Gao J, Lin M, Zhao J, Bi S, Ni Z, Shang X. Different interventions for post-ischaemic stroke depression in different time periods: a single-blind randomized controlled trial with stratification by time after stroke. Clin Rehabil. 2017;31(1):71–81. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.