")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Changes in the number of CD31−CD45−Sca-1+ cells and Shh signaling pathway involvement in the lungs of mice with emphysema and relevant effects of acute adenovirus infection
Authors Deng MH , Li JH, Gan Y, Chen Y, Chen P
Received 9 December 2016
Accepted for publication 30 January 2017
Published 14 March 2017 Volume 2017:12 Pages 861—872
DOI https://doi.org/10.2147/COPD.S129871
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Minhua Deng,1,2 Jinhua Li,2 Ye Gan,3 Yan Chen,2 Ping Chen2
1Respiratory Medicine Department, PLA Rocket Force General Hospital, Beijing, 2Respiratory Medicine Department, 3Rehabilitation Department, Second Xiangya Hospital, Central South University, Changsha, Hunan Province, People’s Republic of China
Background: COPD is a leading cause of mortality worldwide, and cigarette smoke is a pivotal risk factor. Adenovirus is a common cause of acute exacerbations of COPD and expedites COPD progression. Lung stem/progenitor cells play an important role in the development of COPD, while the relevant mechanism remains elusive. Here, we investigated the number of lung CD31-CD45-Sca-1+ cells and sonic hedgehog (Shh) signaling pathway expression levels in cigarette smoke extract (CSE)-induced emphysema mice, as well as the relevant effects of acute adenovirus infection (AAI).
Materials and methods: BALB/c mice were treated with CSE by intraperitoneal injection and/or adenovirus endotracheal instillation at different time points for 28 days. Lung function, lung histomorphology, CD31-CD45-Sca-1+ cell count, and expression levels of major components in the Shh signaling pathway in the lungs were measured.
Results: CSE intraperitoneal injection and adenovirus endotracheal instillation successfully induced emphysema and AAI in mice, respectively. In the lungs of emphysema mice, both the number of CD31-CD45-Sca-1+ cells and expression levels of Shh signaling pathway molecules were reduced. However, AAI increased the number of inhibited CD31-CD45-Sca-1+ cells and activated the suppression of the Shh signaling pathway.
Conclusion: Both CD31-CD45-Sca-1+ cell numbers and Shh signaling pathway expression levels were downregulated in the lungs of emphysema mice induced by CSE intraperitoneal injection, which likely contributes to the pathogenesis of emphysema. Additionally, these inhibited lung CD31-CD45-Sca-1+ cells and Shh signaling pathway molecules were upregulated during AAI, indicating that they play a protective role in the epithelial repair process after AAI injury.
Keywords: emphysema, stem cells, lung CD31-CD45-Sca-1+ cells, sonic hedgehog signaling pathway, adenovirus
Introduction
COPD is a common preventable and treatable disease characterized by persistent airflow limitation that is typically progressive1 and is expected to be the third leading cause of mortality globally by 2020.2 Cigarette smoke (CS) is a pivotal COPD risk factor. A major form of COPD is emphysema (parenchymal destruction). Pathologically, COPD mainly includes chronic inflammation, mucosa metaplasia, alveolar destruction, and parenchyma cell apoptosis, which are associated with chronic and repetitive injury and aberrant repair of pulmonary epithelial cells.3,4
Lung stem/progenitor cells contribute to epithelial maintenance and injury repair. In general, stem cells exhibit a broader capacity of differentiation and higher potential for longer term renewal than progenitor cells. Accumulating evidence has shown that different populations of lung stem/progenitor cells occupy different niches and function in a region-specific manner in homeostasis and injury repair.5 Bronchioalveolar stem cells (BASCs) reside in the bronchioalveolar duct junction. In vitro studies have revealed that BASCs can self-renew over multiple passages and generate both bronchiolar and alveolar lineages.6 BASCs can be enriched by sorting cells positive for the expression of the stem cell marker Sca-1 and negative for endothelial and hematopoietic cell markers (CD31−CD45−Sca-1+). Studies have also demonstrated that the marker profile of CD31−CD45−Sca-1+ can be utilized to select tumor-propagating cells in murine lung cancer. Lung CD31−CD45−Sca-1+ cells contribute to regeneration and repair following naphthalene bronchiolar injury or bleomycin alveolar injury.6–8 These findings suggest that lung CD31−CD45−Sca-1+ cells play key roles in epithelial repair.
The sonic hedgehog (Shh) signaling pathway plays a critical role in stem cell maintenance, likely by stimulating stem cell proliferation.9 The canonical process of this pathway is that when Shh is secreted and reaches its target cell, it binds to the transmembrane protein Patched (Ptch1), relives repression of the transmembrane protein Smoothened (Smo) by Ptch1, induces the activity of three Gli transcription factors (Gli1, 2, 3), and eventually regulates transcription. Gli1 is a transcriptional activator and Gli3 is a repressor, whereas Gli2 has both activating and repressing effects. The Shh signaling pathway is important not only in embryonic lung development and branching morphogenesis but also in tumorigenesis and regeneration and repair after injury to adult lungs.10–12 Additionally, the expression of Shh pathway members, including Shh, Ptch1, and Gli2, is significantly decreased in lung tissues from COPD patients compared to that in control smokers with normal lung function.13 This suggests that the Shh pathway is involved in COPD pathogenesis and may be related to lung stem cells.
Acute exacerbations of COPD (AECOPD) are associated with high morbidity and mortality, increased health care expenditures, reduced health status, and expedited disease progression among COPD patients.14 Up to 60% of AECOPD cases are triggered by respiratory viruses; these AECOPD cases tend to be more severe, require longer recovery times, and increase the risk of hospitalization.15 Recently, researchers have given increasing attention to the initiation and maintenance of proper and active repair responses, particularly those related to stem cell activation, rather than passive inhibition of inflammation during and after respiratory virus infection.16 Adenovirus is a common respiratory virus that causes AECOPD. Latent adenovirus infections during which the adenoviral genome constantly expresses protein in the absence of replicating virus have been suggested to be involved in COPD pathogenesis, probably by upregulating the inflammation induced by smoke and promoting the process of airway remodeling.17 However, CD31−CD45−Sca-1+ cell numbers and Shh signaling pathway expression levels in emphysema and the epithelial repair process of COPD during acute adenovirus infection (AAI) have not been investigated.
In this study, we established a CS extract (CSE)-induced emphysema mouse model18,19 that can be used to investigate the pathobiological mechanisms in COPD patients to explore the number of lung CD31−CD45−Sca-1+ cells and Shh signaling pathway expression levels in emphysema mice. Our data indicate that both lung CD31−CD45−Sca-1+ cell numbers and Shh signaling pathway expression levels were downregulated in this emphysema mouse model. Moreover, lung CD31−CD45−Sca-1+ cell numbers were increased, and Shh signaling pathway was stimulated in emphysema mice with AAI.
Materials and methods
Animals
Six- to eight-week-old male inbred BALB/C mice (20–22 g) were obtained from SJA Laboratories Animal (Hunan, China). The animals were housed and fed at the Xiangya Hospital BioResources Centre under a quiet and controlled condition with temperature between 21°C and 22°C and humidity between 50% and 60% in a 12:12-h light–dark cycle.
Mouse model establishment
The emphysema mouse model induced by CSE intraperitoneal injection was established as previously described.18,19 Five cigarettes (China Tobacco Hunan Industrial Co. Ltd: tar, 12 mg; nicotine, 1.1 mg; carbon monoxide, 14 mg) were burned, and the smoke was collected into 10 mL phosphate-buffered saline (PBS) using a vacuum pump at negative 300 mmHg. The CSE/PBS adjusted to pH 7.2–7.4 was freshly prepared and filtered through a 0.22 μm filter. The adenovirus containing a gene encoding red fluorescent protein (RFP) was purchased from Invitrogen (Carlsbad, CA, USA). Mice were randomized into three groups (n=20 per group): PBS group (intraperitoneal injection of 0.3 mL PBS at days 1, 11, and 22 and endotracheal instillation at day 15, respectively), CSE group (intraperitoneal injection of 0.3 mL CSE at days 1, 11, and 22 and endotracheal instillation of 0.3 mL PBS on day 15), and CSE + adenovirus group (intraperitoneal injection of 0.3 mL CSE at days 1, 11, and 22 and endotracheal instillation of 0.3 mL adenovirus [1010 ifu/mL] at day 15). One mouse from the CSE group and one mouse from the CSE + adenovirus group died within 2 min after intraperitoneal injection of CSE. One mouse from the CSE + adenovirus group died immediately after anesthesia with intraperitoneal injection of 10% chlorine hydrate. At day 21, two mice were randomly selected from each group and sacrificed. Their lung tissues were processed into frozen sections (6 mm), and then RFP expression levels were determined to examine whether adenovirus had successfully infected the lung tissues. The remaining mice were sacrificed at day 28. The experimental protocol was approved by the ethics committee of Xiangya Hospital, Central South University, and all animal care and procedures were performed according to the recommendations in the Guide for the Care and Use of Laboratory Animals.
Lung function
Ten, nine, and eight mice were randomly selected from the PBS group, CSE group, and CSE + adenovirus group, respectively, to measure lung function at day 28. The number of mice from each group varied because one mouse in the CSE group and two in the CSE + adenovirus group died during the study. A plethysmograph chamber (Buxco Respiratory Products, Wilmington, NC, USA) was utilized to evaluate the frequency (bpm), tidal volume (mL), dynamic compliance (Cdyn, mL/cmH2O), and airway resistance (RI, cmH2O mL−1·min−1) in the mice.
Lung tissue sample preparation
After assessing lung function, the left lung tissues of the mouse were inflated with 10% formalin at a constant pressure of 25 cmH2O, allowing the homogenous expansion of lung parenchyma. Next, the left lung tissues were removed and immersed in 10% formalin for 24-h fixation, followed by paraffin embedding using standard procedures. The paraffin sections (5 μm) were prepared and used for subsequent histopathological and immunohistochemical studies. The right lung tissues were stored in liquid nitrogen. The right middle lobes and right lower lobes were used for Western blotting analysis and real-time quantitative reverse transcription polymerase chain reaction (RT-PCR) analysis, respectively.
Flow cytometry quantification of lung CD31−CD45−Sca-1+ cells
At day 28, the remaining mice (n=8 per group) were anesthetized with intraperitoneal injection of 10% chlorine hydrate (3 mL/kg), followed by right atrium perfusion with 10 mL PBS and endotracheal instillation of 3 mL collagenase IV (150 U/mL; Sigma-Aldrich, St Louis, MO, USA) and 0.3 mL 1% low melting point agarose. After 2–3 min, whole lung tissues were removed and 1 mL collagenase IV was added. After incubation at 37°C for 1 h, lung tissues were minced in a Petri dish and then washed with DNase I (20 U/μL; Roche, Basel, Switzerland). The lung emulsion mixed with the used washing buffer was again transferred to collagenase IV solution, incubated at 37°C for 15 min, filtered through 100 μm, 70 μm, and 40 μm cell strainers (BD Biosciences, Franklin Lakes, NJ, USA), and centrifuged at 300× g at 4°C for 5 min. Cells were resuspended in flow cytometry buffer (2% fetal bovine serum, 1 mM ethylene diamine tetraacetic acid, 0.01% NaN3 in PBS) at 1×106/100 μL and incubated at 4°C for 30 min with CD31-APC (eBioscience, San Diego, CA, USA; 0.2 μg/μL), CD45-APC (0.2 μg/μL; eBioscience), and Sca-1-fluorescein isothiocyanate (0.5 μg/μL; eBioscience). Flow cytometry was performed using a BD FACSCanto II flow cytometry machine. Data were analyzed with FlowJo 8.7.3 (FlowJo, Ashland, OR, USA) software.
Histological examination
The extent of alveolar destruction was assessed by measuring the mean linear intercept (MLI) and destructive index (DI) as previously described, and pulmonary emphysema was semi-quantitatively evaluated.18 Peribronchial inflammation was estimated by a semi-quantitative score calculated from the number of peribronchial inflammatory cells.20 Three sections per mouse and >20 bronchi per section were randomly selected at high magnification. A value of 0 was scored when no inflammation was detected, a value of 1 when occasional inflammatory cells were detected, a value of 2 when the bronchus was enclosed by one to five layers of inflammatory cells, and a value of 3 when the bronchus was enclosed by more than five layers of inflammatory cells. The semi-quantitative score was equal to the ratio of the total value of all assessed bronchi over the number of assessed bronchi. The extent of perivascular inflammation was assessed similarly. All assessment calculations were performed blindly by two pathologists from the Second Xiangya Hospital.
Real-time RT-PCR for messenger RNA (mRNA) expression of Shh, Ptch1, and Gli1
Total RNA from lung tissues was prepared using RNAiso Plus reagent (Takara, Shiga, Japan). The complementary DNAs (cDNAs) were synthesized using the PrimeScript™ RT reagent kit with gDNA Eraser (Perfect Real Time) (Takara). Real-time quantitative PCR was performed using SYBR® Premix Ex Taq™ II (Takara) on a CFX96™ PCR machine (Bio-Rad, Hercules, CA, USA). All procedures were conducted according to the manufacturer’s instructions. The results were normalized against the housekeeping gene β-actin in the same sample. Primers used included: Shh(+), 5′-GTTTATTCCCAACGTAGCCGAGA-3′; Shh(−), 5′-CAGAGATGGCCAAGGCATTTA-3′; Ptch1(+), 5′-CGAGACAAGCCCATCGACATTA-3′; Ptch1(−), 5′-AGGGTCGTTGCTGACCCAAG-3′; Gli1(+), 5′-TGAGCATTATGGACAAGTGCAGGTA-3′; Gli1(−), 5′-ATTGAGGCAGGGTGCCAATC-3′; β-actin(+), 5′-CATCCGTAAAGACCTCTATGCCAAC-3′; β-actin(−), 5′-ATGGAGCCACCGATCCACA-3′. Each experiment was performed twice in triplicate.
Western blotting detection of Shh, Ptch1, and Gli1
Lung tissues were homogenized and lysed in 250 μL of 2× sodium dodecyl sulfate (SDS) loading buffer (62.5 mM Tris–HCl, pH 6.8, 2% SDS, 25% glycerol, 0.01% bromophenol blue, 5% 2-mercaptoethanol) for 10 min at 95°C. Equal amounts of proteins from each sample were separated on a 10% SDS-polyacrylamide gel and then transferred on to a polyvinylidene difluoride microporous membrane (Millipore, Billerica, MA, USA). The membrane was incubated with one of the following primary antibodies for 1 h at 25°C: rabbit anti-Shh polyclonal antibody (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA), goat anti-Ptch1 polyclonal antibody (1:500; Santa Cruz Biotechnology); rabbit anti-Gli1 polyclonal antibody (1:400; Santa Cruz Biotechnology), and rabbit anti-actin monoclonal antibody (1:5000; Santa Cruz Biotechnology), followed by washing and incubation with horseradish peroxidase-conjugated goat anti-rabbit and donkey anti-goat IgG (1:2000; Santa Cruz Biotechnology) for 2 h at 25°C. Protein bands were detected using the GE Healthcare ECL kit (Little Chalfont, UK).
Immunohistochemistry for localization and expression of Shh, Ptch1, and Gli1
Lung sections were incubated for 18 h at 4°C with one of the following primary antibodies: anti-Shh (1:200; Santa Cruz Biotechnology), anti-Ptch1 (1:100; Santa Cruz Biotechnology), and anti-Gli1 (1:200; Santa Cruz Biotechnology). The secondary biotinylated anti-immunoglobulin antibody and horseradish peroxidase-conjugated streptavidin were then sequentially added and detected using the Polink-2 HRP Plus Rabbit Detection System (Beijing Zhongshan Goldebridge Biotechnology Co., Ltd, Beijing, China). Brown 3,3′-diaminobenzidine (DAB) staining (DAB Detection Kit; Beijing Zhongshan Goldebridge Biotechnology Co., Ltd) indicated the presence of Shh/Ptch1/Gli1-positive cells in the membrane/cytoplasm/nuclei. In negative control slides, the primary antibody was replaced with PBS. Three different sections were used from each lung specimen, and five random fields were selected from each section. Optical density (OD) per area was analyzed using Image-Pro Plus 6.0 system (Media Cybernetics, Inc., Rockville, MD, USA). OD calculations were performed blindly by two pathologists from the Second Xiangya Hospital.
Statistical analysis
Results are expressed as the mean ± standard deviation. Variances among the three groups were assessed using one-way analysis of variance. A P-value <0.05 was considered statistically significant. Data were analyzed using SPSS version 18.0 for Windows (SPSS Inc., Chicago, IL, USA).
Results
Expression of RFP
Adenovirus contained a gene encoding RFP. The lungs of two mice from each group were excised to detect RFP expression 6 days post-adenovirus infections. As illustrated in Figure 1, frozen sections showing red fluorescence with relatively homogeneous distribution under fluorescence microscope were only observed in lung tissues in the CSE + adenovirus group, suggesting that adenoviruses successfully infected the lung tissues in the CSE + adenovirus group.
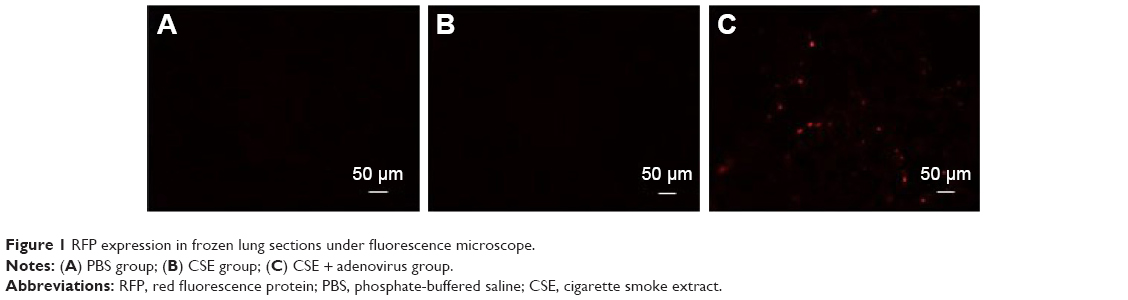 | Figure 1 RFP expression in frozen lung sections under fluorescence microscope. |
Lung function
Compared to the PBS group, CSE intraperitoneal injection had significantly adverse impacts on Cdyn and RI, which were not statistically affected by AAI (Table 1).
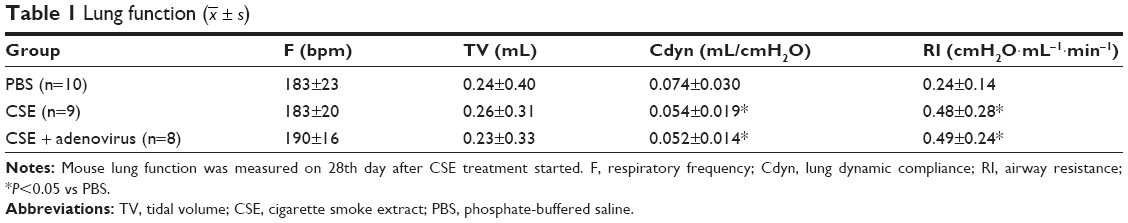 | Table 1 Lung function ( |
 ± s)
± s)Morphological observations
Intraperitoneal injection of CSE in mice caused emphysematous lung destruction within 28 days. Compared to the normal alveolar architecture (PBS group; Figure 2A), enlargement of alveolar air spaces and destruction of lung parenchyma were observed in the CSE group (Figure 2B) and CSE + adenovirus group (Figure 2C). The MLI and DI were also significantly increased following CSE exposure compared to the PBS group, but AAI had no significant influence (Table 2). To assess the extent of lung tissue inflammation, we calculated the semi-quantitative inflammatory scores in peribronchial and perivascular sections (Figures 3 and 4). The peribronchial semi-quantitative inflammatory score in CSE + adenovirus group was higher than that in the CSE group (1.81±0.30 vs 0.18±0.08, P<0.05; Figure 4A), and there were no significant differences in scores between the CSE group and PBS group (0.18±0.08 vs 0.12±0.15, P>0.05; Figure 4A). Similar results were observed for perivascular semi-quantitative inflammatory scores (Figure 4B). Therefore, acute lung inflammation only occurred in the CSE + adenovirus group.
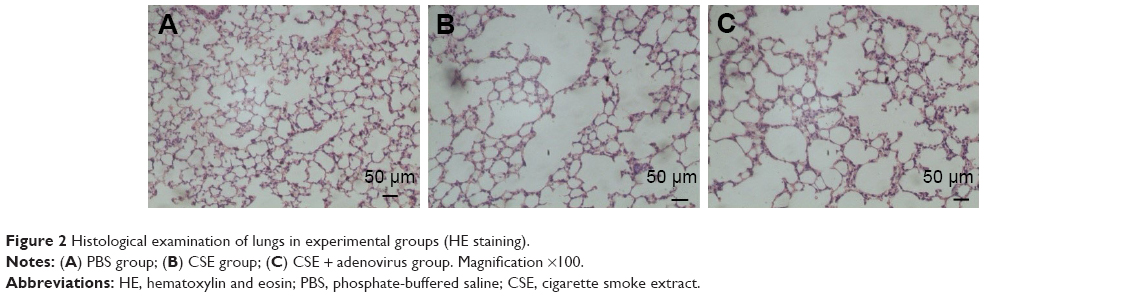 | Figure 2 Histological examination of lungs in experimental groups (HE staining). |
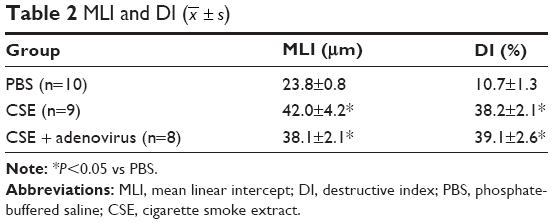 | Table 2 MLI and DI ( |
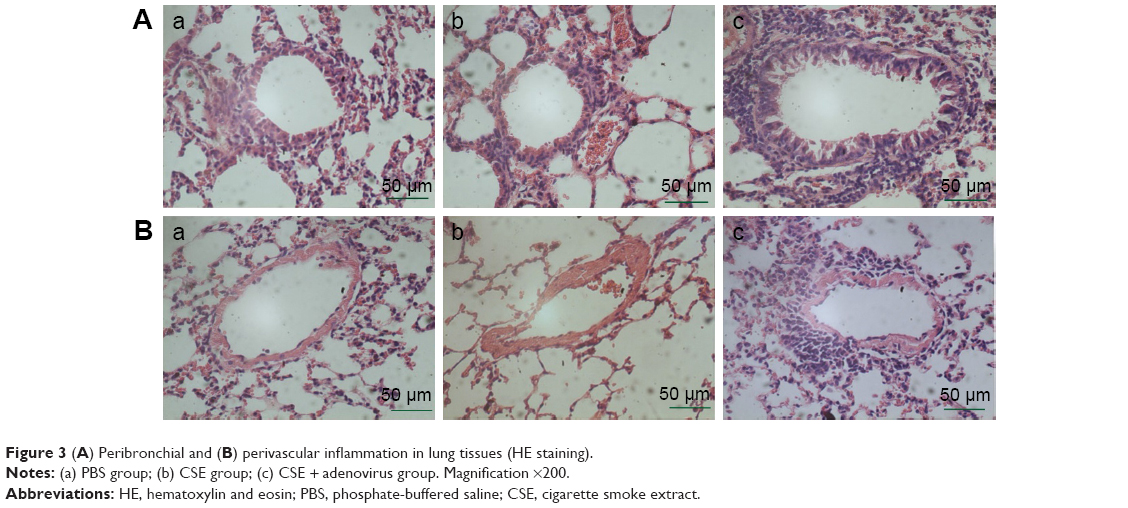 | Figure 3 (A) Peribronchial and (B) perivascular inflammation in lung tissues (HE staining). |
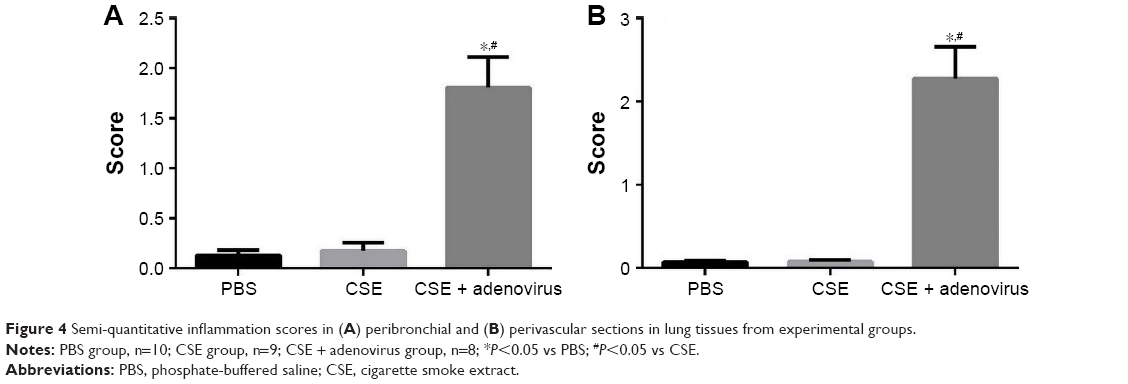 | Figure 4 Semi-quantitative inflammation scores in (A) peribronchial and (B) perivascular sections in lung tissues from experimental groups. |
Quantification of lung CD31−CD45−Sca-1+ cells
We determined the percentage of lung CD31−CD45−Sca-1+ cells in the single-cell suspensions of the whole lung (Figures 5 and 6). The plot revealed clear autofluorescence in the lung tissue (Figure 5), consistent with the report by Ikeda et al.21 As illustrated in Figure 6, the percentage of lung CD31−CD45−Sca-1+ cells was lower in the CSE group compared to the PBS group (0.87%±0.16% vs 1.26%±0.18%, P<0.05) but higher in the CSE + adenovirus group compared to the CSE group (1.30%±0.34% vs 0.87%±0.16%, P<0.05).
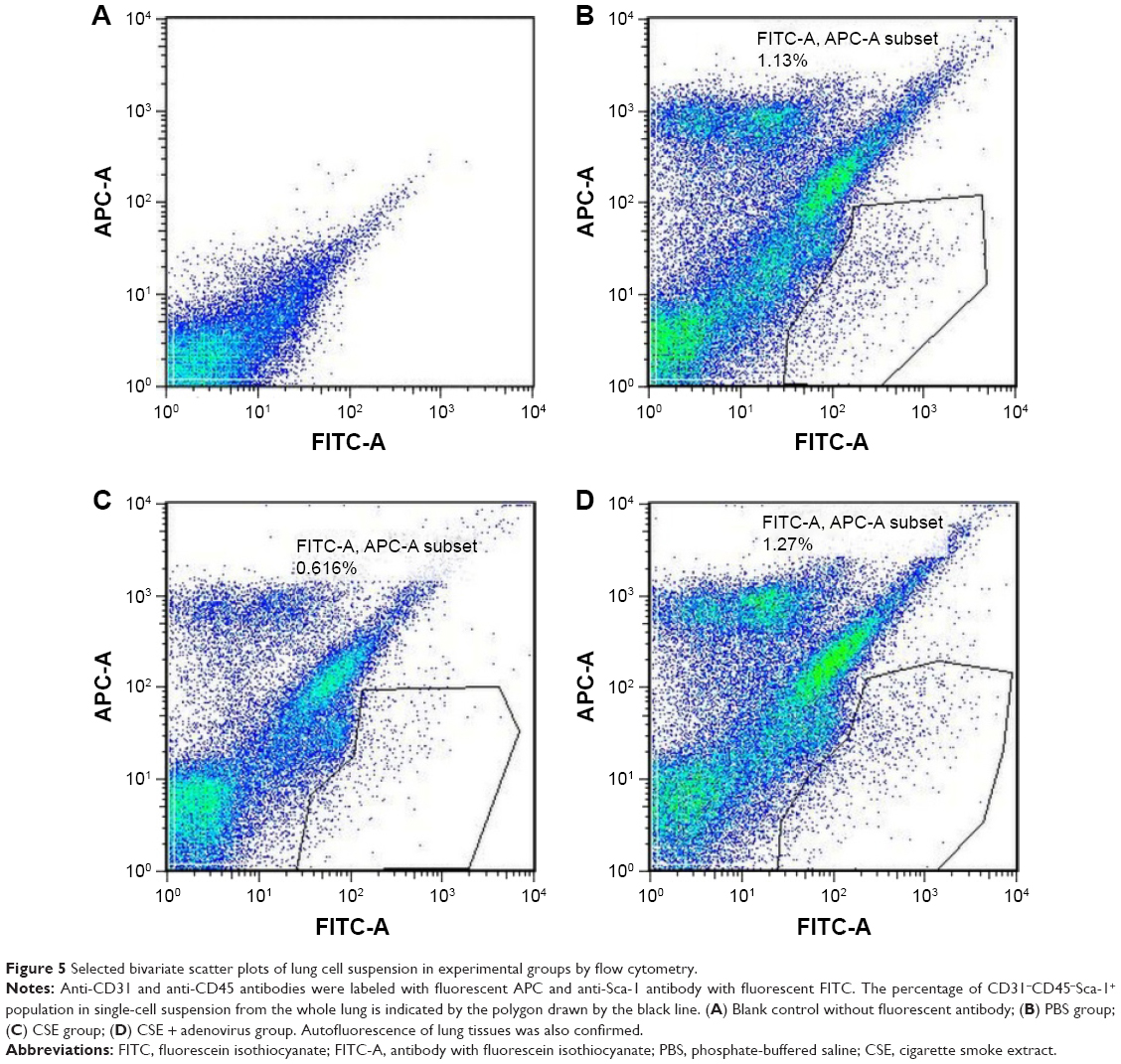 | Figure 5 Selected bivariate scatter plots of lung cell suspension in experimental groups by flow cytometry. |
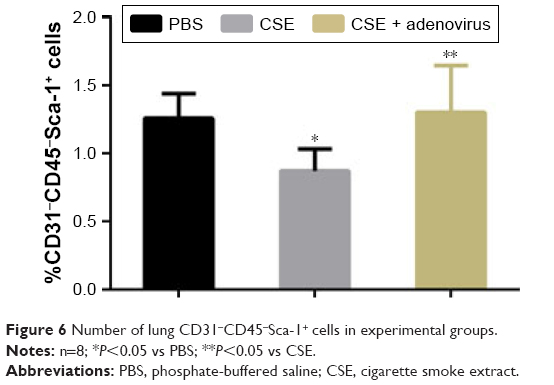 | Figure 6 Number of lung CD31−CD45−Sca-1+ cells in experimental groups. |
mRNA and protein levels of the major Shh signaling pathway components
The gene expression levels of Shh, Ptch1 (receptor of Shh), and Gli1 (transcriptional activator) in lung tissues were determined using real-time RT-PCR. As shown in Figure 7, there were no significant differences in their mRNA levels between the CSE group and PBS group; however, their levels were upregulated in the CSE + adenovirus group compared to the CSE group. Quantitative Western blotting results demonstrated that the protein levels of Shh, Ptch1, and Gli1 were decreased in the CSE group compared to the PBS group (0.28±0.15 vs 0.52±0.02, 0.12±0.05 vs 0.36±0.05, 0.17±0.04 vs 0.45±0.11, P<0.05; Figure 8) but increased in the CSE + adenovirus group compared to the CSE group (0.73±0.16 vs 0.28±0.15, 0.62±0.08 vs 0.12±0.05, 0.56±0.22 vs 0.17±0.04, P<0.05; Figure 8).
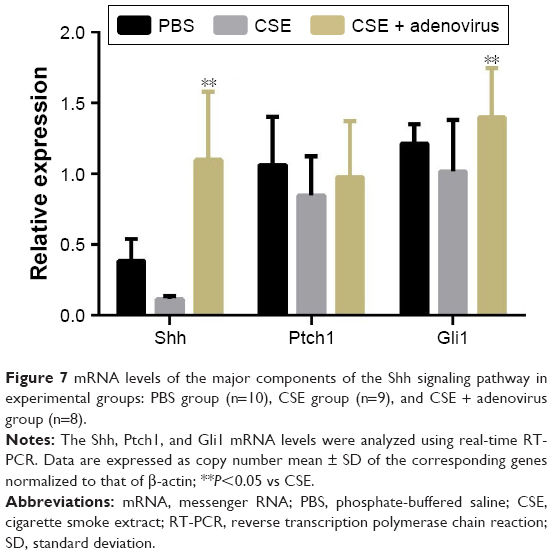 | Figure 7 mRNA levels of the major components of the Shh signaling pathway in experimental groups: PBS group (n=10), CSE group (n=9), and CSE + adenovirus group (n=8). |
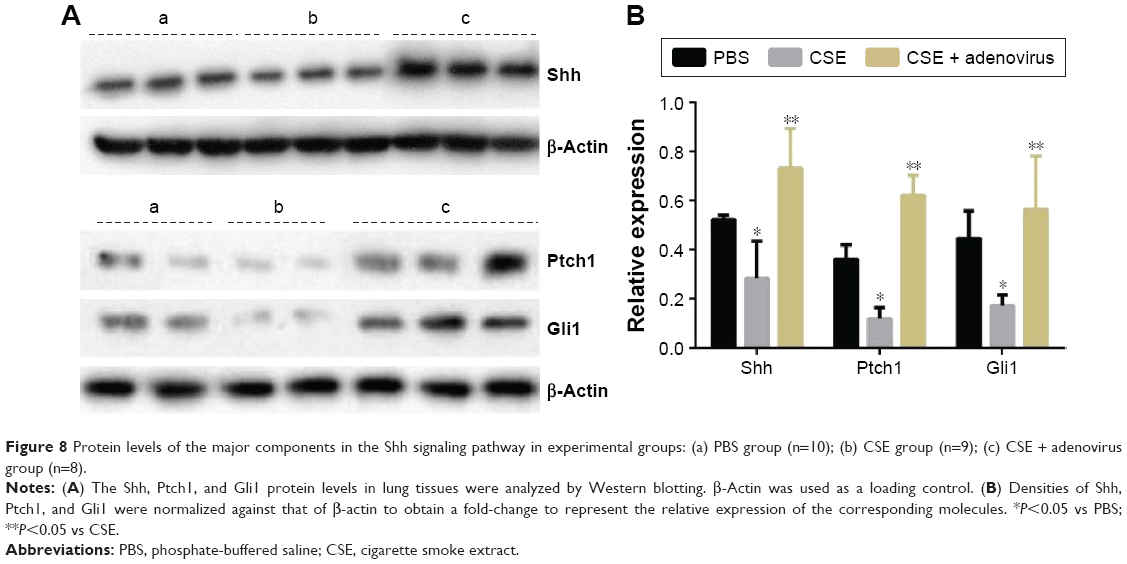 | Figure 8 Protein levels of the major components in the Shh signaling pathway in experimental groups: (a) PBS group (n=10); (b) CSE group (n=9); (c) CSE + adenovirus group (n=8). |
Immunohistochemical detection of Shh signaling pathway molecules
Immunohistochemical staining revealed that Shh, Ptch1, and Gli1 were mainly expressed in bronchioalveolar, bronchiolar, and bronchial epithelial cells and occasionally in alveolar epithelial cells (Figure 9). Shh and Ptch1 were subcellularly localized to the cell membrane and cytoplasm, whereas Gli1 was localized to the cytoplasm and nuclei. Gli1 was predominantly present in the nucleus in the CSE + adenovirus group (Figure 9). OD analysis showed that Gli1 levels were decreased in the CSE group compared to the PBS group; rather, the levels of Shh, Ptch1, and Gli1 were increased in the CSE + adenovirus group compared to the CSE group (P<0.05; Figure 10).
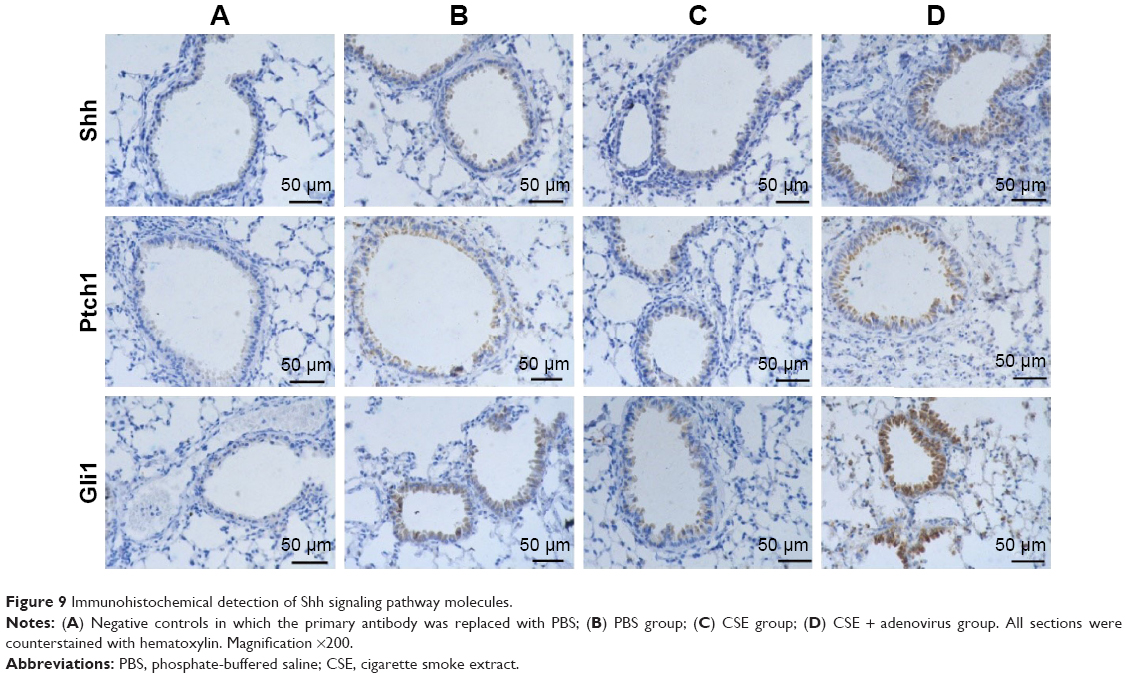 | Figure 9 Immunohistochemical detection of Shh signaling pathway molecules. |
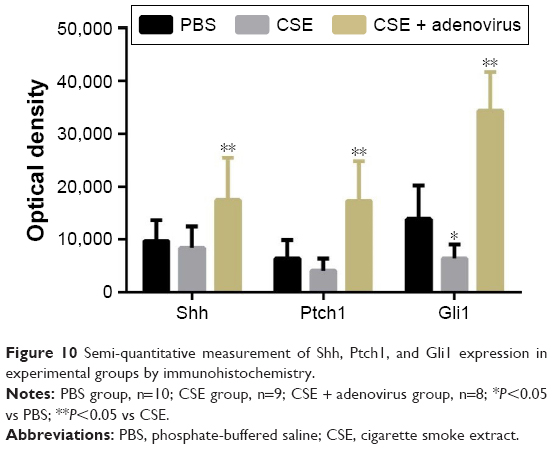 | Figure 10 Semi-quantitative measurement of Shh, Ptch1, and Gli1 expression in experimental groups by immunohistochemistry. |
Discussion
In this study, intraperitoneal administration of CSE caused enlargement of alveolar air spaces, destruction of lung parenchyma, airflow limitation, and decrease in lung Cdyn in mice, confirming the efficiency and efficacy of intraperitoneal CSE injection for establishing a smoking-related rodent emphysema model.18,19,22 Respiratory viral infections target epithelial cells of the lung and are major causes of COPD exacerbation. Adenovirus is one of the most common viruses causing AECOPD. In this study, red fluorescence was only observed in lungs of CSE + adenovirus group, indicating that adenovirus successfully infected mice with emphysema by endotracheal instillation. Adenovirus infections triggered acute inflammation in the lung tissues of CSE-exposed mice, which was in accordance with clinical manifestations of patients with AECOPD. Foronjy et al23 reported that respiratory syncytial viral infections accentuated alveolar destruction in CS-exposed mice. In their study, respiratory syncytial virus was administrated monthly for 6 months, and CS exposure was conducted for 4 h per day, 5 days per week for 6 months. However, adenovirus infections failed to significantly enhance alveolar destruction and lung function in our CSE-exposed mouse model. This may be because the frequency of adenovirus administration and viral load were insufficient to cause significant changes in alveolar destruction and lung function. Additionally, this finding illustrated that lung repair was initiated after AAI. In this study, we analyzed lung CD31−CD45−Sca-1+ cells and Shh signaling pathway expression in our emphysema mouse model and evaluated the effects of AAI in these mice.
Cells expressing CD31−CD45−Sca-1+ were originally isolated using fluorescence-activated cell sorting and identified as BASCs.6 However, the exact phenotype of BASCs remains controversial. Some studies demonstrated that CD31−CD45−Sca-1+ cells resided in side populations, which are considered to possess stem cell characteristics and comprise a heterogeneous population of progenitors.24,25 Later, CD34 served as a positive selection marker for BASCs; however, different antibodies obtained from different companies showed significant variability in the cell populations enriched using this marker. Nevertheless, CD31−CD45−Sca-1+ cells at least comprise epithelial stem/progenitor cells and can be used to reflect the epithelial regeneration and repair process to some extent. Our study revealed that the number of lung CD31−CD45−Sca-1+ cells was reduced in emphysema mice, which is consistent with another finding of our group, indicating decreased Sca-1 gene and protein expression in the lung tissues (unpublished data). This result suggests that the repair capacity of lung CD31−CD45−Sca-1+ cells was impaired in emphysema mice, which may be partially attributable to the aberrant repair process of emphysema/COPD. Recent studies supported that the primary cause of COPD was lung injury, most commonly from CS.1 Apart from damaging lung structure, CS inhibits repair processes. CS exposure may adversely affect the chemotaxis, proliferation, and extracellular matrix production of lung fibroblasts,26 inhibit attachment and migration of airway epithelial cells,27 and negatively impact the proliferation of endothelial progenitor cells.28 Additionally, CS broadly perturbs the metabolism of airway basal stem/progenitor cells (BCs).29 Increasing evidence suggests that smoking plays a critical role in reprogramming of the airway BC phenotype and function.30 An ex vivo experiment showed that BCs from COPD smokers had a diminished capacity to regenerate a fully differentiated airway epithelium.31 These findings indicate that CSE is involved in reducing the number of lung CD31−CD45−Sca-1+ cells and further adversely influences their repair responses.
In addition, we found that AAI could still upregulate inhibited lung CD31−CD45−Sca-1+ cells in emphysema mice. AAI can damage lung epithelial cells through cytopathic effects resulting from the direct infection and/or host-mediated soluble and cell-associated pro-inflammatory mediators produced by both innate and adaptive immune cells. Lung tissues possess a specific repair capacity, and different lung stem/progenitor cells may have different roles in the injury and repair response depending on the exact nature or extent of injury to maintain homeostasis. Two recent studies reported that induced Krt5+ stem/progenitor cells emerged or expanded to participate in the repair process of alveolar and bronchiolar epithelium after severe influenza virus infection in the mouse lung.32,33 Collectively, the inhibited lung CD31−CD45−Sca-1+ cells in emphysema mice may have been triggered to proliferate after AAI, contributing to the repair process. As described earlier, this may partially explain why AAI did not aggravate lung function in CSE-exposed mice in our study.
The Shh signaling pathway has well-established roles in directing the patterning of embryonic tissues and structures and was recently implicated in the maintenance of stem/progenitor cells.9 The Shh signaling pathway plays an important role in ventricular epicardium regeneration in response to cardiac damage as well as adult liver repair and regeneration after liver injury.34,35 The Shh pathway was also shown to be involved in regeneration and repair after injury to lung tissues. Extensive activation of the Shh signaling pathway of the airway epithelium was reported in mice subjected to unilateral pneumonectomy or airway naphthalene injury.11,12 Overexpression of Shh can promote lung epithelial cell proliferation and alleviate lung injury.36,37 To determine the relationship between the canonical Shh signaling pathway and emphysema, we investigated the expression levels of the relevant molecules in this pathway in emphysema mice. There was a little discrepancy between the mRNA and protein levels of Shh, Ptch1, and Gli1 in lung tissues. This may be because the mRNAs of Shh, Ptch1, and Gli1 were posttranscriptionally modified, affecting their stability and translation activity. Differences in Shh and Ptch1 protein levels between immunohistochemistry and Western blot analysis were also observed, which might have resulted from a lower sensitivity of the former technique than the latter. Our findings indicate significant suppression of the Shh signaling pathway in emphysema mice induced by intraperitoneal CSE injection. Hedgehog interacting protein (Hhip) is known to competitively bind with Shh protein to repress the Shh pathway. Hhip is also necessary for lung branching.38 Recently, genome-wide association studies found that Hhip is a genetic risk factor for COPD.39,40 Gene expression of Hhip in lung tissues from patients with COPD was reduced by ~32% compared to ex-smokers with normal lung function.39 More importantly, mice with Hhip haploinsufficiency (Hhip±) are highly susceptible to not only more severe airspace enlargement induced by CS but also age-related emphysema compared to Hhip+/+ mice.41,42 Thus, Hhip plays a critical role in COPD pathogenesis. However, the underlying mechanism and whether the mechanism is linked to the canonical Shh signaling pathway remain unclear. A significant relationship between Hhip and the canonical Shh signaling pathway failed to reveal that Hhip protected against age-related emphysema in mice.42 However, the expression of Gli1 and Gli2 was increased in CS-exposed Hhip± mice compared to that in CS-exposed Hhip+/+ mice.41 Zhou et al13 demonstrated that the major canonical Shh signaling pathway components, including Shh, Ptch1, Gli1, Gli2, and Gli3, were significantly reduced in patients with COPD compared to those in control smokers, which was consistent with our results. We speculate that the decreased canonical Shh pathway activity in lung tissues of our emphysema mouse models is primarily mediated via an Hhip-independent mechanism, but this requires further comprehensive investigation. A recent study reported that overexpression of Shh resulted in an increased number of lung CD31−CD45−Sca-1+CD34+ cells, which were successfully engrafted in the lung by endotracheal administration.12 Therefore, inhibition of the Shh pathway may be associated with the aberrant repair of epithelial cells and decreased number of lung CD31−CD45−Sca-1+ cells in emphysema mice.
Similar to the effect on the reduced lung CD31−CD45−Sca-1+ cells, AAI resulted in widespread activation of the airway intraepithelial Shh signaling pathway in our study. Activation of the Shh signaling pathway can result from sustained increased endogenous Shh expression (ligand-dependent), mutations in Patched and Smo (ligand-independent), or other infrequent mechanisms such as epigenetic modification.43 Our results suggest that AAI-induced Shh expression was likely responsible for Shh pathway activation. Whether the overexpression of Shh ligand is sufficient for pathway activation requires further analysis. We hypothesized that AAI upregulated Shh expression to activate the Shh signaling pathway in lung epithelial compartments, which then promoted the proliferation of lung epithelial cell and/or lung CD31−CD45−Sca-1+ cells to repair the AAI injury in the lung tissues of emphysema mice. Whether Hhip takes part in this process should be further evaluated.
Conclusion
Our findings demonstrated that both the number of lung CD31−CD45−Sca-1+ cells and the expression levels of the Shh signaling pathway were downregulated in the lung tissues of emphysema mice induced by intraperitoneal CSE injection, likely contributing to the pathogenesis of emphysema. Additionally, the inhibited lung CD31−CD45−Sca-1+ cells and Shh signaling pathway were reactivated during AAI, suggesting a possible protective role in the epithelial repair process after AAI injury. Our results provide insight into the development of CSE-mediated COPD and AECOPD caused by AAI.
Acknowledgment
This work was supported by the China National Science Foundation (Nos 81200026, 81370143, 81270100).
Author contributions
Minhua Deng contributed to the study design and most of the research work. Jinhua Li contributed to the animal model establishment and morphological assessment. Ping Chen and Ye Gan coordinated the study. Yan Chen contributed to the flow cytometry and data analysis. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Vestbo J, Hurd SS, Agusti AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187(4):347–365. | ||
Buist AS, McBurnie MA, Vollmer WM, et al. International variation in the prevalence of COPD (the BOLD Study): a population-based prevalence study. Lancet. 2007;370(9589):741–750. | ||
Beers MF, Morrisey EE. The three R’s of lung health and disease: repair, remodeling, and regeneration. J Clin Invest. 2011;121(6):2065–2073. | ||
Rennard SI, Wachenfeldt K. Rationale and emerging approaches for targeting lung repair and regeneration in the treatment of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2011;8(4):368–375. | ||
Leeman KT, Fillmore CM, Kim CF. Lung stem and progenitor cells in tissue homeostasis and disease. Curr Top Dev Biol. 2014;107:207–233. | ||
Kim CF, Jackson EL, Woolfenden AE, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121(6):823–835. | ||
Curtis SJ, Sinkevicius KW, Li D, et al. Primary tumor genotype is an important determinant in identification of lung cancer propagating cells. Cell Stem Cell. 2010;7(1):127–133. | ||
Nolen-Walston RD, Kim CF, Mazan MR, et al. Cellular kinetics and modeling of bronchioalveolar stem cell response during lung regeneration. Am J Physiol Lung Cell Mol Physiol. 2008;294(6):L1158–L1165. | ||
Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature. 2004;432(7015):324–331. | ||
Unger S, Copland I, Tibboel D, Post M. Down-regulation of sonic hedgehog expression in pulmonary hypoplasia is associated with congenital diaphragmatic hernia. Am J Pathol. 2003;162(2): 547–555. | ||
Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422(6929):313–317. | ||
Krause A, Xu Y, Joh J, et al. Overexpression of sonic hedgehog in the lung mimics the effect of lung injury and compensatory lung growth on pulmonary Sca-1 and CD34 positive cells. Mol Ther. 2010;18(2):404–412. | ||
Zhou X, Qiu W, Sathirapongsasuti JF, et al. Gene expression analysis uncovers novel hedgehog interacting protein (HHIP) effects in human bronchial epithelial cells. Genomics. 2013;101(5):263–272. | ||
Wedzicha JA, Seemungal TA. COPD exacerbations: defining their cause and prevention. Lancet. 2007;370(9589):786–796. | ||
Almansa R, Socias L, Andaluz-Ojeda D, et al. Viral infection is associated with an increased proinflammatory response in chronic obstructive pulmonary disease. Viral Immunol. 2012;25(4):249–253. | ||
Gorski SA, Hufford MM, Braciale TJ. Recent insights into pulmonary repair following virus-induced inflammation of the respiratory tract. Curr Opin Virol. 2012;2(3):233–241. | ||
Hayashi S, Hogg JC. Adenovirus infections and lung disease. Curr Opin Pharmacol. 2007;7(3):237–243. | ||
Yan Z, Jun C, Yan C, et al. Intraperitoneal injection of cigarette smoke extract induced emphysema, and injury of cardiac and skeletal muscles in BALB/C mice. Exp Lung Res. 2013;39(1):18–31. | ||
He ZH, Chen P, Chen Y, et al. Comparison between cigarette smoke-induced emphysema and cigarette smoke extract-induced emphysema. Tob Induc Dis. 2015;13(1):6. | ||
Gueders MM, Bertholet P, Perin F, et al. A novel formulation of inhaled doxycycline reduces allergen-induced inflammation, hyperresponsiveness and remodeling by matrix metalloproteinases and cytokines modulation in a mouse model of asthma. Biochem Pharmacol. 2008;75(2):514–526. | ||
Ikeda N, Hayashi A, Iwasaki K, et al. Comprehensive diagnostic bronchoscopy of central type early stage lung cancer. Lung Cancer. 2007;56(3):295–302. | ||
Taraseviciene-Stewart L, Douglas IS, Nana-Sinkam PS, et al. Is alveolar destruction and emphysema in chronic obstructive pulmonary disease an immune disease? Proc Am Thorac Soc. 2006;3(8):687–690. | ||
Foronjy RF, Dabo AJ, Taggart CC, Weldon S, Geraghty P. Respiratory syncytial virus infections enhance cigarette smoke induced COPD in mice. PLoS One. 2014;9(2):e90567. | ||
Martin J, Helm K, Ruegg P, Varella-Garcia M, Burnham E, Majka S. Adult lung side population cells have mesenchymal stem cell potential. Cytotherapy. 2008;10(2):140–151. | ||
McQualter JL, Yuen K, Williams B, Bertoncello I. Evidence of an epithelial stem/progenitor cell hierarchy in the adult mouse lung. Proc Natl Acad Sci U S A. 2010;107(4):1414–1419. | ||
Nakamura Y, Romberger DJ, Tate L, et al. Cigarette smoke inhibits lung fibroblast proliferation and chemotaxis. Am J Respir Crit Care Med. 1995;151(5):1497–1503. | ||
Cantral DE, Sisson JH, Veys T, Rennard SI, Spurzem JR. Effects of cigarette smoke extract on bovine bronchial epithelial cell attachment and migration. Am J Physiol. 1995;268(5 pt 1):L723–L728. | ||
He S, He Z, Chen Y, et al. C-Kit/c-Kit ligand interaction of bone marrow endothelial progenitor cells is influenced in a cigarette smoke extract-induced emphysema model. Exp Lung Res. 2013;39(6):258–267. | ||
Deeb RS, Walters MS, Strulovici-Barel Y, Chen Q, Gross SS, Crystal RG. Smoking-associated disordering of the airway basal stem/progenitor cell metabotype. Am J Respir Cell Mol Biol. 2016;54(2):231–240. | ||
Shaykhiev R, Crystal RG. Early events in the pathogenesis of chronic obstructive pulmonary disease. Smoking-induced reprogramming of airway epithelial basal progenitor cells. Ann Am Thorac Soc. 2014;11(suppl 5):S252–S258. | ||
Staudt MR, Buro-Auriemma LJ, Walters MS, et al. Airway basal stem/progenitor cells have diminished capacity to regenerate airway epithelium in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;190(8):955–958. | ||
Zuo W, Zhang T, Wu DZ, et al. p63(+)Krt5(+) distal airway stem cells are essential for lung regeneration. Nature. 2015;517(7536):616–620. | ||
Vaughan AE, Brumwell AN, Xi Y, et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature. 2015;517(7536):621–625. | ||
Wang J, Cao J, Dickson AL, Poss KD. Epicardial regeneration is guided by cardiac outflow tract and hedgehog signalling. Nature. 2015;522(7555):226–230. | ||
Michelotti GA, Xie G, Swiderska M, et al. Smoothened is a master regulator of adult liver repair. J Clin Invest. 2013;123(6):2380–2394. | ||
Bellusci S, Furuta Y, Rush MG, Henderson R, Winnier G, Hogan BL. Involvement of Sonic hedgehog (Shh) in mouse embryonic lung growth and morphogenesis. Development. 1997;124(1):53–63. | ||
Chen X, Jin Y, Hou X, Liu F, Wang Y. Sonic hedgehog signaling: evidence for its protective role in endotoxin induced acute lung injury in mouse model. PLoS One. 2015;10(11):e0140886. | ||
Chuang PT, Kawcak T, McMahon AP. Feedback control of mammalian hedgehog signaling by the hedgehog-binding protein, Hip1, modulates Fgf signaling during branching morphogenesis of the lung. Genes Dev. 2003;17(3):342–347. | ||
Zhou X, Baron RM, Hardin M, et al. Identification of a chronic obstructive pulmonary disease genetic determinant that regulates HHIP. Hum Mol Genet. 2012;21(6):1325–1335. | ||
Wang B, Zhou H, Yang J, et al. Association of HHIP polymorphisms with COPD and COPD-related phenotypes in a Chinese Han population. Gene. 2013;531(1):101–105. | ||
Lao T, Glass K, Qiu W, et al. Haploinsufficiency of hedgehog interacting protein causes increased emphysema induced by cigarette smoke through network rewiring. Genome Med. 2015;7(1):12. | ||
Lao T, Jiang Z, Yun J, et al. Hhip haploinsufficiency sensitizes mice to age-related emphysema. Proc Natl Acad Sci U S A. 2016;113(32):E4681–E4687. | ||
Teglund S, Toftgard R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim Biophys Acta. 2010;1805(2):181–208. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.