")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 13
Challenges in Detection of Serum Oncoprotein: Relevance to Breast Cancer Diagnostics
Authors Lengfeld J, Zhang H, Stoesz S, Murali R, Pass F, Greene MI, Goel PN, Grover P
Received 30 July 2021
Accepted for publication 2 October 2021
Published 14 October 2021 Volume 2021:13 Pages 575—593
DOI https://doi.org/10.2147/BCTT.S331844
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pranela Rameshwar
Justin Lengfeld,1 Hongtao Zhang,2 Steven Stoesz,1 Ramachandran Murali,3 Franklin Pass,1 Mark I Greene,2 Peeyush N Goel,2 Payal Grover2
1Martell Diagnostic Laboratories, Inc., Roseville, MN, 55113, USA; 2Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, 19104, USA; 3Department of Biomedical Sciences, Research Division of Immunology; Cedars-Sinai Medical Center, Los Angeles, CA, 90048, USA
Correspondence: Justin Lengfeld
Martell Diagnostic Laboratories, Inc., Roseville, MN, 55113, USA
Email [email protected]
Payal Grover
Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, 19104, USA
Email [email protected]; [email protected]
Abstract: Breast cancer is a highly prevalent malignancy that shows improved outcomes with earlier diagnosis. Current screening and monitoring methods have improved survival rates, but the limitations of these approaches have led to the investigation of biomarker evaluation to improve early diagnosis and treatment monitoring. The enzyme-linked immunosorbent assay (ELISA) is a specific and robust technique ideally suited for the quantification of protein biomarkers from blood or its constituents. The continued clinical relevancy of this assay format will require overcoming specific technical challenges, including the ultra-sensitive detection of trace biomarkers and the circumventing of potential assay interference due to the expanding use of monoclonal antibody (mAb) therapeutics. Approaches to increasing the sensitivity of ELISA have been numerous and include employing more sensitive substrates, combining ELISA with the polymerase chain reaction (PCR), and incorporating nanoparticles as shuttles for detection antibodies and enzymes. These modifications have resulted in substantial boosts in the ability to detect extremely low levels of protein biomarkers, with some systems reliably detecting antigen at sub-femtomolar concentrations. Extensive utilization of mAb therapies in oncology has presented an additional contemporary challenge for ELISA, particularly when both therapeutic and assay antibodies target the same protein antigen. Resolution of issues such as epitope overlap and steric hindrance requires a rational approach to the design of diagnostic antibodies that takes advantage of modern antibody generation pipelines, epitope binning techniques and computational methods to strategically target biomarker epitopes. This review discusses technical strategies in ELISA implemented to date and their feasibility to address current constraints on sensitivity and problems with interference in the clinical setting. The impact of these recent advancements will depend upon their transformation from research laboratory protocols into facile, reliable detection systems that can ideally be replicated in point-of-care devices to maximize utilization and transform both the diagnostic and therapeutic monitoring landscape.
Keywords: breast cancer, diagnosis, ELISA, detection, plasmonics, sensitivity
Introduction
Breast cancer is the second most prevalent type of cancer to afflict women, behind only cancers of the skin. The American Cancer Society estimates that 268,000 women were diagnosed with invasive breast cancer in 2019 in the United States, along with 3.8 million survivors with a previous diagnosis.1 While prognoses vary among breast cancer subtypes, it has been well established that patient outcomes are significantly improved with earlier diagnosis. Current 5-year survival rates for localized and regional breast cancer at the time of diagnosis are 99% and 86%, respectively, which drops precipitously to 28% when a patient is diagnosed late-stage with distant metastases.2 Screening methods such as mammography and ultrasonography have increased the early detection of breast cancers, but the limitations of these methods3,4 have driven efforts to identify biomarkers that could be utilized for early-stage diagnosis as well as evaluation of treatment efficacy.
The most desirable method for diagnosing and monitoring therapeutic effectiveness is to measure key molecular characteristics in biofluids, specifically blood and its constituents. An immunoassay of protein biomarkers in biological samples by enzyme-linked immunosorbent assay (ELISA) is ideally suited for this purpose. ELISA is a highly specific, minimally invasive, and cost-effective technique to better understand the pathological and biological status of a patient. ELISA has long been the gold standard immunoassay for quantifying proteins in biofluid, and the ever-expanding list of clinically relevant biomarkers for disease diagnosis and treatment management drives the need for next-generation assays that are specific and sensitive enough to accurately quantify trace amounts of protein. While ELISA is a specific, sensitive, and reliable assay methodology, significant technical challenges have evolved with its modern clinical application.
As proteins are the main drivers of biological function and phenotype, the evaluation of pertinent protein biomarkers can provide the most relevant patient status assessment regarding the diagnosis of disease or evaluation of therapeutic success. Targeted protein quantification from biofluids for biomarkers of interest offers a snapshot in time of a patient’s health status, as the molecular profile of blood is directly associated with their physiological state. ELISA analysis has historically been applied to a large collection of pathologies, ranging from the identification of blood-borne infectious agents to the detection of markers for various cancers and neurological diseases.5–8 This is a challenging pursuit for a single assay format, as the broad range of clinically relevant concentrations of disease-related biomarkers in blood can range from the pg mL−1 (eg, Interleukin-6 in oral cancer) to the ng mL−1 range as is the case for the extracellular domain (ECD) of the human epidermal growth factor receptor-2 (HER2) in breast cancer.7 The limit of detection for a traditional colorimetric ELISA however, which is dependent on several factors, can range from 0.01 to 1.5 ng mL−1,9,10 highlighting the need for more sensitive assays.
Many iterations of the traditional ELISA have been developed that significantly improve the sensitivity of the assay and continue to maintain its clinical relevancy. Because protein targets cannot be amplified to enhance their signal like nucleic acids, alternative signal-based amplification strategies have been developed to improve the sensitivity of ELISA. Approaches include utilizing more sensitive nanoparticle-based substrates,11 combining ELISA with the polymerase chain reaction (PCR),12 and incorporating nanoparticles as shuttles for detection antibodies and enzymes.13,14 These modifications have resulted in substantial boosts in the ability to detect extremely low levels of protein biomarkers, with some systems reliably detecting antigen at sub-femtomolar concentrations.15
The explosion of monoclonal antibody (mAb) therapies, currently the largest class of biopharmaceuticals, has presented an additional contemporary challenge for ELISA. Biomarker monitoring for evaluation of treatment efficacy is a powerful concept but is potentially impeded by the existence of therapeutic antibodies that may be present in biofluid samples. Particularly in oncology, where targeted antibody therapies have been increasingly employed, the accurate quantification of target proteins can be complicated by interfering therapeutic antibodies. Resolution of issues such as epitope overlap and steric hindrance requires a rational approach to the design of diagnostic antibodies that takes advantage of modern antibody generation pipelines, epitope binning techniques and computational methods to strategically target biomarker epitopes.16
This review covers strategies implemented to date that address two key technical areas crucial to the exact quantification of protein targets in biofluids with ELISA in the modern oncology era: amplification of signal to improve assay sensitivity and rational design of assay antibodies and their fragments to circumvent steric issues present when an antibody therapy and diagnostic assay target the same protein. Implementation of these strategies will facilitate improved clinical outcomes through earlier diagnosis and more accurate therapeutic monitoring.
Traditional ELISA
ELISA, a biochemical immunoassay that takes advantage of the specificity of antibody–antigen interactions to capture and detect antigen on a solid substrate, was first described in 1971.17 Variations of the assay have resulted in several different commonly employed methods to capture and detect antigen targets: Direct, Indirect, Competitive and Sandwich ELISA as represented in Figure 1.
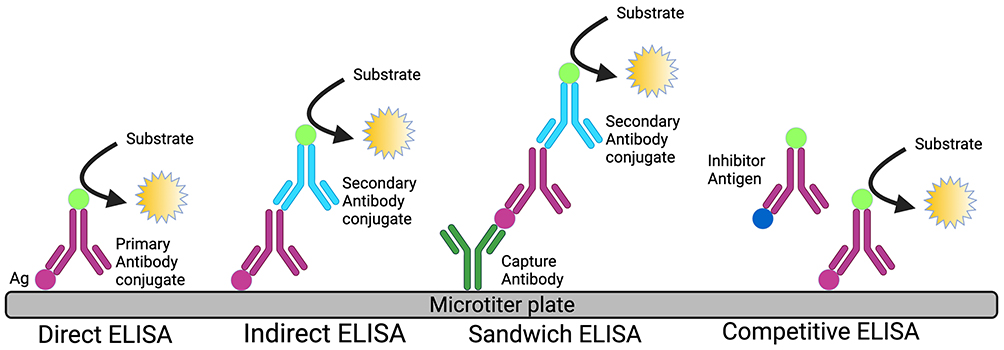 |
Figure 1 Diagrammatic representation of different types of ELISA. |
Direct and Indirect ELISA rely on direct adsorption of antigen to the plate surface for immobilization of target proteins from biological samples. The captured antigen of interest is specifically targeted for detection with either an enzyme-labeled primary antibody (Direct ELISA) or by employing an unlabeled primary antibody matched to an enzyme-labeled secondary antibody (Indirect ELISA). Competitive ELISA determines antigen concentration by evaluating the interference a patient sample has on an expected signal. Plate wells are coated with reference antigen, while test samples are incubated with labeled antibody specific to the antigen of interest. Resulting antibody/antigen complexes in the sample are thus unable to bind the reference antigen upon addition to plate wells, resulting in reduced signal compared to control wells lacking test sample.17,18
Direct, Indirect, and Competitive ELISA variants utilize a sole antibody specific to the protein target and thus rarely reach the specificity of the Sandwich ELISA, which employs primary and secondary antibodies that are reactive to the protein of interest. This increased specificity makes the Sandwich variant the preferred diagnostic ELISA format. In this format, the primary antibody is adsorbed to the surface of the test plate well and used to capture antigen, which is then recognized by a secondary enzyme-labeled detection antibody that is targeted to a different epitope on the captured protein.18 Table 1 describes the advantages and disadvantages of different ELISA formats.
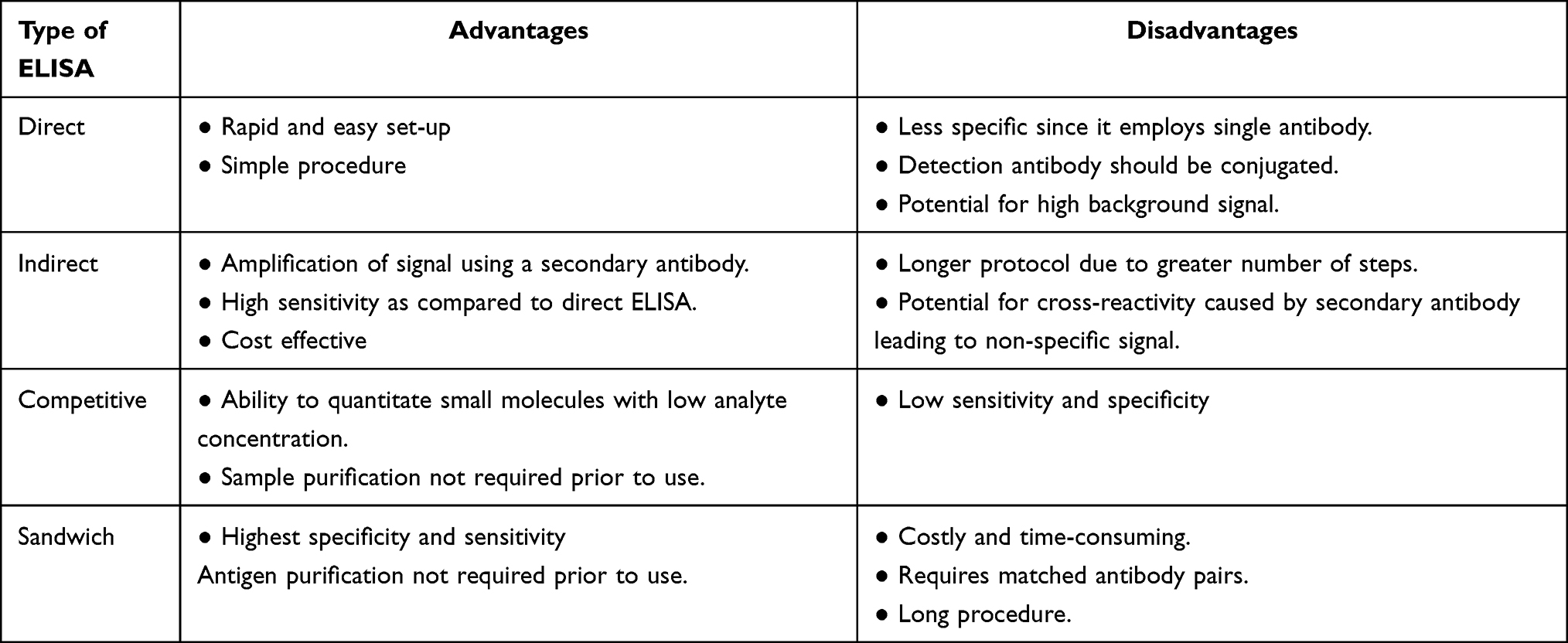 |
Table 1 Advantages and Disadvantages of Different Types of ELISA |
A key element of ELISA sensitivity relates to the affinity of the assay antibody for its antigen target, particularly the capture antibody in the Sandwich ELISA.9,10 Both mAb and polyclonal antibodies (pAb) have been applied to ELISA and fully optimizing the Sandwich ELISA format may result in the employment of both. Over the last 20 years, mAbs, which target a single epitope on an antigen, have come to the forefront of diagnostic testing due to their high specificity and affinity.19 Selection of host species for generating diagnostic antibodies is also a crucial decision for diagnostic assays. While the mouse was often the earliest host for producing ELISA antibodies, rabbits have become increasingly utilized due to several advantages over mice, including affinities up to 100 times greater, superior specificity, and higher stability.20–22 The rabbit immune response to foreign antigen also has a broader repertoire, enabling the recognition of epitopes that are not immunogenic in mice, especially in small molecules and peptide antigens.23
Signal amplification methods are required to detect and quantify the bound protein of interest. This has traditionally been achieved by conjugating one of several different detection enzymes to primary (Direct ELISA) or secondary antibody (Indirect or Sandwich ELISA), most commonly horse-radish peroxidase (HRP) or alkaline phosphatase (AP), to generate signal through catalysis of substrate added in the terminal steps of the ELISA procedure.24 HRP, an oxidoreductase that catalyzes the oxidation of ELISA substrates by hydrogen peroxide, is generally favored over AP for this role due to its higher stability and smaller size (40kDa vs 86kDa),25 which enables more enzyme conjugates per detection antibody and thus more potent signal amplification.24 The increased size of AP can also cause steric hindrance issues due to the proximity of the antigen–antibody complex, potentially resulting in lower enzyme activity.
A multitude of substrates produce different forms of quantifiable signal after catalysis by HRP or AP, with the most well-established substrate categories including, colorimetric, fluorescent, and chemiluminescent. Colorimetric substrates are the most leveraged and have several advantages in ELISA including low cost, simplicity, and a signal that is visible to the naked eye.26 Fluorescent substrate catalysis results in the generation of a product that fluoresces when excited by light at the appropriate wavelength. While fluorescent substrates are marginally more sensitive than colorimetric, the dynamic range of the assay is also wider, allowing higher optical density (OD) measurement than the 4.0 OD limit imposed by most colorimetric plate readers. Chemiluminescent substrates produce photons of light when catalyzed by either HRP or AP, and can reach levels of sensitivity several orders of magnitude higher than fluorescent substrates.26
Interference in ELISA
Biological samples analyzed by immunoassay are complex matrices that may contain components that interfere with the accurate quantification of a target analyte. In the clinic, interfering elements can be endogenous and inherent to the patient, or exogenous and due to the administration of a substance or therapy. Excessive levels of certain serum components, namely lipids and bilirubin, are the most reported causes of endogenous interference.27,28 Lipemia, or excessive levels of triglycerides in the blood, is usually visible to the naked eye and can be caused by recent dietary fat consumption or abnormal lipid metabolism and interferes due to light scattering or absorbance during signal quantification at the end of the ELISA procedure.27 Icterus or jaundice is a condition in which blood concentrations of bilirubin are elevated, which can cause spectrophotometric interference due to bilirubin’s strong absorbance of light between 340 and 500nm.29 This absorbance range overlaps with the absorbance of products from ELISA substrate catalysis, obfuscating the true signal.30 Caution must also be used in interpreting ELISA results from patients with rheumatoid arthritis, as this condition results in the generation of autoantibodies against the fragment crystallizable (Fc) portion of Immunoglobulin G (IgG) class of antibodies potentially skewing assay results through non-specific interaction with ELISA antibodies.31,32
Regardless of the source, the distorting of assay results can occur via direct or indirect mechanisms. Bias through direct interference is induced when the interferent is quantified instead of the target analyte, through cross-reaction with antibody components of the testing assay. Indirect interference manifests when components interact with either a reagent or the target analyte, altering the affinity of the analyte for assay antibodies. The result of these off-target effects can be either false-positive (an increase) or false-negative (a decrease) in the quantified signal, potentially leading to misdiagnosis or a misleading treatment evaluation.
Diagnosing ELISA interference can be achieved through spike-and-recovery experiments and analysis of serially diluted samples. Spiking biological samples with a known amount of target analyte enables the quantification of interference, as an assay free of interference should accurately quantify the total amount of analyte added. Frequently, interfering components will dilute out of a sample, which will be apparent when the dilution-adjusted concentration is calculated from the same sample across a range of dilutions. Testing for parallelism, the ability of an assay to correctly measure an analyte across a dilutional range, can implicate serum matrix interference when analyte measurements display non-parallelism at low dilution which disappears as dilution increases. Implacable non-parallelism at higher dilutions likely indicates either a high concentration of interferent that dilution cannot remedy or an exceptionally high affinity of interferent for the analyte or assay components. In cases of high-affinity interferents such as antibodies, several approaches can be utilized to overcome or minimize the interference. Such techniques include acid-dissociation, which can disrupt the binding of the analyte to interferent,33 choosing an optimal blocking agent or buffer,34,35 or targeted design of immunoassay antibodies that bind to free epitopes on the analyte in the presence of the impeding agent.
Interference through both endogenous and exogenous antibodies has been documented. Endogenous interference can occur in patients who have been exposed to murine antibodies through chance environmental exposure or deliberate antibody therapy, resulting in the production of human anti-mouse antibodies (HAMA) that potentially interfere in immunoassays built on mouse antibodies.36 With the continuing growth of mAb therapies, which display high affinity for their targets, exogenous interference poses an increasing challenge for immunoassays, particularly for therapy-monitoring immunoassays used to analyze patient samples likely to contain high concentrations of these therapeutics. False-positive interference was reported with early antibody therapy for treating immunodeficient states.37–41 This likely under-reported phenomenon has the potential to skew immunoassay results and negatively affect patient outcomes that rely on ELISA for treatment guidance, highlighting the ongoing need for strategies that ensure the validity of results in diagnostic and therapy-tracking immunoassays.
Domain-Specific Targeting for ELISA Antibodies to Prevent Interference
Serum biomarker monitoring by ELISA to evaluate targeted therapy effectiveness is frequently reliant on quantifying the same protein that the mAb therapy targets.42 For instance, in breast cancer, human epidermal growth factor receptor-2 (HER2; also known as ErbB2), a transmembrane tyrosine kinase receptor that is overexpressed in ~20% of breast cancer patients, drives a pro-oncogenic program of unchecked cell growth and apoptosis resistance associated with poor prognosis.43 The HER2 extracellular domain (ECD), the target antigen for current anti-HER2 mAb therapies such as trastuzumab and pertuzumab, is comprised of four subdomains (I–IV) and can be cleaved from the surface of breast cancer cells by extracellular-matrix degrading matrix metalloproteases (MMPs).44–46 A growing body of literature has demonstrated that a reduction in HER2 ECD concentrations in patient serum during therapy is predictive of pathologic complete response (pCR) and improved outcomes7,47–51 while rising or refractory serum HER2 ECD levels indicate resistance to therapy and the need for alternative treatments.35 While the therapeutic binding domains of trastuzumab and pertuzumab have been characterized as within subdomain IV and subdomain II (Figure 2),52 respectively, commercially available ELISAs utilize antibodies that often bind to undefined or undisclosed epitopes on the HER2 ECD. This is potentially problematic, as inherent in therapeutic monitoring programs is the fact that the patient will have the mAb therapeutic in their serum during ELISA testing, potentially obfuscating the true level of HER2 ECD present.
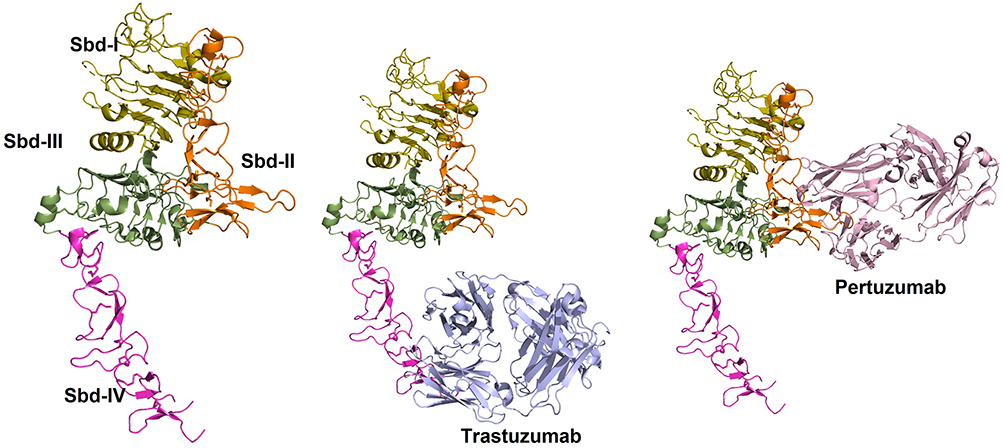 |
Figure 2 Three-dimensional structure of Her2-antibody complexes. (Left) Structure of the extracellular domain of Her2 is shown in ribbon representation. Subdomains I–IV are shown in pale-green, orange, green and purple colors, respectively. (Middle) Structure of Her2-Trastuzumab complex is shown. Trastuzumab (blue) binds to Sbd-IV. (Right) Structure of Her2-Pertuzumab complex is shown. Pertuzumab (pink) binds to Sbd-II (PDB: 1N8Z 159 and 1S78 160). |
When mAb therapy and immunoassays share an antigenic target, two mechanisms may bias attempts at quantification. The therapeutic antibody and immunoassay antibodies may have direct epitope overlap, binding to the same antigenic amino acid sequences, resulting in competition for the same physical location. Sharing of epitopes is not necessary for therapeutic antibodies to interfere, however, as antibody bound to neighboring epitopes can also provoke steric hindrance that physically blocks access of assay antibodies to their target epitopes. Overcoming these issues requires mapping of therapeutic mAb antigen binding sites as well as rational immunoassay antibody approaches that produce high affinity, epitope-specific antibodies to targeted domains that remain unbound in the presence of mAb therapeutics.
Various methods have been employed to generate mAbs for both therapeutic and immunoassay applications, including hybridoma and phage display technology. Hybridoma technology has historically been the predominant method of isolating mAbs and is a favored method due to the in vivo generation of highly specific antibodies that have undergone the natural affinity maturation process. Immunization of the host animal, predominantly mice or rabbits, with antigen provokes the production of antibody in splenic plasma cells. Extracted spleen cells are fused with multiple myeloma-derived cells forming immortalized hybridoma cells that are sorted through dilution to produce cultures generating antibodies against a sole epitope.
Guiding this system to produce mAb against a specific domain can begin with immunizing the host animal against a truncated peptide fragment from a region of interest in the full-size antigen. However, this approach can have limited success45 due to the variable nature of epitopes. Epitopes can be sequential and correspond directly to the antigen peptide sequence, or conformational and result from disparate residues brought into proximity in the folded protein that is not represented by linear peptide fragments. Furthermore, cysteine-rich proteins, like the HER2 ECD, form specific intramolecular disulphide bonds that mediate proper folding of the full-length protein and are difficult to recapitulate when recombinantly expressed in truncated form in vitro. For example, Rockberg et al immunized rabbits with recombinant proteins that were restricted to one of the four subdomains of the HER2 ECD to generate antibodies against individual subdomains.45 Immunogens that contained entire isolated subdomains did not generate antibodies that recognized the native full-length protein, while an immunogen that overlapped subdomains II and III, that the authors speculate contained linear epitopes, produced functional antibody against native HER2 ECD.
The immunogenicity of a peptide fragment is not evenly distributed, as hydrophilic and N- or C-terminal residues are inherently more immunogenic than hydrophobic and central amino acid residues.53,54 This uneven distribution can produce antibodies to epitopes biased to the terminal ends of peptide fragments that may not exist in the full-length antigen. Strategies to target the immune response to specific epitopes on a truncated immunogen have been developed based on observations made about cell surface receptor glycosylation. Work by Peiris et al and others have demonstrated that the HER2 ECD is variably glycosylated depending on oncogenic status and epitope masking due to heavy glycosylation on the ECD reduces the binding and effectiveness of trastuzumab.55 Applications of this concept in recent reports demonstrated that masking the terminal amino acids of peptide antigens with weakly immunogenic glycosylation can shift the immune response toward central residues, providing a path toward the generation of effective domain-specific mAbs using peptide fragments.56,57
Screening of immunized animal antiserum or hybridoma cell cultures for antibodies produced from full-length antigen can also isolate mAb against a targeted domain. Production of domain-specific recombinant forms of antigen by Escherichia coli or other systems, through the expression of plasmids with the antigenic gene restricted to individual domains, can serve as controls to define mAb binding sites. Western blot analysis of these individual domains against candidate mAbs can be rapidly performed and verification against the non-denatured domain-specific antigen can be confirmed by immunocytochemistry in transfected cell lines expressing domain-restricted forms of the target antigen.58,59 More high-throughput, systematic methods of epitope mapping have classically utilized overlapping peptide arrays that enable sequential epitope mapping.60–63 Conformational epitopes, however, require screening with the full three-dimensional structure of the intact antigen. The site-directed epitope masking technique, which utilizes mutants of the full-size targeted antigen tethered to a solid phase, effectively blocks the binding of the antibody to the corresponding surface of the antigen.64 Antibodies that can still access their corresponding epitopes and bind are subsequently depleted from serum, allowing the enrichment of antibodies reactive to the masked surface and identification of their binding sites. This technique was successfully used to generate A21, a mAb against HER2 ECD that exhibited proliferation inhibition activity on HER2 overexpressing cancer cell lines and has been effectively employed in ELISAs to quantify HER2 ECD.65
In the context of an interfering mAb therapy bound to an ELISA target antigen, surface plasmon resonance (SPR) serves as a tool for selecting immunoassay mAbs that react to epitopes free of therapeutic antibody interference. SPR is an optical sensing technology in which antigen is immobilized on the surface of a probe to which different mAbs are serially exposed through microfluidic channels. The binding of the antibody to the antigen causes a shift in SPR signal and if subsequent antibodies react to the same epitope, they will be unable to bind the antigen and there will be no subsequent shift in SPR signal. This enables the sorting and grouping, or binning, of candidate assay antibodies based on whether they compete for the same epitope on their target antigen. Agnolon et al employed SPR to characterize and epitope bin antibodies used in an ELISA sandwich assay for the quantification of serum levels of HER2 ECD.66 SPR was used to verify that both assay antibodies do not share epitopes with trastuzumab by exposing probe bound HER2 ECD to trastuzumab prior to the assay antibodies’ exposure, confirming that all three can simultaneously bind their epitopes. These results were also validated with displacement testing, in which HER2 ECD was pre-incubated with trastuzumab at two concentrations (50 and 200ug/mL) prior to running the sandwich ELISA assay and compared to trastuzumab-naïve samples. Interference due to trastuzumab was minimal, with a decrease in the signal of 7.9%, and interestingly was independent of drug concentration suggesting an interference mechanism driven by steric hindrance, as opposed to competition for overlapping epitopes. As mAb therapies are increasingly employed, the need for simple and economical characterization of assay antibodies that SPR enables is essential to ensure accurate biomarker quantification.
While hybridoma technology has generated a plethora of assay and therapeutic antibodies used today, in vitro techniques based on recombinant DNA technology have rapidly become the fastest and most economical targeted antibody development method. Phage display technology, the most common in vitro technique for producing antibody, facilitates the screening of large libraries of antibodies or antibody fragments against a target antigen by forcing their expression on the surface of bacteriophages. Phage display libraries containing a pool of antibody genes are constructed by extracting mRNA from B-cells isolated from animals that have been immunized with the antigen of interest. Integration of B-cell antibody genes with a phage coat gene results in the display of fusion antibody-coat proteins on the surface of the phage, enabling characterization through repeated rounds of panning against the antigen of interest.67 Phages that display antibody that binds the target antigen with high affinity adhere to the solid phase-bound antigen and are then eluted for sequencing and identification. While this is a rapid method to screen large numbers of antibodies, the panning process identifies only the highest affinity antibodies, limiting diversity and biasing “hits” away from potential epitopes or domains of interest with lower affinity to the target antigen. One recent approach to solving this issue involves the incorporation of noncanonical amino acids (ncAAs) into epitopes of interest in the antigen used for panning phage display libraries.68 The advantage that ncAAs such as p-benzoyl-L-phenylalanine and p-azido-L-phenylalanine confer is their propensity to covalently cross-link proteins in the vicinity when exposed to UV light. Upon panning of phage, the photocrosslinker that forms between the epitope-specific ncAA and reactive antibody on the surface of phage after UV exposure enables the selection of antibodies specific to the target epitope containing the ncAA, regardless of affinity. This promising method allows for the creation of phage libraries from full-size antigen, thus preserving conformational epitopes, while still enabling the selection of epitope-specific mAbs.
Phage display libraries can also be vehicles for displaying peptide fragment libraries to epitope map an existing antibody. The mAb A21, generated against HER2 ECD65 was partially epitope mapped to subdomain I of the HER2 ECD by bio panning a random 12-mer peptide fragment phage library against A21. Peptides that bound A21 were then isolated and sequenced for alignment with the full-length HER2 molecule to identify the location of the epitope.69 Further resolution on the exact amino acids that comprise the A21 epitope was obtained by evaluating A21 binding to individual recombinant subdomains of the ECD in ELISA format, which resulted in a candidate epitope site in subdomain I. Subsequent mutagenesis analysis within subdomain I revealed several surface-exposed residues that were most impactful to the affinity of A21 for HER2 ECD. Characterization of ELISA mAb epitopes to HER2 ECD is vital to ensure non-overlap with mAb therapies and accurate quantification, but is also essential for anti-HER2 mAb therapeutics in particular, as has been demonstrated by the large body of data confirming that downstream cellular effects of mAb therapeutics against the HER2 ECD on cancer cells are variable depending on epitope location.70–75
Computational methods have also been utilized to design and generate complementarity-determining regions (CDRs) of antibodies against specific domains of antigen targets, especially against weakly immunogenic epitopes. Historically, this method has relied on the three-dimensional crystal structure of the antibody, the antigen and the antibody-antigen complex to then model and design antibodies complementary to targeted epitopes.76–78 The success of these computationally designed antibodies relies heavily on precise models for accurate in silico optimization and continues to be a challenge,79 though recent advances have been made in de novo design of antibody without an initial model structure. Specific de novo approaches vary and can involve epitope prediction based on specific antibodies,80–82 and simulating the gene recombination process within B-cells to generate novel CDRs.83,84 These de novo approaches have continuously improved in producing antibodies with high affinity and specificity and, given the difficulty in accurately modeling the structure of antigen-antibody complexes, are likely to be the main driver in expanding the ability of computational methods to design future epitope-targeted assay antibodies.
Antibody Fragments in ELISA
Designing ELISA antibodies to circumvent interfering therapeutics can ultimately be successful. However, due to their large molecular weight (~150 kDa) limiting access to target epitopes, the physical constraints of multiple antibodies binding to the same antigen can reduce the efficacy of assay mAbs. Fortunately, the modular nature of antibodies has enabled the creation of antibody fragments with significantly reduced size which aids in accessing cryptic epitopes and reduces interference from select serum matrix components. These fragments are also more easily genetically manipulated and expressed in bacterial systems for faster cultivation and higher yields.85–88
The gross structure of IgG, the most frequently utilized class of antibody for immunoassays, can be subdivided into the fragment antigen-binding (Fab) and fragment crystallizable (Fc) portions. The Fab region contains the constant and variable domain of the heavy and light chain of the antibody and is responsible for binding antigen, while the Fc portion is located on the tail section and interacts with Fc receptors on the surface of cells to modulate the activation of the immune system. The Fab fragment is the most established antibody fragment in both diagnostics and therapeutics89,90 and can be generated either recombinantly or enzymatically by utilizing papain to cleave IgG into two monovalent Fab fragments and a single Fc fragment. Besides this, reduced size with increased avidity over the single Fab fragment can be achieved by digesting IgG with pepsin, which cleaves below the IgG hinge region and produces a single bivalent F(ab’)2 fragment, retaining both paratopes of the full-size IgG.91 Fab and F(ab’)2 fragments retain the specificity of the full-size antibody while reducing the size to ~50 kDa and 110 kDa, respectively, potentially enhancing accessibility to partially blocked epitopes and allowing deeper penetration into tissue when utilized for immunohistochemistry. Furthermore, the employment of Fab/ F(ab’)2 fragments eliminates the risk of assay interference due to rheumatoid factors, which bind the Fc portion of IgG.92
Single chain variable fragments (scFv) are the smallest fragment of IgG that retains a complete antigen binding domain and are composed of a variable region from the heavy chain (VH) and light chain (VL) connected by a flexible synthetic linker molecule, effectively reducing its size to ~27 kDa. This small size makes them ideal as screening candidates for phage display as well as large-scale production in microbial systems.93 While ELISA assays have been constructed from scFv fragments,94 drawbacks to this format include decreased thermostability as well as dimerization and fragment aggregation due to the synthetic linker molecule.95 These shortcomings have driven the search for additional antibody fragments similarly reduced in size that are not reliant on a synthetic component.
Further size reduction of ELISA antibody components can be achieved by utilizing an atypical class of antibodies found to be naturally occurring in camelids and several different types of sharks. These so-called heavy chain antibodies (HCAbs) lack the light chain variable domain typically found in IgG antibodies, with specificity for antigenic targets determined solely through the heavy chain variable domain. The cleavage of HCAbs to isolate the variable domain yields a fragment known as a single domain antibody (sdAb). Generation of sdAb from camelid antibody generates VHH (Variable domain of the Heavy chain of HCAbs) fragments, or nanobodies, which have a molecular weight of just 15kDa. HCAbs in sharks, termed immunoglobulin new antigen receptors (IgNARs), and the isolated variable domain, termed variable new antigen receptors (vNARs), are the smallest known antibody fragment at ~12kDA. Both VHH and vNARs possess advantages in solubility and stability over equivalent VH domains from human IgG.96,97 Each also has a uniquely long complementarity-determining region 3 (CDR3) that confers a structural advantage, enabling the binding of antigen cleft regions that are not as easily accessed by full size IgG.98,99 The small size and ability to access cryptic epitopes make VHH and vNARs ideal candidates for ELISA, especially in the context of circumventing an interfering component, such as a mAb therapeutic contained in a patient sample. Camelid VHH, in particular, have been employed to build ELISAs against antigens across a wide array of research fields, including oncology (HER2 extracellular domain), virology (HIV, rotavirus),100,101 and toxicology (ricin, cholera).102
The large body of work on antibody structure and function has provided ELISA designers a plethora of antibody fragments to choose from for assay optimization. These fragments offer great promise in solving the multiple issues inherent to biomarker monitoring. However, careful vetting of candidate antibody fragments is essential, as the advantages of smaller size for epitope access and easier production can be outweighed by the increased risk of aggregation and decreased thermal stability.
Signal Amplification
While antibody affinity and specificity are major limiting factors in ELISA, the accurate detection of bound antibody is also a critical determinant in assay sensitivity. Unlike systems that increase signal through target-based amplification methods, as PCR does with DNA, ELISA relies on signal-based amplification to catalytically increase the signal generated from a single binding event between assay antibody and target antigen. The colorimetric ELISA is the most common method employed in the clinical setting, due to its simplicity and low cost. However, this direct approach is offset by lower sensitivity for trace analytes compared with fluorescent and chemiluminescent platforms. Novel methods of signal amplification have been developed to increase ELISA sensitivity, across all signal transduction platforms, to address the clinical challenge of detecting minute concentrations of relevant biomarkers for diagnosing early-stage disease and therapeutic monitoring.
The most utilized method of ELISA signal amplification is the enzyme-mediated process catalyzed by HRP and AP to convert chromogenic substrates such 3,3ʹ,5,5ʹ-tetramethylbenzidine (TMB) and 2,2ʹ-Azinobis [3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt (ABTS) into colored products due to their high catalytic activity and specificity.103–106 While each detection enzyme can amplify the signal by transforming 103−104 molecules of the substrate into product per minute24 the binding of HRP or AP directly to detection antibody results in only a 1:1 stoichiometric ratio between enzyme and detection antibody.107 Increasing this ratio for enhancement of signal produced per detection antibody bound has been achieved by exploiting the relationship between the molecules streptavidin and biotin, which act as a bridging system to enable multiple signal producing enzymes to bind to a single detection antibody. Streptavidin is a bacterial protein tetramer that contains four high-affinity binding sites for biotin, a B vitamin. Detection antibody that has been biotinylated at multiple sites will tightly bind multiple enzyme-streptavidin conjugates, thus amplifying the signal produced from a single bound detection antibody. While this classically used system is successful in measuring analytes as low as 0.01 ng mL−1,9,10 there is a clinical need to reliably detect disease associated biomarkers that typically exist below the limits of this method. Modifications of this approach have generally involved increasing detection enzymes per detection antibody bound to further amplify the signal. This is achieved with the Tyramide amplification system which processes biotinylated Tyramide in the proximity of detection antibody to adhere more HRP enzymes and thus boost the amount of substrate processed per detection antibody.108,109 In general, Tyramide amplification and other related approaches do provide appreciable boosts in sensitivity but fall short of reliable detection at sub-nanomolar concentrations.
Nanoparticles, particularly gold (GNP) and magnetic nanoparticles (MNP), have been incorporated into ELISA platforms in several different roles, including both as substrate replacements and as multivalent shuttles for detection antibody and enzymes to enhance the signal. GNPs have become the preferred nanoparticle due to their simpler synthesis and highly modifiable surface chemistry.110 Integration of nanoparticles in various roles within ELISA has been shown to increase sensitivity and has been a source of great interest in the pursuit of reliable assays for the detection of trace analytes that lie below the limits of detection for traditional ELISA. Plasmonic ELISA takes advantage of the propensity for the surface electrons of noble metal nanoparticles such as gold and silver to oscillate and produce light absorption and reflection properties in the visible and near-infrared range when excited by light111–114 as shown in Figure 3. This characteristic oscillation, known as localized surface plasmon resonance (LSPR), and its visible color output can vary by nanoparticle size, shape, and interparticle proximity.115–117
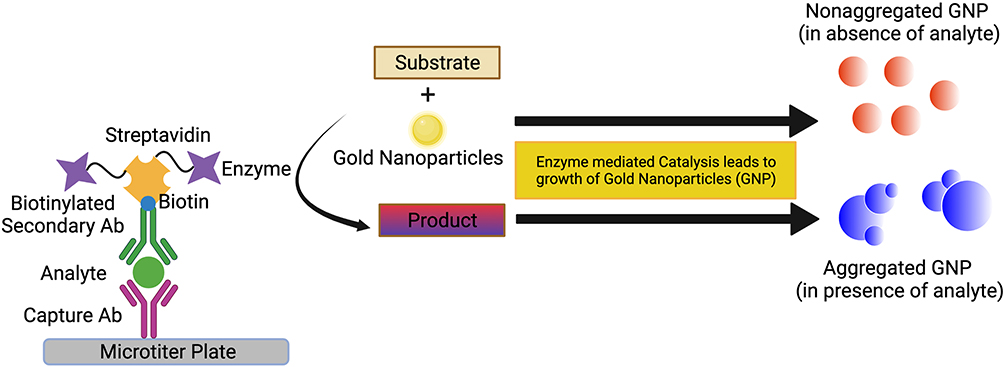 |
Figure 3 Schematic representation of plasmonic ELISA. |
Nanoparticles have been integrated into ELISA as detection systems by using enzyme-driven reactions to alter the proximity, physical attributes, or environment of nanoparticles in solution. This drives a change in LSPR and thus a visible color change that is a function of the analyte concentration.114 For example, acetylcholinesterase enzyme (AChE) has been utilized to alter the LSPR of GNPs to induce their aggregation in proportion to the bound analyte.118,119 During detection steps, the substrate for AChE, acetylthiocholine, is added to the sample plate well along with citrate-capped GNPs, and the subsequent hydrolysis of acetylthiocholine by AChE produces cationic thiocholine. This positively charged thiocholine covalently binds to the negatively charged GNPs, causing a shift in charge distribution and subsequent aggregation and color change from red to blue reflecting analyte concentration. Aggregation-based formats can be highly sensitive, up to a thousand-fold more sensitive than traditional ELISA.118 However, a major limitation is the sensitivity of this system to spontaneous aggregation due to environmental factors, which has limited its real-world application.120 Additional detection approaches that alter LSPR on the nanoparticle surface have included altering the formation and growth dynamics of gold nanoparticles from gold salt, which is driven by the concentration of reducing agent, which is consumed by the bound detection enzyme. This strategy was used to develop an ELISA against prostate-specific antigen (PSA) and human immunodeficiency virus-1 (HIV-1) capsid protein with the detection enzyme catalase, which consumes the reducing agent H2O2. After the formation of the immunosandwich, a solution of H2O2 and gold salt are added and in the absence of analyte the H2O2 concentration remains high, rapidly reducing the gold salt into non-aggregated GNPs, resulting in red color. Growth is subsequently slowed when analyte and thus detection antibody-bound catalase are present, resulting in reduced H2O2, slow growth of aggregated nanoparticles, and a blue color.11 This ultrasensitive technique provided a limit of detection as low as 1 attogram mL−1 of PSA or HIV-1 capsid, which could be observed with the naked eye. Plasmonic ELISA is a highly sensitive detection and signal amplification approach and although it is not without its drawbacks, namely environmentally induced aggregation producing non-specific signal, the ability to observe the presence of antigen with the naked eye makes it a promising platform for point-of-care (POC) devices.
Utilizing GNPs as scaffolds to conjugate multiple signal-generating enzymes per detection antibody takes advantage of the high surface area to volume ratio and tunable surface chemistries inherent to nanoparticles. For example, Duan et al directly adsorbed HRP-labeled detection antibody onto GNPs to quantify the amount of nucleocapsid protein (NP) of the novel bunyavirus for diagnostic purposes. The subsequent increase in sensitivity (limit of detection 0.9 pg mL−1) was significantly higher than traditional ELISA and could even detect protein NP more sensitively than quantitative real time-PCR (qRT-PCR) could with NP mRNA.13 This concept has been taken even further with the coupling of polymers and dendrimers to the surface of the GNPs to serve as additional scaffolding to add yet more binding sites for enzymes and antibodies. Modifying GNPs with polyamidoamine (PAMAM) dendrimer has been used to conjugate up to 20 HRP enzymes on a single particle, producing significant signal amplification that results in double the sensitivity over traditional ELISA in detecting human chorionic gonadotropin.14
Significant signal amplification over traditional ELISA has also been attained by utilizing MNPs as vessels for the delivery of antibodies and HRP enzymes. Wu et al used this approach to magnetically enrich Escherichia coli from river water samples by employing MNPs coated in poly-L-lysine (PLL) brushes, which have copious amine groups for the binding of antibody and HRP.121 The magnetic purification coupled with the signal enhancement of multi-HRP enzyme binding to each MNP resulted in a limit of detection 40-fold lower than traditional ELISA.
Nanoparticles have also been investigated as replacements for enzymes such as HRP and AP in ELISA. Nanoparticles with enzyme-like properties, so-called nanozymes,122 were first described in 2007 with the reporting of a Fe3MnO4 NP that has peroxidase-like activity and since then nanozymes that mimic the activity of oxidase, catalase, and superoxide dismutase have also been discovered.122–128 Nanoparticles often have a lower cost and higher stability than traditional ELISA signal amplification enzymes and have been shown to be more sensitive. Ye et al demonstrated that an enzyme-free ELISA that instead utilized gold vesicles encapsulated with Palladium–Iridium bimetallic nanoparticles as peroxidase mimics was able to achieve a limit of detection in the femtogram mL−1 range, 103 lower than traditional ELISA.129
DNA-based amplification strategies have had success in producing ultrasensitive ELISAs that can detect the antigen in the sub-femtomolar range. Immuno-PCR hybrid assays link the specificity of ELISA immunoassays with the amplification capability of PCR by employing a DNA-tag to mark the ELISA detection antibody which is subsequently amplified and detected through either gel electrophoresis or more commonly qRT-PCR analysis.12 The DNA-tag is typically linked to the detection antibody directly or through a linker such as the commonly utilized biotin-streptavidin or several different covalent conjugates. Linking of DNA-tag and antibody, regardless of method, can be a complicated procedure and a thorough evaluation must be carried out to ensure that antibody affinity has not been compromised.130 The signal can be further enhanced by biotinylating the DNA-tag at both ends, resulting in the formation of supramolecular complexes linked through streptavidin, with potentially hundreds of DNA-tags per detection antibody to further increases sensitivity.131
While immuno-PCR is 105-fold more sensitive than standard enzyme-conjugated amplification, the background signal is increased due to the ability of PCR to amplify even a single DNA molecule that has adhered due to a non-specific interaction. Reducing the background signal of the immuno-PCR platform is a crucial optimization point for the assay to reach its full sensitivity potential and several approaches have been developed, including the proximity ligation assay (PLA) to mitigate this issue.132–135 PLA employs two antibodies reactive to two different epitopes on the same antigen target, which have been conjugated with different DNA-tags. When the antigen target is present in a patient sample, the two antibodies along with their DNA-tags, are brought into proximity of one another enabling their ligation by added DNA ligase. This newly fused DNA-tag then acts as a template for rolling circle amplification, producing a highly sensitive assay with exceptionally low background due to the low odds of non-specific interactions bringing the two different DNA-tags close enough for ligation.136
PCR amplification of the DNA-tag requires temperature fluctuations that, while essential for successful transcription, can be detrimental to the stability of protein components in the immunoassay, as evidenced by the observation that when all ELISA and PCR steps are carried out in the same physical space, there is a 1000-fold decrease in sensitivity.137–139 One solution is to physically segregate the initial immunoassay and ensuing PCR steps by using a DNA-tag that can be cleaved from bound detection antibody at the end of the ELISA procedure, enabling the isolation of the relevant DNA-tag and performing subsequent PCR steps in a separate tube, significantly improving sensitivity.139 Isothermal amplification systems have also been developed to address this reliability issue and enable the transcription of DNA template without temperature cycling. Fluorescent amplification catalyzed by T7 polymerase technique (FACTT) isothermally generates RNA from a DNA-tag consisting of a tetrameric streptavidin molecule with 3 of 4 biotin-binding sites occupied by the DNA template, leaving the fourth to adhere to biotinylated detection antibody as shown in Figure 4.15 RNA transcribed by T7 polymerase is then visualized by the fluorescent RNA intercalating dye RiboGreen and quantified by a fluorometer, which improves sensitivity 105 over traditional ELISA and has enabled detection of protein targets as low as 0.08fM.15
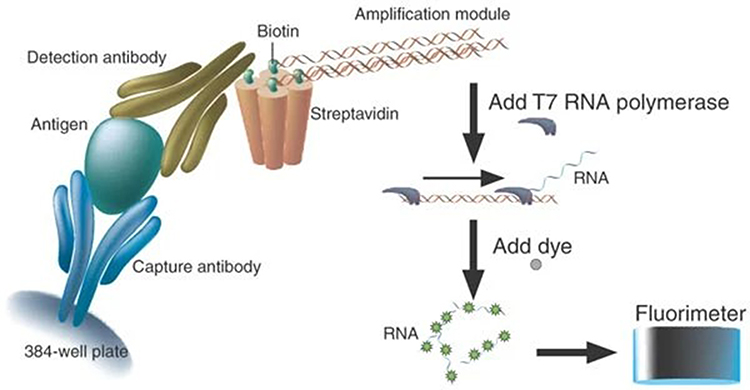 |
Figure 4 Schematic representation of FACTT. Reproduced from Zhang H, Cheng X, Richter M, Greene MI. A sensitive and high-throughput assay to detect low-abundance proteins in serum. Nat Med. 2006;12(4):473–477.15 |
Like FACTT, the recently developed immunoassay coupled with isothermal exponential amplification reaction (IMEXPAR) employs a detection antibody labeled with a DNA primer using a biotin-streptavidin linker.140 After the immunosandwich is formed, a DNA amplification template, DNA polymerase and nicking enzyme are added to mediate an exponential cycle of replication that is proportional to bound detection antibody and can be quantified with the use of the fluorescent nucleic acid dye SYBER Green I and qRT-PCR to a lower limit of detection of 1.63pM.
Enhancing immuno-PCR with GNPs or MNPs has also been shown to be a highly sensitive method of signal amplification. The employment of these particles in a bifunctional manner, in which they are coated with both detection antibody and DNA-tag, is an effective way to avoid the negative effects on antibody affinity of direct detection antibody/DNA-tag conjugation.130 Furthermore, GNPs have even been manipulated into a “polyclonal” status, wherein the nanoparticle has been coated with detection antibodies that react to different epitopes on the same antigenic target, enhancing their ability to bind the antigen target.141 Nam et al developed a biobarcode system that incorporates both GNPs and MNPs, resulting in a highly specific and sensitive immuno-PCR assay that has a reported limit of detection as low as 0.1 fg mL−1.142,143 MNPs coated with capture antibody are used to magnetically isolate the immunosandwich that is subsequently formed with detection antibody and DNA-tag coated GNPs, after which the DNA-tag is dissociated from their GNP scaffold and analyzed by qPCR. Table 2 summarizes different methods for detection of analytes and signal amplification methods used.
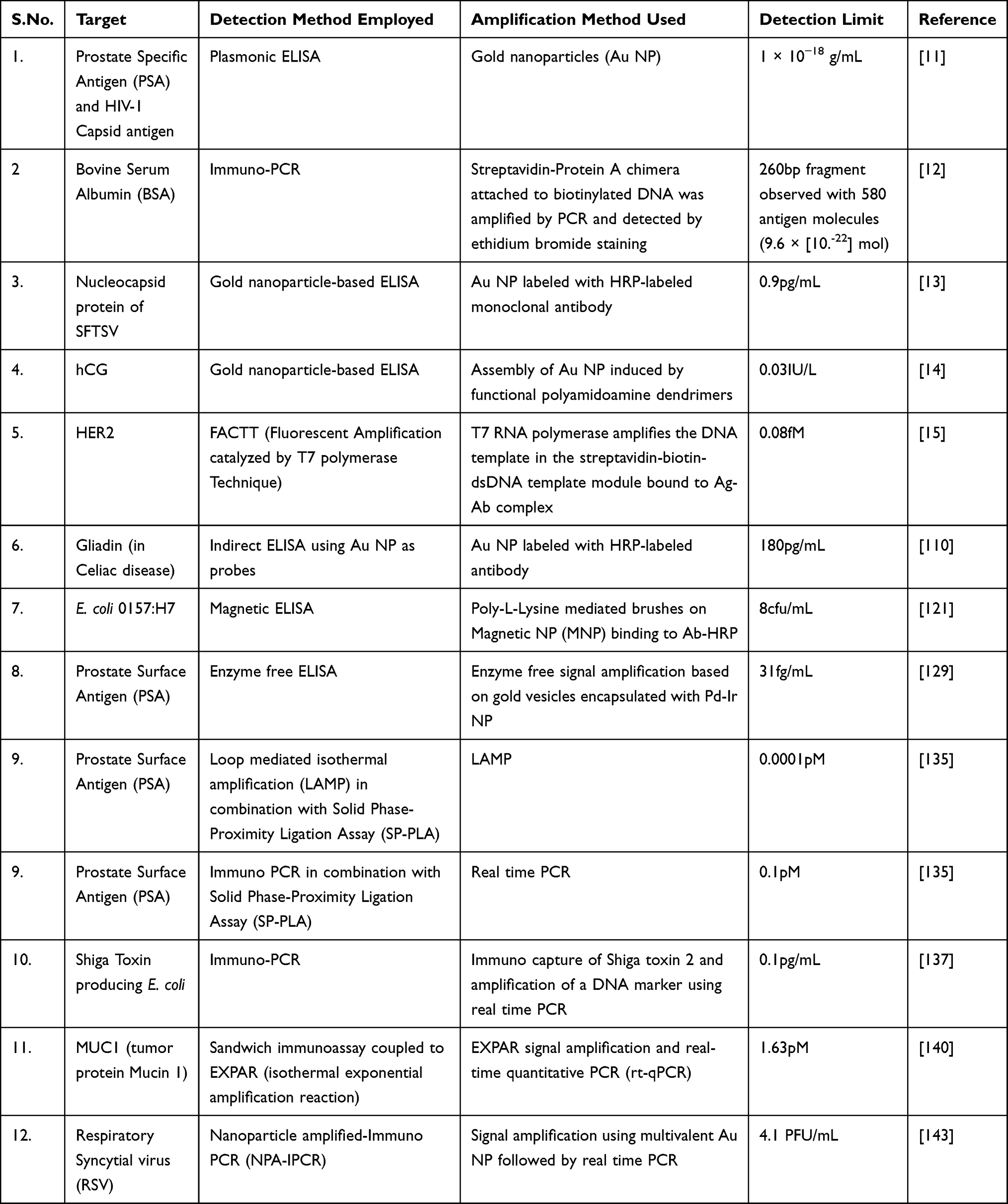 |
Table 2 Different Methods for Detection of Analytes and Signal Amplification |
Improving the amplification of ELISA signal has been a decades long pursuit and has resulted in both modifications to traditional ELISA as well as entirely new assays that combine previously uncoupled techniques to detect biomarkers in trace amounts. Undoubtedly, some of the lowest detection limits have been recorded using nucleic acid-based amplification techniques. However, these immuno-PCR platforms are not without drawbacks, including the length of the procedure, which can take up to two days.144 Continued improvements in ready-made components of the immuno-PCR system, as well as the incorporation of nanoparticles, are now needed to reduce the total assay time from start to finish.
Discussion/Conclusion
ELISA has been extensively used to assay for proteins of interest in both clinical and research applications and improvements to the traditional ELISA platform will be necessary to maintain clinical relevance. The sensitivity of immunoassays is primarily dependent on the characteristics of the assay antibodies and evolving techniques to generate epitope-targeted antibodies and their constituent fragments provide the pathway to develop highly sensitive assays free from any interfering components. Furthermore, as more disease-related biomarkers are discovered to be clinically relevant in the sub-nanomolar range, ultra-sensitive amplification techniques such as those reviewed here will need to be employed.
Typical antibody validation protocols seek to establish candidate antibodies as specific, selective, and reproducible in the intended application and the continued growth of mAb therapy provides a strong justification to incorporate epitope mapping in validation protocols for diagnostic antibodies. Characterization of ELISA antibodies has traditionally been relatively opaque, with commercial diagnostic antibodies rarely having defined or disclosed epitopes. Academic literature is rife with reports on the inadequacy of commercial antibody validation,145–147 and this is particularly troubling when immunoassays are designed to evaluate antigens that may already have a therapeutic antibody bound, as is often the case during HER2 ECD quantification by ELISA in HER2-positive breast cancer. Since the cloning of the rat HER2 gene neu148 and subsequent generation of a mouse mAb reactive to its ECD,149 highly impactful targeted antibody therapies with defined epitopes such as trastuzumab in subdomain IV and pertuzumab in subdomain II have been generated, characterized, and extensively administered in HER2-positive breast cancer patients.52 The majority of diagnostic HER2 antibodies have not been similarly classified, however. Recent reports are recognizing the potential peril of targeted HER2 mAb therapy interference in ELISA,39 while justifiably advocating for the mapping of diagnostic antibody epitopes using a characterization pipeline that includes testing for therapeutic mAb interference. These approaches are essential for the development of an unbiased, reliable evaluation of therapeutic efficacy to inform clinical treatment decisions during targeted mAb therapy.
As disease-related biomarkers continue to be discovered at widely varying concentrations within biofluid, the need for signal amplification techniques will become ever more important to meet the challenge of ultra-sensitive detection. Biomarkers such as programmed death-ligand 1 (PD-L1), a transmembrane ligand that suppresses the adaptive immune system through binding to its receptor PD-1,150 have been implicated in non-small cell lung carcinoma151,152 and breast cancer153 at sub-ng mL-1 concentrations, below the reliable detection limits of a traditional colorimetric ELISA. Similarly, earlier detection of biomarkers such as cardiac troponin I, which are only detectable in blood during pathological conditions such as acute myocardial infarction,154–156 are enabled by more sensitive techniques that have lower limits of detection, providing opportunities for earlier intervention and potentially improved clinical outcomes.
Ideally, amplification techniques should be facile and transferable into POC devices such as lateral-flow assays (LFA) or microfluidics platforms. Indeed, commercial LFA devices are already employing gold plasmonics and magnetic nanoparticles as stable and sensitive methods of signal amplification.157 These devices have the potential to be particularly transformative in underserved regions of the world, where the World Health Organization estimates only 35% of low-income countries have accessible pathology services for diagnosis.158 Targeted treatment for breast cancer is dependent upon biomarker analysis that is traditionally performed by pathologists on tumor biopsies. For example, qualification for targeted mAb therapies trastuzumab and pertuzumab relies upon demonstration of HER2 overexpression by either IHC or fluorescent in situ hybridization (FISH), analyses often unavailable in resource constrained locales. While tissue HER2 analysis is the gold standard for diagnosing HER2-positive breast cancer, soluble HER2 ECD quantification could effectively bring underserved patients off the sidelines by diagnosing those that have an elevated serum HER2 ECD level at the time of presentation and enabling access to targeted therapies that have proven outcome benefits.
Much progress has been made in enhancing the sensitivity, and thus utility, of the traditional ELISA format. As more proof-of-concept improvements migrate from the research laboratory to the clinical diagnostic setting, reliable sub-nanomolar detection capabilities will enable the rapid quantification of trace biomarkers for earlier diagnosis and effective treatment management.
Author Contributions
All authors contributed to drafting or revising the article, gave final approval of the version to be published, agreed to the submitted journal, and agreed to be accountable for all aspects of the work.
Funding
This work was supported by grants from the National Institutes of Health (R01CA219034 and R21AI35359) and Breast Cancer Research Foundation (BCRF-17-061) to MIG.
Disclosure
Dr Hongtao Zhang owns stocks in Martell Diagnostic Laboratories, outside the submitted work; In addition, Dr Hongtao Zhang has patents Greene, MI, Eberwine, J, Kacharmina, JE, and Zhang, H-T. Methods, systems and kits for immuno-detection of epitopes expressed on molecules. US 6,743,592); Greene, M. I., Zhang, H. Methods of detecting molecules expressing selected epitopes via fluorescent dyes. US 7,045,286; Greene, MI, Eberwine, J, Kacharmina, JE, and Zhang, H-T. Methods for immuno-detection of epitopes. US 7,341,831 issued May 16, 2006; Greene, MI, Eberwine, J, Kacharmina, JE, and Zhang, H-T. Method, system and kits for immuno-detection of epitopes expressed on molecules. US 7,361,464; M. I., Zhang, H. Methods of detecting molecules expressing selected epitopes via fluorescent dyes. US 7,524,628 issued related to FACTT mentioned in the review. Dr Franklin Pass is an employee of Martell Diagnostic Laboratories. Dr Mark I Greene reports grants from Martell, during the conduct of the study; grants from the NIH and BCRF, outside the submitted work; In addition, Dr Mark I Greene has a patent on erbB2 therapy owned by the University of Pennsylvania licensed to Roche-Genentech. The authors report no other conflicts of interest in this work.
References
1. Society AC. 2021 Surveillance research. 2021.
2. Society AC. Facts and figures. 2021.
3. Force USPST. Screening for breast cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2009;151(10):
4. Yip CH, Cazap E, Anderson BO, et al. Breast cancer management in middle-resource countries (MRCs): consensus statement from the Breast Health Global Initiative. Breast. 2011;20(Suppl 2):S12–S19. doi:10.1016/j.breast.2011.02.015
5. Yoshihara N. [ELISA for diagnosis of infections by viruses]. Nihon Rinsho. 1995;53(9):2277–2282. Japanese.
6. Dwyer R. The ADVIA centaur infectious disease assays: a technical review. J Clin Virol. 2004;30(Suppl 1):S1–S5. doi:10.1016/j.jcv.2004.02.002
7. Perrier A, Gligorov J, Lefevre G, Boissan M. The extracellular domain of Her2 in serum as a biomarker of breast cancer. Lab Invest. 2018;98(6):696–707. doi:10.1038/s41374-018-0033-8
8. Huppi P, Bologa L, Herschkowitz N. Serum antibodies to central nervous system antigens: an analysis of their relation with different human neurologic disorders. Neurochem Res. 1987;12(7):659–665. doi:10.1007/BF00971016
9. Zhang S, Garcia-D’Angeli A, Brennan JP, Huo Q. Predicting detection limits of enzyme-linked immunosorbent assay (ELISA) and bioanalytical techniques in general. Analyst. 2014;139(2):439–445. doi:10.1039/c3an01835k
10. Porstmann T, Kiessig ST. Enzyme immunoassay techniques. An overview. J Immunol Methods. 1992;150(1–2):5–21. doi:10.1016/0022-1759(92)90061-w
11. de la Rica R, Stevens MM. Plasmonic ELISA for the ultrasensitive detection of disease biomarkers with the naked eye. Nat Nanotechnol. 2012;7(12):821–824. doi:10.1038/nnano.2012.186
12. Sano T, Smith CL, Cantor CR. Immuno-PCR: very sensitive antigen detection by means of specific antibody-DNA conjugates. Science. 1992;258(5079):120–122. doi:10.1126/science.1439758
13. Duan Y, Wu W, Zhao Q, et al. Enzyme-antibody-modified gold nanoparticle probes for the ultrasensitive detection of nucleocapsid protein in SFTSV. Int J Environ Res Public Health. 2020;17(12):4427. doi:10.3390/ijerph17124427
14. Wang W, Li J, Dong C, et al. Ultrasensitive ELISA for the detection of hCG based on assembled gold nanoparticles induced by functional polyamidoamine dendrimers. Anal Chim Acta. 2018;1042:116–124. doi:10.1016/j.aca.2018.08.038
15. Zhang H, Cheng X, Richter M, Greene MI. A sensitive and high-throughput assay to detect low-abundance proteins in serum. Nat Med. 2006;12(4):473–477. doi:10.1038/nm1378
16. Norman RA, Ambrosetti F, Bonvin A, et al. Computational approaches to therapeutic antibody design: established methods and emerging trends. Brief Bioinform. 2020;21(5):1549–1567. doi:10.1093/bib/bbz095
17. Engvall E, Perlmann P. Enzyme-linked immunosorbent assay, Elisa. 3. Quantitation of specific antibodies by enzyme-labeled anti-immunoglobulin in antigen-coated tubes. J Immunol. 1972;109(1):129–135.
18. Kohl TO, Ascoli CA. Indirect competitive enzyme-linked immunosorbent assay (ELISA). Cold Spring Harb Protoc. 2017;2017(7):pdbprot093757. doi:10.1101/pdb.prot093757
19. Ducancel F, Muller BH. Molecular engineering of antibodies for therapeutic and diagnostic purposes. MAbs. 2012;4(4):445–457. doi:10.4161/mabs.20776
20. Feng L, Wang X, Jin H. Rabbit monoclonal antibody: potential application in cancer therapy. Am J Transl Res. 2011;3(3):269–274.
21. Rossi S, Laurino L, Furlanetto A, et al. Rabbit monoclonal antibodies: a comparative study between a novel category of immunoreagents and the corresponding mouse monoclonal antibodies. Am J Clin Pathol. 2005;124(2):295–302. doi:10.1309/NR8H-N08G-DPVE-MU08
22. Vilches-Moure JG, Ramos-Vara JA. Comparison of rabbit monoclonal and mouse monoclonal antibodies in immunohistochemistry in canine tissues. J Vet Diagn Invest. 2005;17(4):346–350. doi:10.1177/104063870501700407
23. Rief N, Waschow C, Nastainczyk W, Montenarh M, Gotz C. Production and characterization of a rabbit monoclonal antibody against human CDC25C phosphatase. Hybridoma. 1998;17(4):389–394. doi:10.1089/hyb.1998.17.389
24. Blake MS, Johnston KH, Russell-Jones GJ, Gotschlich EC. A rapid, sensitive method for detection of alkaline phosphatase-conjugated anti-antibody on Western blots. Anal Biochem. 1984;136(1):175–179. doi:10.1016/0003-2697(84)90320-8
25. Coleman JE. Structure and mechanism of alkaline phosphatase. Annu Rev Biophys Biomol Struct. 1992;21:441–483. doi:10.1146/annurev.bb.21.060192.002301
26. Wild D. The Immunoassay Handbook: Theory and Applications of Ligand Binding, ELISA and Related Techniques. Newnes; 2013.
27. Dimeski G. Interference testing. Clin Biochem Rev. 2008;29(Suppl 1):S43–8.
28. Emerson JF, Lai KKY. Endogenous antibody interferences in immunoassays. Lab Med. 2013;44(1):69–73. doi:10.1309/lmmurcfqhksb5yec
29. Diagnostics R. Serum indices: reduction of clinical errors in laboratory medicine. 2012.
30. Spain MA, Wu AH. Bilirubin interference with determination of uric acid, cholesterol, and triglycerides in commercial peroxidase-coupled assays, and the effect of ferrocyanide. Clin Chem. 1986;32(3):518–521. doi:10.1093/clinchem/32.3.518
31. Berth M, Bosmans E, Everaert J, et al. Rheumatoid factor interference in the determination of carbohydrate antigen 19-9 (CA 19-9). Clin Chem Lab Med. 2006;44(9):1137–1139. doi:10.1515/CCLM.2006.205
32. Xu L, Yu Z, Fan W, et al. Negative interference in serum HBsAg ELISA from rheumatoid factors. PLoS One. 2013;8(11):e80620. doi:10.1371/journal.pone.0080620
33. Lassarre C, Binoux M. Measurement of intact insulin-like growth factor-binding protein-3 in human plasma using a ligand immunofunctional assay. J Clin Endocrinol Metab. 2001;86(3):1260–1266. doi:10.1210/jcem.86.3.7362
34. Zhang H, Fu T, McGettigan S, et al. IL-8 and cathepsin B as melanoma serum biomarkers. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. Int J Mol Sci. 2011;12(3):1505–1518. doi:10.3390/ijms12031505
35. Lam L, Czerniecki BJ, Fitzpatrick E, et al. Interference-free HER2 ECD as a serum biomarker in breast cancer. J Mol Biomark Diagn. 2014;4(3):151. doi:10.4172/2155-9929.1000151
36. Klee GG. Human anti-mouse antibodies. Arch Pathol Lab Med. 2000;124(6):921–923. doi:10.1043/0003-9985(2000)124<0921:HAMA>2.0.CO;2
37. Lichtiger B, Rogge K. Spurious serologic test results in patients receiving infusions of intravenous immune gammaglobulin. Arch Pathol Lab Med. 1991;115(5):467–469.
38. Lichtiger B. Laboratory serologic problems associated with administration of intravenous IgG. Curr Iss Transfus Med. 1994;3:1–7.
39. Agnolon V, Contato A, Meneghello A, et al. ELISA assay employing epitope-specific monoclonal antibodies to quantify circulating HER2 with potential application in monitoring cancer patients undergoing therapy with trastuzumab. Sci Rep. 2020;10(1):1–12. doi:10.1038/s41598-020-59630-y
40. Mills JR, Murray DL. Identification of friend or foe: the laboratory challenge of differentiating M-proteins from monoclonal antibody therapies. J Appl Lab Med. 2017;1(4):421–431. doi:10.1373/jalm.2016.020784
41. van de Donk NW, Moreau P, Plesner T, et al. Clinical efficacy and management of monoclonal antibodies targeting CD38 and SLAMF7 in multiple myeloma. Blood. 2016;127(6):681–695. doi:10.1182/blood-2015-10-646810
42. Lam L, McAndrew N, Yee M, Fu T, Tchou JC, Zhang H. Challenges in the clinical utility of the serum test for HER2 ECD. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review. Biochim Biophys Acta. 2012;1826(1):199–208. doi:10.1016/j.bbcan.2012.03.012
43. Sturgeon CM, Duffy MJ, Stenman UH, et al. National Academy of Clinical Biochemistry laboratory medicine practice guidelines for use of tumor markers in testicular, prostate, colorectal, breast, and ovarian cancers. Clin Chem. 2008;54(12):e11–e79. doi:10.1373/clinchem.2008.105601
44. Molina R, Auge JM, Escudero JM, et al. Evaluation of tumor markers (HER-2/neu oncoprotein, CEA, and CA 15.3) in patients with locoregional breast cancer: prognostic value. Tumour Biol. 2010;31(3):171–180. doi:10.1007/s13277-010-0025-9
45. Rockberg J, Schwenk JM, Uhlen M. Discovery of epitopes for targeting the human epidermal growth factor receptor 2 (HER2) with antibodies. Mol Oncol. 2009;3(3):238–247. doi:10.1016/j.molonc.2009.01.003
46. Carpenter G. Receptors for epidermal growth factor and other polypeptide mitogens. Annu Rev Biochem. 1987;56:881–914. doi:10.1146/annurev.bi.56.070187.004313
47. Perrier A, Boelle PY, Chretien Y, et al. An updated evaluation of serum sHER2, CA15.3, and CEA levels as biomarkers for the response of patients with metastatic breast cancer to trastuzumab-based therapies. PLoS One. 2020;15(1):e0227356. doi:10.1371/journal.pone.0227356
48. Eppenberger-Castori S, Klingbiel D, Ruhstaller T, et al. Plasma HER2ECD a promising test for patient prognosis and prediction of response in HER2 positive breast cancer: results of a randomized study - SAKK 22/99. BMC Cancer. 2020;20(1):114. doi:10.1186/s12885-020-6594-0
49. Reix N, Malina C, Chenard MP, et al. A prospective study to assess the clinical utility of serum HER2 extracellular domain in breast cancer with HER2 overexpression. Breast Cancer Res Treat. 2016;160(2):249–259. doi:10.1007/s10549-016-4000-z
50. Mazouni C, Hall A, Broglio K, et al. Kinetics of serum HER-2/neu changes in patients with HER-2-positive primary breast cancer after initiation of primary chemotherapy. Cancer. 2007;109(3):496–501. doi:10.1002/cncr.22418
51. Witzel I, Loibl S, von Minckwitz G, et al. Monitoring serum HER2 levels during neoadjuvant trastuzumab treatment within the GeparQuattro trial. Breast Cancer Res Treat. 2010;123(2):437–445. doi:10.1007/s10549-010-1030-9
52. Hao Y, Yu X, Bai Y, McBride HJ, Huang X. Cryo-EM structure of HER2-trastuzumab-pertuzumab complex. PLoS One. 2019;14(5):e0216095. doi:10.1371/journal.pone.0216095
53. Lee BS, Huang JS, Jayathilaka LP, Lee J, Gupta S. Antibody production with synthetic peptides. Methods Mol Biol. 2016;1474:25–47. doi:10.1007/978-1-4939-6352-2_2
54. Liang TC, Luo W, Hsieh JT, Lin SH. Antibody binding to a peptide but not the whole protein by recognition of the C-terminal carboxy group. Arch Biochem Biophys. 1996;329(2):208–214. doi:10.1006/abbi.1996.0210
55. Peiris D, Spector AF, Lomax-Browne H, et al. Cellular glycosylation affects Herceptin binding and sensitivity of breast cancer cells to doxorubicin and growth factors. Sci Rep. 2017;7:43006. doi:10.1038/srep43006
56. Manceur AP, Zou W, Marcil A, et al. Generation of monoclonal pan-hemagglutinin antibodies for the quantification of multiple strains of influenza. PLoS One. 2017;12(6):e0180314. doi:10.1371/journal.pone.0180314
57. Pon R, Marcil A, Chen W, et al. Masking terminal neo-epitopes of linear peptides through glycosylation favours immune responses towards core epitopes producing parental protein bound antibodies. Sci Rep. 2020;10(1):18497. doi:10.1038/s41598-020-75754-7
58. Chai L, Mao Q, Liu S, Xia H. Domain-specific monoclonal antibodies produced against human PGRN. Hybridoma (Larchmt). 2011;30(3):271–278. doi:10.1089/hyb.2010.0112
59. Dai X, Li Y, Sun X, Cai K, Mao Q, Xia H. Generation of domain-specific monoclonal antibodies against human glutaredoxin3. Monoclon Antib Immunodiagn Immunother. 2016;35(6):285–292. doi:10.1089/mab.2016.0032
60. Geysen HM, Rodda SJ, Mason TJ, Tribbick G, Schoofs PG. Strategies for epitope analysis using peptide synthesis. J Immunol Methods. 1987;102(2):259–274. doi:10.1016/0022-1759(87)90085-8
61. Radford AJ, Wood PR, Billman-Jacobe H, Geysen HM, Mason TJ, Tribbick G. Epitope mapping of the Mycobacterium bovis secretory protein MPB70 using overlapping peptide analysis. J Gen Microbiol. 1990;136(2):265–272. doi:10.1099/00221287-136-2-265
62. Tribbick G, Triantafyllou B, Lauricella R, Rodda SJ, Mason TJ, Geysen HM. Systematic fractionation of serum antibodies using multiple antigen homologous peptides as affinity ligands. J Immunol Methods. 1991;139(2):155–166. doi:10.1016/0022-1759(91)90185-i
63. Reineke U, Kramer A, Schneider-Mergener J. Antigen sequence- and library-based mapping of linear and discontinuous protein-protein-interaction sites by spot synthesis. Curr Top Microbiol Immunol. 1999;243:23–36. doi:10.1007/978-3-642-60142-2_2
64. Paus D, Winter G. Mapping epitopes and antigenicity by site-directed masking. Proc Natl Acad Sci U S A. 2006;103(24):9172–9177. doi:10.1073/pnas.0600263103
65. Li P, Li Y, Li JY, Liu J. Characterization and utilization of two novel anti-erbB-2 monoclonal antibodies in detection of soluble ErbB-2 for breast cancer prognosis. Cancer Lett. 2003;193(2):139–148. doi:10.1016/s0304-3835(03)00017-x
66. Agnolon V, Contato A, Meneghello A, et al. ELISA assay employing epitope-specific monoclonal antibodies to quantify circulating HER2 with potential application in monitoring cancer patients undergoing therapy with trastuzumab. Sci Rep. 2020;10(1):3016. doi:10.1038/s41598-020-59630-y
67. Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228(4705):1315–1317. doi:10.1126/science.4001944
68. Chen L, Zhu C, Guo H, et al. Epitope-directed antibody selection by site-specific photocrosslinking. Sci Adv. 2020;6(14):eaaz7825. doi:10.1126/sciadv.aaz7825
69. Hu S, Zhu Z, Li L, et al. Epitope mapping and structural analysis of an anti-ErbB2 antibody A21: molecular basis for tumor inhibitory mechanism. Proteins. 2008;70(3):938–949. doi:10.1002/prot.21551
70. Xu F, Lupu R, Rodriguez GC, et al. Antibody-induced growth inhibition is mediated through immunochemically and functionally distinct epitopes on the extracellular domain of the c-erbB-2 (HER-2/neu) gene product p185. Int J Cancer. 1993;53(3):401–408. doi:10.1002/ijc.2910530310
71. Boyer CM, Pusztai L, Wiener JR, et al. Relative cytotoxic activity of immunotoxins reactive with different epitopes on the extracellular domain of the c-erbB-2 (HER-2/neu) gene product p185. Int J Cancer. 1999;82(4):525–531. doi:10.1002/(sici)1097-0215(19990812)82:4<525::aid-ijc10>3.0.co;2-j
72. Klapper LN, Vaisman N, Hurwitz E, Pinkas-Kramarski R, Yarden Y, Sela M. A subclass of tumor-inhibitory monoclonal antibodies to ErbB-2/HER2 blocks crosstalk with growth factor receptors. Oncogene. 1997;14(17):2099–2109. doi:10.1038/sj.onc.1201029
73. Yip YL, Novotny J, Edwards M, Ward RL. Structural analysis of the ErbB-2 receptor using monoclonal antibodies: implications for receptor signalling. Int J Cancer. 2003;104(3):303–309. doi:10.1002/ijc.10951
74. Wang JN, Feng JN, Yu M, et al. Structural analysis of the epitopes on erbB2 interacted with inhibitory or non-inhibitory monoclonal antibodies. Mol Immunol. 2004;40(13):963–969. doi:10.1016/j.molimm.2003.09.012
75. Lewis Phillips G, McMurtrey A, Schroeder K, Fendly B. Diverse activities of anti-HER2 monoclonal antibodies: from growth inhibition to induction of apoptosis. Proc Am Assoc Cancer Res. 1998;39:143.
76. Pantazes RJ, Maranas CD. OptCDR: a general computational method for the design of antibody complementarity determining regions for targeted epitope binding. Protein Eng Des Sel. 2010;23(11):849–858. doi:10.1093/protein/gzq061
77. Lapidoth GD, Baran D, Pszolla GM, et al. AbDesign: an algorithm for combinatorial backbone design guided by natural conformations and sequences. Proteins. 2015;83(8):1385–1406. doi:10.1002/prot.24779
78. Adolf-Bryfogle J, Kalyuzhniy O, Kubitz M, et al. RosettaAntibodyDesign (RAbD): a general framework for computational antibody design. PLoS Comput Biol. 2018;14(4):e1006112. doi:10.1371/journal.pcbi.1006112
79. Sevy AM, Meiler J, Crowe Jr. JE, Boraschi D, Rappuoli R. Antibodies: computer-aided prediction of structure and design of function. Microbiol Spectr. 2014;2(6). doi:10.1128/microbiolspec.AID-0024-2014
80. Sela-Culang I, Ashkenazi S, Peters B, Ofran Y. PEASE: predicting B-cell epitopes utilizing antibody sequence. Bioinformatics. 2015;31(8):1313–1315. doi:10.1093/bioinformatics/btu790
81. Krawczyk K, Liu X, Baker T, Shi J, Deane CM. Improving B-cell epitope prediction and its application to global antibody-antigen docking. Bioinformatics. 2014;30(16):2288–2294. doi:10.1093/bioinformatics/btu190
82. Hua CK, Gacerez AT, Sentman CL, Ackerman ME, Choi Y, Bailey-Kellogg C. Computationally-driven identification of antibody epitopes. Elife. 2017;6. doi:10.7554/eLife.29023.
83. Baran D, Pszolla MG, Lapidoth GD, et al. Principles for computational design of binding antibodies. Proc Natl Acad Sci U S A. 2017;114(41):10900–10905. doi:10.1073/pnas.1707171114
84. Chowdhury R, Allan MF, Maranas CD. OptMAVEn-2.0: de novo design of variable antibody regions against targeted antigen epitopes. Antibodies (Basel). 2018;7(3). doi:10.3390/antib7030023
85. Ahmad ZA, Yeap SK, Ali AM, Ho WY, Alitheen NB, Hamid M. scFv antibody: principles and clinical application. Clin Dev Immunol. 2012;2012:980250. doi:10.1155/2012/980250
86. Tu B, Ziemann RN, Tieman BC, et al. Generation and characterization of chimeric antibodies against NS3, NS4, NS5, and core antigens of hepatitis C virus. Clin Vaccine Immunol. 2010;17(6):1040–1047. doi:10.1128/CVI.00068-10
87. Karn AE, Bell CW, Chin TF. Recombinant antibody technology. ILAR J. 1995;37(3):132–141. doi:10.1093/ilar.37.3.132
88. Fernandes JC. Therapeutic application of antibody fragments in autoimmune diseases: current state and prospects. Drug Discov Today. 2018;23(12):1996–2002. doi:10.1016/j.drudis.2018.06.003
89. Nelson AL. Antibody fragments: hope and hype. MAbs. 2010;2(1):77–83. doi:10.4161/mabs.2.1.10786
90. Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9(10):767–774. doi:10.1038/nrd3229
91. Bazin-Redureau MI, Renard CB, Scherrmann JM. Pharmacokinetics of heterologous and homologous immunoglobulin G, F(ab’)2 and Fab after intravenous administration in the rat. J Pharm Pharmacol. 1997;49(3):277–281. doi:10.1111/j.2042-7158.1997.tb06795.x
92. Mannik M, Nardella FA, Sasso EH. Rheumatoid factors in immune complexes of patients with rheumatoid arthritis. Springer Semin Immunopathol. 1988;10(2–3):215–230. doi:10.1007/BF01857226
93. Montoliu-Gaya L, Esquerda-Canals G, Bronsoms S, Villegas S. Production of an anti-Abeta antibody fragment in Pichia pastoris and in vitro and in vivo validation of its therapeutic effect. PLoS One. 2017;12(8):e0181480. doi:10.1371/journal.pone.0181480
94. Mechaly A, Zahavy E, Fisher M. Development and implementation of a single-chain Fv antibody for specific detection of Bacillus anthracis spores. Appl Environ Microbiol. 2008;74(3):818–822. doi:10.1128/AEM.01244-07
95. Holt LJ, Herring C, Jespers LS, Woolven BP, Tomlinson IM. Domain antibodies: proteins for therapy. Trends Biotechnol. 2003;21(11):484–490. doi:10.1016/j.tibtech.2003.08.007
96. Barthelemy PA, Raab H, Appleton BA, et al. Comprehensive analysis of the factors contributing to the stability and solubility of autonomous human VH domains. J Biol Chem. 2008;283(6):3639–3654. doi:10.1074/jbc.M708536200
97. Streltsov VA, Carmichael JA, Nuttall SD. Structure of a shark IgNAR antibody variable domain and modeling of an early-developmental isotype. Protein Sci. 2005;14(11):2901–2909. doi:10.1110/ps.051709505
98. De Genst E, Silence K, Decanniere K, et al. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc Natl Acad Sci U S A. 2006;103(12):4586–4591. doi:10.1073/pnas.0505379103
99. Desmyter A, Transue TR, Ghahroudi MA, et al. Crystal structure of a camel single-domain VH antibody fragment in complex with lysozyme. Nat Struct Biol. 1996;3(9):803–811. doi:10.1038/nsb0996-803
100. Strokappe N, Szynol A, Aasa-Chapman M, et al. Llama antibody fragments recognizing various epitopes of the CD4bs neutralize a broad range of HIV-1 subtypes A, B and C. PLoS One. 2012;7(3):e33298. doi:10.1371/journal.pone.0033298
101. Vanlandschoot P, Stortelers C, Beirnaert E, et al. Nanobodies(R): new ammunition to battle viruses. Antiviral Res. 2011;92(3):389–407. doi:10.1016/j.antiviral.2011.09.002
102. Goldman ER, Anderson GP, Liu JL, et al. Facile generation of heat-stable antiviral and antitoxin single domain antibodies from a semisynthetic llama library. Anal Chem. 2006;78(24):8245–8255. doi:10.1021/ac0610053
103. Perfezou M, Turner A, Merkoci A. Cancer detection using nanoparticle-based sensors. Chem Soc Rev. 2012;41(7):2606–2622. doi:10.1039/c1cs15134g
104. Aragay G, Pino F, Merkoci A. Nanomaterials for sensing and destroying pesticides. Chem Rev. 2012;112(10):5317–5338. doi:10.1021/cr300020c
105. Gao Z, Xu M, Hou L, Chen G, Tang D. Magnetic bead-based reverse colorimetric immunoassay strategy for sensing biomolecules. Anal Chem. 2013;85(14):6945–6952. doi:10.1021/ac401433p
106. Deshpande S. Enzyme Immunoassays: From Concept to Product Development. Springer Science & Business Media; 1996.
107. Malashikhina N, Garai-Ibabe G, Pavlov V. Unconventional application of conventional enzymatic substrate: first fluorogenic immunoassay based on enzymatic formation of quantum dots. Anal Chem. 2013;85(14):6866–6870. doi:10.1021/ac4011342
108. Bobrow MN, Harris TD, Shaughnessy KJ, Litt GJ. Catalyzed reporter deposition, a novel method of signal amplification. Application to immunoassays. J Immunol Methods. 1989;125(1–2):279–285. doi:10.1016/0022-1759(89)90104-x
109. Bobrow MN, Shaughnessy KJ, Litt GJ. Catalyzed reporter deposition, a novel method of signal amplification. II. Application to membrane immunoassays. J Immunol Methods. 1991;137(1):103–112. doi:10.1016/0022-1759(91)90399-z
110. Ciaurriz P, Fernandez F, Tellechea E, Moran JF, Asensio AC. Comparison of four functionalization methods of gold nanoparticles for enhancing the enzyme-linked immunosorbent assay (ELISA). Beilstein J Nanotechnol. 2017;8:244–253. doi:10.3762/bjnano.8.27
111. Willets KA, Van Duyne RP. Localized surface plasmon resonance spectroscopy and sensing. Annu Rev Phys Chem. 2007;58:267–297. doi:10.1146/annurev.physchem.58.032806.104607
112. Mayer KM, Hafner JH. Localized surface plasmon resonance sensors. Chem Rev. 2011;111(6):3828–3857. doi:10.1021/cr100313v
113. Hutter E, Fendler JH. Exploitation of localized surface plasmon resonance. Adv Mater. 2004;16(19):1685–1706. doi:10.1002/adma.200400271
114. Satija J, Punjabi N, Mishra D, Mukherji S. Plasmonic-ELISA: expanding horizons. RSC Adv. 2016;6(88):85440–85456. doi:10.1039/C6RA16750K
115. Petryayeva E, Krull UJ. Localized surface plasmon resonance: nanostructures, bioassays and biosensing–a review. Anal Chim Acta. 2011;706(1):8–24. doi:10.1016/j.aca.2011.08.020
116. Anker JN, Hall WP, Lyandres O, Shah NC, Zhao J, Van Duyne RP. Biosensing with plasmonic nanosensors. Nat Mater. 2008;7(6):442–453. doi:10.1038/nmat2162
117. Satija J, Bharadwaj R, Sai V, Mukherji S. Emerging use of nanostructure films containing capped gold nanoparticles in biosensors. Nanotechnol Sci Appl. 2010;3:171–188. doi:10.2147/NSA.S8981
118. Liu D, Wang Z, Jin A, et al. Acetylcholinesterase-catalyzed hydrolysis allows ultrasensitive detection of pathogens with the naked eye. Angew Chem Int Ed Engl. 2013;52(52):14065–14069. doi:10.1002/anie.201307952
119. Nie XM, Huang R, Dong CX, Tang LJ, Gui R, Jiang JH. Plasmonic ELISA for the ultrasensitive detection of Treponema pallidum. Biosens Bioelectron. 2014;58:314–319. doi:10.1016/j.bios.2014.03.007
120. Lin S, Cheng Y, Liu J, Wiesner MR. Polymeric coatings on silver nanoparticles hinder autoaggregation but enhance attachment to uncoated surfaces. Langmuir. 2012;28(9):4178–4186. doi:10.1021/la202884f
121. Wu J, Chen Y, Wang Y, et al. Poly-L-lysine brushes on magnetic nanoparticles for ultrasensitive detection of Escherichia coli O157: H7. Talanta. 2017;172:53–60. doi:10.1016/j.talanta.2017.05.035
122. Wei H, Wang E. Nanomaterials with enzyme-like characteristics (nanozymes): next-generation artificial enzymes. Chem Soc Rev. 2013;42(14):6060–6093. doi:10.1039/c3cs35486e
123. Lien CW, Huang CC, Chang HT. Peroxidase-mimic bismuth-gold nanoparticles for determining the activity of thrombin and drug screening. Chem Commun (Camb). 2012;48(64):7952–7954. doi:10.1039/c2cc32833j
124. Dong YL, Zhang HG, Rahman ZU, et al. Graphene oxide-Fe3O4 magnetic nanocomposites with peroxidase-like activity for colorimetric detection of glucose. Nanoscale. 2012;4(13):3969–3976. doi:10.1039/c2nr12109c
125. Song Y, Qu K, Zhao C, Ren J, Qu X. Graphene oxide: intrinsic peroxidase catalytic activity and its application to glucose detection. Adv Mater. 2010;22(19):2206–2210. doi:10.1002/adma.200903783
126. Quan H, Zuo C, Li T, et al. Electrochemical detection of carcinoembryonic antigen based on silver nanocluster/horseradish peroxidase nanocomposite as signal probe. Electrochim Acta. 2015;176:893–897. doi:10.1016/j.electacta.2015.07.086
127. Huang Y, Ding Y, Li T, Yang M. Redox hydrogel based immunosensing platform for the label-free detection of a cancer biomarker. Analytical Methods. 2015;7(2):411–415. doi:10.1039/C4AY02640C
128. Guo Q, Li X, Shen C, et al. Electrochemical immunoassay for the protein biomarker mucin 1 and for MCF-7 cancer cells based on signal enhancement by silver nanoclusters. Microchimica Acta. 2015;182(7–8):1483–1489. doi:10.1007/s00604-015-1471-2
129. Ye H, Yang K, Tao J, et al. An enzyme-free signal amplification technique for ultrasensitive colorimetric assay of disease biomarkers. ACS Nano. 2017;11(2):2052–2059. doi:10.1021/acsnano.6b08232
130. Nikitina IG, Sabirova E, Solopova ON, et al. [A new immuno-PCR format for serological diagnosis of colon cancer]. Mol Biol (Mosk). 2014;48(1):117–123. Russian. doi:10.1134/S0026893313060095
131. Niemeyer CM, Adler M, Pignataro B, et al. Self-assembly of DNA-streptavidin nanostructures and their use as reagents in immuno-PCR. Nucleic Acids Res. 1999;27(23):4553–4561. doi:10.1093/nar/27.23.4553
132. Gullberg M, Gustafsdottir SM, Schallmeiner E, et al. Cytokine detection by antibody-based proximity ligation. Proc Natl Acad Sci U S A. 2004;101(22):8420–8424. doi:10.1073/pnas.0400552101
133. Soderberg O, Gullberg M, Jarvius M, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3(12):995–1000. doi:10.1038/nmeth947
134. Gehwolf R, Band E, Trost A, et al. TaqManR proximity ligation technology for the detection of heterodimeric adhesion receptors on lymphocytes. J Immunol Methods. 2014;404:81–86. doi:10.1016/j.jim.2013.11.024
135. Jiang X, Cheng S, Chen W, Wang L, Shi F, Zhu C. Comparison of oligonucleotide-labeled antibody probe assays for prostate-specific antigen detection. Anal Biochem. 2012;424(1):1–7. doi:10.1016/j.ab.2012.02.004
136. Tong QH, Tao T, Xie LQ, Lu HJ. ELISA-PLA: a novel hybrid platform for the rapid, highly sensitive and specific quantification of proteins and post-translational modifications. Biosens Bioelectron. 2016;80:385–391. doi:10.1016/j.bios.2016.02.006
137. He X, Qi W, Quinones B, McMahon S, Cooley M, Mandrell RE. Sensitive detection of Shiga Toxin 2 and some of its variants in environmental samples by a novel immuno-PCR assay. Appl Environ Microbiol. 2011;77(11):3558–3564. doi:10.1128/AEM.02205-10
138. He X, McMahon S, McKeon TA, Brandon DL. Development of a novel immuno-PCR assay for detection of ricin in ground beef, liquid chicken egg, and milk. J Food Prot. 2010;73(4):695–700. doi:10.4315/0362-028x-73.4.695
139. Sawada T, Nishihara T, Yamamoto A, et al. Preoperative clinical radioimmunodetection of pancreatic cancer by 111 in-labeled chimeric monoclonal antibody Nd2. Jpn J Cancer Res. 1999;90(10):1179–1186. doi:10.1111/j.1349-7006.1999.tb00693.x
140. Liu H, Zhang L, Xu Y, et al. Sandwich immunoassay coupled with isothermal exponential amplification reaction: an ultrasensitive approach for determination of tumor marker MUC1. Talanta. 2019;204:248–254. doi:10.1016/j.talanta.2019.06.001
141. Ding YZ, Liu YS, Zhou JH, et al. A highly sensitive detection for foot-and-mouth disease virus by gold nanoparticle improved immuno-PCR. Virol J. 2011;8:148. doi:10.1186/1743-422X-8-148
142. Nam JM, Park SJ, Mirkin CA. Bio-barcodes based on oligonucleotide-modified nanoparticles. J Am Chem Soc. 2002;124(15):3820–3821. doi:10.1021/ja0178766
143. Perez JW, Vargis EA, Russ PK, Haselton FR, Wright DW. Detection of respiratory syncytial virus using nanoparticle amplified immuno-polymerase chain reaction. Anal Biochem. 2011;410(1):141–148. doi:10.1016/j.ab.2010.11.033
144. Niemeyer CM, Adler M, Wacker R. Detecting antigens by quantitative immuno-PCR. Nat Protoc. 2007;2(8):1918–1930. doi:10.1038/nprot.2007.267
145. Goodman SL. The antibody horror show: an introductory guide for the perplexed. N Biotechnol. 2018;45:9–13. doi:10.1016/j.nbt.2018.01.006
146. Schonbrunn A. Editorial: antibody can get it right: confronting problems of antibody specificity and irreproducibility. Mol Endocrinol. 2014;28(9):1403–1407. doi:10.1210/me.2014-1230
147. Taussig MJ, Fonseca C, Trimmer JS. Antibody validation: a view from the mountains. N Biotechnol. 2018;45:1–8. doi:10.1016/j.nbt.2018.08.002
148. Schechter AL, Stern DF, Vaidyanathan L, et al. The neu oncogene: an erb-B-related gene encoding a 185,000-Mr tumour antigen. Nature. 1984;312(5994):513–516. doi:10.1038/312513a0
149. Drebin JA, Link VC, Weinberg RA, Greene MI. Inhibition of tumor growth by a monoclonal antibody reactive with an oncogene-encoded tumor antigen. Proc Natl Acad Sci U S A. 1986;83(23):9129–9133. doi:10.1073/pnas.83.23.9129
150. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173(2):945–954. doi:10.4049/jimmunol.173.2.945
151. Grizzi F, Castello A, Qehajaj D, et al. Independent expression of circulating and tissue levels of PD-L1: correlation of clusters with tumor metabolism and outcome in patients with non-small cell lung cancer. Cancer Immunol Immunother. 2019;68(9):1537–1545. doi:10.1007/s00262-019-02387-9
152. Vecchiarelli S, Passiglia F, D’Incecco A, et al. Circulating programmed death ligand-1 (cPD-L1) in non-small-cell lung cancer (NSCLC). Oncotarget. 2018;9(25):17554–17563. doi:10.18632/oncotarget.24785
153. Moku P, Shepherd L, Ali SM, et al. Higher serum PD-L1 level predicts increased overall survival with lapatinib versus trastuzumab in the CCTG MA.31 phase 3 trial. Cancer. 2020;126(22):4859–4866. doi:10.1002/cncr.33149
154. Eriksson S, Wittfooth S, Pettersson K. Present and future biochemical markers for detection of acute coronary syndrome. Crit Rev Clin Lab Sci. 2006;43(5–6):427–495. doi:10.1080/10408360600793082
155. Melanson SE, Tanasijevic MJ, Jarolim P. Cardiac troponin assays: a view from the clinical chemistry laboratory. Circulation. 2007;116(18):e501–e504. doi:10.1161/CIRCULATIONAHA.107.722975
156. McDonnell B, Hearty S, Leonard P, O’Kennedy R. Cardiac biomarkers and the case for point-of-care testing. Clin Biochem. 2009;42(7–8):549–561. doi:10.1016/j.clinbiochem.2009.01.019
157. Moyano A, Serrano-Pertierra E, Salvador M, Martinez-Garcia JC, Rivas M, Blanco-Lopez MC. Magnetic lateral flow immunoassays. Diagnostics (Basel). 2020;10(5). doi:10.3390/diagnostics10050288
158. World Health Organization. Early Cancer Diagnosis Saves Lives, Cuts Treatment Costs. World Health Organization; 2017.
159. Cho HS, Mason K, Ramyar KX, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421(6924): 756–760
160. Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, Sliwkowski MX. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. 2004;5(4): 317–328.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.