")
Back to Archived Journals » Clinical Oncology in Adolescents and Young Adults » Volume 6
Challenges faced in the treatment of acute lymphoblastic leukemia in adolescents and young adults
Authors Levine S, McNeer J, Isakoff M
Received 27 August 2015
Accepted for publication 13 November 2015
Published 15 February 2016 Volume 2016:6 Pages 11—20
DOI https://doi.org/10.2147/COAYA.S61424
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Mark Kieran
Selena R Levine,1 Jennifer L McNeer,2 Michael S Isakoff1
1Center for Cancer and Blood Disorders, Connecticut Children’s Medical Center and University of Connecticut School of Medicine, Hartford, CT, 2Section of Pediatric Hematology/Oncology, University of Chicago Comer Children's Hospital, Chicago, IL, USA
Abstract: The survival rate for children with acute lymphoblastic leukemia (ALL) has dramatically improved over the last 50 years. However, for those in the adolescent and young adult (AYA) age-group of 15–30 years with ALL, there has not been the same degree of improvement. Historically, pediatric and adult providers have utilized different treatment approaches based on clinical trials. However, studies that have compared the outcome of AYA patients with ALL treated on pediatric or adult clinical trials have generally shown substantially better outcomes for this patient population treated with the pediatric trials. Additionally, hematopoietic stem cell transplantation has been considered as part of intensified therapy for AYA patients with ALL. Herein, we review the outcomes with chemotherapy alone and with hematopoietic stem cell transplantation, and explore the challenges faced in determining the ideal therapy for the AYA population of patients.
Keywords: adolescent young adult oncology, leukemia, hematopoietic stem cell transplantation
Introduction
Acute lymphoblastic leukemia (ALL) is a multifaceted disease in its biological pathogenesis, with variety of populations affected and numerous epidemiological factors.1 The treatment regimens are typically intense and involve many chemotherapeutic agents that carry a multitude of toxicity risks. However, the improvement in the prognosis for children diagnosed with ALL has been one of the great success stories for the 20th and early 21st centuries for cancer care. While only 30% of children diagnosed with ALL survived in 1970, more than 85% survive today, with some populations of patients having a >95% survival at 5 years. Unfortunately, the same cannot be said for young adults between 15 and 30 years of age with ALL, which, for the purposes of this review, we will consider to be the adolescent and young adult (AYA) population. For the patients in this age-group, the prognosis has been less promising and can vary from 35% to 75% disease-free survival at 5 years, depending on the leukemic subtype and therapy utilized.2–4 Efforts to improve the prognosis for this group of patients have been challenging, and controversy between pediatric and adult oncologists regarding the optimal treatment has contributed to a lack of clarity over a standard approach to care. This includes a discrepancy among some providers regarding the utilization of hematopoietic stem cell transplantation (HSCT). Here, we review many of the issues that need to be considered in determining the ideal treatment course for AYAs with ALL.
Background
ALL develops as a result of deregulation in various pathways in the cell cycle that serve to control cell survival and proliferation.1,5 ALL in particular is characterized by an uncontrollable proliferation of malignant cells with an arrest of normal lymphoid progenitor cell development.6 As a result, normal bone marrow cells are replaced by malignant lymphoblasts, thereby inhibiting homeostatic hematogenous and immune functions.7,8 In ALL, examination of the bone marrow will most likely reveal a hypercellular and homogenous population of blasts.9
Epidemiology and pathogenesis
The B lymphoblastic leukemia immunophenotype (B-ALL) occurs most frequently in children and accounts for approximately 80%–85% of ALL in the pediatric population. T-cell ALL (T-ALL) makes up 10%–15% of leukemia seen in children but is more prevalent in the older AYA population, accounting for 25% of cases in patients 16–21 years of age. Compared to the B-cell phenotype, T-cell ALL is more likely to present with extramedullary disease, such as lymphadenopathy (including the presence of a mediastinal mass) or hepatosplenomegaly.10 While the presentation of ALL can be insidious or occur acutely, cases can also be discovered as an incidental finding on routine laboratory blood work. Some of the more common presenting symptoms of ALL include bruising, pallor, petechiae, and fever. Moreover, approximately 25% of patients who initially present with ALL may have bone pain or vertebral compression fractures, both of which are thought to be due to the bone’s periosteal enlargement from leukemic cell infiltration.11,12
The exact pathogenesis involved in the development of ALL remains under investigation, and there are a variety of theories to consider. One such theory has suggested that pediatric cancer tends to result from genetic defects during the growth and development phases of organs and tissues during times of proliferative stress. This seems to correlate with the fact that the peak incidence of ALL in children is between the ages of 2–5 years, a time when lymphocyte progenitor cells, particularly of the B-cell lineage, are actively proliferating and rearranging their genetic material for immunoglobulins.6 Furthermore, genetic mutations and chromosomal translocations that are responsible for activating and/or deactivating specific genes are the driving force of most cases of ALL.5,13 For instance, in 25% of cases of B-cell ALL, the t(12;21) translocation leads to the ETV6-RUNX1 fusion gene (formerly TEL-AML1), which leads to the proliferation and self-renewal of an immature B lymphocyte.5,13 In T-ALL, greater than 50% of the cases result from a mutation in the NOTCH1 gene, which codes for a transmembrane receptor that regulates normal cell development, and produces an activating effect.13 Moreover, in many adult cases, the most common chromosomal translocation seen is t(9;22), or the Philadelphia chromosome, which produces the BCR-ABL protein, resulting in constitutive tyrosine kinase activity.
Retrospective analysis of leukemic fusion genes from neonatal blood spots (ie, Guthrie cards) revealed that a prenatal origin may explain some cases of childhood ALL.14 However, the 10% concordance rate of leukemia in identical twins points to the notion that postnatal environmental influences must also be responsible for the full malignant transformation of the cells.13,14 In addition, it is well established that several inherited genetic syndromes, such as trisomy 21, Bloom syndrome, Shwachman–Diamond syndrome, neurofibromatosis type 1, and ataxia-telangiectasia, predispose an individual to having a higher likelihood of developing ALL.13 Other proposed risk factors for the development of ALL include pre- and postnatal radiation exposure, as well as male sex. There is male predominance in all age-groups, but in the AYA population, particularly ages 15–29 years, the male-to-female ratio of cases of ALL is 2:1.5 The extent to which the risk factors noted earlier affect those in the AYA population is not clear.
Prognosis
Disease prognosis and patient outcomes in ALL are largely dependent upon a patient’s age at the time of diagnosis, as there is a strong correlation between age at presentation and specific biological and/or molecular characteristics of the disease. AYAs traditionally have worse survival rates compared to children aged 1–10 years, as AYA patients tend to have less favorable disease characteristics, such as an increased incidence of t(9;22) and the T-cell immunophenotype. They also present less often with the favorable t(12;21) or hyperdiploidy.1,15 Thus, the focus of disease-risk stratification differs between pediatric and adult treatment approaches. Adult risk stratification is based upon the patient’s perceived risk of relapse secondary to the presence or absence of poor biological prognostic factors, whereas in pediatric cases, risk stratification is based not only upon age and biology, but also upon the patient’s response to therapy.1
With ALL being the most common cancer in children, cure rates for pediatric patients have improved substantially over the past 50 years. Progress in treatment modalities for pediatric patients has lead to better survival rates in children, with the prognosis for pediatric ALL being among the most favorable of all malignancies. The same, however, cannot be said for adolescents and adults, in whom outcomes have not improved as substantially.6 In the USA, it is estimated that approximately 25,000 individuals between the ages of 15 and 30 years are diagnosed with cancer each year,16 and in the 16–25-year-old age-group, cancer remains the fourth leading cause of death, after accidents, suicides, and homicides.17 All AYA patients with cancer have suffered as a whole as a result of the lack of progress in survival outcomes. This is due, in part, to the decreased recognition of the need for specific clinical trials for AYA patients, and lack of medical insurance and access to proper medical care in this population, as well as the inferior psychosocial support available to AYAs when compared to the pediatric population. This in turn has only widened the gap between the care provided to pediatric versus adult patients.
While the cure rate for children with ALL under the age of 10 years remains one of the most favorable of all childhood malignancies, the same cannot be said for those 15 years and older.6 The most substantial decreases in survival begin to become apparent at approximately 15 years of age.18 Current chemotherapy protocols for children aged 1–10 years have led to a complete first remission rate of 98% and a 5-year event-free survival (EFS) rate of 80%–85%.19 In contrast, the complete remission rate for adults is approximately 80%, with overall survival (OS) rates reported in the range of 35%–70%.19 The US Surveillance Epidemiology and End Results (SEER) data has demonstrated that the 5- and 10-year survival rates for children aged 1–14 years are 87.5% and 83.8%, respectively, while they drop to 61.4% and 60.4% for adolescents between the ages of 15 and 19 years. Survival rates decline even further for young adults between the ages of 20 and 29 years with their 5- and 10-year EFS rates being 44.8% and 30.8%, respectively.16,20
Reasons for why the survival rate of AYAs with ALL is about one-half that of younger children are multifactorial. The AYA population has the lowest rate of primary care utilization. In addition, they represent the highest proportion of the population that is uninsured or underinsured.21,22 These two factors may contribute to the fact that AYA patients with ALL tend to present with more advanced disease at the time of diagnosis, and this in addition to their inherently higher risk biology predispose them to unfavorable outcomes.23 Another host characteristic contributing to the poor survival outcome in this age-group includes decreased compliance with the long and complex treatment regimens that are necessary. Treatment failure as a result of poor compliance in this age-group has not only been an issue for patients with ALL, but also for other AYAs with chronic diseases such as asthma, cystic fibrosis, and diabetes.19,24
Increasing age is accompanied by biological differences in the disease itself, tending to favor higher risk and less favorable prognostic indicators. Childhood features of the disease that are usually associated with a favorable prognosis, such as B-cell immunophenotype, t(12;21), and hyperdiploidy (>47 chromosomes), are less common in the AYA population and mostly absent in older adults.1 The t(12;22) chromosomal translocation responsible for the ETV6-RUNX1 fusion protein is present in about a quarter of young children with ALL.15 This fusion protein makes the malignant cells particularly susceptible to the effects of chemotherapy, resulting in an extremely high OS rate of >95% in those with rapid early response to therapy. This translocation and its favorable prognostic factors, however, are virtually absent in patients over the age of 18 years.25 Hyperdiploidy is only present in approximately 6% of AYA patients, as compared to approximately 35% of pediatric ones with pre-B-cell ALL.15 In children, hyperdiploidy is often associated with a very low risk of disease relapse. In adults on the other hand, the presence of hypodiploidy confers a higher risk for relapse and decreased OS.26
With increasing age, the biologic features of the disease itself are oftentimes more complex and ominous. The adverse prognostic features of the disease that are more common in AYAs include a high white blood cell count, T-cell immunophenotype, and the t(9;22) translocation.27 Historically, the T-cell immunophenotype of the disease has had a worse prognostic outcome than B-ALL regardless of age, though current treatment strategies are leading to improved outcomes than were seen historically, and are now approaching the survival seen in B-ALL.28–31 Furthermore, in some adult trials, patients with T-cell disease had even better outcomes than those with B-cell disease.32,33 Moreover, the t(9;22) translocation responsible for the BCR-ABL1 fusion gene is an unfavorable cytogenetic variation regardless of age, but with the incorporation of tyrosine kinase inhibitors into multiagent chemotherapy regimens, the outcome for these patients is drastically improving.34 In contrast to the t(12;21), the prevalence of t(9;22) increases with age, occurring in 2% of patients aged 1–9 years and 8% in adolescents. Its presence in adults is as high as 20%–25%, making it the most common chromosomal abnormality in adult ALL.35,36
There are other factors that were historically believed to contribute to a worse outcome for the AYA population, including high rates of death during induction therapy.18,37 However, more recent data have indicated that the induction death rate in recent protocols is quite low at approximately 2% and is not significantly different between AYA and younger patients.38,39 In addition, increased rates of disease relapse, particularly within the central nervous system (CNS), are as high as 30%–40% in the AYA population.13 Moreover, increasing age is believed to place AYA patients at a higher risk for treatment-related morbidity and mortality, which has in part been proposed to be as a result of delayed drug clearance and preexisting medical comorbidities.13,18
Current treatment strategies
Traditional pediatric and adult treatment approaches for AYAs have differed, not only in terms of the chemotherapy agents used but also in the phases of therapy applied. Medical versus pediatric oncologists have had different philosophies regarding induction, intensity, and duration of overall therapy, as well as the role of targeted CNS prophylaxis.40 Most pediatric treatment regimens are based on a backbone developed by the Berlin-Frankfurt-Münster (BFM) group in the 1970s. This model of therapy, used by most cooperative groups, is based on the Goldie–Coldman hypothesis that postulates that in order to cure ALL, early intensification is a necessity, in conjunction with the Norton–Simon hypothesis that states that a later delayed intensification, consisting of new or analogous drugs, is needed to destroy drug-resistant malignant cells that survived the initial round of chemotherapy.18 As a result, the treatment of ALL involves a multistep approach that is both complex and intense. This therapy consists of both initial and delayed aggressive phases of therapy (induction/consolidation and delayed intensification) with an intervening less intense phase of treatment known as interim maintenance. Following the completion of these blocks of more aggressive therapy, which generally takes between 6 and 9 months, there is a prolonged maintenance therapy which lasts for 2–3 years, depending on the specific protocol.
Thus, the overall theme of pediatric protocols is to treat ALL with high cumulative doses of nonmyelosuppressive agents, such as vincristine, asparaginase, and corticosteroids, especially during periods of myelosuppression induced by alkylators, anthracyclines, and antimetabolies. Asparaginase, in particular, serves as a hallmark component of most pediatric ALL treatment regimens. Its mechanism of action is to induce apoptosis of leukemic cells as a result of the cells’ inability to undergo proper protein biosynthesis due to the lack of extracellular asparagine.41,42 Studies in which intensive rounds of asparaginase are used in induction and intensification have demonstrated significant benefits in EFS and overall disease cure rates.42 In addition, a series of clinical trials conducted through the Dana Farber Cancer Institute (DFCI) has clearly demonstrated the range of toxicities, the importance of frequent exposure, and that more doses improve outcomes.43–45 Moreover, pediatric regimens include a focus on CNS-directed chemotherapy, generally in the form of intrathecal methotrexate, as the CNS is a sanctuary site for leukemic cells.6
In contrast to the pediatric regimens, standard adult ALL treatment protocols typically comprised blocks of myelosuppressive therapies followed by periods of rest, such as is given with hyper-CVAD therapy. This treatment approach utilizes smaller fractions of cyclophosphamide, vincristine, Adriamycin®, and dexamethasone, alternating with courses of methotrexate and cytarabine. As compared to classic pediatric treatment regimens, adult protocols are generally shorter, though they often require longer inpatient hospitalizations. For instance, hyper-CVAD therapy typically lasts 6 months, though patients must be hospitalized for chemotherapy.46 This is followed by maintenance therapy with prednisone, vincristine, mercaptopurine, and methotrexate, commonly given for 24–30 months of therapy.47
In part, the rationale for this type of therapy is that older patients are believed to be more susceptible to the side effects from chemotherapy, and in particular vincristine-induced peripheral neuropathy, as well as the multitude of asparaginase toxicities, such as pancreatitis, hyperglycemia, avascular necrosis, thromboembolism, hepatic toxicity, and allergic hypersensitivity reactions. The incidence of most, if not all of these, increases with age.18,42 As a result of these toxicity concerns and general lack of use in traditional adult ALL regimens, the benefit of vincristine and asparaginase in ALL outcomes is not as appreciated in adults as in children.48 It should be noted that when treated with typical adult protocols, the AYA population represents only a small number of total patients being treated. As such, the side effect profiles and precautions taken in treating older patients versus younger ones with different therapeutic modalities may not be applicable to this specific group. On the other hand, the excellent tolerance of chemotherapy by the population <18 years of age may also not fully represent the degree to which chemotherapy may be tolerated in the AYA population composed of 18–30 year olds.
Depending on local referral patterns and to whom the patient first presents, both pediatric and adult oncologists treat patients with ALL who fall into the AYA age-group.18,20 Pediatricians tend to refer their AYA patients to pediatric oncologists who are affiliated with universities and large research-based groups, whereas internists usually refer to medical oncologists who might be affiliated with a large hospital, but many adult oncologists practice within the community and not within an academic center. In the USA, only approximately 34%–66% of AYA patients are treated by pediatric oncologists at academic centers, and this difference in referral and care patterns may influence prognosis as well.18–20,36
It is believed that the caregiver’s overall approach and experience with a particular disease is important when comparing survival rates and treatment success. ALL remains the most common cancer in children and is commonly encountered by pediatric oncologists. Schiffer has argued that adult oncologists tend to be more familiar with the treatment of solid tumors and infrequently care for patients with acute leukemia.49 As such, it is believed that “disparities in treatment attitudes between pediatric and adult departments” may contribute significantly to the different outcomes. Patients referred to pediatric centers often interact with experienced support teams, in addition to their doctor, who aid in their recovery.49 As such, the most drastic decline in survival rates occurs approximately at the age of 18 years, which is the same time that the transition of care from pediatric to adult practitioners occurs.46
Consideration for the potential differences in therapeutic practices between pediatric and adult practitioners may be equally important when comparing treatment outcomes. The chemotherapy historically given in typical adult protocols may lead to longer chemotherapy administration delays as compared to the chemotherapy given in pediatric protocols, and it has been suggested by some that adult oncologists tend to give patients additional recovery time between cycles.20 Pediatric oncologists, on the other hand, are known to be trained to administer their therapies with a greater adherence to schedules and dose intensity.49
Furthermore, there is a growing body of evidence that an increased rate of clinical trial participation has a positive correlation with treatment success and cure rates.50 In the USA, approximately 5% of 15–25 year olds with cancer are entered onto US clinical trials, as compared to 60%–65% of children.17,51 A review of SEER data showed that clinical trial enrollment was likely to be decreased for patients older than 19 (10% vs 34%) years, not treated by a pediatric oncologist (9% vs 53%), and uninsured (3% vs 14%).52 Excluding infants less than 12 months, AYAs with ALL serve as the smallest group of patients represented on clinical trials.18 The lack of clinical trial enrollment in the AYA population is an international problem, as a study from England demonstrated that 80% of patients younger than 14 years with the diagnosis of ALL were enrolled on a clinical trial, while only 36% of patients between the ages of 15 and 29 years did.53 This lack of clinical trial enrollment has contributed to the difficulty in establishing a “standard of care” for patients in the AYA age-group. In addition, AYA-specific data from clinical trials have historically been extrapolated from data collected within larger trials that include AYAs, but may not have been designed to assess AYA outcomes specifically. Therefore, although challenges exist in comparing one trial to another, there have been many attempts to assess outcomes for AYA patients treated with adult-inspired therapy versus pediatric-inspired therapy.
In the USA, the Children’s Oncology Group (COG) has recognized the gap in clinical trial participation, lack of improvement in outcomes, and possible barriers that may be preventing AYAs from participating in clinical trials. To this end, the upper age limits for many COG clinical trials have been increased, with almost 30 trials currently open that have upper age limits between the ages of 25 and 50 years.21
Comparison of treatment protocols
In the USA, an analysis of SEER data revealed a rapid decline in survival for patients with ALL from 75% at age 16 to 48% at age 18–21 years, which has been postulated to be partly related to the transition of patients from pediatric treatment centers to adult treatment centers, along with the switch from pediatric to adult treatment regimens.17 However, several studies from around the world have demonstrated improved outcomes for AYA patients when treated with pediatric-inspired regimens as compared to traditional adult-inspired regimens. In the USA, a retrospective analysis of 321 patients aged 16–20 years with newly diagnosed ALL between the years 1988 and 2001 – who were treated on trials from either the Children’s Cancer Group (CCG) or Cancer and Leukemia Group B (CALGB) – was performed. This study demonstrated favorable outcomes for those treated on the pediatric trials, with the CCG protocols resulting in a 7-year EFS of 63%, compared to that of 34% for those treated with the CALGB protocol. In addition, the OS at 7 years for those treated by the CCG was 67%, while that of the CALGB was 46%. In a subsequent trial, the CCG reported an even higher EFS of 71.5% for patients aged 16–21 years.40,54 The CALGB also reported more favorable outcomes for AYAs when intensified, postremission protocols modeled after pediatric-inspired trials were used.40,55 Additionally, a significant difference in the rates of relapse between the groups was reported. The CCG reported two isolated CNS relapses versus nine in the CALGB group. This in turn resulted in a 7-year overall CNS relapse rate of 1% for those patients treated with the CCG protocol and 11% for the CALGB group.40 This finding raises concern for the low degree of CNS-directed treatment in typical adult regimens. Unlike the CALGB group, the CCG patients received intrathecal therapy starting at the beginning of the induction phase and continuing throughout all phases of therapy, including maintenance, whereas the patients in the CALGB group failed to receive any CNS-directed therapy during their final phase of treatment. As a result, it is believed that use of earlier and more frequent intrathecal therapies played a role in the lower rate of relapse seen in the CCG patient population as compared to that of the CALGB group. This supports the notion that the pediatric practice of aggressive CNS-targeted therapy may help to prevent CNS relapse and also that higher cumulative doses of nonmyelosuppressive agents such as glucocorticoids, vincristine, and asparaginase may reduce the occurrence of systemic disease relapse.56,57
Similar results have been demonstrated by investigators at the DFCI who assessed patients aged 15–18 years treated between the years 1991 and 2000. They found that treating adolescents with newly diagnosed ALL on the pediatric-inspired DFCI-ALL consortium protocol led to a favorable 5-year EFS of 78%, compared to previous rates of 46%–68% for a similar population when using adult therapies.58 Once again, investigators speculated that the superior outcome may have been attributed to the types and intensity of chemotherapy agents used, including the frequent and high doses of corticosteroids, vincristine, and asparaginase.58
Studies from various countries in Europe regarding the AYA ALL population have generated very similar results. In France, 77 patients aged 15–20 years were treated on the pediatric-based FRALLE-93 trial, while 100 patients aged 15–20 years were treated on the adult LALA-94 protocol.59 A comparison of the two groups highlighted that AYA patients who were treated on the pediatric protocol had significantly higher rates of disease remission and EFS. The FRALLE-93 protocol resulted in a complete remission rate of 94% and a 5-year EFS rate of 67%. Both the complete remission and 5-year EFS rates were lower for the adult LALA-94 trial, at 83% and 41%, respectively. Moreover, of the 72 FRALLE patients who achieved complete remission, only eleven relapsed, whereas 38 of the 83 patients who achieved remission in the LALA-94 trial ultimately suffered from disease relapse.59 When comparing the two treatment regimens, it was once again found that the major difference was that patients on the pediatric-inspired FRALLE protocol received greater quantities of steroids, vincristine, and asparaginase. When comparing the chemotherapy agents and dosing of the FRALLE-93 and LALA-94 protocols, it is notable that approximately five times more prednisone and 20 times more asparaginase are used in the pediatric regimen versus the adult regimen, including no doses of asparaginase in the induction phase of the LALA-94 protocol.59 Thus, investigators in France concluded that AYAs should either be included on the intensive pediatric ALL protocols, or that new trials based heavily upon the established pediatric protocols should be utilized for treating AYAs with ALL.
In Italy, investigators conducted a similar study in which patients aged 14–18 years were either treated by the Italian Association of Paediatric Haematology and Oncology (AIEOP) or the adult-based Italian Group Italiano Malattie Ematologiche Maligne dell’ Adulte (GIMEMA ALL). The AIEOP group used BFM-based therapy consisting of intensive induction therapy, high-dose methotrexate postremission therapy, and standard continuation therapy for a minimum of 2 years. The GIMEMA group utilized induction with high-dose anthracycline followed by consolidation therapy with high-dose cytarabine and did not use high-dose methotrexate or delayed intensification. Moreover, cranial radiation was provided to all patients in the GIMEMA group, but reserved only for those in the high-risk category in the pediatric AIEOP trials, where there was higher use of intrathecal chemotherapy.60 Among the two groups, the AYA patients treated with the AIEOP pediatric regimen demonstrated a 2-year EFS of 78%, whereas the patients treated on the GIMEMA ALL trial showed a 2-year EFS of 47%.60
In the Netherlands, a retrospective analysis compared the outcomes of 15–18 year olds treated on either by the pediatric Dutch Childhood Oncology Group (DCOG) or the adult Dutch-Belgian Hemato-Oncology Cooperative Study Group (HOVON). Participants in the DCOG had a 5-year EFS of 69% and 5-year OS of 79%, compared to a 5-year EFS of 34% and 5-year OS of 38% for those on the HOVON study.61 Perhaps the greatest explanation for such varied results is the different treatment modalities utilized by both groups. In the DCOG, treatment consisted of induction therapy, followed by high-dose methotrexate, a reintensification phase, and then maintenance chemotherapy, whereas the HOVON trial primarily focused on intensified chemotherapy followed by HSCT. The HOVON group did not utilize high-dose methotrexate nor did it provide a reintensification phase for its participants. In addition, the DCOG protocol included a maintenance phase, whereas the HOVON regimen did not, resulting in significantly reduced doses of 6-mercaptopurine. Moreover, an additional major difference between the two groups was that the HOVON protocol was based upon intensified chemotherapy followed by HSCT, whereas in the DCOG, HSCT was reserved for a select subset of high-risk patients, such as those with the Philadelphia Chromosome or MLL rearrangement. As such, only 4% of the DCOG patients received an HSCT in their first complete remission, in comparison to the 25% of HOVON patients. Five of nine patients in the HOVON group who received an HSCT did not survive secondary to transplant-related toxicities, contributing to the higher mortality rate in the HOVON group.61
While we have outlined the details of some critical studies earlier, it is important to note that similar results have emerged from a variety of other countries where it has been demonstrated that patients in the AYA age-group had better outcomes when treated with pediatric-inspired therapy (Table 1). Of note, the adult Finnish Leukemia Group utilizes therapy that is similar to the pediatric Nordic (NOPHO) study group in Finland, and found there was not a significant difference when comparing the outcomes of these groups, illustrating the benefit of pediatric-inspired therapy.62 Additionally, single institutional data from Rytting et al reported the outcome of BFM-based therapy in their young adult population and did not appreciate an improvement when compared to historical controls.63
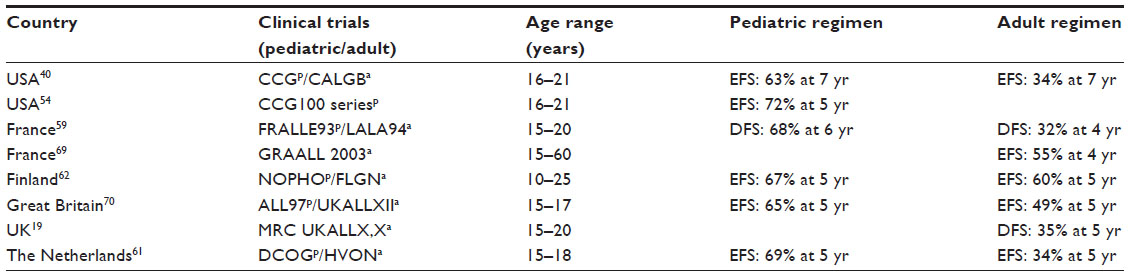 | Table 1 Outcomes for adolescent and young adult patients treated on pediatric-based versus adult-based clinical trials |
Role of hematopoietic stem cell transplantation
Generally, HSCT in the pediatric population is reserved for those with refractory disease, particular very-high-risk biology, or those with early bone marrow relapse, occurring within the first 36 months of remission. For those with extramedullary relapse within 18 months of remission, HSCT is also typically utilized. However, for those with extramedullary relapse >18 months from remission and those with bone marrow relapse >36 months from remission, treatment does not always require HSCT, since outcomes are similar (40%–50% 5-year EFS) with transplant or chemotherapy alone.64 For those with relapse, encouraging new research has emerged regarding targeted immunotherapy utilizing chimeric antigen receptor T-cell therapy (CAR-T).65 This therapy utilizes genetically engineered chimeric antigen receptors that have an anti-CD19 single-chain Fv domain connected to intracellular T-cell signaling domains of the T-cell receptor, directing cytotoxic T lymphocytes to cells expressing this antigen.66 In children, adolescents, and adults, clinical trials with CAR-T therapy have demonstrated that between 67% and 90% of patients with relapsed or refractory disease can successfully achieve remission.66–68 Although promising, the manner in which this therapy will change the treatment algorithm for those with relapse who have historically undergone HSCT is not yet known. Likewise, for those patients with newly diagnosed leukemia, it is not yet known what role CAR-T therapy will play and further research will be needed.
Controversy continues regarding the role of HSCT in first remission for the AYA ALL population. In a meta-analysis comparing HSCT in first remission for adult patients with a matched sibling donor (MSD) to autologous transplant or chemotherapy alone for those without a donor, there was a statistically significant improvement in outcome for those who underwent HSCT, with a 49.9% 5-year EFS, compared to 42.7% 5-year EFS for those who received chemotherapy alone.69 In a joint trial between the MRC and ECOG, outcomes were compared for patients between the ages of 15 and 59 years, all of whom were initially treated with chemotherapy, and then those with an MSD underwent HSCT.70 Those without a donor were randomized to autologous transplant or chemotherapy alone. Patients with a donor had a 5-year OS of 54%, versus 44% for those with no donor (P=0.007), but on subanalysis, this survival benefit was only observed in patients with what was considered standard-risk disease.
These data should be interpreted with caution for the AYA population, however, as the chemotherapy regimens used were classic “adult”-type chemotherapy, especially given the growing evidence of survival benefits afforded to younger adults who are treated with “pediatric”-type chemotherapy. In fact, two recent studies suggest that HSCT does not benefit young adult patients with ALL who are treated according to pediatric regimens.71,72 The International Bone Marrow Transplant Registry (IBMTR) conducted a study that compared outcomes for patients 18–50 years of age who were treated with pediatric-inspired chemotherapy regimens or who underwent related or unrelated HSCT.71 Relapse rates were similar between the groups, but with the higher treatment-related morbidity associated with transplant, and 4-year OS was higher in the group treated with chemotherapy alone (73% [63%–81%] vs 45% [40%–50%], P<0.0001). In a study conducted by the Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL), investigators evaluated the role of HSCT in adults (15–55 years) with ALL treated with a pediatric-inspired regimen and considered to have high-risk disease.72 There was a lower cumulative incidence of relapse in the HSCT, but higher nonrelapse mortality, leading to similar survival outcomes. However, there did appear to be a benefit with HSCT for patients with poor disease response, defined as residual disease ≥10−3 after induction chemotherapy, with a hazard ratio of 0.37 for relapse-free survival (RFS) (95% CI 0.2–0.69; P=0.001) and 0.41 for OS (95% CI 0.22–0.76; P=0.005).
Thus, while some medical oncologists may advocate for HSCT in CR1 for AYAs with ALL, the survival outcomes for these patients treated with pediatric-inspired therapy ranges from 60% to 75%, higher than the EFS reported in several of the chemotherapy-alone arms in studies comparing MSD transplant to chemotherapy alone. Additionally, the pediatric-inspired outcomes are quite a bit higher than the EFS described in several of the HSCT arms. Furthermore, factors influencing outcomes following HSCT are complex and include the presence/absence of MRD prior to and immediately after transplant, the donor source, and the development of graft versus host disease both in terms of its harmful toxicity as well as its potentially beneficial graft versus leukemia effects.73–77 Whether the influence of these factors is age-dependent remains to be seen. Similar to the aforementioned study from the Netherlands, there may not be a general benefit to HSCT in CR1 for all AYAs with ALL. It will be important, however, to identify subgroups for whom the value of transplant may remain.
Acknowledgment
The authors express their gratitude to Lea’s Foundation for Leukemia Research, Inc. for the funding support provided to Dr Isakoff.
Disclosure
The authors report no conflicts of interest in this work.
References
Mattison R, Stock W. Approaches to treatment for acute lymphoblastic leukemia in adolescents and young adults. Curr Hematol Malig Rep. 2008;3(3):144–151. | |
Hunger SP, Lu X, Devidas M, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group. J Clin Oncol. 2012; 30(14):1663–1669. | |
Nachman JB, La MK, Hunger SP, et al. Young adults with acute lymphoblastic leukemia have an excellent outcome with chemotherapy alone and benefit from intensive postinduction treatment: a report from the children’s oncology group. J Clin Oncol. 2009;27(31):5189–5194. | |
Thomas X, Boiron JM, Huguet F, et al. Outcome of treatment in adults with acute lymphoblastic leukemia: analysis of the LALA-94 trial. J Clin Oncol. 2004;22(20):4075–4086. | |
Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004;350(15):1535–1548. | |
Plasschaert SL, Kamps WA, Vellenga E, de Vries EG, de Bont ES. Prognosis in childhood and adult acute lymphoblastic leukaemia: a question of maturation? Cancer Treat Rev. 2004;30(1):37–51. | |
Eguiguren JM, Pui CH. Bone marrow necrosis and thrombotic complications in childhood acute lymphoblastic leukemia. Med Pediatr Oncol. 1992;20(1):58–60. | |
Hann IM, Evans DI, Marsden HB, Jones PM, Palmer MK. Bone marrow fibrosis in acute lymphoblastic leukaemia of childhood. J Clin Pathol. 1978;31(4):313–315. | |
Raney RB, McMillan CW. Simultaneous disparity of bone marrow specimens in acute leukemia. Am J Dis Child. 1969;117(5):548–552. | |
Hann IM, Richards SM, Eden OB, Hill FG. Analysis of the immunophenotype of children treated on the Medical Research Council United Kingdom Acute Lymphoblastic Leukaemia Trial XI (MRC UKALLXI). Medical Research Council Childhood Leukaemia Working Party. Leukemia. 1998;12(8):1249–1255. | |
Ribeiro RC, Pui CH, Schell MJ. Vertebral compression fracture as a presenting feature of acute lymphoblastic leukemia in children. Cancer. 1988;61(3):589–592. | |
Rogalsky RJ, Black GB, Reed MH. Orthopaedic manifestations of leukemia in children. J Bone Joint Surg Am. 1986;68(4):494–501. | |
Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371(9617):1030–1043. | |
Wiemels JL, Cazzaniga G, Daniotti M, et al. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet. 1999;354(9189):1499–1503. | |
Harrison CJ. Cytogenetics of paediatric and adolescent acute lymphoblastic leukaemia. Br J Haematol. 2009;144(2):147–156. | |
Ries L, Eisner M, Kosary C, et al. SEER Cancer Statistics Review, 1975–2001. Bethesda, MD: National Cancer Institute; 2004. Available form: http://seer.cancer.gov/csr/1975_2001/. Accessed August 2, 2015. | |
Bleyer A. The adolescent and young adult gap in cancer care and outcome. Curr Probl Pediatr Adolesc Health Care. 2005;35(5):182–217. | |
Nachman J. Clinical characteristics, biologic features and outcome for young adult patients with acute lymphoblastic leukaemia. Br J Haematol. 2005;130(2):166–173. | |
Ramanujachar R, Richards S, Hann I, Webb D. Adolescents with acute lymphoblastic leukaemia: emerging from the shadow of paediatric and adult treatment protocols. Pediatr Blood Cancer. 2006;47(6):748–756. | |
Schafer ES, Hunger SP. Optimal therapy for acute lymphoblastic leukemia in adolescents and young adults. Nat Rev Clin Oncol. 2011; 8(7):417–424. | |
Burke ME, Albritton K, Marina N. Challenges in the recruitment of adolescents and young adults to cancer clinical trials. Cancer. 2007; 110(11):2385–2393. | |
Collins SR, Schoen C, Kriss JL, Doty MM, Mahato B. Rite of passage? Why young adults become uninsured and how new policies can help. Issue Brief (Commonw Fund). 2006;20:1–14. | |
Ibrahim A, Ali A, Mohammed MM. Outcome of adolescents with acute lymphoblastic leukemia treated by pediatrics versus adults protocols. Adv Hematol. 2014;2014:697675. | |
Fielding D, Duff A. Compliance with treatment protocols: interventions for children with chronic illness. Arch Dis Child. 1999; 80(2):196–200. | |
Ramakers-van Woerden NL, Pieters R, Loonen AH, et al. TEL/AML1 gene fusion is related to in vitro drug sensitivity for L-asparaginase in childhood acute lymphoblastic leukemia. Blood. 2000;96(3):1094–1099. | |
Pui CH, Evans WE. Acute lymphoblastic leukemia. N Engl J Med. 1998;339(9):605–615. | |
Moricke A, Zimmermann M, Reiter A, et al. Prognostic impact of age in children and adolescents with acute lymphoblastic leukemia: data from the trials ALL-BFM 86, 90, and 95. Klin Padiatr. 2005;217(6):310–320. | |
Gaynon PS, Angiolillo AL, Carroll WL, et al. Long-term results of the children’s cancer group studies for childhood acute lymphoblastic leukemia 1983–2002: a Children’s Oncology Group Report. Leukemia. 2010;24(2):285–297. | |
Moricke A, Zimmermann M, Reiter A, et al. Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia. 2010; 24(2):265–284. | |
Salzer WL, Devidas M, Carroll WL, et al. Long-term results of the pediatric oncology group studies for childhood acute lymphoblastic leukemia 1984–2001: a report from the children’s oncology group. Leukemia. 2010;24(2):355–370. | |
Silverman LB, Stevenson KE, O’Brien JE, et al. Long-term results of Dana-Farber Cancer Institute ALL Consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1985–2000). Leukemia. 2010;24(2):320–334. | |
Larson RA, Dodge RK, Burns CP, et al. A five-drug remission induction regimen with intensive consolidation for adults with acute lymphoblastic leukemia: cancer and leukemia group B study 8811. Blood. 1995; 85(8):2025–2037. | |
Stock W, Johnson JL, Stone RM, et al. Dose intensification of daunorubicin and cytarabine during treatment of adult acute lymphoblastic leukemia: results of Cancer and Leukemia Group B Study 19802. Cancer. 2013;119(1):90–98. | |
Schultz KR, Carroll A, Heerema NA, et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children’s Oncology Group study AALL0031. Leukemia. 2014;28(7):1467–1471. | |
Arico M, Valsecchi MG, Camitta B, et al. Outcome of treatment in children with Philadelphia chromosome-positive acute lymphoblastic leukemia. N Engl J Med. 2000;342(14):998–1006. | |
Burke PW, Douer D. Acute lymphoblastic leukemia in adolescents and young adults. Acta Haematol. 2014;132(3–4):264–273. | |
Rubnitz JE, Lensing S, Zhou Y, et al. Death during induction therapy and first remission of acute leukemia in childhood: the St Jude experience. Cancer. 2004;101(7):1677–1684. | |
Advani AS, Sanford B, Luger S, et al. Frontline-Treatment Of Acute Lymphoblastic Leukemia (ALL) In Older Adolescents and Young Adults (AYA) Using a Pediatric Regimen Is Feasible: Toxicity Results of the Prospective US Intergroup Trial C10403 (Alliance). 2013;122. | |
Larsen EC, Salzer W, Nachman J, et al. Treatment toxicity in adolescents and young adult (AYA) patients compared with younger patients treated for high risk B-precursor acute lymphoblastic leukemia (HR-ALL): a report from the Children’s Oncology Group Study AALL0232 [ASH abstract 1510]. Blood. 2012;120(suppl 21):1510a. | |
Stock W, La M, Sanford B, et al. What determines the outcomes for adolescents and young adults with acute lymphoblastic leukemia treated on cooperative group protocols? A comparison of Children’s Cancer Group and Cancer and Leukemia Group B studies. Blood. 2008; 112(5):1646–1654. | |
Muller HJ, Boos J. Use of L-asparaginase in childhood ALL. Crit Rev Oncol Hematol. 1998;28(2):97–113. | |
Rizzari C, Putti MC, Colombini A, et al. Rationale for a pediatric-inspired approach in the adolescent and young adult population with acute lymphoblastic leukemia, with a focus on asparaginase treatment. Hematol Rep. 2014;6(3):5554. | |
Moghrabi A, Levy DE, Asselin B, et al. Results of the Dana-Farber Cancer Institute ALL Consortium Protocol 95-01 for children with acute lymphoblastic leukemia. Blood. 2007;109(3):896–904. | |
Silverman LB, Gelber RD, Dalton VK, et al. Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91-01. Blood. 2001;97(5):1211–1218. | |
Vrooman LM, Stevenson KE, Supko JG, et al. Postinduction dexamethasone and individualized dosing of Escherichia coli L-asparaginase each improve outcome of children and adolescents with newly diagnosed acute lymphoblastic leukemia: results from a randomized study – Dana-Farber Cancer Institute ALL Consortium Protocol 00-01. J Clin Oncol. 2013;31(9):1202–1210. | |
Shaw PH, Reed DR, Yeager N, Zebrack B, Castellino SM, Bleyer A. Adolescent and young adult (AYA) oncology in the United States: a specialty in its late adolescence. J Pediatr Hematol Oncol. 2015; 37(3):161–169. | |
Thomas DA, O’Brien S, Faderl S, et al. Chemoimmunotherapy with a modified hyper-CVAD and rituximab regimen improves outcome in de novo Philadelphia chromosome-negative precursor B-lineage acute lymphoblastic leukemia. J Clin Oncol. 2010;28(24):3880–3889. | |
Stock W. Adolescents and Young Adults with Acute Lymphoblastic Leukemia. Hematology Am Soc Hematol Educ Program. 2010;2010:21–29. | |
Schiffer CA. Differences in outcome in adolescents with acute lymphoblastic leukemia: a consequence of better regimens? Better doctors? Both? J Clin Oncol. 2003;21(5):760–761. | |
Bleyer A, Barr R. Cancer in young adults 20 to 39 years of age: overview. Semin Oncol. 2009;36(3):194–206. | |
Bleyer A. Older adolescents with cancer in North America deficits in outcome and research. Pediatr Clin North Am. 2002;49(5):1027–1042. | |
Parsons HM, Harlan LC, Seibel NL, Stevens JL, Keegan TH. Clinical trial participation and time to treatment among adolescents and young adults with cancer: does age at diagnosis or insurance make a difference? J Clin Oncol. 2011;29(30):4045–4053. | |
Stiller CA, Benjamin S, Cartwright RA, et al. Patterns of care and survival for adolescents and young adults with acute leukaemia – a population-based study. Br J Cancer. 1999;79(3–4):658–665. | |
Nachman J, Sather HN, Buckley JD, et al. Young adults 16–21 years of age at diagnosis entered on Childrens Cancer Group acute lymphoblastic leukemia and acute myeloblastic leukemia protocols. Results of treatment. Cancer. 1993;71(Suppl 10):3377–3385. | |
Larson RA. The US trials in adult acute lymphoblastic leukemia. Ann Hematol. 2004;83(Suppl 1):S127–S128. | |
Bostrom BC, Sensel MR, Sather HN, et al. Dexamethasone versus prednisone and daily oral versus weekly intravenous mercaptopurine for patients with standard-risk acute lymphoblastic leukemia: a report from the Children’s Cancer Group. Blood. 2003;101(10):3809–3817. | |
Mitchell CD, Richards SM, Kinsey SE, Lilleyman J, Vora A, Eden TO. Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: results of the UK Medical Research Council ALL97 randomized trial. Br J Haematol. 2005; 129(6):734–745. | |
Barry E, DeAngelo DJ, Neuberg D, et al. Favorable outcome for adolescents with acute lymphoblastic leukemia treated on Dana-Farber Cancer Institute Acute Lymphoblastic Leukemia Consortium Protocols. J Clin Oncol. 2007;25(7):813–819. | |
Boissel N, Auclerc MF, Lheritier V, et al. Should adolescents with acute lymphoblastic leukemia be treated as old children or young adults? Comparison of the French FRALLE-93 and LALA-94 trials. J Clin Oncol. 2003;21(5):774–780. | |
Testi A. Difference in outcome of adolescents (14–17 years) with acute lymphoblastic leukemia (ALL) enrolled in the Italian pediatric (AIEOP) and adult (GIMEMA) multicenter protocols. J Clinical Oncol, 2006 ASCO Annual Meeting Proceedings Part I. 2006; 24(18S):9024. | |
de Bont JM, Holt B, Dekker AW, van der Does-van den Berg A, Sonneveld P, Pieters R. Significant difference in outcome for adolescents with acute lymphoblastic leukemia treated on pediatric vs adult protocols in the Netherlands. Leukemia. 2004;18(12):2032–2035. | |
Usvasalo A, Raty R, Knuutila S, et al. Acute lymphoblastic leukemia in adolescents and young adults in Finland. Haematologica. 2008; 93(8):1161–1168. | |
Rytting ME, Thomas DA, O’Brien SM, et al. Augmented Berlin- Frankfurt-Munster therapy in adolescents and young adults (AYAs) with acute lymphoblastic leukemia (ALL). Cancer. 2014;120(23):3660–3668. | |
Nguyen K, Devidas M, Cheng SC, et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children’s Oncology Group study. Leukemia. 2008;22(12):2142–2150. | |
Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015;125(26):4017–4023. | |
Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014; 371(16):1507–1517. | |
Annesley CE, Brown P. Novel agents for the treatment of childhood acute leukemia. Ther Adv Hematol. 2015;6(2):61–79. | |
Tasian SK, Gardner RA. CD19-redirected chimeric antigen receptor-modified T cells: a promising immunotherapy for children and adults with B-cell acute lymphoblastic leukemia (ALL). Ther Adv Hematol. 2015;6(5):228–241. | |
Gupta V, Richards S, Rowe J. Allogeneic, but not autologous, hematopoietic cell transplantation improves survival only among younger adults with acute lymphoblastic leukemia in first remission: an individual patient data meta-analysis. Blood. 2013;121(2):339–350. | |
Goldstone AH, Richards SM, Lazarus HM, et al. In adults with standard-risk acute lymphoblastic leukemia, the greatest benefit is achieved from a matched sibling allogeneic transplantation in first complete remission, and an autologous transplantation is less effective than conventional consolidation/maintenance chemotherapy in all patients: final results of the International ALL Trial (MRC UKALL XII/ECOG E2993). Blood. 2008;111(4):1827–1833. | |
Seftel MD, Neuberg D, Zhang M-J, et al. Superiority of Pediatric Chemotherapy over Allogeneic Hematopoietic Cell Transplantation for Philadelphia Chromosome Negative Adult ALL in First Complete Remission: A Combined Analysis of Dana-Farber ALL Consortium and CIBMTR Cohorts. 2014;124:310. | |
Dhedin N, Huynh A, Maury S, et al. Role of allogeneic stem cell transplantation in adult patients with Ph-negative acute lymphoblastic leukemia. Blood. 2015;125(16):2486–2496; quiz 2586. | |
Bar M, Wood BL, Radich JP, et al. Impact of minimal residual disease, detected by flow cytometry, on outcome of myeloablative hematopoietic cell transplantation for acute lymphoblastic leukemia. Leuk Res Treatment. 2014;2014:421723. | |
Holtick U, Albrecht M, Chemnitz JM, et al. Bone marrow versus peripheral blood allogeneic haematopoietic stem cell transplantation for haematological malignancies in adults. Cochrane Database Syst Rev. 2014;4:CD010189. | |
Pulsipher MA, Langholz B, Wall DA, et al. Risk factors and timing of relapse after allogeneic transplantation in pediatric ALL: for whom and when should interventions be tested? Bone Marrow Transplant. 2015;50(9):1173–1179. | |
Pulsipher MA, Langholz B, Wall DA, et al. The addition of sirolimus to tacrolimus/methotrexate GVHD prophylaxis in children with ALL: a phase 3 Children’s Oncology Group/Pediatric Blood and Marrow Transplant Consortium trial. Blood. 2014;123(13):2017–2025. | |
Zhang H, Chen J, Que W. A meta-analysis of unrelated donor umbilical cord blood transplantation versus unrelated donor bone marrow transplantation in acute leukemia patients. Biol Blood Marrow Transplant. 2012;18(8):1164–1173. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.